

Abstract
Neocortical circuits use a family of homeostatic plasticity mechanisms to stabilize firing, including excitatory and inhibitory synaptic scaling and homeostatic intrinsic plasticity (Turrigiano and Nelson, 2004). All three mechanisms can be induced in tandem in cultured rat neocortical pyramidal neurons by chronic manipulations of firing, but it is unknown whether they are coinduced by the same activity-sensors and signaling pathways, or whether they are under independent control. Calcium/calmodulin-dependent protein kinase type IV (CaMKIV) is a key sensory/effector in excitatory synaptic scaling that senses perturbations in firing through changes in calcium influx, and translates this into compensatory changes in excitatory quantal amplitude (Ibata et al., 2008; Goold and Nicoll, 2010). Whether CaMKIV also controls inhibitory synaptic scaling and intrinsic homeostatic plasticity was unknown. To test this we manipulated CaMKIV signaling in individual neurons using dominant-negative (dn) or constitutively-active (ca) forms of nuclear-localized CaMKIV and measured the induction of all three forms of homeostatic plasticity. We found that excitatory synaptic scaling and intrinsic plasticity were bidirectionally coinduced by these manipulations. In contrast, these cell-autonomous manipulations had no impact on inhibitory quantal amplitude. Finally, we found that spontaneous firing rates were shifted up or down by dnCaMKIV or caCaMKIV, respectively, suggesting that uncoupling CaMKIV activation from activity generates an error signal in the negative feedback mechanism that controls firing rates. Together, our data show that excitatory synaptic scaling and intrinsic excitability are tightly coordinated through bidirectional changes in the same signaling pathway, whereas inhibitory synaptic scaling is sensed and regulated through an independent control mechanism.
SIGNIFICANCE STATEMENT Maintaining stable function in highly interconnected neural circuits is essential for preventing circuit disorders, and is accomplished through a set of negative feedback mechanisms that sense and compensate for perturbations in activity. These “homeostatic” mechanisms can target synaptic excitation, synaptic inhibition, and intrinsic excitability, but whether they are independently controlled is not known. We find that synaptic excitation and intrinsic excitability are coregulated in individual neurons through CaMKIV signaling, which is tightly controlled by neuronal activity. In contrast, synaptic inhibition is unaffected by changes in firing or CaMKIV signaling in individual neurons. These results show that circuit stability is controlled both through cell-autonomous mechanisms that regulate some aspects of excitability, as well as circuit-level mechanisms that adjust inhibition.
Keywords: CaMKIV, excitatory synaptic scaling, firing rate set point, homeostasis, inhibitory synaptic scaling, intrinsic excitability
Introduction
Unconstrained neuronal activity can lead to serious neurological disorders, such as epilepsy, chronic pain, and spasticity (Du et al., 2005; Kullmann and Waxman, 2010). Neocortical circuits contain extensively recurrent excitatory connections which must be actively balanced by appropriate levels of inhibition to prevent the development of hyper or hypoactivity. To achieve this, neurons and circuits use a variety of homeostatic negative feedback mechanisms to (1) sense activity, (2) compare this activity to a target firing rate (FR), and (3) make compensatory adjustments in their own excitability to return to this preferred FR “set point” (Davis and Bezprozvanny, 2001; Marder and Prinz, 2002; Turrigiano and Nelson, 2004; Hengen et al., 2016). Neurons stabilize firing by exploiting a family of homeostatic plasticity mechanisms that bidirectionally tune synaptic strength and intrinsic excitability (Turrigiano et al., 1998; Desai et al., 1999). Although manipulating activity can trigger synaptic and intrinsic homeostatic mechanisms in tandem (Turrigiano, 2012; Lambo and Turrigiano, 2013; Cannon and Miller, 2016), it is currently unknown whether they are coordinately regulated by the same upstream signaling pathways.
The calcium/calmodulin-dependent protein kinase type IV (CaMKIV) pathway has emerged as a candidate for sensing and homeostatically adjusting neuronal activity. It exhibits steep dependence on neuronal Ca2+ levels to mediate transcription through phosphorylation of cAMP responsive element binding protein (CREB) at Ser 133 (Matthews et al., 1994; Finkbeiner et al., 1997; Chawla et al., 1998). One important form of homeostatic plasticity, synaptic scaling (SS), is transcription-dependent (Ibata et al., 2008; Benevento et al., 2016; Schaukowitch et al., 2017), and a role for CaMKIV signaling in SS has been firmly established (Ibata et al., 2008; Goold and Nicoll, 2010; Pratt et al., 2011). Expression of either a dominant-negative (dn) or a constitutively-active (ca) form of CaMKIV was able to scale up (dnCaMKIV; Ibata et al., 2008), or scale down (caCaMKIV; Goold and Nicoll, 2010) excitatory synaptic strengths, respectively, and these changes occluded further scaling. Together, these studies suggest that deviations in neural activity away from an internal FR set point lead to a corresponding change in the activation state of CaMKIV (phosphorylated CaMKIV) in the nucleus. CaMKIV subsequently regulates transcription in a manner that results in altered trafficking and accumulation of synaptic AMPA receptors such that synaptic strength scales in the appropriate direction to move FR back toward a target value (Turrigiano, 2008).
Although activity-dependent excitatory SS is mediated through CaMKIV signaling, it remains unknown whether this pathway coordinately regulates homeostatic inhibitory synaptic scaling (Kilman et al., 2002; Vale and Sanes, 2002; Hartman et al., 2006) and intrinsic excitability (Turrigiano et al., 1994; Desai et al., 1999). Here we directly tested whether CaMKIV is a “master regulator” of multiple homeostatic mechanisms. We expressed nuclear-localized dnCaMKIV or caCaMKIV in individual neocortical pyramidal neurons in vitro, and probed for bidirectional changes in synaptic currents and intrinsic excitability onto affected neurons. We found that changes in CaMKIV signaling coordinately regulate excitatory synaptic strengths and intrinsic excitability, with both changes in the right direction to stabilize firing: caCaMKIV reduced intrinsic excitability and excitatory synaptic strength, whereas dnCaMKIV had the opposite effect. In contrast, inhibitory synaptic strength was unaffected by these cell-autonomous manipulations of CaMKIV signaling. The cooperative changes in intrinsic excitability and excitatory synaptic strength (coupled with a lack of change in inhibition) prompted us to ask whether basal FRs were also affected. Indeed, we found that altered CaMKIV signaling was able to bidirectionally and cell-autonomously regulate the average FR of pyramidal neurons. Together, our results demonstrate that two fundamental forms of homeostatic plasticity are coordinately and cell-autonomously induced through changes in CaMKIV signaling. In contrast, the lack of effect on inhibition indicates that cell-autonomous changes in CaMKIV signaling, and the resultant effect on firing rates, do not trigger inhibitory scaling. This suggests that inhibitory scaling is mediated through an independent, possibly circuit-level mechanism. These results have important implications for understanding how multiple homeostatic mechanisms are organized within neurons and circuits.
Materials and Methods
All experiments were performed in vitro and approved by the Animal Care and Use Committee of Brandeis University and complied with the guidelines of the National Institutes of Health.
Neuronal cultures and transfection.
Dissociated neocortical cultures were prepared from postnatal day (P)0–P1 Long–Evans rat pups of either sex, and plated onto glass bottomed dishes with confluent astrocytes as described previously (Pratt et al., 2003). Experiments were performed between 7 and 14 d in vitro on morphologically identified pyramidal neurons as described previously (Pratt et al., 2003). Transfections were done at low efficiency for 24 h using Lipofectamine2000 (Invitrogen) following the manufacturer's instructions. To visualize transfected neurons, constructs were cotransfected with a soluble fluorescent protein (XFP; mCherry or GFP as appropriate). Experimental conditions were compared within age-matched sister cultures, as described previously (Pratt et al., 2003).
Constructs.
CaMKIV mutant constructs were gifts of Prof. Thomas Soderling. dnCAMKIV is constructed with three point mutations 1) Lys75Ala in the ATP-binding site mutated to Ala, 2) the activation loop phosphorylation site (Thr196) mutated to Ala, and 3) the autoinhibitory domain inactivated by the triple mutation HMDT308 to DEDD (Wayman et al., 2011); caCaMKIV is constructed with a mutation in the autoinhibitory domain inactivated by the triple mutation HMDT308 to DEDD (Wayman et al., 2011); and Empty Vector (EV) encodes a green fluorescent protein (GFP; Clontech Laboratories). All constructs have three copies of a nuclear localization signal (NLS) attached. The triplicate NLS significantly increases the efficiency of translocation into the nucleus of mammalian cells (Fischer-Fantuzzi and Vesco, 1988). Additionally, our constructs are GFP-fused for easy identification of protein expression.
In vitro electrophysiology.
Whole-cell recordings were obtained from pyramidal neurons using an Axopatch 200B amplifier (Molecular Devices); cultures were continuously perfused with oxygenated artificial CSF (ACSF) containing the following (in mm):126 NaCl, 5.5 KCl, 2 MgSO4, 1 NaH2PO4, 25 NaHCO3, 2 CaCl2, and 14 dextrose at 25°C. Recordings with Vm > −55 mV, Rs > 20 MΩ, and Rin < 100 MΩ and <25 mEPSC events were excluded. In-house Igor Pro (Wavemetrics) and MATLAB software was used to detect and measure events.
To record AMPA-mediated mEPSCs neurons were voltage-clamped to −70 mV as described previously (Turrigiano et al., 1998; Wierenga et al., 2005) and perfused in standard ACSF containing 0.1 μm TTX, 50 μm APV, and 25 μm picrotoxin. The internal recording solution contained the following (in mm): 120 KMeSO4, 10 KCl, 2 MgSO4, 10 K-HEPES, 0.5 EGTA, 3 K2ATP, 0.3 Na-GTP, and 10 Na2 phosphocreatine. To record mIPSCs neurons were voltage-clamped to −80 mV in standard ACSF containing 0.1 μm TTX, 50 μm APV, and 20 μm DNQX, using a “symmetric Cl−” internal recording solution containing the following (in mm): 120 KCl, 10 K-HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 10 Na-phosphocreatine, 10 sucrose. f–I curves were recorded in ACSF containing APV (50 μm), DNQX (25 μm), and picrotoxin (50 μm) in current-clamp; a small dc bias current was injected to maintain resting Vm at ∼−60 mV. Spontaneous action potentials were recorded in standard ACSF and a small dc bias current was injected to maintain resting Vm at ∼−55 mV.
Immunostaining.
To visualize phospho-CREB ser133 we performed immunostaining as described previously (Steinmetz et al., 2016) using an anti-Phospho Creb antibody (Cell Signaling Technology; catalog #9198S RRID:AB_2561044, 1/500). To quantify the nuclear localization of CaMKIV-XFP expression DAPI was used as a nuclear marker. Images were acquired using an oil-immersion 60× objective on a Leica SP5 confocal microscope under sequential scanning settings for multichannel fluorescence image acquisition. Digital images were quantified with MetaMorph software (Molecular Devices).
Statistical analysis.
Each experiment was repeated on at least three separate dissociations, and n represents the number of neurons/condition. Data are presented as mean ± SEM. There were no significant differences in untransfected and EV-transfected neurons in any of the experimental conditions, so these conditions were pooled and are reported as control (ctrl). To determine statistical significance, unpaired two-tailed t tests were performed for single comparisons; for multiple-comparisons single-factor ANOVAs followed by an appropriate post hoc test were used. A test for homogeneity/normality was used to determine whether a nonparametric (Games–Howell) or parametric (Tukey) post hoc test was appropriate. For cumulative distributions we performed a Kolmogorov–Smirnov (KS) test. p values ≤0.05 were considered significant.
Results
Homeostatic plasticity is crucial for preventing hypoactivity or hyperactivity within neocortical networks. Nuclear CaMKIV is a key signaling element in homeostatic scaling of excitatory synapses, but it is not known whether it concurrently induces other forms of homeostatic plasticity. Here we bidirectionally manipulated nuclear CaMKIV signaling in individual pyramidal neurons in vitro and assayed for induction of three major forms of homeostatic plasticity—excitatory and inhibitory synaptic scaling and intrinsic excitability. This approach allowed us to assess whether CaMKIV signaling coordinates the induction of multiple homeostatic mechanisms.
Cell-autonomous, bidirectional manipulation of nuclear CaMKIV signaling
CaMKIV resides predominantly in the nucleus (Jensen et al., 1991; Nakamura et al., 1995) where it regulates transcription (Bito et al., 1996; Impey et al., 2002). To reduce nuclear CaMKIV signaling we used a dnCaMKIV, containing three mutations in its catalytic and autoinhibitory domains that render it unphosphorylatable and kinase-dead (Lemrow et al., 2004; Wayman et al., 2011). To enhance nuclear CaMKIV signaling we used a constitutively active CaMKIV (caCaMKIV) containing a mutation in its autoinhibitory domain, thereby allowing activity in the absence of Ca2+/CaM-binding (Wayman et al., 2011). To specifically manipulate nuclear CaMKIV signaling (and avoid off-target effects due to interaction with other CaMK pathways outside the nucleus) all constructs used throughout this study had a NLS and were GFP-tagged. We first verified the nuclear expression pattern of these CaMKIV mutants by transfecting cultured neocortical pyramidal neurons with GFP alone (no NLS), GFP-dnCaMKIV-NLS (dn), GFP-NLS (EV), or GFP-caCaMKIV-NLS (ca) and measured the cytosolic-to-nuclear GFP signal; neurons were cotransfected with mCherry to identify neuronal morphology. Whereas GFP without an NLS was approximately evenly distributed between soma and nucleus, GFP-NLS, dnCaMKIV-NLS, and caCaMKIV-NLS expression was largely restricted to the nucleus (Fig. 1A,B; GFP n = 8, 0.86 ± 0.06, NLS n = 9, 0.09 ± 0.02; p < 0.001; NLS conditions were not different so were combined). To verify that these CaMKIV constructs were able to increase (caCaMKIV) or decrease (dnCaMKIV) nuclear CaMKIV signaling, we monitored CREB phosphorylation levels 24 h after transfection (using soluble GFP as a marker of pyramidal morphology), using a phosphospecific antibody that recognizes P-Ser-133. The nuclear Phospho-CREB signal was significantly decreased (dnCaMKIV) or increased (caCaMKIV) compared with EV. EV transfected and untransfected neurons were not different so here and in subsequent experiments were combined into one control group (Fig. 1C,D; dn n = 20, 0.64 ± 0.08, ctrl n = 47, 1 ± 0.11; ca n = 27, 5.39 ± 1.37; p < 0.001). These data verify that our CaMKIV constructs remain in the nucleus and effectively alter CaMKIV signaling in a cell-autonomous manner.
Figure 1.

Cell-autonomous, bidirectional manipulation of nuclear CaMKIV signaling. A, Example pyramidal neurons transfected for 24 h with NLS tagged construct (top) or soluble GFP without NLS (bottom). The fluorescent signal of GFP (green) and DAPI (magenta) are shown. Scale bar, 25 μm; white arrows indicate nucleus. B, Quantification of the cytoplasmic-to-nuclear fluorescent intensity ratio for NLS and non-NLS-expressing neurons (GFP n = 8, 0.86 ± 0.06; NLS n = 9, 0.09 ± 0.02; t test, p < 0.001). C, Example pyramidal neurons transfected for 24 h with dnCaMKIV (dn), control (ctrl) or caCaMKIV (ca), along with soluble GFP to visualize cell morphology; fixed and stained with an antibody against endogenous phospho-CREB Ser133 (grayscale). The fluorescent signal of GFP (green) and DAPI (magenta) are also shown. D, Quantification of the nuclear fluorescent intensity of phospho-CREB Ser133 (normalized to ctrl; dn, n = 20, 0.64 ± 0.08; ctrl, n = 47, 1 ± 0. 11; ca, n = 27, 5.39 ± 1.37; *p < 0.05, ANOVA followed by post hoc Games–Howell test).
Excitatory synaptic strength is bidirectionally regulated by CaMKIV activity
Previous work from our lab showed that synaptic scaling up is mediated through a drop in somatic calcium, reduced activation of CaMKIV, and an increase in transcription (Ibata et al., 2008). However, this and other studies implicating CaMKIV signaling in synaptic scaling used manipulations that were not restricted to the nucleus (Ibata et al., 2008; Goold et al., 2010; Pratt et al., 2011). To unambiguously determine the role of nuclear CaMKIV in synaptic scaling we transfected cortical cultures at low efficiency for 24 h with dnCaMKIV, EV, or caCaMKIV. All experiments were performed on neurons with a pyramidal morphology as described previously (Rutherford et al., 1998; Watt et al., 2000; Pratt et al., 2003). We recorded AMPA-mediated miniature EPSCs (mEPSCs) from transfected neurons in each condition as described previously (Turrigiano et al., 1998; Thiagarajan et al., 2005; Sun and Turrigiano, 2011). The amplitude of mEPSCs were significantly larger in dnCaMKIV-expressing neurons, and significantly smaller in caCaMKIV-expressing neurons compared with sister controls (Fig. 2A,B,E; dn n = 20, 16.7 ± 1 pA; ctrl n = 3 8, 13.2 ± 0.3 pA; ca n = 14, 11.6 ± 0.5 pA; p < 0.001). Analysis of the cumulative histogram of mEPSC amplitudes showed that these distributions were shifted significantly to the right for dnCaMKIV (Fig. 2D; p < 0.001, KS test), and to the left for caCaMKIV (Fig. 2D; p < 0.05, KS test). There were no significant differences in the mean (Fig. 2C) or distribution (KS test, p > 0.05) of mEPSC frequencies, or in passive neuronal properties or mEPSC kinetics between conditions (Table 1; Fig. 2C; p > 0.05, NS for all parameters). Together, these data show that nuclear CaMKIV signaling bidirectionally regulates the quantal amplitude of excitatory synapses.
Figure 2.

Excitatory synaptic strength is bidirectionally regulated by CaMKIV cell-autonomous modulation of CaMKIV signaling. A, Representative raw recordings of whole-cell AMPA mediated mEPSCs from pyramidal neurons expressing (top) dnCaMKIV, (middle) control (UT), and (bottom) caCaMKIV. B, Average mEPSC waveform for each condition. C, Peak-scaled average mEPSCs waveforms for each condition, overlaid to illustrate average kinetics. D, Cumulative histograms of mEPSC amplitudes (dn p < 0.001; ca p < 0.05, KS test). E, Mean mEPSC amplitude (dn, n = 20, 16.7 ± 1 pA; ctrl, n = 38, 13.2 ± 0.3 pA; ca, n = 14, 11.6 ± 0.5 pA; *p < 0.05 ANOVA followed by post hoc Games–Howell test). F, Mean frequency (dn, 0.56 ± 0.05 Hz; ctrl, 0.62 ± 0.07 Hz; ca, 0.42 ± 0.07 Hz; ANOVA, p = 0.21).
Table 1.
Passive neuronal properties
| Property | dn | ctrl | ca |
|---|---|---|---|
| Vm | 59.7 ± 0.4 | 61.1 ± 0.5 | 59.3 ± 0.5 |
| Rin, MΩ | 263.2 ± 15.3 | 263.1 ± 15.1 | 238.4 ± 16.7 |
| mEPSC decay, τ | 3.61 ± 0.2 | 3.91 ± 0.2 | 3.97 ± 0.2 |
| mEPSC rise, ms | 0.69 ± 0.02 | 0.70 ± 0.03 | 0.72 ± 0.03 |
| mIPSC decay, τ | 19.75 ± 1.1 | 18.76 ± 0.82 | 19.28 ± 0.86 |
| mIPSC rise, ms | 1.86 ± 0.10 | 1.86 ± 0.23 | 1.47 ± 0.09 |
Resting potential (Vm), resting input resistance (Rin) mEPSC, and mIPSC rise and decay time (exponential fit) for all experimental conditions. None of the differences across conditions were significant (ANOVA, p > 0.05).
Inhibitory synaptic strength is not affected by cell-autonomous changes in CaMKIV signaling
The amplitude and frequency of miniature IPSCs (mIPSCs) are also homeostatically regulated; they are reduced by activity-deprivation (Kilman et al., 2002; Hartman et al., 2006) and enhanced when firing is elevated (Pribiag et al., 2014). Thus, manipulations of neuronal activity have opposite effects on mEPSCs and mIPSCs, but whether these two forms of synaptic homeostasis are driven by the same upstream signaling pathways is unclear. To determine whether CaMKIV signaling coordinately regulates excitation and inhibition we recorded mIPSCs using the same CaMKIV manipulations described above. Surprisingly, we found no significant changes in inhibitory quantal events in neurons with altered CaMKIV signaling (Fig. 3A–C; dn n = 18, 21.4 ± 0.9 pA; ctrl n = 15, 21.6 ± 1 pA; ca n = 11, 11.6 ± 2 pA; p = 1). Similarly, there were no significant differences in frequency (dn, n = 18, 0.8 ± 0.2 Hz; ctrl, n = 15, 1.0 ± 0.4 Hz; ca, n = 11, 0.6 ± 0.3 Hz; p = 0.6), passive properties (Table 1), or kinetics of the events (Fig. 3D). Thus inhibitory synaptic scaling is mediated through an independent signaling pathway from excitatory synaptic scaling.
Figure 3.

Inhibitory synaptic strength is unaffected by cell-autonomous modulation of CaMKIV signaling. A, Representative raw recordings of whole-cell mIPSCs from pyramidal neurons expressing (top) dnCaMKIV, (middle) control (UT), and (bottom) caCaMKIV. B, Mean mIPSC amplitude (dn, n = 18, 21.4 ± 0.9 pA; ctrl, n = 15, 21.6 ± 1 pA; ca, n = 11, 21.6 ± 2 pA; ANOVA, p = 1). C, Average mIPSC waveforms for each condition. D, Peak scaled average mIPSCs waveforms for each condition to illustrate average kinetics.
Intrinsic excitability is bidirectionally regulated by CaMKIV signaling
In addition to synaptic scaling, chronic perturbations in firing also induce homeostatic changes in intrinsic excitability (Turrigiano et al., 1994; Desai et al., 1999; Nataraj et al., 2010). Regulation of intrinsic excitability can be achieved through a variety of mechanisms, such as relocation of the axon initial segment or changes to inward and outward current densities (Golowasch et al., 1999; Nelson et al., 2005; Grubb and Burrone, 2010). Despite the fact that the same activity-manipulations trigger both synaptic and intrinsic plasticity it is unknown whether these two forms of homeostatic compensation are mediated through the same upstream signaling pathways, which then target both ligand and voltage-gated ion channels.
To test whether CaMKIV signaling coordinately regulates excitatory synaptic scaling and intrinsic excitability, we manipulated CaMKIV signaling as described above, and measured the frequency of action potential firing elicited by dc current injections to generate firing frequency versus current (f–I) curves; synaptic transmission was blocked with DNQX, APV, and picrotoxin in these experiments. We found an increase in firing for dnCaMKIV transfected neurons (Fig. 4A, blue traces) relative to control neurons (black traces). This was reflected in a leftward shift in the f–I curve which was especially prominent at lower current steps; the apparent convergence of the f–I curves at higher current steps was due to pronounced spike inactivation of dnCaMKIV neurons at current steps >100 pA (Fig. 4A, top right trace). In addition, there was a decrease of ∼60% in the threshold (rheobase) current for evoking action potentials (Fig. 4B, inset). Thus reducing CaMKIV signaling significantly enhances intrinsic excitability of neocortical pyramidal neurons. Conversely, caCaMKIV expression produced a rightward and downward shift in the f–I curve, particularly prominent at higher current steps (Fig. 4A,B). Although there was a trend toward an increased rheobase, this difference was not significant (Fig. 4B, inset). The overall bidirectional changes in firing properties observed in response to changes in CaMKIV activity were not accompanied by any significant differences in passive membrane properties (as confirmed in other recording conditions; Table 1), suggesting that CaMKIV modulates voltage-dependent conductances to regulate intrinsic excitability. Together, these data show that CaMKIV signaling can bidirectionally regulate intrinsic excitability in tandem with excitatory quantal amplitude.
Figure 4.
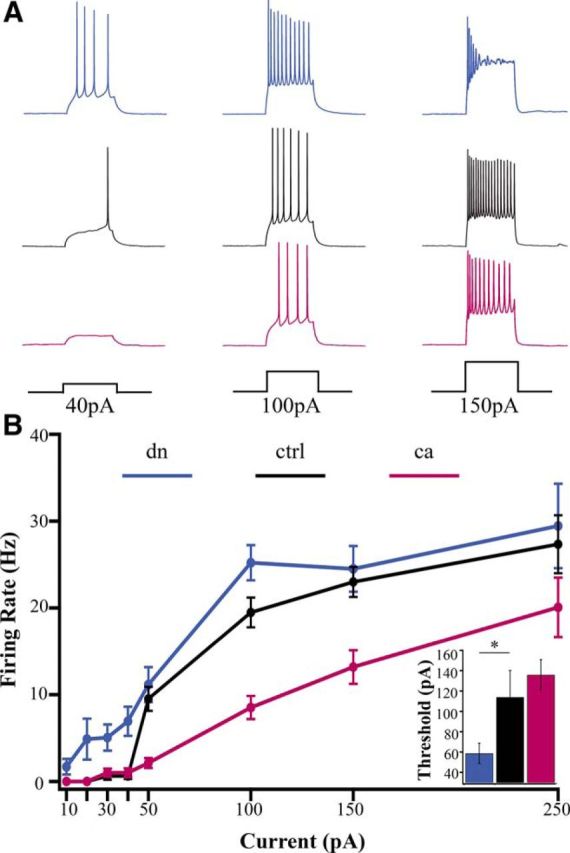
Intrinsic excitability is bidirectionally regulated by CaMKIV signaling. A, Examples of responses evoked by current steps of 40, 100, and 150 pA, from neurons expressing (top) dnCaMKIV, (middle) control (EV), and (bottom) caCaMKIV. B, Average f–I curves for the three conditions. Inset, Average rheobase (dn, n = 17, 58.8 ± 9.9 pA; ctrl, n = 10, 114.0 ± 26.2 pA; ca, n = 22, 135.9 ± 14.9 pA; *p < 0.05, ANOVA followed by post hoc Tukey test).
Spontaneous firing rate is bidirectionally regulated by CaMKIV activity
Cortical neurons in our culture system form functioning excitatory and inhibitory networks that generate spontaneous electrical activity (Turrigiano and Nelson, 2004). Homeostatic plasticity in these networks is implemented through a combination of synaptic and intrinsic mechanisms to stabilize their firing output (Nelson and Turrigiano, 2008; Feldman, 2009). Our findings thus far demonstrate a bidirectional and coordinated change in both intrinsic excitability and excitatory synaptic strength. The implication of such cooperative changes (with no change in inhibition) is that basal firing rates should also change in a cell-autonomous manner. To determine whether manipulating CaMKIV signaling is able to adjust spontaneous firing rates we recorded spontaneous firing from neurons transfected with either dnCaMKIV or caCaMKIV constructs. We compared this to firing of nearby control neurons in the same cultures, under conditions in which synaptic and network activity was intact. We found that dnCaMKIV expression increased spontaneous firing to ∼260% of sister control neurons (Fig. 5A,B). In contrast, caCaMKIV expression decreased spontaneous firing frequency to ∼30% of sister control neurons (Fig. 5C,D). As previously observed in other recording conditions, these changes in spontaneous firing were not accompanied by any significant change in membrane potential or input resistance. Together, these data show that directly manipulating nuclear CaMKIV signaling is able to induce bidirectional changes in basal neuronal firing rates.
Figure 5.

Spontaneous firing rate is bidirectionally regulated by CaMKIV signaling. A, Examples of spontaneous firing recorded from control (black) and dnCaMKIV (blue) neurons from the same dish. B, Average firing frequency for sister dnCaMKIV transfected or control neurons, normalized to control (ctrl, n = 21, 100 ± 23.7%; dn, n = 27, 262.7 ± 66.1%; t test, p < 0.05), individual data points plotted in green circles. C, Examples of spontaneous firing recorded from control (black) and caCaMKIV (magenta) neurons from the same dish. D, Average firing frequency for sister caCaMKIV transfected or control neurons, normalized to control (ctrl, n = 28, 100 ± 24.0%; ca, n = 18, 27.9 ± 7.1%; t test, p < 0.05), individual data points plotted in green circles.
Discussion
The stability of neuronal firing is maintained by a set of homeostatic mechanisms that include reciprocal synaptic scaling of excitatory and inhibitory synapses and changes in intrinsic neuronal excitability. Although much progress has been made in uncovering the signaling pathways underlying excitatory synaptic scaling, the other two mechanisms remain poorly explored, and in particular it was not known whether the same upstream sensors of perturbed activity were able to trigger all three mechanisms in tandem, or whether each mechanism uses unique sensors and effectors. Here we show that a key activity sensor and effector of excitatory synaptic scaling, CaMKIV, is able to induce excitatory synaptic scaling and homeostatic intrinsic excitability changes in parallel. In contrast, cell-autonomous manipulations of CaMKIV did not induce inhibitory synaptic scaling. Thus, excitatory synaptic scaling and intrinsic excitability are tightly coordinated, whereas inhibitory synaptic scaling is sensed and regulated through an independent control mechanism.
Like synaptic scaling, pyramidal neuron intrinsic excitability is bidirectionally modulated by manipulations of activity (Desai et al., 1999; Driscoll et al., 2013; Lee et al., 2015). Here we show for the first time that manipulations of nuclear CaMKIV signaling bidirectionally coregulate synaptic scaling and intrinsic excitability. However, the effects of increasing and decreasing CaMKIV signaling on the f–I curves were not completely symmetrical. Increasing CaMKIV signaling shifts the f–I curve down and to the right without much impact on current threshold, as described for chronic elevations in activity (Driscoll et al., 2013; Lee et al., 2015). In contrast, unlike activity blockade or sensory deprivation, which shift the f–I curve uniformly to the left (Desai et al., 1999; Lambo and Turrigiano, 2013), the effects of reduced CaMKIV signaling are most pronounced at low current levels. This nonuniform effect on the f–I curve is likely due to the tendency of these neurons to undergo very pronounced spike inactivation at higher current values (Fig. 4A), so that they cannot sustain high firing rates. Such spike inactivation would be expected if dnCaMKIV strongly downregulates K+ channels necessary for spike repolarization, as activity-blockade is known to do (Driscoll et al., 2013; Lee et al., 2015); it is possible that dnCaMKIV is more effective at reducing CaMKIV activation than activity-blockade, and so has a more powerful effect on channel expression. Alternatively, it may be that not all effects of activity-blockade on intrinsic excitability are mediated through the CaMKIV pathway. The coinduction of intrinsic and excitatory synaptic plasticity by manipulations of CaMKIV raises the interesting question of whether these two forms of homeostatic plasticity are purely redundant, or whether they might cooperate to stabilize distinct aspects of neuronal firing (Turrigiano, 2012; Cannon and Miller, 2016).
Intriguingly, inhibitory synaptic scaling was not induced by cell-autonomous manipulations of CaMKIV signaling. This is especially interesting because the mean firing of these manipulated neurons was strongly and bidirectionally modulated by CaMKIV (Fig. 5). This implies that, unlike excitatory synaptic scaling (Ibata et al., 2008; Goold and Nicoll, 2010; but see Fong et al., 2015), inhibitory synaptic scaling is not induced by changes in firing of an individual postsynaptic neuron. This result is at odds with recent studies showing that cell-autonomous suppression of firing in visual cortical pyramidal neurons reduced evoked inhibition (Xue et al., 2014), and that mIPSC frequency was rapidly increased by elevating postsynaptic firing (Peng et al., 2010). On the other hand, our results are consistent with other experiments showing that, whereas global suppression of firing efficiently induced inhibitory synaptic scaling, suppression of firing in individual presynaptic or postsynaptic neurons did not (Hartman et al., 2006; Peng et al., 2010). What accounts for these discrepancies between studies is not clear, but it is possible that distinct forms of inhibitory plasticity are being induced in different studies due to differences in preparation, time course, or extremity of the manipulation; for example, strong hyperpolarization and complete suppression of firing (Xue et al., 2014) versus a 70% reduction in firing here. In any case, our data show that cell-autonomous manipulations that can induce bidirectional excitatory synaptic scaling are not sufficient to induce inhibitory synaptic scaling.
This uncoupling of the signaling pathways that mediate excitatory and inhibitory synaptic scaling suggests that homeostatic regulation of inhibition responds to distinct activity-perturbations and likely controls distinct aspects of circuit function and homeostasis. This is further emphasized by the heterogeneity of neocortical GABAergic cell types and their targets (Tremblay et al., 2016), and emerging evidence that the transcriptional pathways that homeostatically regulate inhibition are cell-type-specific (Bloodgood et al., 2013; Spiegel et al., 2014). One important signaling pathway known to mediate inhibitory homeostasis is the activity-dependent production and release of brain-derived neurotrophic factor (BDNF), which enhances inhibitory tone onto pyramidal neurons (Rutherford et al., 1997, 1998; Peng et al., 2010; Bloodgood et al., 2013; Saiepour et al., 2015). However, the extent to which BDNF serves as a diffusible measure of circuit-wide changes in activity, as opposed to a locally released signal that mediates local synaptic changes, remains controversial (Edelmann et al., 2014). Further, BDNF may homeostatically regulate only some forms of pyramidal inhibition (Bloodgood et al., 2013). Better understanding of how different classes of GABAergic synapses sense and respond to perturbations in circuit activity (Maffei et al., 2004; Karayannis et al., 2012) will be critical in the future for understanding how inhibitory homeostasis interacts with and complements the cell-autonomous regulation of excitatory synaptic strengths and intrinsic excitability.
When the firing of neocortical neurons is chronically perturbed by activity or sensory deprivation, firing rates are restored to baseline over time (Turrigiano et al., 1998; Burrone and Murthy, 2001; Hengen et al., 2013; Slomowitz et al., 2015) and recently we found that individual pyramidal neurons in vivo return very precisely to an individual, presumably cell-autonomous, firing rate set point (Hengen et al., 2016). An important unanswered question is how these individual set points are generated and maintained. The data we present here suggest that CaMKIV is one key signaling element that contributes to the maintenance of this set point. Under normal conditions, changes in firing alter the activation state of nuclear CaMKIV (Ibata et al., 2008); these then induce bidirectional changes in mEPSC amplitude and intrinsic excitability, and these changes are in the correct direction to restore firing in response to these activity-perturbations. If CaMKIV indeed contributes to the maintenance of a firing rate set point, then artificially changing the coupling between calcium influx and CaMKIV signaling should generate an error signal that is predicted to move the firing rate to a new value (Turrigiano, 2008), as we observe here. Together, our results suggest that manipulating CaMKIV signaling might be an effective means of adjusting the firing rate set point of neocortical neurons.
Footnotes
This work was supported by NIH Grants R37NS092635 and P01NS079419 to G.G.T.
The authors declare no competing financial interests.
References
- Benevento M, Iacono G, Selten M, Ba W, Oudakker A, Frega M, Keller J, Mancini R, Lewerissa E, Kleefstra T, Stunnenberg HG, Zhou H, van Bokhoven H, Nadif Kasri N (2016) Histone methylation by the Kleefstra syndrome protein EHMT1 mediates homeostatic synaptic scaling. Neuron 91:341–355. 10.1016/j.neuron.2016.06.003 [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87:1203–1214. 10.1016/S0092-8674(00)81816-4 [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sharma N, Browne HA, Trepman AZ, Greenberg ME (2013) The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503:121–125. 10.1038/nature12743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Murthy VN (2001) Synaptic plasticity: rush hour traffic in the AMPA lanes. Curr Biol 11:R274–R277. 10.1016/S0960-9822(01)00137-3 [DOI] [PubMed] [Google Scholar]
- Cannon J, Miller P (2016) Synaptic and intrinsic homeostasis cooperate to optimize single neuron response properties and tune integrator circuits. J Neurophysiol 116:2004–2022. 10.1152/jn.00253.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla S, Hardingham GE, Quinn DR, Bading H (1998) CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science 281:1505–1509. 10.1126/science.281.5382.1505 [DOI] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I (2001) Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol 63:847–869. 10.1146/annurev.physiol.63.1.847 [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG (1999) Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci 2:515–520. 10.1038/9165 [DOI] [PubMed] [Google Scholar]
- Driscoll HE, Muraro NI, He M, Baines RA (2013) Pumilio-2 regulates translation of Nav1.6 to mediate homeostasis of membrane excitability. J Neurosci 33:9644–9654. 10.1523/JNEUROSCI.0921-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Lüders HO, Shi J, Cui J, Richerson GB, Wang QK (2005) Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet 37:733–738. 10.1038/ng1585 [DOI] [PubMed] [Google Scholar]
- Edelmann E, Lessmann V, Brigadski T (2014) Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 76:610–627. 10.1016/j.neuropharm.2013.05.043 [DOI] [PubMed] [Google Scholar]
- Feldman DE. (2009) Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci 32:33–55. 10.1146/annurev.neuro.051508.135516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19:1031–1047. 10.1016/S0896-6273(00)80395-5 [DOI] [PubMed] [Google Scholar]
- Fischer-Fantuzzi L, Vesco C (1988) Cell-dependent efficiency of reiterated nuclear signals in a mutant simian virus 40 oncoprotein targeted to the nucleus. Mol Cell Biol 8:5495–5503. 10.1128/MCB.8.12.5495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong MF, Newman JP, Potter SM, Wenner P (2015) Upward synaptic scaling is dependent on neurotransmission rather than spiking. Nat Commun 6:6339. 10.1038/ncomms7339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golowasch J, Casey M, Abbott LF, Marder E (1999) Network stability from activity-dependent regulation of neuronal conductances. Neural Comput 11:1079–1096. 10.1162/089976699300016359 [DOI] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA (2010) Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron 68:512–528. 10.1016/j.neuron.2010.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J (2010) Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465:1070–1074. 10.1038/nature09160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman KN, Pal SK, Burrone J, Murthy VN (2006) Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat Neurosci 9:642–649. 10.1038/nn1677 [DOI] [PubMed] [Google Scholar]
- Hengen KB, Lambo ME, Van Hooser SD, Katz DB, Turrigiano GG (2013) Firing rate homeostasis in visual cortex of freely behaving rodents. Neuron 80:335–342. 10.1016/j.neuron.2013.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengen KB, Torrado Pacheco A, McGregor JN, Van Hooser SD, Turrigiano GG (2016) Neuronal firing rate homeostasis is inhibited by sleep and promoted by wake. Cell 165:180–191. 10.1016/j.cell.2016.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG (2008) Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57:819–826. 10.1016/j.neuron.2008.02.031 [DOI] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH (2002) Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron 34:235–244. 10.1016/S0896-6273(02)00654-2 [DOI] [PubMed] [Google Scholar]
- Jensen KF, Ohmstede CA, Fisher RS, Sahyoun N (1991) Nuclear and axonal localization of Ca2+/calmodulin-dependent protein kinase type Gr in rat cerebellar cortex. Proc Natl Acad Sci U S A 88:2850–2853. 10.1073/pnas.88.7.2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayannis T, De Marco García NV, Fishell GJ (2012) Functional adaptation of cortical interneurons to attenuated activity is subtype-specific. Front Neural Circuits 6:66. 10.3389/fncir.2012.00066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG (2002) Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. J Neurosci 22:1328–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Waxman SG (2010) Neurological channelopathies: new insights into disease mechanisms and ion channel function. J Physiol 588:1823–1827. 10.1113/jphysiol.2010.190652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambo ME, Turrigiano GG (2013) Synaptic and intrinsic homeostatic mechanisms cooperate to increase L2/3 pyramidal neuron excitability during a late phase of critical period plasticity. J Neurosci 33:8810–8819. 10.1523/JNEUROSCI.4502-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Royston SE, Vest MO, Ley DJ, Lee S, Bolton EC, Chung HJ (2015) N-methyl-d-aspartate receptors mediate activity-dependent down-regulation of potassium channel genes during the expression of homeostatic intrinsic plasticity. Mol Brain 8:4. 10.1186/s13041-015-0094-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemrow SM, Anderson KA, Joseph JD, Ribar TJ, Noeldner PK, Means AR (2004) Catalytic activity is required for calcium/calmodulin-dependent protein kinase IV to enter the nucleus. J Biol Chem 279:11664–11671. 10.1074/jbc.M312613200 [DOI] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG (2004) Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci 7:1353–1359. 10.1038/nn1351 [DOI] [PubMed] [Google Scholar]
- Marder E, Prinz AA (2002) Modeling stability in neuron and network function: the role of activity in homeostasis. Bioessays 24:1145–1154. 10.1002/bies.10185 [DOI] [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS (1994) Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol 14:6107–6116. 10.1128/MCB.14.9.6107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Okuno S, Sato F, Fujisawa H (1995) An immunohistochemical study of Ca2+/calmodulin-dependent protein kinase IV in the rat central nervous system: light and electron microscopic observations. Neuroscience 68:181–194. 10.1016/0306-4522(95)00092-W [DOI] [PubMed] [Google Scholar]
- Nataraj K, Le Roux N, Nahmani M, Lefort S, Turrigiano G (2010) Visual deprivation suppresses L5 pyramidal neuron excitability by preventing the induction of intrinsic plasticity. Neuron 68:750–762. 10.1016/j.neuron.2010.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Gittis AH, du Lac S (2005) Decreases in CaMKII activity trigger persistent potentiation of intrinsic excitability in spontaneously firing vestibular nucleus neurons. Neuron 46:623–631. 10.1016/j.neuron.2005.04.009 [DOI] [PubMed] [Google Scholar]
- Nelson SB, Turrigiano GG (2008) Strength through diversity. Neuron 60:477–482. 10.1016/j.neuron.2008.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YR, Zeng SY, Song HL, Li MY, Yamada MK, Yu X (2010) Postsynaptic spiking homeostatically induces cell-autonomous regulation of inhibitory inputs via retrograde signaling. J Neurosci 30:16220–16231. 10.1523/JNEUROSCI.3085-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Watt AJ, Griffith LC, Nelson SB, Turrigiano GG (2003) Activity-dependent remodeling of presynaptic inputs by postsynaptic expression of activated CaMKII. Neuron 39:269–281. 10.1016/S0896-6273(03)00422-7 [DOI] [PubMed] [Google Scholar]
- Pratt KG, Zimmerman EC, Cook DG, Sullivan JM (2011) Presenilin 1 regulates homeostatic synaptic scaling through Akt signaling. Nat Neurosci 14:1112–1114. 10.1038/nn.2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H, Peng H, Shah WA, Stellwagen D, Carbonetto S (2014) Dystroglycan mediates homeostatic synaptic plasticity at GABAergic synapses. Proc Natl Acad Sci U S A 111:6810–6815. 10.1073/pnas.1321774111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG (1997) Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci 17:4527–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG (1998) BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 21:521–530. 10.1016/S0896-6273(00)80563-2 [DOI] [PubMed] [Google Scholar]
- Saiepour MH, Chakravarthy S, Min R, Levelt CN (2015) Competition and homeostasis of excitatory and inhibitory connectivity in the adult mouse visual cortex. Cereb Cortex 25:3713–3722. 10.1093/cercor/bhu245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaukowitch K, Reese AL, Kim SK, Kilaru G, Joo JY, Kavalali ET, Kim TK (2017) An intrinsic transcriptional program underlying synaptic scaling during activity suppression. Cell Rep 18:1512–1526. 10.1016/j.celrep.2017.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomowitz E, Styr B, Vertkin I, Milshtein-Parush H, Nelken I, Slutsky M, Slutsky I (2015) Interplay between population firing stability and single neuron dynamics in hippocampal networks. Elife 4. 10.7554/eLife.04378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, Greenberg ME (2014) Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 157:1216–1229. 10.1016/j.cell.2014.03.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz CC, Tatavarty V, Sugino K, Shima Y, Joseph A, Lin H, Rutlin M, Lambo M, Hempel CM, Okaty BW, Paradis S, Nelson SB, Turrigiano GG (2016) Upregulation of mu3A drives homeostatic plasticity by rerouting AMPAR into the recycling endosomal pathway. Cell Rep 16:2711–2722. 10.1016/j.celrep.2016.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Turrigiano GG (2011) PSD-95 and PSD-93 play critical but distinct roles in synaptic scaling up and down. J Neurosci 31:6800–6808. 10.1523/JNEUROSCI.5616-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW (2005) Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47:725–737. 10.1016/j.neuron.2005.06.037 [DOI] [PubMed] [Google Scholar]
- Tremblay R, Lee S, Rudy B (2016) GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91:260–292. 10.1016/j.neuron.2016.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. (2012) Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4:a005736. 10.1101/cshperspect.a005736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135:422–435. 10.1016/j.cell.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5:97–107. 10.1038/nrn1327 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391:892–896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- Turrigiano G, Abbott LF, Marder E (1994) Activity-dependent changes in the intrinsic properties of cultured neurons. Science 264:974–977. 10.1126/science.8178157 [DOI] [PubMed] [Google Scholar]
- Vale C, Sanes DH (2002) The effect of bilateral deafness on excitatory and inhibitory synaptic strength in the inferior colliculus. Eur J Neurosci 16:2394–2404. 10.1046/j.1460-9568.2002.02302.x [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG (2000) Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26:659–670. 10.1016/S0896-6273(00)81202-7 [DOI] [PubMed] [Google Scholar]
- Wayman GA, Tokumitsu H, Davare MA, Soderling TR (2011) Analysis of CaM-kinase signaling in cells. Cell Calcium 50:1–8. 10.1016/j.ceca.2011.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG (2005) Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci 25:2895–2905. 10.1523/JNEUROSCI.5217-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Atallah BV, Scanziani M (2014) Equalizing excitation-inhibition ratios across visual cortical neurons. Nature 511:596–600. 10.1038/nature13321 [DOI] [PMC free article] [PubMed] [Google Scholar]