

Abstract
Deep brain stimulation (DBS) of the subthalamic nucleus (STN) is the most common neurosurgical treatment for Parkinson's disease motor symptoms. In preclinical models, STN DBS provides neuroprotection for substantia nigra (SN) dopamine neurons and increases BDNF in the nigrostriatal system and primary motor cortex. However, whether BDNF signaling in the SN participates in the neuroprotective effects of DBS remains unknown. We demonstrate that STN DBS in male rats activates signaling downstream of tropomyosin receptor kinase type B (trkB), namely, phosphorylation of Akt and ribosomal protein S6, in SN neurons. Long-term trkB blockade abolished STN DBS-mediated neuroprotection of SN neurons following progressive 6-hydroxydopamine lesion and was associated with decreased phosphorylated ribosomal protein S6 immunoreactivity. Acute trkB blockade in rats with stable nigrostriatal denervation attenuated the forelimb akinesia improvement normally induced by STN DBS. These results suggest that STN DBS increases BDNF-trkB signaling to contribute to the neuroprotective and symptomatic efficacy of STN DBS.
SIGNIFICANCE STATEMENT Subthalamic nucleus deep brain stimulation (STN DBS) is increasingly used in mid- to late-stage Parkinson's disease (PD) but with an incomplete knowledge of its molecular mechanisms. STN DBS is neuroprotective against neurotoxicants in animal models and increases BDNF. This study is the first to show that BDNF signaling through the cognate tropomyosin receptor kinase type B (trkB) receptor occurs in substantia nigra pars compacta neurons and is required for neuroprotection. In addition, blockade of trkB unexpectedly reduced the functional benefit of STN DBS on a short timescale that is inconsistent with canonical trkB signaling pathways, suggesting a noncanonical role for trkB in STN DBS-mediated behavioral effects. Together, these data implicate trkB signaling in the symptomatic efficacy and disease-modifying potential of STN DBS.
Keywords: AKT, ANA-12, BDNF, neuroprotection, ribosomal protein S6, STN DBS
Introduction
Deep brain stimulation (DBS) is currently the gold-standard neurosurgical therapy for Parkinson's disease (PD) with the subthalamic nucleus (STN) as the most commonly targeted and studied site. Apart from its symptomatic benefits, STN DBS has been investigated as a disease-modifying therapy, although these clinical studies arrived at opposing conclusions, presumably because they exclusively used late-stage PD subjects (Krack et al., 2003; Rodriguez-Oroz et al., 2004, 2005; Hilker et al., 2005; Tagliati et al., 2010). However, it is well established that the overwhelming majority of dopamine (DA) neurons of the substantia nigra pars compacta (SNpc) and dopaminergic (DAergic) innervation to the putamen already have succumbed to degeneration at later stages of the disease (Cheng et al., 2010; Kordower et al., 2013). As a result, understanding potential disease-modifying effects of DBS is an important step in justifying early-stage PD interventions when a larger portion of the SNpc can be saved.
Preclinical studies in parkinsonian animal models can provide insight into the disease-modifying potential of STN DBS. STN DBS or lesions of the STN can prevent SNpc DA neuron loss when initiated before or following 6-hydroxydopamine (6-OHDA) or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in rodents and nonhuman primates in addition to showing clinically relevant alleviation of motor impairments (Piallat et al., 1996; Meissner et al., 2002; Chang et al., 2003; Darbaky et al., 2003; Maesawa et al., 2004; Shi et al., 2004, 2006; Fang et al., 2006; Temel et al., 2006; Vlamings et al., 2007; Wallace et al., 2007; Harnack et al., 2008; Spieles-Engemann et al., 2010a, b, 2011). However, the mechanism for STN DBS-mediated neuroprotection in preclinical models of PD is currently unknown. Identification of the neuroprotective mechanism(s) in preclinical PD models would inform a strategy to optimize the disease-modifying potential of STN DBS in the parkinsonian brain.
Evidence suggests a role for BDNF in the neuroprotection associated with STN DBS in preclinical models. Previously, our laboratory demonstrated that STN DBS increases BDNF in the nigrostriatal system and the primary motor (M1) cortex in a stimulation-dependent manner (Spieles-Engemann et al., 2009, 2011), and a role for BDNF has been revealed in other studies examining high-frequency stimulation of other brain circuitries (Friedman et al., 2009; Gersner et al., 2010; Hamani et al., 2012). STN DBS likely increases BDNF release in an activity-dependent manner, similar to that observed with high-frequency stimulation in vitro (Hartmann et al., 2001; Matsuda et al., 2009). Activity-dependent release of BDNF promotes survival, growth, and synaptic plasticity via activation of its high-affinity receptor tropomyosin-related kinase type B (trkB) (Yoshii and Constantine-Paton, 2010; Lewin et al., 2014). trkB activation of the mitogen-activated protein kinase/extracellular signal related-kinase (MAPK/ERK) cascade or the phosphatidylinositol 3-kinase/Akt (PI3K/Akt) cascade is linked to survival signaling (Cobb and Cobb, 1999; Airaksinen et al., 2002; Yoshii and Constantine-Paton, 2010; Lewin et al., 2014). Therefore, it is reasonable to suggest that an increase in BDNF-trkB survival signaling could promote the survival of SNpc neurons.
Using our long-term stimulation platform and our well-characterized 6-OHDA rat model of PD (Spieles-Engemann et al., 2010b), we examined SNpc neurons in situ for evidence supporting stimulation-dependent trkB activation. Phosphorylation of both Akt (protein kinase B) and ribosomal protein S6 (rpS6) phosphorylation (p-rpS6), downstream readouts of BDNF-trkB signaling through the converging pathways PI3K-Akt-mTOR and ras-ERK-mTOR (Mendoza et al., 2011), was significantly increased in SNpc neurons receiving chronic stimulation. Using long-term treatment with the potent and specific trkB receptor antagonist N-[2-[[(Hexahydro-2-oxo-1H-azepin-3-yl)amino]carbonyl]phenyl]benzo[b]thiophene-2-carboxamide (ANA-12) (Cazorla et al., 2011) concurrently with stimulation, we then examined whether trkB antagonism attenuated STN DBS-mediated neuroprotection. Furthermore, we examined whether acute trkB blockade attenuates the ability of STN DBS to reverse forelimb akinesia induced by unilateral nigrostriatal denervation. Our data implicate trkB signaling in both the symptomatic and disease-modifying potential of STN DBS.
Materials and Methods
Experimental design.
In Experiment 1, rats underwent surgery for STN electrode implantation only (i.e., no 6-OHDA administration) and received either stimulation or no stimulation starting 1 week after implantation for a period of 1 week until death on day 14. In Experiment 2, all rats underwent intrastriatal 6-OHDA neurotoxicant lesioning and electrode placement into the STN during the same surgical session. Stimulation and drug (or vehicle) administration were both begun on the 10th day after surgery and continued until the end of the experiment. A subset of rats underwent behavioral testing on days 26 and 27 when “on” or “off” stimulation, respectively. Rats were sacrificed on day 28. In Experiment 3, all rats were administered 6-OHDA and implanted with electrodes during the same surgical session, and the lesion was allowed to progress until stable at 6 weeks (Spieles-Engemann et al., 2010b) with the cylinder task used as detailed below. Timelines for all three experiments are depicted in Figures 1A, 2A, and 4A.
Figure 1.

Chronic stimulation increases phosphorylation of Akt and rpS6 in SNpc neurons in unlesioned rats. A, The timeline for Experiment 1 depicting electrode implantation surgery, stimulation, and death. SNpc of a stimulated, unlesioned rat ipsilateral to the electrode at high magnification, respectively, showing immunofluorescence for TH (B, C), p-rpS6 (F, G), and merged images (J, K). To control for the potential effects of unilateral electrode implantation, data were normalized to percentage expression relative to the identical hemisphere in Inactive rats. SNpc neuron immunoreactivity for Inactive and Active groups was quantified for TH, p-Akt, total Akt, and p-rpS6. No differences were observed in TH immunoreactivity due to stimulation (D, E, p = 0.186), though there was a trend for increased TH immunoreactivity in the contralateral hemisphere (E). STN DBS did result in a statistically significant increase in pAkt (H) and p-rpS6 (I) immunoreactivity in SNpc neurons (p < 0.04). Scale bars: low magnification, 200 μm; high magnification, 50 μm. Graphs represent mean ± SEM.*p < 0.05.
Figure 2.

ANA-12 does not affect SNpc lesion paradigm and abolishes STN DBS-mediated neuroprotection. A, The timeline for Experiment 2. All rats included in the present work were successfully implanted with electrodes in the STN as shown by Kluver–Barrera histochemistry at low (B) and high magnification (C). The percentages of remaining THir SNpc neurons as measured by unbiased stereology are shown (D) for 6-OHDA-injected rats with implanted but not activated electrodes receiving injections of Vehicle or ANA-12 from day 10 to day 28. There is no significant difference between the two groups (p = 0.260). The lesioned hemisphere for 6-OHDA-injected rats with activated electrodes receiving Vehicle or ANA-12 injections, respectively, from day 10 to day 28 at low magnification (E, H) and high magnification (F, I) compared with the intact hemisphere shown at high magnification (G, J). K, The percentages of remaining THir SNpc neurons as measured by unbiased stereology are shown for rats with Active electrodes. Active groups received either Vehicle or ANA-12, and there is a significant difference between them (p = 0.031). Scale bars: low magnification, 200 μm; high magnification, 50 μm. Graphs represent mean ± SEM.*p < 0.05.
Figure 4.
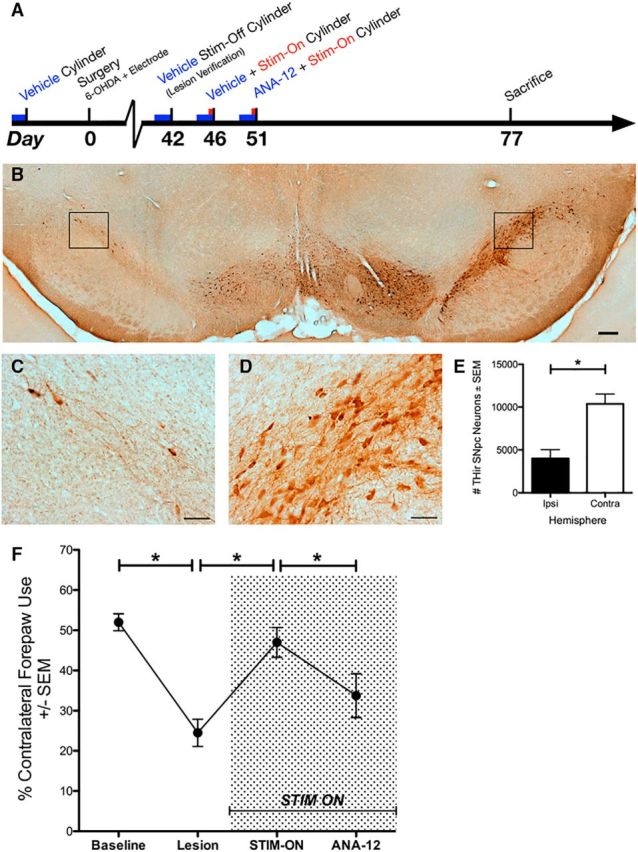
trkB signaling contributes to the functional efficacy of STN DBS. A, The timeline for Experiment 3. A stable lesion of the SNpc by 6 weeks is shown at low (B) and high magnification (C, D, for the lesioned and intact hemispheres, respectively) with a statistical difference in number of neurons on the lesioned/ipsilateral side (p < 0.001, E). F, Rats were assessed for forelimb asymmetery using the cylinder task before surgery with vehicle (Baseline), 6 weeks after intrastriatal 6-OHDA with vehicle (Lesion), receiving acute STN stimulation with vehicle (STIM-ON), and receiving stimulation with ANA-12 (ANA-12). Significant differences were observed for Baseline versus Lesion (p < 0.001), Lesion versus STIM-ON (p < 0.001), STIM-ON versus ANA-12 (p = 0.015), and Baseline versus ANA-12 (data not shown, p = 0.012). Scale bars: low magnification, 200 μm; high magnification, 50 μm. Graphs represent mean ± SEM.*p < 0.05.
Animals.
A total of 42, male, Sprague Dawley rats (Harlan, ∼250 g) were used in Experiments 1 (n = 6), 2 (n = 26), and 3 (n = 10). Rats were only included in the final analysis if they successfully completed the study with electrode placement adequately targeting the STN, as described previously (Spieles-Engemann et al., 2010a). Animals were allowed food and water ad libitum and were housed in reverse dark-light cycle conditions in the Association for Assessment and Accreditation of Laboratory Animal Care-approved facility at the Van Andel Research Institute. The Michigan State University Institutional Animal Care and Use Committee approved this study.
Electrode implantation.
All rats were implanted with stimulating electrodes. In Experiments 2 and 3, rats also received intrastriatal 6-OHDA injections before electrode implantation during the same surgical session. Rats were unilaterally implanted (ipsilateral to 6-OHDA injections) with a bipolar, concentric microelectrode (inner electrode projection 1.0 mm, inner insulated electrode diameter 0.15 mm, outer electrode gauge 26, Plastics One) targeted to the dorsal border of the STN (anteroposterior −3.4 mm, mediolateral 2.5 mm, relative to bregma and dorsoventral −7.7 mm, relative to the dura mater). The dorsal STN border placement site was selected to minimize damage to the nucleus, as has been described previously (Spieles-Engemann et al., 2010b). Burr holes were drilled in the skull; the electrode was fixed in place using bone screws and dental acrylic.
Intrastriatal 6-OHDA injections.
Intrastriatal 6-OHDA injections were conducted as described previously (Spieles-Engemann et al., 2010b). Rats were anesthetized before surgery with Equithesin (0.3 ml/100 g body weight i.p.; chloral hydrate 42.5 mg/ml + sodium pentobarbital 9.72 mg/ml). They received two unilateral, intrastriatal injections (anteroposterior 1.6 mm, mediolateral 2.4 mm, dorsoventral −4.2 mm and anteroposterior 0.2 mm, mediolateral 2.6 mm, dorsoventral −7.0 mm relative to bregma) of 6-OHDA (MP Biomedicals; 5.0 μg/μl 6-OHDA in 0.02% ascorbic acid, 0.9% saline solution, injection rate 0.5 μl/min, 2.0 μl per site). Drill holes were filled with gel foam or bone wax to prevent entry of cement during electrode placement. These 6-OHDA lesion parameters result in ∼50% loss of SNpc TH-immunoreactive (THir) neurons after 2 weeks that progresses to ∼75% loss after 4 weeks and is stable at 6 weeks (Spieles-Engemann et al., 2010b).
Continuous stimulation platform.
Rats received STN stimulation that was continuously delivered in a freely moving setup as previously described (Spieles-Engemann et al., 2010b). Stimulation was generated by an Accupulser Signal Generator (World Precision Instruments) via a battery-powered Constant Current Bipolar Stimulus Isolator (World Precision Instruments). Stimulation parameters consisted of a frequency of 130 Hz, a pulse width of 60 μs and an intensity of 30–50 μA. At the onset of stimulation, intensity settings were increased until orofacial or contralateral forepaw dyskinesias were observed to confirm stimulation delivery. Immediately following a positive dyskinetic response, the intensity was set below the lower limit of dyskinesias, such that no rat was functionally impaired by stimulation. When rats were not being stimulated, they were still physically connected within their specially designed stimulator cage to a commutator.
ANA-12 and vehicle preparation.
ANA-12 (catalog #4781, Tocris Bioscience) was dissolved in DMSO (Fisher Scientific BP231-1, lot #072871) and added to a solution of polyethylene glycol 400 (PEG-400, Fluka 81172, lot #BCBK4383V) and normal saline (Teknova, S5812, lot #S581204D1301) for a final solution of 5% DMSO, 40% normal saline, and 55% PEG-400. Vehicle was prepared under identical parameters, except for the addition of ANA-12. For each injection of ANA-12, the compound was dissolved and a new solution made immediately before use.
Experiment 2: behavioral testing, stimulation, and drug administration timeline.
Rats were randomly assigned to receive stimulation (Active) or not (Inactive). Spontaneous forelimb use was assessed using the cylinder task as described previously (Schallert and Tillerson, 2000; Schallert, 2006; Spieles-Engemann et al., 2011; Fischer et al., 2015). Other behavioral measures were not used due to their incompatibility with the external hardware required for continuous stimulation in an awake animal.
During the dark cycle, rats were placed in a clear Plexiglas cylinder and videotaped until 20, weight-bearing forelimb placements on the side of the cylinder occurred, or until a maximum trial time of 5 min had elapsed. To determine whether forelimb preference was present, the number of contralateral, ipsilateral, and simultaneous paw placements was quantified. Data are reported as the percentage of contralateral (to 6-OHDA and electrode) forelimb use: [(contralateral + ½ both)/(ipsilateral + contralateral + both)] × 100%. Rats with a unilateral nigrostriatal lesion will show a bias toward using the ipsilateral limb. A forelimb deficit was defined as possessing a minimum of a 20% relative reduction in contralateral forepaw use compared with baseline. Inclusion in this study required a forelimb deficit at the second behavioral task 6 weeks after surgery.
Within the Active and Inactive arms of Experiment 2, rats were randomly assigned to receive ANA-12 or vehicle twice per day (0.5 mg/kg, i.p.) (Cazorla et al., 2011; Ambrogini et al., 2013) from day 10 to day 28 after surgery, resulting in four total treatment groups: Active + ANA-12, Inactive + ANA-12, Active + Vehicle, Inactive + Vehicle.
Experiment 3: behavioral testing, stimulation, and drug administration timeline.
Within 2 h of the start of the dark cycle, rats received an injection of ANA-12 (0.5 mg/kg, i.p.) or vehicle and were immediately connected to the stimulation hardware. Stimulation was turned on 3.5–4.0 h after injection, and behavior was assessed using the cylinder task (described above) at least 4 h after injection (i.e., forelimb use was assessed after a 30 min stimulation acclimation period), as intraperitoneal administration of ANA-12 results in peak attenuation of trkB phosphorylation 4 h after injection (Cazorla et al., 2011). Stimulation was discontinued after cylinder task testing was complete. The cylinder task was conducted at the following time points for all rats under the conditions listed: (1) before surgery, vehicle injected; (2) 6 weeks following surgery, off stimulation, vehicle injected; (3) 6.5 weeks following surgery, on stimulation, vehicle injected; and (4) 7.5 weeks following surgery, on stimulation, ANA-12 injected.
Euthanasia.
At 2 weeks (Experiment 1), 4 weeks (Experiment 2), or 11 weeks (Experiment 3) after surgery, rats were deeply anesthetized (60 mg/kg, pentobarbital, i.p.) and perfused intracardially with heparinized normal saline at 37°C followed by ice-cold normal saline. Care was taken to minimize the tissue damage resulting from removing the skull with the electrode still intact. All brains were placed in ice-cold normal saline for 1 min and then hemisected on the coronal plane at the optic chiasm. The caudal half was fixed in 4% PFA for 1 week and transferred to 30% sucrose in 0.1 m phosphate buffer. The rostral half was immediately flash-frozen in 3-methyl butane on dry ice and stored at −80°C.
Kluver–Barrera histology.
Saline-perfused/PFA-postfixed brains (caudal half after hemisection) were frozen on dry ice and sectioned at 40 μm thickness using a sliding microtome in six series. Every sixth section of the STN was stained using Kluver–Barrera histochemistry (Kluver and Barrera, 1953) to evaluate for appropriate targeting of the electrode to the STN. Only rats with correctly positioned electrodes were included in the data analysis (see, e.g., Fig. 2B,C). Electrode location was considered to be appropriate if the tip of the electrode was observed within 250 μm of the border of the STN within any of the sections based on previous estimations of current spread conducted using similar stimulation parameters (Spieles-Engemann et al., 2010a).
Brightfield tyrosine hydroxylase immunohistochemistry for SNpc neurons.
One series (i.e., every sixth section) was stained with antisera for TH using the free-floating method. Tissue was blocked in normal goat serum and incubated overnight in primary antisera directed against TH (Millipore catalog #MAB318 RRID:AB_2201528, mouse anti-TH 1:4000). Cell membranes were permeabilized with the addition of Triton-X (0.3%) to the 0.1 m Tris buffer during incubations. Sections were then incubated in biotinylated secondary antisera against mouse IgG (Millipore catalog #AP124B RRID:AB_92458, 1:400) followed by the Vector ABC detection kit using HRP (Vector Laboratories). THir neurons were visualized upon exposure to 0.5 mg/ml DAB and 0.03% H2O2 in Tris buffer. Sections were mounted on subbed slides, dehydrated with ethanol followed by xylenes, and coverslipped with Cytoseal (Richard-Allan Scientific). Images shown in figures were processed in Photoshop (Adobe) using the automatic levels function.
Unbiased stereology of THir neurons in the SNpc.
The number of THir neurons in the SNpc ipsilateral and contralateral to 6-OHDA injection was quantified using unbiased stereology with the optical fractionator principle. Using a Nikon Eclipse 80i microscope, Retiga 4000R camera (QImaging) and Microbrightfield StereoInvestigator software (Microbrightfield Bioscience), THir neuron quantification was completed by drawing a contour around the SNpc borders at 4× and counting THir neurons according to stereological principles at 60× (NA 1.4) with parameters chosen for Schmitz-Hof Coefficients of Error to be ≤0.15. The percentage of remaining THir neurons of the ipsilateral, lesioned SNpc relative to the contralateral, intact SNpc was calculated.
Dissection of striatum for HPLC analysis.
Frozen brains were equilibrated at a temperature of −20°C before dissection. Coronal slabs of 1–2 mm thickness were blocked from each brain using a brain blocker (Zivic). Striatal tissue from both hemispheres was microdissected while being held at a constant −12°C on a cold plate (Teca). Frozen, dissected structures were placed individually in cryovials and stored at −80°C until analysis.
HPLC.
Homogenized samples were analyzed as described previously (Perez et al., 2005; Spieles-Engemann et al., 2010b; Kanaan et al., 2015). Briefly, samples were sonicated into an antioxidant solution (0.4 N perchlorate, 1.34 mm EDTA, and 0.53 mm sodium metabisulfite), and protein concentration was determined on a small aliquot using a bicinchoninic acid assay (Pierce). Clarified samples were separated on a 250 × 4.6 mm Microsorb MV C8 100–5 column (Varian) and simultaneously examined for DA, homovanillic acid (HVA) 3,4-dihydroxyphenylacetic acid (DOPAC), 5-hydroxytryptophan (5-HT), 5-hydroxyindoleacetic acid (5-HIAA), and norepinephrine (NE). Compounds were detected using a 12-channel coulometric array detector (CoulArray 5200, ESA) attached to a Waters 2695 Solvent Delivery System (Waters) under the following conditions: flow rate of 1 ml/min; detection potentials of 50, 175, 350, 400, and 525 mV; and scrubbing potential of 650 mV. The mobile phase consisted of a 15% methanol solution in distilled water containing 100 mm citric acid, 75 mm Na2HPO4, 80 μm heptanesulfonate monohydrate, and sodium salt, pH 4.25. Data are expressed as ng/mg protein. Striatal DA depletion of 90% was used as a criterion for inclusion in Experiment 2 based on previous determination that this intrastriatal 6-OHDA paradigm results in near-complete depletion of dopaminergic innervation within 2 weeks (Spieles-Engemann et al., 2010b).
Dual-label immunofluorescence for TH/phosphorylated ribosomal protein S6 or Akt/phosphorylated Akt.
One series (i.e., every sixth section) was double labeled either with antisera for TH and phosphorylated ribosomal protein S6 (p-rpS6, Ser235/236) or Akt/phosphorylated Akt using the free-floating method. Tissue was washed and blocked in normal goat serum for 1 h and incubated overnight in primary antisera directed against TH (Millipore catalog #MAB318 RRID:AB_2201528, mouse anti-TH 1:4000), p-rpS6 (Cell Signaling Technology catalog #2211 also 2211L, 2211S RRID:AB_331679, rabbit anti-p-rpS6 1:400), total Akt (R&D Systems catalog #MAB2055 RRID:AB_2224581, mouse anti-Akt, 1:2000), and phospho-AKT (R&D Systems catalog #AF887 RRID:AB_355685, rabbit anti-phospho-Akt, 1:1000). Cell membranes were permeabilized with the addition of Triton-X (0.5%) to the 0.1 m Tris buffer during incubations. Sections were then incubated in secondary antisera conjugated to fluorophores against mouse IgG (AlexaFluor-488, Thermo Fisher Scientific catalog #A-11001 RRID:AB_2534069, 1:500) and rabbit IgG (AlexaFluor-568, Thermo Fisher Scientific catalog #A11077 RRID:AB_10562719, 1:500) for 1 h and rinsed. Sections were mounted on subbed slides and coverslipped with Vectashield Hardset Mounting Medium (Vector Laboratories H1400). All treatment groups were processed simultaneously.
The three SNpc sections in which the fibers of the medial terminal nucleus of the accessory optic tract could be identified were photographed at 20× magnification at a single focal plane using a Nikon Eclipse microscope, and images were stitched using Nikon Elements software. Identical exposure times and settings within each channel were used across all sections that are statistically compared. For TH/p-rpS6 measurements, regions of interest (ROIs) were drawn around THir SNpc neurons with identifiable cell boundaries and evidence of nuclear pallor and with the p-rpS6 channel not viewable to the investigator. For total Akt/phosphorylation (activation) of Akt (pAkt) intensity measurements, an ROI contour was drawn encompassing the SNpc. ROI mean intensity data were exported for both channels. Each ROI/neuron across the three tissue sections was treated as a technical replicate and averaged. Immunoreactivity values were converted to percentages of the control group average within each comparison. Images shown in figures were processed in Photoshop using the automatic levels function.
Statistical analysis.
All statistical analyses were performed using SPSS Statistics (IBM). The effect of 6-OHDA on THir neuron survival was determined by a two-tailed, independent-samples t test by comparing the intact to the lesioned hemisphere. The impact of chronic ANA-12 administration on the extent of the 6-OHDA lesion was determined by a two-tailed, independent-samples t test comparing the percentage lesion between the two Inactive groups. A two-tailed, independent-samples t test was conducted to assess differences in THir neuron survival due to ANA-12 administration in rats receiving stimulation. A one-tailed, independent-samples t test was conducted to validate the neuroprotective effect of stimulation on the number of THir SNpc neurons in the Active-Vehicle group compared with the other groups combined. Comparisons in TH, p-rpS6, Akt, and p-Akt immunoreactivity were made using independent-samples t tests. Levene's test for equality of variances was used as appropriate. A repeated-measures (RM)-ANOVA followed by a least significant difference post hoc analysis was conducted to confirm the presence of forelimb asymmetry, the functional/behavioral response to DBS, and the effect of ANA-12 on spontaneous forelimb use. Statistical significance was set at p < 0.05. Statistical outliers were assessed using the Absolute Deviation from the Median method using the “very conservative” criterion (Leys et al., 2013).
Results
Chronic stimulation results in phosphorylation of both Akt and rpS6 in SNpc neurons
In Experiment 1, unlesioned/intact rats with implanted electrodes were continuously stimulated (Active) or not (Inactive) for 1 week and were assessed for pAkt and rpS6 (p-rpS6), two downstream signaling molecules of trkB, used here as readouts of trkB activation, in THir SNpc neurons (Fig. 1). To control for the potential effects of unilateral electrode implantation, data were normalized to percentage expression relative to the identical hemisphere in Inactive rats. No differences were observed in TH immunoreactivity due to STN stimulation (ipsilateral t(4) = −0.805, p = 0.466; contralateral t(4) = −1.597, p = 0.186; Fig. 1D,E), although there was a trend for increased TH immunoreactivity in the contralateral hemisphere. STN stimulation was associated with a bilateral increase in p-Akt in the SNpc compared with Inactive rats (t(10) = 2.364, p = 0.0397; Fig. 1H) with no significant difference observed between hemispheres or in total Akt immunoreactivity. STN stimulation also was associated with an increase in p-rpS6 in SNpc neurons compared with Inactive rats (t(10) = 4.072, p = 0.002; Fig. 1I). These results suggest that STN stimulation results in trkB activation in SNpc neurons using measurements of p-Akt and p-rpS6 as readouts.
Chronic ANA-12 administration does not exacerbate nigral degeneration
Rats receiving intrastriatal 6-OHDA and Inactive stimulation, regardless of Vehicle or ANA-12 treatment, possessed significantly fewer SNpc THir neurons ipsilateral to the injections/electrode compared with the contralateral SNpc (t(31.364) = 5.778, p < 0.001). Specifically, the intact SNpc in Inactive rats possessed 10,115 ± 941 THir neurons, whereas the lesioned SNpc contained 3750 ± 572, or ∼63% fewer THir neurons than the unlesioned SNpc, as expected from this lesion paradigm (Spieles-Engemann et al., 2010b; Gombash et al., 2014). No significant difference was observed in the magnitude of degeneration measured in Inactive + Vehicle versus Inactive + ANA-12 rats (t(18) = 1.164, p = 0.260). These data are illustrated in Figure 2D and support the conclusion that ANA-12 does not have a neuroprotective or an additive neurotoxic effect to 6-OHDA.
ANA-12 abolishes STN DBS-mediated neuroprotection
Active STN DBS in the absence of ANA-12 resulted in a neuroprotective effect, as we have reported previously (Spieles-Engemann et al., 2010b). Specifically, vehicle-treated Active STN DBS rats exhibited an average THir SNpc neuron survival rate of 43.3 ± 6.0% compared with an average 32.3 ± 2.7% survival in all other groups (t(31) = 1.800, p = 0.041).
ANA-12 administration blocked the neuroprotective effect of STN DBS. Specifically, the Active + ANA-12 group had 23.4 ± 5.1% and the Active + Veh group 43.3 ± 6.0% remaining THir SNpc neurons, comparing the lesioned hemisphere to the intact one. A statistically significant difference was observed between the groups (t(11) = −2.478, p = 0.031). These data are illustrated in Figure 2E–K and implicate trkB signaling in the neuroprotection afforded by long-term STN DBS.
ANA-12 does not alter striatal monoamines
As described previously, in our lesion paradigm (Spieles-Engemann et al., 2010b), nigrostriatal terminals and DA levels are almost immediately depleted. Striatal DA levels were used as a criterion for inclusion in this study to verify 6-OHDA lesion efficacy. Striatal punches were analyzed for monoamines and their metabolites to verify that ANA-12 had no effect, particularly on the intact hemisphere. A one-way ANOVA comparing the levels of monoamines in the intact striatum of the Inactive Vehicle, Inactive ANA-12, Active Vehicle, and Active ANA-12 groups showed no between-group differences on any measure (p > 0.05), as expected. Data are presented in Table 1.
Table 1.
HPLC on striatal punchesa
| Laterality | Inactive + vehicle | Inactive + ANA-12 | Active + vehicle | Active + ANA-12 | |
|---|---|---|---|---|---|
| DA | Ipsilateral | 2.3 ± 1.0 | BDL | 2.3 ± 2.3 | BDL |
| Contralateral | 174.2 ± 24.4 | 221.8 ± 14.1 | 194.5 ± 26.4 | 202.8 ± 48.7 | |
| HVA | Ipsilateral | 3.4 ± 1.3 | 3.8 ± 0.8 | 2.4 ± 1.3 | 2.2 ± 1.6 |
| Contralateral | 11.0 ± 1.7 | 12.0 ± 1.6 | 10.7 ± 2.9 | 14.1 ± 3.4 | |
| DOPAC | Ipsilateral | 1.9 ± 0.5 | 0.8 ± 0.4 | 2.0 ± 0.5 | 0.7 ± 0.4 |
| Contralateral | 15.0 ± 2.0 | 15.1 ± 1.4 | 17.1 ± 1.5 | 17.2 ± 4.1 | |
| HVA/DA | Contralateral | 0.085 ± 0.029 | 0.053 ± 0.006 | 0.050 ± 0.010 | 0.068 ± 0.004 |
| DOPAC/DA | Contralateral | 0.135 ± 0.061 | 0.067 ± 0.004 | 0.095 ± 0.012 | 0.090 ± 0.010 |
| 5-HT | Ipsilateral | 4.3 ± 1.3 | 6.0 ± 0.7 | 4.0 ± 1.9 | 2.5 ± 1.6 |
| Contralateral | 5.2 ± 1.4 | 8.0 ± 0.8 | 4.1 ± 1.9 | 3.4 ± 2.1 | |
| 5-HIAA | Ipsilateral | 8.0 ± 1.7 | 11.5 ± 1.0 | 9.3 ± 2.9 | 9.4 ± 2.8 |
| Contralateral | 7.4 ± 1.4 | 10.2 ± 1.0 | 7.6 ± 2.1 | 8.9 ± 2.4 | |
| NE | Ipsilateral | 39.2 ± 15.8 | 36.9 ± 6.0 | 14.4 ± 8.3 | 16.2 ± 11.7 |
| Contralateral | 28.7 ± 13.5 | 28.5 ± 4.2 | 10.3 ± 4.8 | 10.6 ± 7.6 |
aBDL, Below detectable limit. Values are mean ± SEM (in ng/mg of protein), except in the cases of DOPAC/DA and HVA/DA because they are unitless.
ANA-12 diminishes phosphorylation of rpS6 associated with STN stimulation
In Experiment 2, lesioned rats “on” or “off” stimulation were assessed for SNpc TH (Fig. 3A,B,G–J) and p-rpS6 immunoreactivity to compare vehicle- and ANA-12-treated rats (Fig. 3C,D,K–N). To control for the potential effects of unilateral electrode implantation and 6-OHDA, data were normalized to percentage expression relative to the identical hemisphere in Vehicle-treated rats. No effect of ANA-12 was observed on TH or p-rpS6 in Inactive rats, in either hemisphere (Fig. 3G,H,K,L), supporting that ANA-12 did not alter basal levels of trkB signaling. In contrast, in rats with Active stimulation, ANA-12 was associated with a significant decrease in TH immunoreactivity in SNpc neurons ipsilateral to the lesion (t(11) = 3.873, p = 0.003, Fig. 3I) but not contralateral, although a trend was observed (Fig. 3J). Of importance, systemic administration of ANA-12 was associated with decreased p-rpS6 immunoreactivity bilaterally in Active rats (ipsilateral SNpc: t(10) = 3.038, p = 0.012, Fig. 3M; contralateral SNpc: t(7.953) = 3.457, p = 0.009, Figure 3N). These results demonstrate ANA-12 engagement of trkB signaling in SNpc neurons in Active STN stimulation rats.
Figure 3.
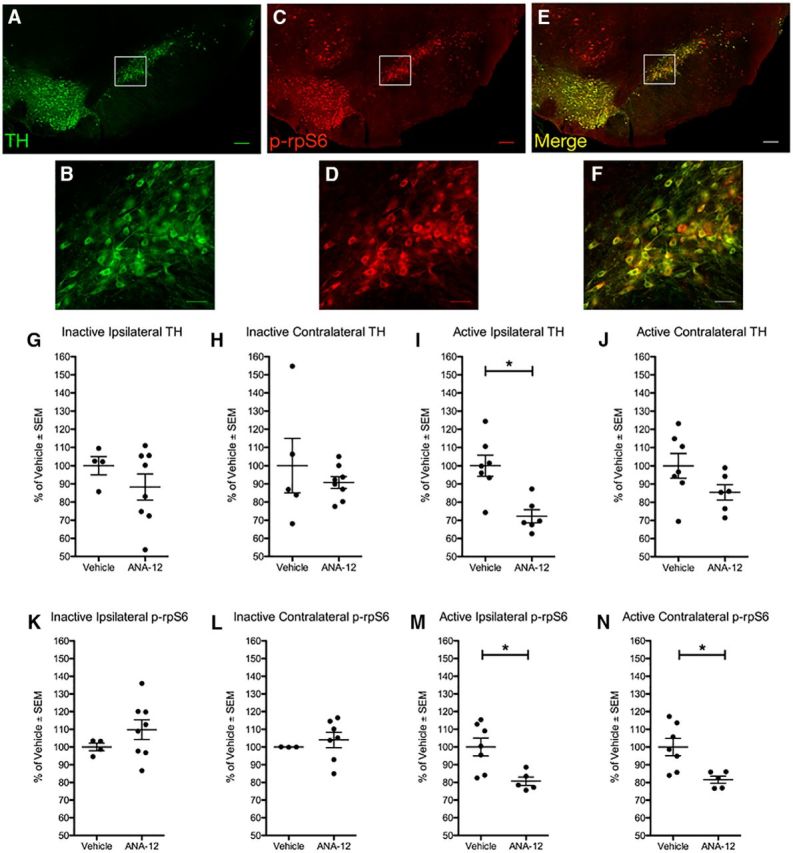
ANA-12 reduces phosphorylation of rpS6 in SNpc neurons in stimulated, lesioned rats. A–F, The SNpc contralateral to the electrode of 6-OHDA-lesioned, stimulated rats receiving chronic injections of ANA-12. Photomicrographs at low and high magnification, respectively, are shown for TH (A, B), p-rpS6 (C, D), and merged images (E, F). To control for the potential effects of unilateral electrode implantation and 6-OHDA, data were normalized to percentage expression relative to the identical hemisphere in Vehicle-treated rats. Quantification for rats not randomized to stimulation (Inactive) is shown for the SNpc ipsilateral and contralateral to the electrode, respectively, for TH (G, H) and p-rpS6 (K, L). Quantification for rats receiving stimulation (Active) is shown for the SNpc ipsilateral and contralateral to the electrode, respectively, for TH (I, J) and p-rpS6 (M, N). Statistically significant differences between Vehicle and ANA-12 groups within the stimulation arms were only observed within the Active arm: the ipsilateral SNpc showed a decrease in TH (p = 0.003, I) and p-rpS6 (p = 0.012, M) immunoreactivity, and the contralateral SNpc showed a decreased in p-rpS6 (p = 0.009, N) immunoreactivity. Scale bars: low magnification, 200 μm; high magnification, 50 μm. Graphs represent mean ± SEM.*p < 0.05.
ANA-12 attenuates functional efficacy of STN DBS
We observed in Experiment 2 that forelimb asymmetry did not show as robust a remediation to chronic stimulation in the rats receiving ANA-12 administration compared with rats receiving vehicle (data not shown). Because these two cohorts also had an underlying difference in SNpc neuron numbers, we performed an additional experiment (Experiment 3) using rats with stable, near-complete 6-OHDA lesions to determine whether blockade of trkB by ANA-12 contributed to this observation.
In Experiment 3, rats that exhibited a ≥20% deficit in contralateral forelimb use after 6-OHDA at the 6 week time point compared with baseline values were considered adequately lesioned and included for acute behavioral analysis (Spieles-Engemann et al., 2011; Fischer et al., 2015). In these animals, 6-OHDA produced a significant loss of THir SNpc neurons on the side ipsilateral to 6-OHDA injection (and electrode implantation) compared with the contralateral SNpc (t(18) = 4.143, p < 0.001; Fig. 4E). There was an ∼61% loss of THir neurons in the lesion versus intact sides (Lesioned = 4001 ± 1035; Intact = 10370 ± 1137). These data are depicted in Figure 4B–F and support that all of the rats in Experiment 3 were comparably lesioned.
All adequately lesioned rats were assessed at two additional time points: 6.5 weeks with stimulation plus vehicle treatment and at 7.5 weeks with stimulation and acute ANA-12 treatment. repeated-measures ANOVA was used to compare spontaneous forelimb use across all four time points. Values for spontaneous forelimb use are expressed as a percentage of contralateral (to the lesion) forelimb use of the total. Results are depicted in Figure 4F. Averages of contralateral forelimb use for each time point were as follows: 52.0 ± 2.1% on week 0 (“Baseline”); 24.5 ± 3.4% on week 6 (“Lesion”); 47.0 ± 3.7% on week 6.5 (“Stim-On”); and 33.8 ± 5.5% on week 7.5 (“ANA-12,” where stimulation was also Active). A statistically significant difference was observed between the time points (F(3,27) = 13.465, p < 0.001). A post hoc analysis showed a significant difference between: Baseline versus Lesion (p < 0.001), Lesion versus Stim-On (p < 0.001), Stim-On versus ANA-12 (p = 0.015), and Baseline versus ANA-12 (comparison not shown in Fig. 4F; p = 0.012). There was a trend toward a significant difference for the Lesion versus ANA-12 comparison (p = 0.108). The number of paw touches used to calculate the forelimb asymmetry value was the full 20 required by the cylinder task (Schallert and Tillerson, 2000) for all time points in 90% of the rats, suggesting that general locomotor activity did not influence the asymmetry values reported. These data implicate BDNF-trkB signaling as a component in the alleviation of symptoms by STN DBS.
Discussion
Canonical BDNF-trkB survival signaling underpins STN DBS-mediated neuroprotection of the SNpc dopamine neurons
We have previously shown that STN DBS increases BDNF protein and transcript in the nigrostriatal system (Spieles-Engemann et al., 2011). In the present study, we demonstrate that chronic STN DBS results in phosphorylation of Akt and rpS6 in SNpc neurons, two readouts downstream of trkB that implicate BDNF-trkB canonical signaling. Further, we hypothesize that when trkB signaling is attenuated, DBS no longer provides neuroprotection from ongoing 6-OHDA-induced DA neuron loss. These findings strongly suggest that STN DBS activates canonical trkB survival signaling pathways in the SNpc.
High-frequency stimulation of cultured glutamatergic hippocampal neurons results in the secretion of BDNF (Hartmann et al., 2001; Chen et al., 2004). As the STN is primarily composed of glutamatergic neurons and expresses BDNF mRNA (Kitai and Kita, 2006; Zhang et al., 2006), it is therefore reasonable to suggest that these neurons respond to high-frequency stimulation by similarly releasing BDNF at target structures, including the SN pars reticulata, SNpc, or striatum (Jackson and Crossman, 1981; Kita et al., 1983; Kita and Kitai, 1987; Parent and Hazrati, 1995; Kitai and Kita, 2006; Zhang et al., 2006; Degos et al., 2008; Watabe-Uchida et al., 2012). The fact that we observe increased Akt and rpS6 phosphorylation within SNpc neurons in rats with lesioned and intact striatal terminals supports an extrastriatal source of BDNF. Our previous observation that nigrostriatal lesion status alters the site of BDNF increase (i.e., striatum vs nigra) (Spieles-Engemann et al., 2011) suggests that nigral dopamine neurons themselves participate in STN DBS-mediated increases in BDNF. Future studies are required to determine which neurons in the basal ganglia circuitry are the sources of the BDNF secreted in response to STN DBS.
Noncanonical BDNF-trkB signaling contributes to STN DBS-mediated reversal of forelimb impairments
Canonical trkB survival signaling involves long-term alterations. Specifically, ligand binding followed by autophosphorylation of trkB leads to activation of PI3K-Akt and Erk that in turn promote cell survival or growth through changes in gene transcription and production of prosurvival proteins (Cobb and Cobb, 1999; Airaksinen et al., 2002; Yoshii and Constantine-Paton, 2010; Lewin et al., 2014). These effects of canonical trkB signaling generally occur on the order of hours to days. The time course of our neuroprotection Experiment 2 is consistent with the time required for canonical trkB survival signaling. However, the acute behavioral effect observed in Experiment 3 suggests a different mechanism. Unexpectedly, we observed that following an acute, single dose of ANA-12 and acute stimulation, the reversal of forelimb impairments normally associated with STN DBS (Meissner et al., 2002; Chang et al., 2003; Darbaky et al., 2003; Maesawa et al., 2004; Shi et al., 2004, 2006; Temel et al., 2005; Fang et al., 2006; Vlamings et al., 2007; Akita et al., 2010; Li et al., 2010; Spieles-Engemann et al., 2011) was significantly attenuated. We hypothesize that noncanonical trkB signaling mechanisms are involved in this effect. In recent years, our understanding of noncanonical trkB signaling mechanisms has grown. trkB activation can potentiate the response of NMDA receptors through phosphorylation of the NMDA receptor 2B subunit (Li et al., 2006; Chen et al., 2014; Nakai et al., 2014). This noncanonical pathway, which takes place over a shorter time course, has been used to explain other behavioral phenomena (Quintana et al., 2012; Nakai et al., 2014). BDNF-trkB signaling also can affect DA reuptake or release in an immediate manner (Bosse et al., 2012). Future studies are needed to examine whether BDNF-enhanced glutamatergic or dopaminergic signaling pathways participate in the functional efficacy of STN DBS.
Considerations for the use of ANA-12
We considered the pharmacokinetics and pharmacodynamics of ANA-12, as detailed previously (Cazorla et al., 2011; Ambrogini et al., 2013), in the design and interpretation of our experiment. Under similar injection parameters, ANA-12 has been shown to reach the brain within 30 min at a level of 400 nm and remains in the brain at least 6 h later at a level of 10 nm. The effects of ANA-12 on trkB are specific, as it does not interact with other members of the receptor family TrkA or TrkC. The ratio of trkB phosphorylation to total levels is inhibited by ∼30% after 4 h; the degree of inhibition after 4 h has not been measured, but the slope from 0 to 4 h was negative. Of note, whether phosphorylation of trkB was measured by whole-brain tissue or by dissection of specific structures (i.e., the cortex, striatum, and hippocampus), the degree of receptor inhibition was essentially equal. As such, ANA-12 is a particularly appropriate molecule for this study. Future studies could examine whether similar inhibition of STN DBS-mediated neuroprotection and attenuation of symptomatic effects are observed using a different compound to target trkB signaling, should one become available.
One shortcoming of using ANA-12 to achieve widespread blockade of trkB receptors is that it does not allow for identification of what circuitry is mediating the BDNF-trkB signaling and functional efficacy of STN DBS. However, there are some likely possibilities. In our previous work, we showed that STN DBS increases BDNF in the primary motor (M1) cortex. Gradinaru et al. (2009) demonstrated using optogenetics that the functional efficacy of STN DBS can be explained through antidromic activation of the hyperdirect pathway, a set of fibers from Layer V neurons in the M1 cortex that project to the STN. As these projections are glutamatergic, they may also release BDNF to act on trkB receptors located on STN neurons. In addition, the rat has a proportion of STN fibers, which are also glutamatergic, that innervate the M1 cortex (Degos et al., 2008). BDNF may then be released by STN neurons and act on trkB receptors within the M1 cortex. However, these reciprocal, subthalamocortical fibers are not present in primates (Parent and Smith, 1987), so it is unlikely that the clinical phenomenon can be explained by this circuitry.
In conclusion, collectively, our results suggest a role for BDNF-trkB signaling in two related, yet distinct, phenomena. Long-term activation of canonical trkB signaling in SNpc neurons is associated with the neuroprotection afforded by STN DBS in this 6-OHDA model. Acute trkB signaling through a noncanonical pathway likely contributes to STN DBS-mediated reversal of forelimb asymmetry in a unilaterally parkinsonian rat. Localizing these phenomena to specific circuitries through future experiments will specify the sources and sites of action for BDNF and may lead to refinements to STN DBS or generation of novel, less-invasive therapies for either symptom or disease-modifying indications.
Footnotes
This work was supported by Spectrum Health to D.L.F., Mercy Health Saint Mary's to D.L.F., and Morris K. Udall Center of Excellence for Parkinson's Disease Research at Michigan State University NS058830 to T.J.C., C.E.S., K.S.-C., and J.W.L.
The authors declare no competing financial interests.
References
- Airaksinen MS, Saarma M (2002) The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3:383–394. 10.1038/nrn812 [DOI] [PubMed] [Google Scholar]
- Akita H, Honda Y, Ogata M, Noda K, Saji M (2010) Activation of the NMDA receptor involved in the alleviating after-effect of repeated stimulation of the subthalamic nucleus on motor deficits in hemiparkinsonian rats. Brain Res 1306:159–167. 10.1016/j.brainres.2009.09.040 [DOI] [PubMed] [Google Scholar]
- Ambrogini P, Lattanzi D, Ciuffoli S, Betti M, Fanelli M, Cuppini R (2013) Physical exercise and environment exploration affect synaptogenesis in adult-generated neurons in the rat dentate gyrus: possible role of BDNF. Brain Res 1534:1–12. 10.1016/j.brainres.2013.08.023 [DOI] [PubMed] [Google Scholar]
- Bosse KE, Maina FK, Birbeck JA, France MM, Roberts JJ, Colombo ML, Mathews TA (2012) Aberrant striatal dopamine transmitter dynamics in brain-derived neurotrophic factor-deficient mice. J Neurochem 120:385–395. 10.1111/j.1471-4159.2011.07531.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, Rognan D (2011) Identification of a low-molecular weight trkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest 121:1846–1857. 10.1172/JCI43992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Shi LH, Luo F, Woodward DJ (2003) High frequency stimulation of the subthalamic nucleus improves treadmill locomotion in unilateral 6-hydroxydopamine lesioned rats. Brain Res 983:174–184. 10.1016/S0006-8993(03)03053-1 [DOI] [PubMed] [Google Scholar]
- Chen W, Walwyn W, Ennes HS, Kim H, McRoberts JA, Marvizón JC (2014) BDNF released during neuropathic pain potentiates NMDA receptors in primary afferent terminals. Eur J Neurosci 39:1439–1454. 10.1111/ejn.12516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, Lee FS (2004) Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci 24:4401–4411. 10.1523/JNEUROSCI.0348-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715–725. 10.1002/ana.21995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb MH. (1999) MAP kinase pathways. Prog Biophys Mol Biol 71:479–500. 10.1016/S0079-6107(98)00056-X [DOI] [PubMed] [Google Scholar]
- Darbaky Y, Forni C, Amalric M, Baunez C (2003) High frequency stimulation of the subthalamic nucleus has beneficial antiparkinsonian effects on motor functions in rats, but less efficiency in a choice reaction time task. Eur J Neurosci 18:951–956. 10.1046/j.1460-9568.2003.02803.x [DOI] [PubMed] [Google Scholar]
- Degos B, Deniau JM, Le Cam J, Mailly P, Maurice N (2008) Evidence for a direct subthalamo-cortical loop circuit in the rat. Eur J Neurosci 27:2599–2610. 10.1111/j.1460-9568.2008.06229.x [DOI] [PubMed] [Google Scholar]
- Fang X, Sugiyama K, Akamine S, Namba H (2006) Improvements in motor behavioral tests during deep brain stimulation of the subthalamic nucleus in rats with different degrees of unilateral parkinsonism. Brain Res 1120:202–210. 10.1016/j.brainres.2006.08.073 [DOI] [PubMed] [Google Scholar]
- Fischer DL, Collier TJ, Cole-Strauss A, Wohlgenant SL, Lipton JW, Steece-Collier K, Manfredsson FP, Kemp CJ, Sortwell CE (2015) High-frequency stimulation of the rat entopeduncular nucleus does not provide functional or morphological neuroprotection from 6-hydroxydopamine. PLoS One 10:e0133957. 10.1371/journal.pone.0133957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Frankel M, Flaumenhaft Y, Merenlender A, Pinhasov A, Feder Y, Taler M, Gil-Ad I, Abeles M, Yadid G (2009) Programmed acute electrical stimulation of ventral tegmental area alleviates depressive-like behavior. Neuropsychopharmacology 34:1057–1066. 10.1038/npp.2008.177 [DOI] [PubMed] [Google Scholar]
- Gersner R, Toth E, Isserles M, Zangen A (2010) Site-specific antidepressant effects of repeated subconvulsive electrical stimulation: potential role of brain-derived neurotrophic factor. Biol Psychiatry 67:125–132. 10.1016/j.biopsych.2009.09.015 [DOI] [PubMed] [Google Scholar]
- Gombash SE, Manfredsson FP, Mandel RJ, Collier TJ, Fischer DL, Kemp CJ, Kuhn NM, Wohlgenant SL, Fleming SM, Sortwell CE (2014) Striatonigral pleiotrophin overexpression provides superior protection compared to overexpression in both striatonigral and nigrostriatal pathways. Gene Ther 21:682–693. 10.1038/gt.2014.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K (2009) Optical deconstruction of parkinsonian neural circuitry. Science 324:354–359. 10.1126/science.1167093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamani C, Machado DC, Hipólide DC, Dubiela FP, Suchecki D, Macedo CE, Tescarollo F, Martins U, Covolan L, Nobrega JN (2012) Deep brain stimulation reverses anhedonic-like behavior in a chronic model of depression: role of serotonin and brain derived neurotrophic factor. Biol Psychiatry 71:30–35. 10.1016/j.biopsych.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnack D, Meissner W, Jira JA, Winter C, Morgenstern R, Kupsch A (2008) Placebo-controlled chronic high-frequency stimulation of the subthalamic nucleus preserves dopaminergic nigral neurons in a rat model of progressive Parkinsonism. Exp Neurol 210:257–260. 10.1016/j.expneurol.2007.10.002 [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V (2001) Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20:5887–5897. 10.1093/emboj/20.21.5887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilker R, Portman AT, Voges J, Staal MJ, Burghaus L, van Laar T, Koulousakis A, Maguire RP, Pruim J, de Jong BM, Herholz K, Sturm V, Heiss WD, Leenders KL (2005) Disease progression continues in patients with advanced Parkinson's disease and effective subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry 76:1217–1221. 10.1136/jnnp.2004.057893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A, Crossman AR (1981) Subthalamic nucleus efferent projection to the cerebral cortex. Neuroscience 6:2367–2377. 10.1016/0306-4522(81)90023-3 [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Collier TJ, Cole-Strauss A, Grabinski T, Mattingly ZR, Winn ME, Steece-Collier K, Sortwell CE, Manfredsson FP, Lipton JW (2015) The longitudinal transcriptomic response of the substantia nigra to intrastriatal 6-hydroxydopamine reveals significant upregulation of regeneration-associated genes. PLoS One 10:e0127768. 10.1371/journal.pone.0127768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Kitai ST (1987) Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol 260:435–452. 10.1002/cne.902600309 [DOI] [PubMed] [Google Scholar]
- Kita H, Chang HT, Kitai ST (1983) The morphology of intracellularly labeled rat subthalamic neurons: a light microscopic analysis. J Comp Neurol 215:245–257. 10.1002/cne.902150302 [DOI] [PubMed] [Google Scholar]
- Kitai S, Kita H (2006) Anatomy and physiology of the subthalamic nucleus: a driving force of the basal ganglia. In: The basal ganglia. Vol II. Structure and function: current concepts (Carpenter M, Jayarama A, ed.), pp 357–373. New York: Plenum. [Google Scholar]
- Kluver H, Barrera E (1953) A method for the combined staining of cells and fibers in the nervous system. J Neuropathol Exp Neurol 12:400–403. 10.1097/00005072-195312040-00008 [DOI] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136:2419–2431. 10.1093/brain/awt192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krack P, Batir A, Van Blercom N, Chabardes S, Fraix V, Ardouin C, Koudsie A, Limousin PD, Benazzouz A, LeBas JF, Benabid AL, Pollak P (2003) Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Engl J Med 349:1925–1934. 10.1056/NEJMoa035275 [DOI] [PubMed] [Google Scholar]
- Lewin GR, Carter BD (2014) Neurotrophic factors. In: Handbook of experimental pharmacology. Berlin: Springer. [PubMed] [Google Scholar]
- Leys C, Ley C, Klein O, Bernard P, Licata L (2013) Detecting outliers: do not use standard deviation around the mean, use absolute deviation around the median. J Exp Soc Psychol 49:764–766. 10.1016/j.jesp.2013.03.013 [DOI] [Google Scholar]
- Li J, McRoberts JA, Ennes HS, Trevisani M, Nicoletti P, Mittal Y, Mayer EA (2006) Experimental colitis modulates the functional properties of NMDA receptors in dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol 291:G219–G228. 10.1152/ajpgi.00097.2006 [DOI] [PubMed] [Google Scholar]
- Li XH, Wang JY, Gao G, Chang JY, Woodward DJ, Luo F (2010) High-frequency stimulation of the subthalamic nucleus restores neural and behavioral functions during reaction time task in a rat model of Parkinson's disease. J Neurosci Res 88:1510–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesawa S, Kaneoke Y, Kajita Y, Usui N, Misawa N, Nakayama A, Yoshida J (2004) Long-term stimulation of the subthalamic nucleus in hemiparkinsonian rats: neuroprotection of dopaminergic neurons. J Neurosurg 100:679–687. 10.3171/jns.2004.100.4.0679 [DOI] [PubMed] [Google Scholar]
- Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, Poo MM (2009) Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J Neurosci 29:14185–14198. 10.1523/JNEUROSCI.1863-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner W, Harnack D, Paul G, Reum T, Sohr R, Morgenstern R, Kupsch A (2002) Deep brain stimulation of subthalamic neurons increases striatal dopamine metabolism and induces contralateral circling in freely moving 6-hydroxydopamine-lesioned rats. Neurosci Lett 328:105–108. 10.1016/S0304-3940(02)00463-9 [DOI] [PubMed] [Google Scholar]
- Mendoza MC, Er EE, Blenis J (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 36:320–328. 10.1016/j.tibs.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai T, Nagai T, Tanaka M, Itoh N, Asai N, Enomoto A, Asai M, Yamada S, Saifullah AB, Sokabe M, Takahashi M, Yamada K (2014) Girdin phosphorylation is crucial for synaptic plasticity and memory: a potential role in the interaction of BDNF/trkB/Akt signaling with NMDA receptor. J Neurosci 34:14995–15008. 10.1523/JNEUROSCI.2228-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A, Hazrati LN (1995) Functional anatomy of the basal ganglia: II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Brain Res Rev 20:128–154. 10.1016/0165-0173(94)00008-D [DOI] [PubMed] [Google Scholar]
- Parent A, Smith Y (1987) Organization of efferent projections of the subthalamic nucleus in the squirrel monkey as revealed by retrograde labeling methods. Brain Res 436:296–310. 10.1016/0006-8993(87)91674-X [DOI] [PubMed] [Google Scholar]
- Perez SE, Lazarov O, Koprich JB, Chen EY, Rodriguez-Menendez V, Lipton JW, Sisodia SS, Mufson EJ (2005) Nigrostriatal dysfunction in familial Alzheimer's disease-linked APPswe/PS1DeltaE9 transgenic mice. J Neurosci 25:10220–10229. 10.1523/JNEUROSCI.2773-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piallat B, Benazzouz A, Benabid AL (1996) Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6-OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci 8:1408–1414. 10.1111/j.1460-9568.1996.tb01603.x [DOI] [PubMed] [Google Scholar]
- Quintana A, Sgambato-Faure V, Savasta M (2012) Effects of l-DOPA and STN-HFS dyskinesiogenic treatments on NR2B regulation in basal ganglia in the rat model of Parkinson's disease. Neurobiol Dis 48:379–390. 10.1016/j.nbd.2012.06.009 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Oroz MC, Obeso JA, Lang AE, Houeto JL, Pollak P, Rehncrona S, Kulisevsky J, Albanese A, Volkmann J, Hariz MI, Quinn NP, Speelman JD, Guridi J, Zamarbide I, Gironell A, Molet J, Pascual-Sedano B, Pidoux B, Bonnet AM, Agid Y, et al. (2005) Bilateral deep brain stimulation in Parkinson's disease: a multicentre study with 4 years follow-up. Brain 128:2240–2249. 10.1093/brain/awh571 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Oroz MC, Zamarbide I, Guridi J, Palmero MR, Obeso JA (2004) Efficacy of deep brain stimulation of the subthalamic nucleus in Parkinson's disease 4 years after surgery: double blind and open label evaluation. J Neurol Neurosurg Psychiatry 75:1382–1385. 10.1136/jnnp.2003.031294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallert T. (2006) Behavioral tests for preclinical intervention assessment. NeuroRx 3:497–504. 10.1016/j.nurx.2006.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallert T, Tillerson JL (2000) Intervention strategies for degeneration of dopamine neurons in parkinsonism: optimizing behavioral assessment of outcome. In: Central nervous system diseases: innovative models of CNS diseases from molecule to therapy (Emerich DF, Sanberg PR, eds), pp 131–151. Totowa, NJ: Humana. [Google Scholar]
- Shi LH, Woodward DJ, Luo F, Anstrom K, Schallert T, Chang JY (2004) High-frequency stimulation of the subthalamic nucleus reverses limb-use asymmetry in rats with unilateral 6-hydroxydopamine lesions. Brain Res 1013:98–106. 10.1016/j.brainres.2004.03.053 [DOI] [PubMed] [Google Scholar]
- Shi LH, Luo F, Woodward DJ, Chang JY (2006) Basal ganglia neural responses during behaviorally effective deep brain stimulation of the subthalamic nucleus in rats performing a treadmill locomotion test. Synapse 59:445–457. 10.1002/syn.20261 [DOI] [PubMed] [Google Scholar]
- Spieles-Engemann AL, Behbehani MM, Collier TJ, Steece-Collier K, Wohlgenant SL, Thompson VB, Lipton JW, Sortwell CE (2009) Deep brain stimulation in a rodent model of Parkinson's disease upregulates BDNF in subthalamic nucleus target structures, 46:1 Chicago: Society for Neuroscience Abstracts. [Google Scholar]
- Spieles-Engemann AL, Collier TJ, Sortwell CE (2010a) A functionally relevant and long-term model of deep brain stimulation of the rat subthalamic nucleus: advantages and considerations. Eur J Neurosci 32:1092–1099. 10.1111/j.1460-9568.2010.07416.x [DOI] [PubMed] [Google Scholar]
- Spieles-Engemann AL, Behbehani MM, Collier TJ, Wohlgenant SL, Steece-Collier K, Paumier K, Daley BF, Gombash S, Madhavan L, Mandybur GT, Lipton JW, Terpstra BT, Sortwell CE (2010b) Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol Dis 39:105–115. 10.1016/j.nbd.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieles-Engemann AL, Steece-Collier K, Behbehani MM, Collier TJ, Wohlgenant SL, Kemp CJ, Cole-Strauss A, Levine ND, Gombash SE, Thompson VB, Lipton JW, Sortwell CE (2011) Subthalamic nucleus stimulation increases brain derived neurotrophic factor in the nigrostriatal system and primary motor cortex. J Parkinsons Dis 1:123–136. [PMC free article] [PubMed] [Google Scholar]
- Tagliati M, Martin C, Alterman R (2010) Lack of motor symptoms progression in Parkinson's disease patients with long-term bilateral subthalamic deep brain stimulation. Int J Neurosci 120:717–723. 10.3109/00207454.2010.518777 [DOI] [PubMed] [Google Scholar]
- Temel Y, Visser-Vandewalle V, Aendekerk B, Rutten B, Tan S, Scholtissen B, Schmitz C, Blokland A, Steinbusch HW (2005) Acute and separate modulation of motor and cognitive performance in parkinsonian rats by bilateral stimulation of the subthalamic nucleus. Exp Neurol 193:43–52. 10.1016/j.expneurol.2004.12.025 [DOI] [PubMed] [Google Scholar]
- Temel Y, Visser-Vandewalle V, Kaplan S, Kozan R, Daemen MA, Blokland A, Schmitz C, Steinbusch HW (2006) Protection of nigral cell death by bilateral subthalamic nucleus stimulation. Brain Res 1120:100–105. 10.1016/j.brainres.2006.08.082 [DOI] [PubMed] [Google Scholar]
- Vlamings R, Visser-Vandewalle V, Koopmans G, Joosten EA, Kozan R, Kaplan S, Steinbusch HW, Temel Y (2007) High frequency stimulation of the subthalamic nucleus improves speed of locomotion but impairs forelimb movement in Parkinsonian rats. Neuroscience 148:815–823. 10.1016/j.neuroscience.2007.06.043 [DOI] [PubMed] [Google Scholar]
- Wallace BA, Ashkan K, Heise CE, Foote KD, Torres N, Mitrofanis J, Benabid AL (2007) Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP-treated monkeys. Brain 130:2129–2145. 10.1093/brain/awm137 [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N (2012) Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74:858–873. 10.1016/j.neuron.2012.03.017 [DOI] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M (2010) Postsynaptic BDNF-trkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 70:304–322. 10.1002/dneu.20765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Andren PE, Svenningsson P (2006) Repeated L-DOPA treatment increases c-fos and BDNF mRNAs in the subthalamic nucleus in the 6-OHDA rat model of Parkinson's disease. Brain Res 1095:207–210. 10.1016/j.brainres.2006.04.019 [DOI] [PubMed] [Google Scholar]