

Abstract
Biased agonism, the ability of different ligands for the same receptor to selectively activate some signaling pathways while blocking others, is now an established paradigm for G protein-coupled receptor signaling. One group of receptors in which endogenous bias is critical is the chemokine system, consisting of over 50 ligands and 20 receptors that bind one another with significant promiscuity. We have previously demonstrated that ligands for the same receptor can cause biased signaling responses. The goal of this study was to identify mechanisms that could underlie biased signaling between different receptor splice variants. The C-X-C motif chemokine receptor 3 (CXCR3) has two splice variants, CXCR3A and CXCR3B, which differ by 51 amino acids at its N-terminus. Consistent with an earlier study, we found that C-X-C motif chemokine ligands 4, 9, 10, and 11 all activated Gαi at CXCR3A, while at CXCR3B these ligands demonstrated no measurable Gαi or Gαs activity. β-arrestin (βarr) was recruited at a reduced level to CXCR3B relative to CXCR3A, which was also associated with differences in βarr2 conformation. βarr2 recruitment to CXCR3A was attenuated by both G protein receptor kinase (GRK) 2/3 and GRK5/6 knockdown, while only GRK2/3 knockdown blunted recruitment to CXCR3B. Extracellular regulated kinase 1/2 phosphorylation downstream from CXCR3A and CXCR3B was increased and decreased, respectively, by βarr1/2 knockout. The splice variants also differentially activated transcriptional reporters. These findings demonstrate that differential splicing of CXCR3 results in biased responses associated with distinct patterns of βarr conformation and recruitment. Differential splicing may serve as a common mechanism for generating biased signaling and provides insights into how chemokine receptor signaling can be modulated post-transcriptionally.
Introduction
Chemokine receptors (CKRs) are a family of G protein-coupled receptors (GPCRs) that bind small cognate peptide ligands, i.e., chemokines. Chemokines are so named for their ability to induce chemotaxis and guide leukocyte migration. Chemokines are produced by a variety of cell types at sites of inflammation. They not only mediate the extravasation and chemotaxis of inflammatory mediators, but they also are involved in cell activation, differentiation, actin polymerization, and direct immune cell function (Thelen, 2001; Koelink et al., 2012). CKRs often interact with multiple chemokines, and chemokines often bind to multiple CKRs, supporting the prevailing sentiment that the chemokine system is both promiscuous and redundant. However, recent work demonstrates nonoverlapping intracellular pathway activation by various chemokines at the same CKR (Kohout et al., 2004; Zidar et al., 2009; Rajagopal et al., 2010; Drury et al., 2011; Zohar et al., 2014), suggesting a unique signaling repertoire may be encoded with each distinct ligand-receptor complex.
GPCRs typically interact with three classes of proteins—heterotrimeric G proteins, β-arrestins (βarrs), and G protein receptor kinases (GRKs). Some chemokines selectively activate certain GPCR signaling pathways, such as the G protein pathway, while blocking others, like the βarr pathway (Kohout et al., 2004; Zidar et al., 2009; Rajagopal et al., 2010, 2013; Karin et al., 2016). This signaling paradigm is known as biased agonism or functional selectivity (Urban et al., 2007; Rajagopal et al., 2011). Many GPCRs, including CKRs, show evidence of biased signaling through G-proteins or βarrs (Kohout et al., 2004; Zidar et al., 2009; Rajagopal et al., 2010, 2013). In addition, some CKRs were thought to act as decoys for ligands given their inability to active classic G protein pathways. However, we now appreciate that some of these receptors, such as C-X-C motif chemokine receptor 7, are βarr-biased receptors that do not signal through G protein pathways but do couple to βarrs and mediate βarr-dependent signaling (Rajagopal et al., 2010). However, the mechanisms underlying biased signaling at CKRs remain only partially characterized (Busillo et al., 2010).
C-X-C motif chemokine receptor 3 (CXCR3) has two seven-transmembrane spanning splice variants, CXCR3A and CXCR3B. CXCR3A and CXCR3B have identical intracellular sequences and only differ by the replacement of the four most distal residues on the N-terminus of CXCR3A with 51 amino acids unique to CXCR3B due to alternative splicing at the 5′ end of the second exon (Lasagni et al., 2003) (Fig. 1). These splice variants are known to signal in response to four chemokines: C-X-C motif chemokine ligand 4 [(CXCL4); platelet factor-4], C-X-C motif chemokine ligand 9 [(CXCL9); monokine induced by interferon-γ], C-X-C motif chemokine ligand 10 [(CXCL10); interferon-induced protein 10], and C-X-C motif chemokine ligand 11 [(CXCL11) interferon-inducible T-cell α chemoattractant] (Hermodson et al., 1977; Taub et al., 1993; Cole et al., 1998; Tensen et al., 1999). CXCR3 was first discovered through its selective recruitment of effector T-cells (Loetscher et al., 1996), and is now known to be a critical mediator of inflammation, vascular disease, and cancer (Van Raemdonck et al., 2015). Aberrant CXCR3 signaling is implicated in inflammatory diseases, inhibition of blood vessel formation, and both tumor repression and tumorigenesis (Kawada et al., 2004; Koch et al., 2009; Villarroel et al., 2014; Peng et al., 2015). Due to differential regulatory promotor elements, the endogenous ligands of CXCR3 are spatially and temporally separated by expression pattern (Groom and Luster, 2011), suggesting distinct functional properties of CXCL4, CXCL9, CXCL10, and CXCL11. CXCR3A is primarily expressed on activated T-lymphocytes (Qin et al., 1998), but expression is also noted on a variety of other cell types including dendritic cells, natural killer cells, B-cells, and macrophages (García-López et al., 2001). In contrast, CXCR3B is expressed predominantly on microvascular endothelial cells (Lasagni et al., 2003), as well as on T-lymphocytes, although at a lower level than CXCR3A. A recent report demonstrated differential effects of endogenous ligands at CXCR3B compared with CXCR3A (Berchiche and Sakmar, 2016); however, the role of β-arrestin recruitment and how recruitment propagates to downstream signaling between CXCR3A and CXCR3B remains unclear. The goal of this study was to understand how the different N-termini of CXCR3A and CXCR3B influence intracellular signaling through G-proteins, βarrs, and GRKs.
Fig. 1.

CXCR3A and CXCR3B splice variants. Schematic representation of CXCR3A (A) and CXCR3B (B). Light pink signifies the additional 47 amino acids present on the N-terminus of CXCR3B. There is no difference in intracellular residues between these CXCR3 splice variants. (C) Sequence alignment of CXCR3A and B.
Materials and Methods
Cell Culture
Human embryonic kidney (HEK) 293 cells stably expressing a modified firefly luciferase enzyme linked to the cAMP sensor Epac (GloSensor, Promega, Madison, WI), active in the presence of cAMP and luciferin (Promega), were used to quantify G protein signaling. HEK 293T cells were used for bioluminescence resonance energy transfer (BRET) experiments to assess βarr recruitment. The cells were maintained in minimum essential media (Corning, NY) containing 1% penicillin/streptomycin and 10% fetal bovine serum (Corning). Cells were grown at 37°C in a humidified 5% CO2 incubator in poly-D-lysine–coated tissue culture plates. Media were changed every 48–72 hours.
Transfections
Luciferase attached to the C-terminus of CXCR3A and CXCR3B was used in donor constructs for BRET assays. Luciferase constructs were encoded either in a Renilla luciferase (Rluc) pN-3 vector (Promega) or Nanoluc Luciferase vector pNL1.1 (Promega). Yellow fluorescent protein (YFP) was attached to the C-terminus of βarr1/2; YFP tags for βarrs and GRKs were used as acceptors in BRET assays. Transient transfections were conducted with calcium phosphate for all BRET assays as previously described (Peterson et al., 2015). Briefly, calcium phosphate transfections were conducted with HEPES buffered saline with 4 μg of the CXCR3-RLuc receptor, 10 μg of βarr2-YFP, 10 μg βarr1-YFP, 4 μg of GRK2-YFP, or 4 μg of GRK6-YFP and 0.25M CaCl2. Here, 50 ng of the Nanoluc βarr2 biosensor was determined to be the optimal concentration for conformational assessment, and was transfected with untagged receptor (4 μg CXCR3A, 5 μg CXCR3B) for biosensor studies. Media were changed 30 minutes prior to transfection, and again 4 hours after CaCl2 transfection. Receptor expression was quantified through CXCR3A-Rluc and CXCR3B-Rluc signal and surface staining. When equal amounts of expression vector were transfected, CXCR3B signal was found to be approximately 80% of CXCR3A (normalized CXCR3A expression 100 ± 2.0, n = 56; normalized CXCR3B expression 79 ± 1.6, n = 41); P < 0.05 by unpaired student’s t test, consistent with prior findings that surface expression of CXCR3B is lower than CXCR3A (Mueller et al., 2008; Korniejewska et al., 2011). For assays dependent on absolute quantity of receptor expression (i.e., non-BRET assays), splice variant expression was normalized by increasing the concentration of CXCR3B by 25% relative to CXCR3A. This resulted in equivalent surface expression levels of CXCR3A and CXCR3B (Supplemental Fig. 1).
Bioluminescence Resonance Energy Transfer
Twenty-four hours after transfection, cells were plated onto a 96-well plate at 50,000–100,000 cells/well. Approximately 44 hours after transfection, media were changed to minimum essential media (Corning) supplemented with 10 mM HEPES, 0.1% bovine serum albumin, and 1% penicillin/streptomycin. After approximately 3 to 4 hours of serum starvation, cells were washed with room temperature phosphate-buffered saline (PBS). Next, 80 μl of a coelenterazine-h/Hanks’ balanced salt solution solution (3 μM coelenterazine-h) was added. Ligands were prepared at 5× concentration and read by a Mithras LB940 instrument (Berthold, Bad Wildbad, Germany) with 485 and 530 nm emission filters. The BRET ratio was calculated using the following equation:
 |
(1) |
where cf represents the BRET ratio in the vehicle control group. For the bystander BRET total internalization assay, titration response experiments determined that 400 ng of myr-palm-mVenus and 2.5 μg of 2xFYVE-mVenus and a constant 4 μg of Rluc labeled CXCR3 maximized signal:noise, and these concentrations were used throughout for BRET internalization experiments. For CXCR3-βarr interaction, the net BRET ratio was quantified 5 minutes following ligand addition. For bystander BRET-based receptor internalization assays [receptor association with FYVE-mVenus or dissociation from a myristoyl-palmitoylated-mVenus (myr-palm)], cells were not serum starved and instead were incubated in assay buffer with ligand for 1 hour before adding coelenterazine-h as described previously. Recombinant human CXCL4, CXCL9, CXCL10, and CXCL11 were obtained from either Peprotech (Rocky Hill, NJ) or BioLegend (San Diego, CA).
Generation of FLAG-Tagged CXCR3 and C-terminal Truncation Mutants.
FLAG-tagged CXCR3 isoform constructs were created by polymerase chain reaction (PCR) amplification with the signal sequence hemagglutinin 5′ to the FLAG-epitope sequence as previously described (Guan et al., 1992). The PCR product was gel purified and inserted into pcDNA3.1 using XbaI endonuclease cleavage sequence 5′ to the hemagglutinin sequence and an EcoRI endonuclease cleavage sequence 3′ to the stop codon. L344X and L391X C-terminal CXCR3 truncation mutants were created by PCR amplification of wild-type (WT) CXCR3A or CXCR3B, introduction of a HindIII endonuclease cleavage sequence 5′ to the start codon on the forward primers (CXCR3A: 5′-CGCGGTAAGCTTATGGTCCTTGAG-3′; CXCR3B: 5′- CGGACCGAAGCTTATGGAGTTGAG-3′) and the introduction of a KpnI endonuclease cleavage sequence 3′ to either the 343rd or 390th amino acid on the reverse primer (5′-ACCCATGGTACCATCCCTCTCTGG-3′). The amplified PCR product was gel purified and inserted in the pRluc-N3 expression vector. All constructs were verified by Sanger DNA sequencing.
GRK Inhibition on βarr Recruitment.
For small inhibitory RNA (siRNA) GRK knockdown studies, HEK 293T cells were transfected with Lipofectamine 3000 (Thermo Fisher, Waltham, MA) per manufacturer specifications within a 96-well plate with 5 ng of either CXCR3A-Rluc or CXCR3B-Rluc, 12.5ng of βarr2-YFP, and 30 ng GRK2/GRK3 siRNA (60 ng total), or 30 ng GRK5/6 siRNA or 60 ng control siRNA per well. Cells were then stimulated with 500 nM of CXCL11 for 5 minutes, and the BRET signal of βarr recruitment was recorded. GRK2, GRK3, GRK5, GRK6, and control siRNA sequences were used as previously validated and described (Kim et al., 2005).
GloSensor cAMP Assay
Lipofectamine 2000 (Thermo Fisher) transfected cells were plated on 96-well plates 24 hours after transfection with either CXCR3A or CXCR3B at a density of 10–25,000 cells/well. At approximately 44 hours post transfection, cells were serum starved for 2 hours. Next, the plate was washed with room temperature PBS, and 60 μl of GloSensor cAMP Reagent (Promega) in Hanks’ balanced salt solution with 20 mM HEPES (Gibco, Gaithersburg, MD) was then added per well. The plate was incubated for 2 hours in the dark at room temperature. For the Gαi pathway assay, the cells were then stimulated with 100 nM isoproterenol and incubated for 5 minutes, and then luminescence over 1 second was quantified by the Mithras LB940 instrument. Vehicle or CXCR3 ligands were then added, incubated for 10 minutes, and the plate was read a second time. Data were normalized to vehicle-treated wells, and to 1 μM CXCL11 for the Gαi studies. For the Gαs studies, baseline luminescence was determined prior to the addition of CXCR3 ligand, and the plate was read a second time after ligand addition.
DiscoveRX Active Internalization Assay
This assay was conducted as previously described (Rajagopal et al., 2013) and in accordance with the manufacturer’s protocols. Briefly, an Enzyme Acceptor-tagged βarr and a ProLink tag localized to endosomes are stably expressed in U2OS cells. Untagged CXCR3A or CXCR3B are transiently expressed. βarr-mediated internalization results in the complementation of the two β-galactosidase enzyme fragments that hydrolyze a substrate (DiscoveRx, Fremont, CA) to produce a chemiluminescent signal.
Serum Response Element/Serum Response Factor Pathway Assay
HEK 293T cells were transiently transfected with serum response element (SRE) or serum response factor (SRF) and either unlabeled CXCR3A (4 μg) or CXCR3B (5 μg). Four hours after transfection, the cells were plated on a 96-well plate at a concentration of 25,000 cells/well. Twenty-four hours after transfection, cells were serum starved overnight. The next day, cells were incubated with ligand for 5 hours and subsequently lysed with passive lysis buffer (Promega) for 10 minutes as previously described (Evron et al., 2014). Luciferin was added to the lysate and luminescence was quantified using the Mithras LB940 instrument, with no wavelength filter between the cells, and the photomultiplier as previously described (Peterson et al., 2015). To observe mitogen-activated protein kinase kinase–dependent effects of CXCL11, the mitogen-activated protein kinase kinase inhibitor PD98059 (Tocris, Minneapolis, MN) was applied to cells at 20 μM for 15 minutes prior to ligand stimulation, similar to a previously described protocol (Gesty-Palmer et al., 2005).
Immunoblot
HEK 293 cells were transiently transfected with 4 μg of CXCR3A or 5 μg of CXCR3B using Lipofectamine 2000. After 48 hours, cells were starved in serum-free Dulbecco’s modified Eagle’s medium >4 hours and treated with the indicated ligand for the indicated duration. Cells were lysed in ice-cold radioimmunoprecipitation assay buffer containing phosphatase and protease inhibitors [Phos-STOP (Roche, Basel, Switzerland), cOmplete EDTA free (Sigma, St. Louis, MO)] for 15 minutes, sonicated, and cleared of insoluble debris by centrifugation at 12,000g (4°C, 15 minutes) after which the supernatant was collected. Protein was resolved on SDS-10% polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with the indicated primary antibody overnight (4°C). Phosphorylated extracellular regulated kinase (ERK) (Cell Signaling Technology, Danvers, MA) and total ERK (Millipore, Danvers, MA) were used to assess ERK activation. GRK knockdown was assessed by immunoblot of GRK2 (Santa Cruz Biotechnology, Dallas, TX), GRK3 (Santa Cruz), GRK5 (Santa Cruz), GRK6 (Santa Cruz), and protein loading assessed with alpha-tubulin (Sigma). β-Arrestin knockout (KO) was assessed using anti-βarr1 and anti-βarr2 antibodies kindly provided by Dr. Robert J. Lefkowitz (Duke University) and previously validated (Attramadal et al., 1992). Horseradish peroxidase–conjugated polyclonal mouse anti-rabbit-IgG or anti-mouse-IgG was used as secondary antibodies. Immune complexes on nitrocellulose membrane were imaged by SuperSignal enhanced chemiluminescent substrate (Thermo Fisher). For quantification, phosphorylated ERK (pERK)1/2 relative intensity was normalized to total ERK1/2 relative intensity using ImageLab (Bio-Rad, Hercules, CA).
Intact Cell Phosphorylation
These assays were performed as described previously (Freedman et al., 2002; Wu et al., 2005). HEK 293 cells were transfected with pcDNA3.1 plasmids encoding N-terminal FLAG-tagged constructs of CXCR3A, CXCR3B, or no protein. Confluent cells in 6-well dishes were aliquoted to metabolic labeling or to cell surface immunofluorescence and flow cytometry. For metabolic labeling, cells were serum starved overnight, rinsed and then labeled with 32Pi (100 μCi/ml, 37°C, 1 hour) in phosphate-free Dulbecco’s modified Eagle’s medium, 20 mM HEPES, pH 7.4, with 0.1% (w/v) fatty acid–free bovine serum albumin (Sigma), 100 μg/ml streptomycin, and 100 units/ml penicillin. Cells were then exposed to the same medium lacking or containing 100 nM CXCL11 for 5 minutes (37°C), and transferred to ice. After 2 washes with Dulbecco’s PBS, cells were solubilized in M2 buffer: 1% (w/v) Triton X-100, 0.05% SDS, 5 mM EDTA, 50 mM Tris-Cl, pH 8.0 (25°C), 200 mM NaCl, 50 mM NaF, 10 mM disodium pyrophosphate, and protease inhibitors (1 mM benzamidine, 5 ug/ml aprotinin, 10 μg/ml leupeptin). After solubilization (4°C, 1 hour), cell lysates were cleared of insoluble debris by two sequential centrifugations at 10,000g (4°C, 30 minutes). Supernatant aliquots were subjected to immunoprecipitation and modified Lowry protein assay (DC Protein Assay Kit, Bio-Rad). The FLAG-CXCR3A and -CXCR3B were immunoprecipitated from 500 μl of cell lysate by inversion mixing for 60 minutes (4°C) with 10 μl of agarose beads conjugated to M2 monoclonal anti-FLAG IgG1 (Sigma-Aldrich, St. Louis, MO) [these beads were suspended in M2 buffer supplemented with 3% (w/v) bovine serum albumin]. Beads were then washed thrice with M2 buffer and finally incubated in 1× Laemmli buffer at 37°C for 2 hours to dissociate immune complexes, which were resolved on SDS-10% polyacrylamide gels. Proteins were transferred to nitrocellulose membranes, as described (Freedman et al., 2002), and subsequently processed for autoradiography with an intensifying screen at −80°C for 2–4 days. After autoradiography, each nitrocellulose membrane was immunoblotted for CXCR3 with mouse IgG (R&D Systems, Minneapolis, MN), as described (Wu et al., 2012). Chemiluminescence was imaged and quantitated with a Bio-Rad ChemiDoc XRS+, which was also used to photograph and quantitate autoradiography films. The 32P signals in each CXCR3 band were normalized to cognate receptor immunoblot signals, after subtracting nonspecific signals obtained from lanes loaded with immunoprecipitations of mock transfected–cell lysates.
Generation of β-Arrestin1/2-Double Knockout (∆ARRB1/2) HEK 293 Cells
∆ARRB1/2 HEK 293 cells were generated by simultaneously targeting the ARRB1 and the ARRB2 genes with a CRISPR/Cas9 genome editing technology using a similar strategy that was employed for Gq/11-double KO cells and as previously described (Schrage et al., 2015; Alvarez-Curto et al., 2016). Cells were transfected with 0.8 μg of CXCR3A or 1μg of CXCR3B with Fugene 6 (Promega) per the manufacturer’s specifications. For β-arrestin rescue studies, either 1 μg of βarr1 and 1 μg βarr2 or 2 μg empty vector (pcDNA3.1) were also transfected. Lysates were collected and immunoblotted as described previously.
Flow Cytometry
HEK 293 cells were transiently transfected with either 4 μg CXCR3A or 5 μg CXCR3B as described previously. After 48 hours, transfected cells and untransfected controls were harvested with trypsin and washed twice with ice-cold PBS supplemented with 1% bovine serum albumin (w/v). Cells were then resuspended in flow cytometry buffer (PBS supplemented with 3% fetal bovine serum and 10 mM EDTA). One million cells were transferred to each flow cytometry tube, spun down, and blocked with flow cytometry buffer supplemented with 5% normal human serum and 5% normal rat serum for 15 minutes at 4°C. Anti-human CXCR3 phycoerythrin conjugated antibody (R&D systems) was applied to cells at a 1:150 dilution (100 mcl final volume). Cells were incubated at 37°C for 30 minutes and then washed with 2 ml of fluorescence-activated cell sorting buffer, fixed for 10 minutes with 0.4% paraformaldehyde, and resuspended in fluorescence-activated cell sorting buffer. Cells were immediately analyzed with a Guava EasyCyte HT cytometer (Millipore). To enlarge axis titles of representative signal, a high-resolution image of the unaltered histogram was placed on identical axis titles and value that were created in adobe illustrator.
Statistical Analyses
Dose-response curves were fitted to a log agonist versus stimulus with three parameters (span, baseline, and EC50) with the minimum baseline corrected to zero using Prism 7.0 (GraphPad, San Diego, CA). For comparing ligands in concentration-response assays, a two-way analysis of variance of ligand and concentration with all four ligands was first conducted. If a significant ligand by concentration interaction was observed (P < 0.05), then comparative two-way analyses of variance between individual ligands were conducted and corrected for multiple comparisons (e.g., for studies with four ligands, P < 0.05/6 was used as the cutoff for statistical significance), with comparisons finding a significant effect of ligand noted. Further details of statistical analysis and replicates are included in the figure legends. These are all figures, we do not believe we need to list specific figures here. In Fig. 10D, one value of eight initial replicates in the CXCL11-treated CXCR3A ARRB 1/2 overexpression rescue group was excluded by the Grubbs outlier test (excluded value 164%; prior to exclusion the mean value ± S.E.M. was 54% ± 18%, after exclusion it was 39% ± 9%). Error bars shown in dose-response analysis signify the S.E.M. unless otherwise noted; * indicates P < 0.05 throughout the paper to indicate statistical significance, unless otherwise noted.
Fig. 10.

β-arrestin overexpression differentially regulates pERK signaling between CXCR3 splice variants at 5 minutes. Either βarr1 and βarr2 or empty vector were transfected in ARRB1/2 KO cells expressing either (A) CXCR3A or (B) CXCR3B as a rescue overexpression experiment and stimulated for 5 minutes with either vehicle or CXCL11 (100 nM). (C) Rescue of β-arrestin was confirmed (nonspecific band in both lanes noted by an arrow). (D) Immunoblots were quantified by calculating pERK/total ERK signal. β-arrestin rescue resulted in decreased pERK signal in CXCR3A-expressing cells, in contrast to an increased signal in CXCR3B-expressing cells, relative to respective vehicle treatments. In (D), #P < 0.05 indicates a significant difference in pERK signal between pcDNA empty vector and β-arrestin rescue within isoform treatment by two-way analysis of variance (ANOVA) followed by Bonferroni post hoc comparison, while *P < 0.05 indicates a significant difference in pERK signal between receptor isoforms treated with CXCL11 by two-way ANOVA followed by Bonferroni post hoc comparison; ± S.E.M., n ≥ 3 biologic replicates per condition. Western blots shown are representative of at least three separate experiments; IB denotes immunoblot.
Results
Confirmation that CXCR3B is βarr Biased Compared with CXCR3A.
After optimizing transfection conditions for equivalent CXCR3 splice variant surface expression (Supplemental Fig. 1), we first examined G protein signaling at CXCR3A and CXCR3B. Prior studies identified selective affinity of CXCL4, CXCL9, CXCL10, and CXCL11 for CXCR3A and CXCR3B through radioligand binding (Lasagni et al., 2003; Mueller et al., 2008). We tested CXCR3A and CXCR3B signaling via Gαi by assessing inhibition of cAMP production through a modified cAMP-dependent firefly luciferase (see the schematic in Supplemental Fig. 2A) with the four known endogenous ligands. In agreement with previous results at CXCR3A (Rajagopal et al., 2013), CXCL10 and CXCL11 were found to be full agonists, while CXCL9 was found to be a partial agonist, in their ability to suppress cAMP production in a highly amplified assay (Fig. 2A; Table 1). Slight inhibition of cAMP signal was observed with CXCL4 at high concentrations. In contrast, none of these four ligands were observed to inhibit cAMP at CXCR3B (Fig. 2B; Table 1). Since CXCR3B has previously been reported to couple to Gαs (Lasagni et al., 2003), we also tested these four ligands’ ability to stimulate cAMP. No signal was observed with CXCL4, CXCL9, CXCL10, or CXCL11 while a positive control of 100 nM isoproterenol demonstrated a ∼15-fold increase in signal from baseline (Supplemental Fig. 2B). We then used BRET to test βarr1 and βarr2 recruitment to CXCR3A and CXCR3B following CXCL4, CXCL9, CXCL10, and CXCL11 stimulation (Fig. 2, C–F). At CXCR3A, we found that all the ligands recruited βarr2 with rank order of efficacy of CXCL11 > CXCL10 > CXCL9 > CXCL4 (Fig. 2C). A bias plot of relative intrinsic activity (Onaran et al., 2017) displayed divergent efficacy of CXCL11 toward the βarr2 pathway relative to the other three endogenous CXCR3 ligands (Supplemental Fig. 3). At CXCR3B, only CXCL11 recruited βarr2, with no significant recruitment noted for the other endogenous ligands (Fig. 2D). The association of βarr2 with CXCR3A was of longer duration than with CXCR3B (Supplemental Fig. 4). CXCL4 displayed inverse agonist characteristics for βarr2 recruitment to CXCR3B, with significantly less signal than CXCL9, which displayed no significant activity. CXCL11 recruited βarr1 to a significantly greater efficacy to CXCR3A than other ligands (Fig. 2E). No statistically significant interaction of ligand and concentration was observed for βarr1 recruitment to CXCR3B (Fig. 2F). These findings are consistent with those of Berchiche and Sakmar (2016) who found that CXCR3B acted as a βarr-biased receptor relative to CXCR3A (Berchiche and Sakmar, 2016).
Fig. 2.

Differential G protein activation and beta-arrestin recruitment by CXCR3 splice variants. (A) cAMP signal following transient CXCR3A expression in HEK 293 cells stably expressing the cAMP activated firefly luciferase. CXCL10 and CXCL11 are full agonists in their ability to inhibit cAMP production through CXCR3A, while CXCL4 and CXCL9 are partial agonists. (B) No Gαi activity is observed after transient transfection with CXCR3B. CXCL11 recruited βarr2-YFP to both CXCR3A-Rluc (C) and CXCR3B-Rluc (D) with higher efficacy than the other endogenous ligands. CXCL11 recruited βarr1-YFP to CXCR3A-Rluc with a significantly greater efficacy than CXCL4, but not significantly greater than CXCL9 or CXCL10 (E). At CXCR3B, no ligands were observed to be significantly different in recruiting βarr1-YFP (F). Best fit calculated by a 3-parameter fit, ± S.E.M., n ≥ 3 biologic replicates per treatment group. *P < 0.05, significant effect of ligand by two-way analysis of variance; n.s denotes not significant.
TABLE 1.
Efficacies and potencies for chemokines at CXCR3A and CXCR3B
The Emax (expressed as percentage of CXCL11 signal) and EC50 values of CXCL4, CXCL9, CXCL10, and CXCL11 at CXCR3A and CXCR3B were calculated from a 3-parameter fit [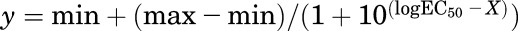 ]. If the 3-parameter fit of ligand-receptor interaction produced a poor fit, then the data were omitted. Radioligand binding data are from Lasagni et al. (2003), and their data were converted to log base 10. The S.E.M. was propagated using the equation S.E.M. = 0.434(log10 EC50 error/log10 EC50).
]. If the 3-parameter fit of ligand-receptor interaction produced a poor fit, then the data were omitted. Radioligand binding data are from Lasagni et al. (2003), and their data were converted to log base 10. The S.E.M. was propagated using the equation S.E.M. = 0.434(log10 EC50 error/log10 EC50).
| CXCR3A | Emax ± S.E.M. | Log EC50 ± S.E.M. | R2 |
|---|---|---|---|
| Gαi | |||
| CXCL4 | 33 ± 21 | −6.9 ± 1.04 | 0.12 |
| CXCL9 | 58 ± 11 | −7.1 ± 0.33 | 0.54 |
| CXCL10 | 85 ± 9 | −7.6 ± 0.21 | 0.66 |
| CXCL11 | 100 ± 7 | −7.4 ± 0.12 | 0.73 |
| βarr1 | |||
| CXCL4 | −16 ± 7 | −7.1 ± 0.81 | 0.41 |
| CXCL9 | 33 ± 5 | −7.5 ± 0.33 | 0.80 |
| CXCL10 | 37 ± 19 | −6.5 ± 0.57 | 0.65 |
| CXCL11 | 100 ± 12 | −6.8 ± 0.17 | 0.94 |
| βarr2 | |||
| CXCL4 | 17 ± 6 | −8.6 ± 0.41 | 0.40 |
| CXCL9 | 24 ± 5 | −7.2 ± 0.42 | 0.54 |
| CXCL10 | 37 ± 4 | −7.3 ± 0.19 | 0.81 |
| CXCL11 | 100 ± 3 | −7.4 ± 0.06 | 0.97 |
| Total Internalization | |||
| CXCL9 | 69 ± 178 | −6.0 ± 1.6 | 0.26 |
| CXCL10 | 88 ± 48 | −6.3 ± 0.56 | 0.77 |
| CXCL11 | 100 ± 18 | −7.0 ± 0.23 | 0.77 |
| Endosome-associated internalization | |||
| CXCL10 | 57 ± 10 | −7.5 ± 0.34 | 0.56 |
| CXCL11 | 100 ± 11 | −8.0 ± 0.19 | 0.73 |
| βarr-associated Internalization | |||
| CXCL11 | 100 ± 7 | −7.8 ± 0.14 | 0.82 |
| CXCR3B | |||
| βarr2 | |||
| CXCL4 | −115 ± 10 | −6.5 ± 0.51 | 0.43 |
| CXCL11 | 100 ± 15 | −7.2 ± 0.30 | 0.47 |
| Total Internalization | |||
| CXCL11 | 100 ± 24 | −6.7 ± 0.56 | 0.80 |
| Endosome-associated internalization | |||
| CXCL11 | 100 ± 20 | −6.9 ± 0.24 | 0.71 |
| Radioligand binding (Lasagni et al. 2003) | Log IC50 ± S.E.M. | ||
| CXCR3A | |||
| CXCL4 | −6.3 ± 0.47 | ||
| CXCL9 | −7.5 ± 0.44 | ||
| CXCL10 | −9.5 ± 0.46 | ||
| CXCL11 | −9.4 ± 0.46 | ||
| CXCR3B | |||
| CXCL4 | −8.1 ± 0.45 | ||
| CXCL9 | −6.9 ± 0.46 | ||
| CXCL10 | −8.2 ± 0.46 | ||
| CXCL11 | −7.5 ± 0.46 |
Both CXCR3A and CXCR3B Undergo Agonist-Dependent Phosphorylation.
We next focused on assessing whether the mechanisms underlying βarr recruitment to CXCR3A and CXCR3B differed from each other. Phosphorylation of intracellular residues is necessary to recruit βarrs. Given the differential isoform trafficking, we examined agonist-induced phosphorylation of both CXCR3A and CXCR3B by 32P metabolic labeling. CXCL11 treatment resulted in phosphorylation of both receptor isoforms to comparable levels (Fig. 3); consistent with the observation that βarr2 is recruited to both receptors. Based on the CXCR3 immunoblot, much of the expressed receptor is not glycosylated; the phosphorylated receptor corresponds to the glycosylated form of the receptor that would be expressed on the plasma membrane (Kobilka, 1990). While a slight difference in mobility of CXCR3B is present in the unglycosylated form, which is likely related to the small predicted shift in molecular weight, there was no significant difference in the mobility of the glycosylated splice variants.
Fig. 3.

CXCR3A and CXCR3B demonstrate equivalent agonist-induced phosphorylation. HEK 293 cells were transfected with plasmids encoding either no protein (Vector) or N-terminal FLAG-tagged constructs of CXCR3A or CXCR3B. After serum starvation and metabolic labeling with 32Pi, cells were exposed to serum-free medium lacking (−) or containing (+) the CXCR3 agonist CXCL11 (100 nM) for 5 minutes (37°C), and then solubilized. CXCR3 isoforms were immunoprecipitated (IP) via their N-terminal FLAG epitope, and immune complexes were resolved by SDS-PAGE. Proteins were transferred to nitrocellulose and then subjected sequentially to autoradiography (32P) and immunoblotting (IB) for CXCR3, as indicated. Shown are an autoradiogram and immunoblot from a single experiment, representative of three experiments performed. The arrow indicates the cell-surface, mature-glycosylated isoform of CXCR3 (which has slower electrophoretic mobility than the cotranslationally glycosylated or immature-glycosylated bands). From the 32P signal for each CXCR3 band we subtracted the (nonspecific) signal in the cognate location of the lane from untransfected cells; the resulting specific 32P CXCR3 signal was normalized to the cognate CXCR3 immunoblot band signal (from which nonspecific pixels in the cognate untransfected cell lane had been subtracted). In each experiment, this ratio of 32P/CXCR3 was normalized to that obtained for CXCR3B immunoprecipitated from unstimulated cells, to obtain fold/control, plotted as individual values and corresponding means ± S.E.M. Compared with unstimulated cells: *P < 0.05 indicates a significant effect of agonist by two-way analysis of variance, with no significant effect of receptor isoform. n = 3 biologic replicates per treatment group.
Activation of CXCR3A and CXCR3B Generate Distinct Patterns of β-Arrestin Recruitment and Conformation.
We then examined CXCL11-induced βarr/green fluorescent protein (GFP) trafficking in cells transfected with CXCR3A or CXCR3B by confocal microscopy (Fig. 4). At CXCR3A, βarr2-GFP was strongly recruited to the plasma membrane immediately following treatment with 100 nM CXCL11, with βarr2-GFP complexes forming in the cytosol in a class B recruitment pattern 30–40 minutes post ligand treatment. In contrast, CXCL11 treatment of CXCR3B-transfected cells only induced weak βarr2-GFP recruitment to the plasma membrane in a class A pattern (Oakley et al., 2000), with maximal βarr2-GFP complex formation at the cell membrane observed 20–30 minutes post stimulation. No appreciable βarr2-GFP complexes in CXCR3B-transfected cells were observed in the cytosol at 40 minutes or longer (data not shown).
Fig. 4.

CXCR3A and CXCR3B induce distinct βarr2 trafficking patterns. Confocal microscopy of HEK 293 cells transiently transfected with unlabeled CXCR3A or CXCR3B and βarr2-GFP pre-CXCL11 (left panels) and post-CXCL11 (three right panels) treatment (100 nM). Three different cells are shown post-treatment of each splice variant.
To assess whether CXCR3A and CXCR3B generate distinct βarr conformations, we designed a βarr2 biosensor consisting of a nanoluc (Promega) donor linked to the N-terminus and a YFP acceptor on the C-terminus of βarr2 (Fig. 5A), which produces a higher intensity signal than a previously designed βarr biosensor (Charest et al., 2005). A significant change in the magnitude of the BRET ratio could be due to a change in distance and/or dipole orientation between the donor and acceptor or a change in avidity in the β-arrestin/receptor interaction. We performed a dose-receptor titration of the biosensor to find the optimal biosensor concentration that attempts to limit avidity effects in our assay by utilizing the lowest concentration of biosensor that produced a reliable signal and reducing excess biosensor remaining in the cellular pool (Supplemental Fig. 5). Therefore, the changes seen in the net BRET ratio primarily imply a conformational change of βarr2. We transiently transfected cells with this biosensor as well as untagged CXCR3A or CXCR3B and subsequently stimulated with CXCL4, CXCL9, CXCL10, or CXCL11. We detected a significant change in BRET signal in the βarr2 biosensor after stimulation of CXCR3A with CXCL11 (Fig. 5B), but not after stimulation of CXCR3B (Fig. 5C). This suggests that stimulation of CXCR3A with CXCL11 leads to a distinct change in βarr conformation.
Fig. 5.

CXCR3A, but not CXCR3B, stimulation with CXCL11 causes a change in βarr2 conformation (A). Schematic of βarr2 biosensor for probing βarrestin-conformational changes following transfection with the biosensor and untagged CXCR3A or CXCR3B. HEK 293 cells were stimulated for 15 minutes with the indicated ligand (250 nM) prior to BRET measurements. (B) CXCL11 stimulation of CXCR3A led to a significant conformational change in βarrestin signified by a change in magnitude of the net BRET ratio compared with all ligands, while CXCL10 stimulation caused a significantly different signal from CXCL4. (C) Stimulation of CXCR3B resulted in no appreciable change in signal across all comparisons. *P < 0.05 by one-way analysis of variance with Tukey’s post hoc comparison between all treatment groups. n ≥ 3 biologic replicates per condition. n.s, not significant.
Differences in Internalization and β-Arrestin Activity between CXCR3 Splice Variants.
Because recruitment does not necessarily correlate with function, we next investigated if CXCL11 stimulation resulted in a functional difference compared with the other three endogenous ligands. The earlier study by Berchiche and Sakmar (2016) demonstrated that both CXCR3A and B undergo similar agonist-induced internalization, although the kinetics for internalization depended on the specific agonist. However, there are distinct mechanisms for GPCR internalization, which can be dependent on or independent of βarrs that can act as clathrin adapters (Shenoy and Lefkowitz, 2011). Therefore, we quantified receptor internalization in three separate assays: 1) a bystander BRET-based membrane dissociation assay, in which receptor-Rluc internalization results in a decrease in the net BRET ratio from myr-palm labeled mVenus (Fig. 6A, middle panel); 2) a bystander BRET-based early endosome association assay, in which receptor-Rluc association with 2x-FYVE labeled mVenus localized to cause an increase in the net BRET ratio (Fig. 6A, right panel); and 3) a βarr-complex mediated internalization using an assay in U2OS cells permanently expressing split β-galactosidase fragments on βarr2 and endosomes (DiscoveRx). In this last assay, when βarr2 and endosomes are in close proximity for a sustained duration, complementation of β-galactosidase fragments in the presence of a substrate produces a chemiluminescent signal. Signal is produced in class B stable endosome βarr2 interactions; however, limited or no signal is produced in class A transient βarr2-endosome interactions. At CXCR3A, total internalization, early endosome association, and βarr-associated internalization at CXCR3A mirrored rank order of efficacy of βarr2 recruitment, with the order of CXCL11 > CXCL10 > CXCL9 > CXCL4. CXCL4 stimulation resulted in negligible internalization and displaying weak inverse agonist activity in the early endosome association assay (Fig. 6, B, D, and F). At CXCR3B, only CXCL11 induced statistically significant total internalization and early endosome association (Fig. 6, C and E). No CXCR3B-mediated chemiluminescent signal in the βarr-mediated internalization assay was observed (Fig. 6G), consistent with the class A pattern of βarr recruitment noted on confocal imaging (Fig. 4). Thus, although both receptors undergo agonist-induced internalization, they appear to do so using different underlying mechanisms.
Fig. 6.

The internalization pattern of CXCR3 splice variants diverge. (A) Confocal images of transiently transfected untagged mVenus (left panel), myr-palm-mVenus (center panel) localized to the plasma membrane, and 2x-FYVE-mVenus (left panel) localized in an endosomal distribution. Internalization efficacy measured by BRET of myr-palm-mVenus transiently transfected cells with (B) CXCR3A-Rluc or (C) CXCR3B-Rluc CXCL11 and treated with the indicated ligand for 1 hour as a measure of total receptor internalization. 2x-FYVE-mVenus transiently transfected cells (D) CXCR3A-Rluc or (E) CXCR3B-Rluc and treated with the indicated ligand for 1 hour as a measure of receptor-endosome association. Treatment CXCL11 resulted in greater βarrestin-mediated internalization efficacy compared with all other ligands in cells transiently transfected with CXCR3A (F), but not CXCR3B (G). Aside from (G), which lacked appreciable signal with any ligand, data are normalized to CXCL11 (1 μM) stimulation and expressed as a percentage of maximal signal. *P < 0.05, significant effect of ligand by two-way analysis of variance. Scale bar is 10 μm. n ≥ 3 biologic replicates per condition.
C-Terminus Truncation, GRK Knockdown, and Pharmacologic GRK2 Inhibition Reduce βarr2 Recruitment.
Given our findings in the GRK BRET experiments, we hypothesized that inhibition of GRKs would attenuate βarr2 recruitment at CXCR3A. We first generated C-terminal truncation mutants of CXCR3A and CXCR3B that lack putative C-terminal phosphorylation sites (Fig. 7A). The mutants’ truncation location is at the identical C-terminal residue given the extended N-terminus of CXCR3B (344th residue on CXCR3A, 391st residue on CXCR3B). Truncation resulted in ∼50% reduction, but not elimination, of βarr2 recruitment efficacy to both CXCR3A and CXCR3B as quantified by BRET (Fig. 7B), similar to other GPCRs such as the angiotensin type 1A receptor (Wei et al., 2004), but unlike other GPCRs, such as CCR5 and the apelin receptor, where βarr2 recruitment is not observed after removal of the C-terminus (Huttenrauch et al., 2002; Chen et al., 2014). Given these data, CXCR3 C-terminal phosphorylation-dependent βarr2 recruitment would not be expected to completely eliminate βarr2 recruitment to the receptor. Indeed, siRNA knockdown of either GRK2/3 or GRK5/6 decreased, but did not eliminate, CXCR3A-βarr2 association following CXCL11 stimulation as measured by BRET (Fig. 7C). Interestingly, GRK2/3, but not GRK5/6, siRNA knockdown prominently attenuated CXCR3B-βarr2 association (Fig. 7D); although both receptor splice variants undergo agonist-dependent phosphorylation, this suggests that different GRKs are playing distinctive roles at each receptor.
Fig. 7.

GRK5/6 knockdown differentially attenuates βarrestin2 recruitment. (A) Illustration of CXCR3 truncation C-terminal mutants. (B) Truncation of either CXCR3A or CXCR3B reduced βarr2 recruitment as measured by BRET in response to CXCL11 (500 nM). (C) siRNA knockdown of either GRK2/3 or GRK5/6 reduced CXCL11(500 nM) induced βarr2 recruitment to CXCR3A; however, as shown in (D) only siRNA knockdown of GRK2/3, but not GRK5/6, reduced βarr2 recruitment to CXCR3B. In (B), *P < 0.05 by unpaired student’s t test between respective WT and truncation mutant; in (C) and (D), *P < 0.05 by one-way analysis of variance with Tukey’s post hoc comparison between all treatment groups. n ≥ 3 biologic replicates per condition.
CXCL11 Activates ERK1/2 at Both CXCR3A and CXCR3B.
G proteins and βarrs can activate similar intracellular pathways, but with different spatial and temporal patterns that result in distinct cellular responses (Shenoy and Lefkowitz, 2011; Smith and Rajagopal, 2016). One well-characterized example is activation and phosphorylation (pERK) of the ERK1/2 mitogen-activated protein kinases (Wei et al., 2003). For many receptors, such as the angiotensin type IA and parathyroid hormone receptors, G proteins, βarr1, and/or βarr2 significantly contribute to early phase pERK (Gesty-Palmer et al., 2006; Lee et al., 2008; Kim et al., 2009), while at others, G protein-mediated pERK includes both a fast and a slow phase (Luo et al., 2008). Thus, kinetics alone cannot distinguish between G protein- and βarr-dependent pERK, and the contributions of these different pathways to the pERK response require more detailed characterization. The earlier study by Berchiche and Sakmar (2016) demonstrated that agonist-induced activation of CXCR3A and CXCR3B results in different intensities of activation with similar kinetics at early time points. We observed a similar response at early time points, since the pattern of pERK activation at 5 minutes by the four endogenous ligands was not different between HEK 293 cells transiently transfected with CXCR3A or CXCR3B (Fig. 8, A and C). Given that differential βarr signaling often occurs at time points beyond 15 minutes, we evaluated signaling up to 1 hour. Interestingly, a differential pattern of signaling emerged between splice variants at the 1-hour time point, since CXCR3A-transfected cells showed greater ERK activation at 60 minutes following stimulation with CXCL11, while CXCR3B transfected cells did not (Fig. 8, B and D).
Fig. 8.

Divergent ERK activation kinetics between CXCR3 splice variants. HEK 293 cells were transfected with either CXCR3A or CXCR3B and stimulated for either 5 minutes (A) or 60 minutes (B) with the indicated ligand (100 nM). The 60-minute, but not 5-minute, pERK1/2 CXCL11 signal in cells transfected with CXCR3A was significantly greater than cells transfected with CXCR3B. (C and D) Quantification of pERK/ERK signal normalized to CXCR3A vehicle treatment. Data were normalized to vehicle treatment of each respective isoform. In (C) and (D), *P < 0.05 indicates a significant effect of transfected isoform at 60 minutes by two-way analysis of variance (ANOVA). #P < 0.05; by one-way ANOVA and Tukey post hoc comparisons within all CXCR3A treatment groups, with * indicating a significant difference between CXCR3A CXCL11 stimulated cells compared with vehicle and CXCL4 at both 5 and 60 minutes; ‡P < 0.05, by one-way ANOVA and Tukey’s post hoc comparisons between all CXCR3B treatment groups, with ‡ indicating a significant difference between CXCR3B CXCL11 stimulated cells compared with all treatment groups at 5 minutes. No significant differences between ligands were observed in CXCR3B transfected cells at 60 minutes; ± S.E.M., n ≥ 3 biologic replicates per condition. Western blots shown are representative of at least three separate experiments.
Given the differential pattern of observed ERK activity, we tested the effects of βarr KO on ERK activation by CXCR3 splice variants. To perform this, we used a different HEK 293 cell line in which both βarr1 and βarr2 (Arrb 1/2 KO) were removed by CRISPR/Cas9 genome editing. βarr1 and βarr2 KO was confirmed by immunoblot with polyclonal anti-βarrestin antibodies recognizing either the C-terminus of βarr1 (anti-βarr1) or the C-terminus of βarr2 (anti-βarr2) (Supplemental Fig. 6). The pERK signal in these Arrb 1/2 KO cells was compared with WT cells following 100 nM CXCL11 stimulation. Both WT and Arrb 1/2 KO cells from this HEK 293 line differed in morphology and growth rate compared with HEK 293 cells used in other experiments. The basal pERK activity of the Arrb 1/2 KO vehicle control group of both CXCR3A- and CXCR3B-transfected cells was greater than in identically treated WT cells (Fig. 9, A and B). In both CXCR3A-transfected WT and Arrb 1/2 KO cells, CXCL11 at 5 minutes caused a significant increase in pERK signal (Fig. 9, A and C). In contrast, at CXCR3B, WT cells displayed a significant increase in pERK signal compared with vehicle while Arrb 1/2 KO cells did not (Fig. 9, B and D). The residual ERK phosphorylation by CXCR3B in Arrb 1/2 KO cells may represent very low residual Gαi signaling not detected in our second messenger assays or coupling to alternative G proteins. To further probe the differential pERK signal observed between WT and Arrb 1/2 KO cells at 5 minutes expressing either CXCR3A or CXCR3B, we rescued βarr1 and βarr2 through transient overexpression in the Arrb 1/2 KO cells and compared pERK signal at 5 minutes to empty vector transfection controls. Similar to the trend observed between WT and Arrb 1/2 KO cells, the 5-minute pERK signal in CXCR3A-transfected cells was significantly decreased by βarr overexpression rescue relative to vehicle (Fig. 10, A and D). Conversely, the pERK signal was significantly potentiated in CXCR3B-transfected cells relative to vehicle (Fig. 10, B and D), with a significant difference also noted between isoforms (Fig. 10D).
Fig. 9.

β-arrestin KO differentially alters pERK signaling between CXCR3 splice variants. WT or ∆ARRB1/2 cells transfected with CXCR3A (A) or CXCR3B (B) and stimulated with CXCL11 (100 nM). A significant increase in pERK1/2 signal was observed in both WT and ∆ARRB1/2 cells transfected with CXCR3A. However, only WT, but not ∆ARRB1/2, cells transfected with CXCR3B displayed a significant increase in pERK1/2 signal. The CXCR3A (C) and CXCR3B (D) phospo-ERK signal was quantified in WT and ∆ARRB1/2 cells [note the change in scale of the y-axis between (C) and (D)]. In (C) and (D), &P < 0.05 indicates a significant effect of cell line by two-way analysis of variance (ANOVA) in cells transfected with CXCR3B; *P < 0.05 indicates a significant difference in the 5-minute time point by two-way ANOVA followed by Bonferroni post hoc comparison (corrected for all time points) in cells transfected with CXCR3B; #P < 0.05 by one-way ANOVA and Dunnett’s post hoc comparison of CXCL11 stimulation to the respective isoform vehicle control at the indicated time point; ± S.E.M., n ≥ 3 biologic replicates per condition. Western blots shown are representative of at least three separate experiments.
Differential Transcriptional Regulation by CXCR3 Splice Variants.
When assessing bias, testing activity distal to direct receptor transducers is helpful in modeling physiologic relevance. For example, if biased signaling is observed at proximal effectors such as G proteins and β-arrestins, but not distal transducers such as regulators of transcription, the significance of the observed bias is less clear. We further probed distal signaling by evaluating transcriptional activation through serum response reporter assays. The SRE is known to respond to the ternary complex factor–dependent ERK/mitogen-activated protein kinase activity, while the SRF is a mutant form of SRE lacking the ternary complex factor binding domain that also responds to SRF-dependent and ternary complex factor–independent signals, such as RhoA (Hill et al., 1995; Cheng et al., 2010). Either SRE or SRF reporters were transiently cotransfected with either CXCR3A or CXCR3B. Transcriptional activity was assessed following incubation with the four CXCR3 endogenous ligands. Stimulation with CXCL11 resulted in robust signal from both SRE and SRF-RE Response Element reporters cotransfected with CXCR3A (Fig. 11, A and C), but not when cotransfected with CXCR3B (Fig. 11, B and D). Both SRE and SRF signals were attenuated, but not eliminated, by mitogen-activated protein kinase kinase inhibition prior to CXCL11 stimulation (Fig. 11, E and F). This is consistent with ERK activation at later time points resulting in some transcriptional regulation of SRE and SRF.
Fig. 11.

CXCL11 robustly activates SRE and SRF response element signaling at CXCR3A, but not CXCR3B. HEK 293T cells were transiently transfected with either the SRE or SRF reporter and CXCR3A or CXCR3B. Prior to acquiring luminescence signal, cells were incubated for 5 hours with ligand (1 μM). CXCL11 incubation caused a significant increase in luminescence signal in both SRE-transfected (A) and SRF-transfected (C) cells. In cells transfected with CXCR3B, none of the endogenous ligands tested resulted in significant SRE (B) or SRF (D) signal, although cells still responded to the positive fetal bovine serum (FBS) control. In (E) and (F), cells were pretreated either with vehicle or with mitogen-activated protein kinase kinase (MEK) inhibitor PD98059 (20 μM). The CXCL11-induced SRE and SRF signals were sensitive to MEK inhibition (E and F); *P < 0.05. (A)–(D) One-way analysis of variance (ANOVA) and Tukey’s post hoc comparisons of treatment groups. The positive control of 10% FBS is included for reference, but not included in statistical analyses. (E) and (F) One-way ANOVA, followed by a directed Bonferroni post hoc comparison of vehicle (Veh) + CXCL11 to PD98059 + CXCL11, was conducted. n ≥ 3 biologic replicates per condition.
Discussion
In this study, we found that CXCR3 receptor splice variants displayed biased signaling that was associated with distinct βarr conformations and signaling to downstream pathways, including ERK and SRE/SRF. Unlike CXCR3A, which demonstrated robust signaling through Gαi, CXCR3B did not display any appreciable Gαi protein signaling in second messenger assays. In rank order of efficacy, CXCL11, CXCL10, and CXCL9, but not CXCL4, stimulation caused internalization of CXCR3A in the same order previously described in human T-lymphocytes and HEK cells (Korniejewska et al., 2011; Berchiche and Sakmar, 2016). This internalization rank order was identical to the rank order of efficacy in βarr2 recruitment to CXCR3A as measured by BRET. Interestingly, only CXCL11, and not the other CXCR3 ligands recruited βarr2 to CXCR3B and resulted in measureable receptor internalization. The CXCR3B-βarr interaction was confined to the plasma membrane in a class A pattern compared with the CXCR3A-βarr interaction displaying an endocytic class B pattern (Tohgo et al., 2003). Notably, a biosensor demonstrated distinct βarr conformations associated with these two patterns of recruitment, with distinct βarr conformations recently to correlate with distinct βarr functions at the β2AR (Cahill et al., 2017). Functional activity of CXCR3B was only observed when stimulated by a ligand that caused βarr2 recruitment, CXCL11. In further support of CXCR3B acting as a βarr-biased receptor, βarr1/2 overexpression in Arrb 1/2 KO cells increased signaling by CXCR3B while decreasing signaling by CXCR3A. Taken in its entirety, these findings suggest that CXCR3B acts as a βarr-biased receptor relative to CXCR3A, albeit with a less stable interaction with βarr compared with CXCR3A (Fig. 12, summary). This bias may be encoded at least in part by differential GRK recruitment, since siRNA knockdown of GRK5/6 attenuated βarr recruitment to CXCR3A, but not to CXCR3B.
Fig. 12.

Summary of observed CXCR3 isoform signaling. Both CXCR3A and CXCR3B recruited β-arrestin2, became phosphorylated, and internalized in response to CXCL11. In contrast, only CXCR3A was observed to signal through Gαi since CXCR3B-transfected cells did not produce appreciable signal in either Gαi or Gαs assays. In further signaling divergence, CXCR3A and CXCR3B show distinct patterns of downstream signaling, with CXCR3A observed to display a stable class B interaction with β-arrestin2 in contrast to CXCR3B, which displayed a transient class A interaction in confocal recruitment assays. GRK2/3 siRNA knockdown attenuated βarr2 recruitment to both receptor isoforms; however, only GRK5/6 knockdown was observed to attenuate βarr2 recruitment to CXCR3A. Only CXCR3A was observed to show significant late phase (1 hour) pERK activity, and overexpression rescue of β-arrestin attenuated early phase pERK activity mediated by CXCR3A while enhanced pERK activity was mediated by CXCR3B. SRE and SRF transcriptional reporter activity was only observed downstream from CXCR3A.
Phosphorylation of mitogen-activated protein kinases is a well-studied signaling response regulated by G proteins and β-arrestins downstream from GPCRs (Shenoy and Lefkowitz, 2011; Smith and Rajagopal, 2016). Although pERK signal was diminished in CXCR3B-transfected Arrb 1/2 KO cells, the signal 5 minutes after stimulation was still higher than vehicle. This suggests that while CXCR3B is biased toward β-arrestin signaling compared with CXCR3A, CXCR3B signaling does not absolutely require β-arrestins. CXCR3B may couple to a different G protein, an unidentified signaling effector, or both. Further investigation will be needed to understand how N-terminal splice variants alter intracellular effector coupling. CXCL11 stimulation resulted in phosphorylation of both receptor isoforms to comparable levels, suggesting that the differential signaling by CXCR3A and CXCR3B is due to the phosphorylation of different residues in the C-terminus and/or different conformational changes induced in the receptors.
In our studies, we focused on receptor coupling to two GRK receptor families, GRK2/3 and GRK5/6, because GRK2- and GRK6-deficient lymphocytes display defective trafficking and because the expression of both CXCR3A and CXCR3B is high in lymphocytes (Fong et al., 2002; Lasagni et al., 2003; Arnon et al., 2011). Phosphorylation by different kinases of serines and/or threonines in GPCR intracellular loops and/or the C-terminus is often necessary for recruitment of βarrs to the receptor (Freedman et al., 1995; Ahn et al., 2002). Our siRNA knockdown studies demonstrate a role for both GRK2/3 and GRK5/6 in CXCL11-mediated βarr2 recruitment to CXCR3A, but a role only for GRK2/3 in βarr2 recruitment to CXCR3B. At other GPCRs, such as the angiotensin II type I receptor (Kim et al., 2005), the β2 adrenergic receptor (Nobles et al., 2011), and CCR7 (Zidar et al., 2009), different GRKs have been demonstrated to perform signaling or desensitization functions. The activity of these different kinases is thought to result in a phosphorylation barcode of the third intracellular loop and C-terminal tail of other GPCRs that regulates βarr activity (Nobles et al., 2011; Reiter et al., 2012). Indeed, the C-terminus and third intracellular loop of CXCR3 is known to be necessary for chemotaxis, calcium flux, and internalization of CXCR3A, processes that are known to be mediated by βarrs (Colvin et al., 2004). Once activated, βarr is thought to form a core scaffolding complex, which activates ERK1/2 by phosphorylation (Eichel et al., 2016). The findings here support a model for a CXCR3 signaling barcode, in which post-translational modifications in the receptor C-terminus regulate the affinity of βarr recruitment and the pattern of GPCR intracellular trafficking (Oakley et al., 2000, 2001). However, future studies that clearly link the phosphorylation of specific sites in the receptor by distinct kinases will be needed to test this hypothesis at CXCR3.
It is somewhat surprising that an N-terminal CKR modification has such an impact on receptor coupling to G proteins, GRKs, and βarrs. This is likely due to the mode of chemokines binding to their CKRs, which is primarily mediated by distinct chemokine recognition sites (CRS) (Qin et al., 2015). Our current understanding of chemokine:CKR binding, which is informed both by earlier biophysical studies (Booth et al., 2002) and now crystal structures (Wu et al., 2010; Tan et al., 2013; Qin et al., 2015), is that the N-terminus of the chemokine inserts itself in the transmembrane regions of the CKR and interacts through an interface termed CRS2. The C-terminal domain of the chemokine interacts with the extracellular N-terminus of the receptor through the CRS1 site. Recent crystal structures have demonstrated that the extracellular loops of the CKR interact with the chemokine through a CRS1.5 site. Post-translational sulfation of two sites on the N-terminus of CXCR3A (Tyr 27 and Tyr 29) is necessary for receptor function following ligand stimulation (Colvin et al., 2006). The extended N-terminus of CXCR3B adds two additional sulfation sites (Tyr 6 and Tyr 40). We speculate that this extended N-terminus of CXCR3B binds to its ligands through the CRS1 site or an alternate surface on the chemokine, thereby allosterically coupling with CRS2 site to generate distinct receptor:ligand:transducer conformations.
In summary, CXCR3 splice variants CXCR3A and CXCR3B, differing by an extended N-terminus at CXCR3B, demonstrated significantly biased signaling responses. Unlike signaling observed at CXCR3A, βarr2, but not Gαi protein signaling, was detected at CXCR3B. Different patterns of receptor-βarr2 interaction were also observed between splice variants, with CXCR3A displaying a more stable interaction with arrestin compared with CXCR3B. siRNA knockdown of GRK5/6 attenuated βarr2 recruitment at CXCR3A, but not CXCR3B. In addition, the splice variants had distinct internalization profiles that correlated with differences in late-phase ERK activation and transcriptional activity. At this time, it is unclear which mechanisms underlying the phosphorylation barcode of the CXCR3 C-terminus and phosphorylation sites are critical for activation of G protein- and βarr-mediated pathways downstream from the receptor. Appreciating the signaling differences between these splice variants may provide clarification to conflicting reports of CXCR3 function and offer a compelling example of how the GPCR extracellular residues can dramatically change intracellular pathway activation.
Acknowledgments
The authors thank Dr. Robert J. Lefkowitz for invaluable insight and review and Dr. Marc Caron, Dr. Sudha Shenoy, and Dr. Robert J. Lefkowitz for use of laboratory equipment. The authors also thank Nour Nazo for administrative assistance and Nour Nazo, Rachel Glenn, Jaimee Gundry, and Alex Antonia for laboratory assistance.
Abbreviations
- βarr
beta-arrestin
- BRET
bioluminescence resonance energy transfer
- CKR
chemokine receptor
- CRS
chemokine recognition site
- CXCL
C-X-C motif chemokine ligand
- ERK
extracellular regulated kinase
- GFP
green fluorescent protein
- GPCR
G protein-coupled receptor
- GRK
G protein receptor kinase
- HEK
human embryonic kidney
- KO
knockout
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- pERK
phosphorylated extracellular regulated kinase
- Rluc
Renilla luciferase
- siRNA
small inhibitory RNA
- SRE
serum response element
- SRF
serum response factor
- WT
wild type
- YFP
yellow fluorescent protein
Authorship Contributions
Participated in research design: Smith, Pack, Freedman, Rajagopal.
Conducted experiments: Smith, Alagesan, Desai, Wu, Rajagopal
Contributed new reagents or analytic tools: Pack, Inoue.
Wrote or contributed to the writing of the manuscript: Smith, Alagesan, Desai, Pack, Freedman, Rajagopal.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant 5T32GM7171], the National Heart, Lung, and Blood Institute [Grants HL121689 and HL114643]; the Burroughs Wellcome Career Award for Medical Scientists; the Duke Pinnell Center for Investigative Dermatology; the Japan Science and Technology Agency; and Precursory Research for Embryonic Science and Technology.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Ahn S, Kim J, Lucaveche CL, Reedy MC, Luttrell LM, Lefkowitz RJ, Daaka Y. (2002) Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J Biol Chem 277:26642–26651. [DOI] [PubMed] [Google Scholar]
- Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, Milligan G. (2016) Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein-coupled receptor signaling. J Biol Chem 291:27147–27159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon TI, Xu Y, Lo C, Pham T, An J, Coughlin S, Dorn GW, Cyster JG. (2011) GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science 333:1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, Lefkowitz RJ. (1992) β-Arrestin2, a novel member of the arrestin/β-arrestin gene family. J Biol Chem 267:17882–17890. [PubMed] [Google Scholar]
- Berchiche YA, Sakmar TP. (2016) CXC chemokine receptor 3 alternative splice variants selectively activate different signaling pathways. Mol Pharmacol 90:483–495. [DOI] [PubMed] [Google Scholar]
- Booth V, Keizer DW, Kamphuis MB, Clark-Lewis I, Sykes BD. (2002) The CXCR3 binding chemokine IP-10/CXCL10: structure and receptor interactions. Biochemistry 41:10418–10425. [DOI] [PubMed] [Google Scholar]
- Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. (2010) Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem 285:7805–7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill TJ, 3rd, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, et al. (2017) Distinct conformations of GPCR–β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci USA 114:2562-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest PG, Terrillon S, Bouvier M. (2005) Monitoring agonist-promoted conformational changes of β-arrestin in living cells by intramolecular BRET. EMBO Rep 6:334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bai B, Tian Y, Du H, Chen J. (2014) Identification of serine 348 on the apelin receptor as a novel regulatory phosphorylation site in apelin-13-induced G protein-independent biased signaling. J Biol Chem 289:31173–31187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Garvin D, Paguio A, Stecha P, Wood K, Fan F. (2010) Luciferase reporter assay system for deciphering GPCR pathways. Curr Chem Genomics 4:84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, Lin W, Boyd JG, Moser B, Wood DE, et al. (1998) Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med 187:2009–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin RA, Campanella GS, Manice LA, Luster AD. (2006) CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol 26:5838–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin RA, Campanella GS, Sun J, Luster AD. (2004) Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem 279:30219–30227. [DOI] [PubMed] [Google Scholar]
- Drury LJ, Ziarek JJ, Gravel S, Veldkamp CT, Takekoshi T, Hwang ST, Heveker N, Volkman BF, Dwinell MB. (2011) Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci USA 108:17655–17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, Jullié D, von Zastrow M. (2016) β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat Cell Biol 18:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evron T, Peterson SM, Urs NM, Bai Y, Rochelle LK, Caron MG, Barak LS. (2014) G Protein and β-arrestin signaling bias at the ghrelin receptor. J Biol Chem 289:33442–33455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong AM, Premont RT, Richardson RM, Yu YR, Lefkowitz RJ, Patel DD. (2002) Defective lymphocyte chemotaxis in β-arrestin2- and GRK6-deficient mice. Proc Natl Acad Sci USA 99:7478–7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman NJ, Kim LK, Murray JP, Exum ST, Brian L, Wu JH, Peppel K. (2002) Phosphorylation of the platelet-derived growth factor receptor-β and epidermal growth factor receptor by G protein-coupled receptor kinase-2. Mechanisms for selectivity of desensitization. J Biol Chem 277:48261–48269. [DOI] [PubMed] [Google Scholar]
- Freedman NJ, Liggett SB, Drachman DE, Pei G, Caron MG, Lefkowitz RJ. (1995) Phosphorylation and desensitization of the human β1-adrenergic receptor. Involvement of G protein-coupled receptor kinases and cAMP-dependent protein kinase. J Biol Chem 270:17953–17961. [DOI] [PubMed] [Google Scholar]
- García-López MA, Sánchez-Madrid F, Rodríguez-Frade JM, Mellado M, Acevedo A, García MI, Albar JP, Martínez C, Marazuela M. (2001) CXCR3 chemokine receptor distribution in normal and inflamed tissues: expression on activated lymphocytes, endothelial cells, and dendritic cells. Lab Invest 81:409–418. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, et al. (2006) Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem 281:10856–10864. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, El Shewy H, Kohout TA, Luttrell LM. (2005) β-Arrestin 2 expression determines the transcriptional response to lysophosphatidic acid stimulation in murine embryo fibroblasts. J Biol Chem 280:32157–32167. [DOI] [PubMed] [Google Scholar]
- Groom JR, Luster AD. (2011) CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XM, Kobilka TS, Kobilka BK. (1992) Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J Biol Chem 267:21995–21998. [PubMed] [Google Scholar]
- Hermodson M, Schmer G, Kurachi K. (1977) Isolation, crystallization, and primary amino acid sequence of human platelet factor 4. J Biol Chem 252:6276–6279. [PubMed] [Google Scholar]
- Hill CS, Wynne J, Treisman R. (1995) The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81:1159–1170. [DOI] [PubMed] [Google Scholar]
- Huttenrauch F, Nitzki A, Lin FT, Höning S, Oppermann M. (2002) β-Arrestin binding to CC chemokine receptor 5 requires multiple C-terminal receptor phosphorylation sites and involves a conserved Asp-Arg-Tyr sequence motif. J Biol Chem 277:30769–30777. [DOI] [PubMed] [Google Scholar]
- Karin N, Wildbaum G, Thelen M. (2016) Biased signaling pathways via CXCR3 control the development and function of CD4+ T cell subsets. J Leukoc Biol 99:857–862. [DOI] [PubMed] [Google Scholar]
- Kawada K, Sonoshita M, Sakashita H, Takabayashi A, Yamaoka Y, Manabe T, Inaba K, Minato N, Oshima M, Taketo MM. (2004) Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res 64:4010–4017. [DOI] [PubMed] [Google Scholar]
- Kim J, Ahn S, Rajagopal K, Lefkowitz RJ. (2009) Independent β-arrestin2 and Gq/protein kinase Cζ pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J Biol Chem 284:11953–11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, Lefkowitz RJ. (2005) Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci USA 102:1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK. (1990) The role of cytosolic and membrane factors in processing of the human β-2 adrenergic receptor following translocation and glycosylation in a cell-free system. J Biol Chem 265:7610–7618. [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. (2009) The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelink PJ, Overbeek SA, Braber S, de Kruijf P, Folkerts G, Smit MJ, Kraneveld AD. (2012) Targeting chemokine receptors in chronic inflammatory diseases: an extensive review. Pharmacol Ther 133:1–18. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. (2004) Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem 279:23214–23222. [DOI] [PubMed] [Google Scholar]
- Korniejewska A, McKnight AJ, Johnson Z, Watson ML, Ward SG. (2011) Expression and agonist responsiveness of CXCR3 variants in human T lymphocytes. Immunology 132:503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, Sagrinati C, Mazzinghi B, Orlando C, Maggi E, et al. (2003) An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med 197:1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, El-Shewy HM, Luttrell DK, Luttrell LM. (2008) Role of β-arrestin-mediated desensitization and signaling in the control of angiotensin AT1a receptor-stimulated transcription. J Biol Chem 283:2088–2097. [DOI] [PubMed] [Google Scholar]
- Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. (1996) Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med 184:963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Busillo JM, Benovic JL. (2008) M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol Pharmacol 74:338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller A, Meiser A, McDonagh EM, Fox JM, Petit SJ, Xanthou G, Williams TJ, Pease JE. (2008) CXCL4-induced migration of activated T lymphocytes is mediated by the chemokine receptor CXCR3. J Leukoc Biol 83:875–882. [DOI] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, et al. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal 4:ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. (2001) Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis. J Biol Chem 276:19452–19460. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. (2000) Differential affinities of visual arrestin, βarrestin1, and βarrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275:17201–17210. [DOI] [PubMed] [Google Scholar]
- Onaran HO, Ambrosio C, Uğur Ö, Madaras Koncz E, Grò MC, Vezzi V, Rajagopal S, Costa T. (2017) Systematic errors in detecting biased agonism: Analysis of current methods and development of a new model-free approach. Sci Rep 7:44247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015) Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SM, Pack TF, Wilkins AD, Urs NM, Urban DJ, Bass CE, Lichtarge O, Caron MG. (2015) Elucidation of G-protein and β-arrestin functional selectivity at the dopamine D2 receptor. Proc Natl Acad Sci USA 112:7097–7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, et al. (2015) Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347:1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. (1998) The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest 101:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, Violin JD, Lefkowitz RJ. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol 80:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Bassoni DL, Campbell JJ, Gerard NP, Gerard C, Wehrman TS. (2013) Biased agonism as a mechanism for differential signaling by chemokine receptors. J Biol Chem 288:35039–35048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, Gerard C, Lefkowitz RJ. (2010) β-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci USA 107:628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. (2012) Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol 52:179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage R, Schmitz AL, Gaffal E, Annala S, Kehraus S, Wenzel D, Büllesbach KM, Bald T, Inoue A, Shinjo Y, et al. (2015) The experimental power of FR900359 to study Gq-regulated biological processes. Nat Commun 6:10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci 32:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Rajagopal S. (2016) The β-arrestins: multifunctional regulators of G protein-coupled receptors. J Biol Chem 291:8969–8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Li J, et al. (2013) Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341:1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ. (1993) Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med 177:1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tensen CP, Flier J, Van Der Raaij-Helmer EM, Sampat-Sardjoepersad S, Van Der Schors RC, Leurs R, Scheper RJ, Boorsma DM, Willemze R. (1999) Human IP-9: A keratinocyte-derived high affinity CXC-chemokine ligand for the IP-10/Mig receptor (CXCR3). J Invest Dermatol 112:716–722. [DOI] [PubMed] [Google Scholar]
- Thelen M. (2001) Dancing to the tune of chemokines. Nat Immunol 2:129–134. [DOI] [PubMed] [Google Scholar]
- Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. (2003) The stability of the G protein-coupled receptor-β-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem 278:6258–6267. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320:1–13. [DOI] [PubMed] [Google Scholar]
- Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. (2015) CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev 26:311–327. [DOI] [PubMed] [Google Scholar]
- Villarroel VA, Okiyama N, Tsuji G, Linton JT, Katz SI. (2014) CXCR3-mediated skin homing of autoreactive CD8 T cells is a key determinant in murine graft-versus-host disease. J Invest Dermatol 134:1552–1560. [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Barnes WG, Lefkowitz RJ. (2004) Stable interaction between β-arrestin 2 and angiotensin type 1A receptor is required for β-arrestin 2-mediated activation of extracellular signal-regulated kinases 1 and 2. J Biol Chem 279:48255–48261. [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA 100:10782–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JH, Goswami R, Kim LK, Miller WE, Peppel K, Freedman NJ. (2005) The platelet-derived growth factor receptor-β phosphorylates and activates G protein-coupled receptor kinase-2. A mechanism for feedback inhibition. J Biol Chem 280:31027–31035. [DOI] [PubMed] [Google Scholar]
- Wu JH, Zhang L, Fanaroff AC, Cai X, Sharma KC, Brian L, Exum ST, Shenoy SK, Peppel K, Freedman NJ. (2012) G protein-coupled receptor kinase-5 attenuates atherosclerosis by regulating receptor tyrosine kinases and 7-transmembrane receptors. Arterioscler Thromb Vasc Biol 32:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ. (2009) Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Natl Acad Sci USA 106:9649–9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohar Y, Wildbaum G, Novak R, Salzman AL, Thelen M, Alon R, Barsheshet Y, Karp CL, Karin N. (2014) CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J Clin Invest 124:2009–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]