

Summary
Establishing a state of transplantation tolerance that leads to indefinite graft survival without the need for lifelong immunosuppression has been achieved successfully in limited numbers of transplant recipients in the clinic. These successes led to studies aimed at identifying potential biomarkers that diagnose allograft tolerance and identify the patients most amenable to drug minimization, and implicated an enriched B cell signature of tolerance. The emergence of a specialized subset of regulatory B cell (Bregs), that possess immune‐modulatory function in inflammation and autoimmune disease, raised the possibility that Bregs play critical roles in the promotion of transplantation tolerance and that Bregs are the underlying explanation for the B cell signature of tolerance. However, B cells are best known to play a key role in humoral immunity, and excessive production of donor specific antibodies has clear deleterious effects in transplantation. Thus, for tolerance to be persistent, alloantibody responses must also be curtailed, either through the suppression of T cell help or the induction of B cell‐intrinsic dysfunction. Recent findings indicate a unique subset of follicular regulatory T cells (Tfr) that can suppress B cell function and induce epigenetic modifications that result in sustained defects in B cell differentiation and function. In this review, we summarize studies in animals and humans that suggest roles for Bregs and dysfunctional B cells in transplantation tolerance, and discuss how these insights may provide a roadmap for new approaches to diagnose, and new therapies to induce allograft tolerance.
Keywords: anergy, Bregs, biomarkers, IL‐10, transplantation tolerance
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Immune tolerance in transplantation. Clinical and Experimental Immunology 2017, 189: 133–4.
Transplantation tolerance: the big picture. Where do we stand, where should we go? Clinical and Experimental Immunology 2017, 189: 135–7.
Operational tolerance in kidney transplantation and associated biomarkers. Clinical and Experimental Immunology 2017, 189: 138–57.
Immune monitoring as prerequisite for transplantation tolerance trials. Clinical and Experimental Immunology 2017, 189: 158–70.
Murine models of transplantation tolerance through mixed chimerism: advances and roadblocks. Clinical and Experimental Immunology 2017, 189: 181–9.
Chimerism‐based tolerance in organ transplantation: preclinical and clinical studies. Clinical and Experimental Immunology 2017, 189: 190–6.
Regulatory T cells: tolerance induction in solid organ transplantation. Clinical and Experimental Immunology 2017, 189: 197–210.
Introduction
High levels of donor‐specific antibodies (DSA), that may either be preformed or develop de novo after transplantation, mediate antibody‐mediated allograft rejection (AMR) that is now considered the leading cause of graft loss in the clinic 1, 2. DSA can be produced by long‐lived plasma cells without further need for antigen stimulation or T cell help, and they bind directly to graft endothelium to mediate acute AMR through the activation of complement and recruitment of FcγR+ natural killer (NK) cells, macrophages and neutrophils 3, 4. The classical histological features of acute AMR include platelet aggregation, thrombotic microangiopathy (TMA) and neutrophilic accumulation, resulting in early cellular necrosis and a relatively rapid decline in allograft function 5. In chronic AMR a repetitive pattern of subacute thrombotic events and inflammatory changes results in cellular and repair, culminating as late transplant glomerulopathy and a gradual decline in renal function 6. Thus, if immune tolerance is to maintain the allograft for the life of the recipient successfully, humoral responses have to be controlled. This control may be achieved by cell extrinsic mechanisms that are dependent upon the control of T follicular helper cells (Tfh), suppression by regulatory T follicular cells (Tfr) or by B cell intrinsic mechanisms (Fig. 1).
Figure 1.
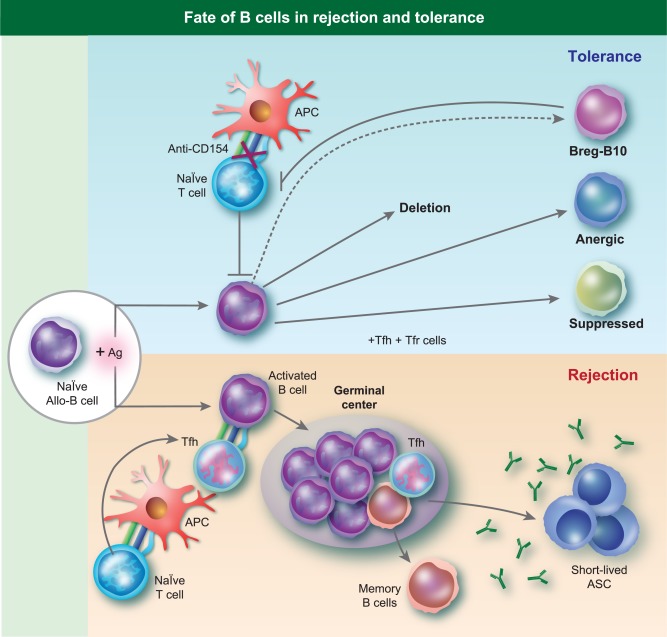
Fate of B cells in rejection and tolerance. The activation and differentiation of naive alloreative B cells during rejection occurs in a T cell‐ and germinal centre‐dependent manner. Under tolerogenic conditions, naive alloreactive B cells encounter antigen, but in the absence of T cell help may be deleted, develop into regulatory B cells (Bregs)/B10 cells that suppress T cell responses, and into anergic or suppressed B cells that fail to develop into antibody‐secreting plasma cells.
In 2010, two cross‐sectional studies on renal transplant tolerance in humans unexpectedly identified an enriched B cell signature 7, 8. The authors hypothesized that this enriched B cell signature of tolerance could, potentially, be used to guide the safe minimization or withdrawal of immunosuppressive therapy in certain transplant recipients. Furthermore, these observations, combined with the identification of B cells producing the T cell immunosuppressive cytokine, interleukin (IL)−10, led to the hypothesis that this signature was indicative of a role for regulatory B cells (Bregs) in clinical tolerance. This hypothesis therefore expands the role for B cells from mediators of rejection to mediators of transplantation tolerance (Fig. 1).
Here we provide an overview on the rapidly evolving area of B cells in transplantation tolerance, discussing the findings of the B cell signature of tolerance, the potential role of IL‐10‐producing B cells as regulators of donor‐specific T cell responses, and the mechanisms that curtail the differentiation of B cells into antibody‐secreting cells during allograft tolerance.
B cell signature and tolerance in humans
When kidney transplant recipients discontinue their immunosuppressive medication, a very small number of patients continue to maintain good graft function for many years 7, 8, 9. These recipients, labelled as ‘operationally tolerant’, have provided a unique opportunity to study the basis of clinical transplant tolerance. Indeed, the peripheral blood of these tolerant patients have been subject to many biomarker discovery investigations to identify a non‐invasive gene signature of tolerance. The investigators reasoned that such a biomarker could be used in clinical trials to evaluate the effectiveness of potential tolerance induction therapies, and identify individuals that may be weaned successfully from immunosuppression and provide insights into the mechanisms of transplantation tolerance 10.
Newell et al. 7 and Sagoo et al. 8 reported on a tolerance signature that comprised enrichment of genes involved in B cell activation and differentiation compared to stable recipients on immunosuppression. This signature from peripheral blood was confirmed with real‐time polymerase chain reaction (PCR) gene expression analyses of urine sedimentary cells, and the three most predictive genes that clearly separated tolerant from immunosuppressed patients were immunoglobulin kappa variable 4–1 (IGKV4–1), immunoglobulin lambda‐like polypeptide 1 (IGLL1) and immunoglobulin kappa chain variable region D‐13 (IGKVD‐13), while flow cytometry analysis confirmed the expansion of total B cells in tolerant compared to immunosuppressed recipients 7, 8. In a separate study, Pallier et al. 11 reported a comparable increase in B cells in the blood of tolerant compared to immunosuppressed patients, especially those with an activated, memory and early memory phenotypes, as well as the up‐regulation of co‐stimulatory and inhibitory [B‐cell scaffold protein with ankyrin repeats 1 (BANK1) and receptors for the Fc region of immunoglobulin G IIB (FcγRIIB)] molecules on those B cells.
One major caveat of those studies was that the B cell signature of tolerance arose from the comparison between tolerant and immunosuppressed recipients, thus the differences observed between these two groups could have been due to the effect of immunosuppression on the B cell differentiation rather than being a unique marker of tolerance that is more likely to be subtle. Indeed, no consistent difference in the gene profile or cell frequencies was detected when tolerant recipients were compared to healthy controls. A number of recent studies have addressed the impact of immune‐suppression on the gene profile and cellular subsets in transplant recipients. Tebbe et al. 12 reported that calcineurin‐inhibitors in renal transplant recipients reduced the numbers of B cells, especially the immature transitional CD19+CD24hiCD38hi regulatory B cells and IL‐10‐producing B cells. Rebollo‐Mesa et al. compared patients on conventional triple immune‐suppression of calcineurin inhibitor with patients on double or monotherapy, and attributed seven of the 10‐gene original tolerance signature to the effects of steroids [CD79b, T cell leukaemia/lymphoma 1A (TCL1A), glucosaminyl 3‐O‐sulphotransferase (H3ST1), Toll‐like receptor (TLR‐5), membrane spanning 4‐domains A1 (MS4A1), Fc receptor‐like 1 (FCRL1), FCRL2] and azathioprine [CD79b, TCL1A, H3ST1, SH2DB1, MS4A1, FCRL1, FCRL2] 13. They showed that patients on azathioprine or prednisone had lower percentages of transitional B cells compared to patients off each of these drugs, with steroids showing a clear dose‐dependent effect. Calcineurin inhibitors had no effect on the tolerant gene signature or B cell frequencies. Finally, when the impact of immunosuppression was accounted for, a gene signature of five differentially expressed biological pathways was identified that differentiated between tolerant, immunosuppressed and healthy controls: nuclear factor kappa B (NF‐κB), CD40, tumour necrosis factor (TNF), granulocyte–macrophage colony‐stimulating factor (GM–CSF) and glucocorticoid receptor regulatory network, with the CD40 pathway preferential to B cells. In addition, Bottomley et al. showed independently that the initial signatures of tolerance were influenced significantly by immunosuppressive agents 14. Specifically, azathioprine decreased transitional and naive B cell numbers, whereas calcineurin inhibition was associated with increased numbers of plasmablast and class‐switched memory B cells. Finally, Leibler et al. reported that in renal transplant patients, treatment with belatacept results in increased transitional and naive B cells, suggesting further that choice of immunosuppression influences B cell populations 15. These data suggest collectively that the previously identified signature of transitional B cells in tolerant recipients is more likely to be a signature of immunosuppression and normalized B cell development in the absence of immunosuppression in tolerant recipients. Nevertheless, these data do not preclude an involvement of regulatory B cells in the development or maintenance of tolerance.
Regulatory B cells as mediators of tolerance
There is accumulating evidence that B cell‐producing immunosuppressive cytokines can curtail T cells responses in autoimmunity, tumour immunity, infectious disease and transplantation tolerance 16, 17, 18, 19, 20. These immunosuppressive B cells have been referred to as Bregs 21, 22, but it remains unclear whether Bregs represent a developmentally specified and stable lineage comparable to forkhead box protein 3 (FoxP3+) regulatory T cells or a differentiated subset of effector cells that secrete immunosuppressive cytokines preferentially, such as IL‐10, IL‐35 and transforming growth factor (TGF)‐β. Despite considerable effort, studies that have performed gene arrays on Breg cells in mice 23 or humans 24 have not identified conclusively a lineage‐specific marker equivalent to FoxP3. In line with the possibility that any B cell might differentiate potentially into a ‘Breg’ cell in response to the right environmental stimuli, B cells that down‐regulate immune responses based on their ability to secrete IL‐10 have now been labelled more conservatively as B10 cells 25.
The lack of a definitive transcription factor or cell surface marker for Bregs has made investigations into their biology challenging, and currently their quantification is dependent upon the expression of IL‐10 either after in‐vitro culture with phorbol myristate acetate (PMA) + ionomycin 25 or with anti‐CD40, TLR agonists and inflammatory cytokines 22, 23. Using both in‐vitro and in‐vivo approaches, Bregs show various surface phenotypes consistent with being splenic marginal zone (MZ) 26, MZ‐precursor (MZ‐P) or transitional 2 (T2) 16, follicular 16, 27 CD1dhiCD5+ B cells 28, pro‐B 29 and plasmablasts/plasma cells 30, 31 (Fig. 2).
Figure 2.
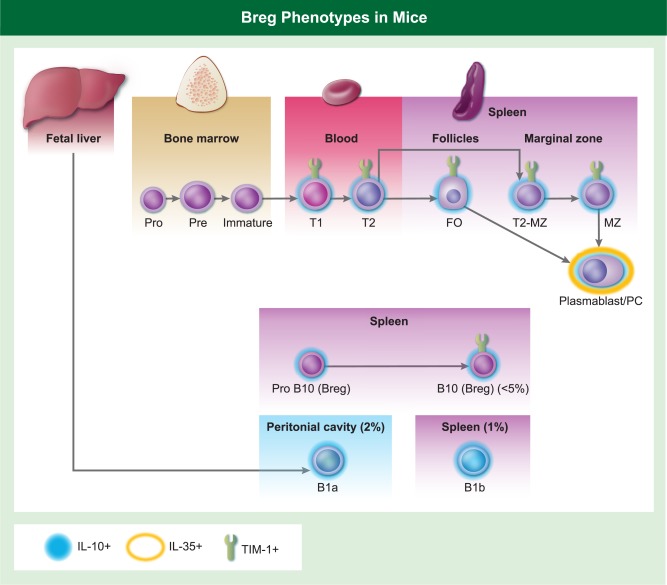
The phenotype of the subsets of murine B cells that have been reported as regulatory B cells (Bregs), as defined by interleukin (IL)−10 or IL‐35 production or T cell immunoglobulin and mucin domain‐1 (TIM‐1) expression under different autoimmune, tumour or transplantation conditions.
In mice, the most widely investigated Breg population comprises the IL‐10‐producing B10 cells expressing CD19+CD5+CD1dhi; these cells have been shown to modulate T cell function in models of experimental autoimmune encephalomyelitis (EAE) 32, inflammatory bowel disease 33, collagen‐induced arthritis 34 and systemic lupus erythematosus (SLE) 35. One way that B10 cells mediate suppression of T helper type 1 (Th1) and Th17 cell differentiation is through the suppression of dendritic cells 22, 36. A different subset of regulatory B cells producing IL‐35 has been reported to protect mouse models of EAE and increase susceptibility to the intracellular pathogen, Salmonella enterica serovar Typhimurium. IL‐35 production by B cells was associated with reduced macrophage activation and inflammatory T cells, as well as decreased ability of B cells to function as antigen‐presenting cells 23. Interestingly, IL‐35 and IL‐10 were produced by immunoglobulin (Ig)M+CD138+CD1dint T cell immunoglobulin and mucin domain 1 (TIM1)int plasma cells expressing the transcription factor B‐lymphocyte‐induced maturation protein 1 (Blimp‐1), whereas CD19+CD138–CD1dhi B cells produced IL‐10 and the proinflammatory cytokine, IL‐6 [23]. Matsumoto et al. also reported that plasmablasts express predominantly IL‐10 during EAE induction, and was essential for the ability of dendritic cells to limit the generation of pathogenic T cells 30. These observations, together with the identification of plasmablasts and plasma cells producing IL‐10, IL‐35, transfer RNA (TNR)‐a, IL‐17 and GM–CSF in various experimental conditions in mice, have led to the hypothesis of distinct subsets of terminally differentiated regulatory and effector B cells regulating and promoting T cell function, independently of antibody secretion 37, 38, 39.
In experimental transplantation models, Ding et al. reported that B cells are required for Th2 cytokine expression induced by anti‐TIM‐1 for prolonged graft acceptance of allogeneic islets 19. They showed that approximately 15% of B cells expressed TIM‐1, and Tim‐1 ligation induced IL‐4 and IL‐10 production by B cells that promoted a Th2 response. In addition, anti‐Tim‐1 treatment increased the percentage of Tim‐1+IL‐10+ B cells with potential regulatory activity. Finally, they showed that TIM‐1 identified the majority of IL‐10‐producing B cells across a wide spectrum of phenotypes, and concluded that TIM‐1 may serve as an inducible and inclusive marker of Bregs. A follow‐up study by Yeung et al. reported that TIM‐1 signalling was necessary for the maintenance and induction of Bregs, and that B cells expressing a mutant form of TIM‐1 that lacked the mucin domain was unable to produce IL‐10 following ligation with anti‐TIM‐1. As anticipated, these mice with the mutant TIM‐1 exhibited accelerated rejection that was prevented with the adoptive transfer of wild‐type (WT) TIM‐1+ B cells 40.
In a model of islet transplantation, allograft tolerance induced by anti‐CD45RB, which targeted a transmembrane protein phosphatase expressed on naive T cells and B cells, required B cells expressing CD40 and CD80/86 41. However, IL‐10 was shown to have an unexpected counter‐regulatory function in an allogeneic heart transplant model where graft survival was extended with anti‐CD45RB 42. By using IL‐10‐deficient recipients, IL‐10 neutralization and adoptive transfer of IL‐10‐deficient B cells into B cell‐deficient recipients, IL‐10‐deficiency enhanced tolerance and improved chronic allograft vasculopathy 42. In a subsequent study, anti‐TIM‐1 was shown to synergize with anti‐CD45RB to induce islet allograft tolerance that was dependent upon B cells, IL‐10, regulatory T cells (Tregs) and TGF‐β 43, 44. These data suggest that the role of IL‐10‐producing B cells may be dependent upon the allograft type as well as the therapeutic agent used to induce allograft tolerance.
IL‐10‐producing B cells from the MZP B cell subset was reported to be necessary for anti‐CD154 plus donor spleen cell‐induced tolerance to cardiac allografts 45. IL‐10 was increased only in the MZP B cells that up‐regulated IL‐21R in tolerant recipients, and that were able to promote graft acceptance in B cell‐specific IL‐10‐deficient recipients. These IL‐10‐producing MZP B cells controlled the differentiation and position of Th17, Tfh and Tfr cells in secondary lymphoid tissues 46, thus providing contrasting mechanistic insights to previous studies on Bregs, and underscoring the diversity in phenotype of IL‐10‐producing B cells that promote transplantation tolerance. Similarly, Durand et al. 47 reported that splenic B cells from rats tolerant to allogeneic hearts were enriched for a CD24intCD38+CD27+IgD‐IgM+/low regulatory subpopulation that expressed granzyme B and interferon regulatory factor 4 (Irf4) as well as inhibitory CD23 and BANK1. Furthermore, the IgD−IgMlow/− but not IgMhi transitional splenic B cells were able to transfer donor‐specific tolerance via IL‐10 and TGF‐β1‐dependent mechanisms, and to suppress in‐vitro TNF‐α.
Bregs within the peripheral blood have been identified in humans to have transitional (immature) and memory phenotypes (Table 1). Blair et al. reported initially that CD19+CD24hiCD38hi B cells from healthy individuals, upon CD40 ligation, have the ability to inhibit the proliferation and differentiation of Th1 cells 48. Their suppressive capacity was mediated in part by IL‐10, and was reversed by anti‐CD80 and CD86, consistent with a cognate‐dependent suppression. In SLE patients, these CD19+CD24hiCD38hi B cells were refractory to CD40 stimulation, produced less IL‐10 and lack suppressive capacity, suggesting that alterations in Breg function contribute to SLE 48. More recently, Flores‐Borja et al. reported that these immature B cells had the additional ability of converting CD4+CD25– conventional T cells (Tconv) into Tregs in an IL‐10‐dependent manner. Of note, Bregs from patients with rheumatoid arthritis failed to convert CD4+CD25– Tconv into Tregs or to curb Th17 differentiation, but maintained their ability to inhibit Th1 development in vitro 49.
Table 1.
Cell surface phenotype of Bregs and Anergic B cells in humans.
| Bregs | Anergic B cells |
|---|---|
|
Transitional: CD19+CD20+CD10+CD27‐CD24hiCD38hi |
Mature naive: IgD+, IgM– |
|
Mature (memory): CD19+CD20+CD10‐CD27+CD24hiCD38‐ |
Mature naive: CD19hiCD40loCD86+CD58hiIL‐4Rlo CD11c+CCR7−/lo CXCR4loCD44loCD62L−/loCD72hiCD32hiCD85j+CD85d+Fas+ |
Bregs = regulatory B cells; Ig = immunoglobulin.
Bregs have also been described for renal transplant recipients. Bregs enriched within the IgM memory and transitional subsets in healthy donors are deficient in chronic graft‐versus‐host disease (GVHD) 50. Chesneau et al. 34 showed that B cells from tolerant patients have lower numbers of plasma cells compared to stable patients, and their activated B cells secreted more IL‐10 compared to healthy subjects and stable immunosuppressed patients. These observations led to the conclusion that the combination of IL‐10 production and defective B cell differentiation into plasma cells favoured the maintenance of transplantation tolerance. Indeed, Bigot et al. reported on the transcriptomic signature of CD24hiCD38hi transitional B cells, and the enrichment of IL‐10 expression in the CD1b+ and inducible T cell co‐stimulatory ligand (ICOS‐L)+ subset 51. More recently, Nova‐Lamperti et al. reported that human transitional B cell production of IL‐10 resulted in down‐regulated CD86 expression and reduced CD4+ T cell responses in vitro 52, whereas higher percentages of B cells producing IL‐10 after CD40 ligation was observed in tolerant kidney recipients compared to healthy controls 53. Finally, Cherukuri et al. demonstrated that while IL‐10 was produced by CD19+CD24hiCD38hi transitional B cells (TrBs), CD24hiCD27+ memory B cells and naive B cells, the TrB subset had the highest IL‐10/TNF‐α ratio and suppressed Th1 cells more potently than memory B cells that expressed similar IL‐10 but more TNF‐α 54. Collectively, it appears that IL‐10 is essential but not a sufficient marker of Bregs, and that reduced production of proinflammatory cytokines by these IL‐10 Bregs is additionally necessary to specify their ability to mediate T cell regulation and transplantation tolerance.
Anergy and regulation as mechanisms of B cell tolerance
There are ample clinical and experimental data showing the ability of alloantibodies to bind to graft endothelium and to mediate antibody‐mediated rejection independently of T cells 55, 56, 57. Therefore, if tolerance is to persist long term, alloantibody production has also to be controlled. Because alloantibody is predominantly T cell‐dependent, it can be argued that restraining the alloreactive T cells, specifically the Tfh subset of cells that provide help to B cells, is sufficient to control B cell responses. However, under infection and inflammatory cytokines, B cells can differentiate into antibody‐secreting plasmablasts and plasma cells with minimal requirement for T cell help, as has been described for autoimmune B cells, so additional B cell intrinsic mechanisms may be essential to reinforce self‐tolerance 58.
Anergy, a condition in which cells persist in the periphery but are unresponsive to antigen, has been shown to be responsible for silencing self‐reactive B cells that comprise more than half the B cell repertoire in mouse and man 59, 60, 61. Multiple transgenic mouse models have been used in the early dissection of mechanisms that underlie anergy and several features that distinguish anergic B cells from naive B cells have been identified, including impaired signal transduction and tyrosine phosphorylation in response to B cell receptor (BCR) aggregation and elevated basal–intracellular–calcium concentration ([Ca2+]i), but no further increase in [Ca2+]i upon BCR stimulation 62, 63, 64, 65, 66. This defective BCR signalling results in reduced lifespan of the anergic B cells, altered migration and anatomical localization, and an inability to interact productively with helper T cells. Continuous inhibitory signalling pathways through tyrosine phosphatase Src homology region 2 1 domain‐containing phosphatase‐1 (SHP‐1) and the SH2‐domain‐containing inositol phosphatase (SHIP‐1) are required to maintain anergy, and inhibition of either of these pathways restored B cell activation, proliferation and generation of short‐lived plasma cells 67. Finally, anergic B cells have been reported as having an immature T3 [IgMmid, CD80hi, CD95hi, major histocompatibility complex (MHC) class IIhi] or a preplasma phenotype depending on the BCR specificity as well as ligand quality and quantity 60.
The fate of endogenous (non‐BCR‐tg) self‐reactive B cells that recognize the ubiquitously expressed transmembrane chicken ovalbumin (OVA) was reported by Taylor et al. 68. They showed that tolerance was maintained by the deletion of BCR‐expressing B cells with high affinity for OVA antigen and by anergy of the remaining B cells expressing low‐affinity BCR to OVA. Despite functional unresponsiveness, the anergic B cells are indistinguishable phenotypically from functional naive OVA‐specific B cells from WT mice based on the expression of CD24 [heat stable antigen (HSA)], CD38, CD40, CD44, CD80, CD86, CD95 (FAS), IgD, IgM and MHC‐II.
Taylor et al. also reported that B cell responses to soluble antigen, glucose 6‐phosphate isomerase, were inhibited as a result of an absence of T cell help 68. T follicular helper (Tfh) CD4+ T cells specialize in providing help to B cells 69 and, more recently, Tfr cells have been identified as a distinct subset of regulatory T cells derived from FoxP3 precursors that express high levels of C‐X‐C chemokine receptor type 5 (CXCR5) [which directs them to the germinal centre (GC)], programmed cell death protein 1 (PD‐1), ICOS 70 and the canonical Tfh transcription factor B cell lymphoma 6 (Bcl6), albeit at a lower level than Tfh cells. Tfr cells have been shown to inhibit multiple states of B cell differentiation, but most importantly, to migrate into the GC to inhibit Tfh‐mediated B cell activation 71, 72, 73, 74, 75. While Linterman et al. reported that Tfr cells limited the outgrowth of non‐antigen‐specific B cells in the GC, thus specifying the GC reaction to antigen‐specific B cells 72, others have reported that Tfr limit antigen‐specific responses 71, 73. CD28 and ICOS are both essential for Tfr cell differentiation, cytotoxic T lymphocyte‐associated protein 4 (CTLA‐4) is essential for their maintenance and function, while PD‐1 on Tfr down‐regulates Tfh differentiation and function 74, 76, 77. Sage et al. 78 showed subsequently that while key transcriptional signatures defining Tfh and its activation were maintained by Tfh from the Tfh–Tfr–B cell co‐cultures, selective effector molecules and metabolic pathways were suppressed in ways that were distinct from T cell anergy or exhaustion. Similarly, Tfr‐suppressed B cells underwent early activation and limited proliferation, but did not express the complete transcriptional programme of fully differentiated antibody‐secreting cells. Specifically, the Myc, mechanistic target of rapamycin (mTOR) and metabolic pathways were suppressed in B cells during co‐culture with Tfr and Tfh, and shown to be durable even in subsequent culture with Tfh cells and in the absence of Tfr cells. This imprinting of Tfr suppression was shown to be due, at least in part, to epigenetic changes that could be overcome with IL‐21 that acted on Tfr to diminish its suppressive capacity and on B cells to restore their differentiation into antibody‐secreting cells. The authors speculated that Tfr suppressed Tfh and B cells by interrupting bidirectional co‐stimulation of T cell–B cell immunological synapses 70, 75. Consistent with defective Tfh promoting B cell tolerance, Chenouard et al. 79 recently compared Tfh cells in operationally tolerant with stable kidney transplant recipients to reveal a defect in their ability to stimulate IgG production by naive B cells, and reduced IL‐21 production compared to Tfh cells from non‐tolerant recipients.
Anergic B cells have also been described in human peripheral blood. Duty et al. described a population of naive IgD+, IgM– phenotype B cells from healthy humans that expressed autoreactive immunoglobulin receptors and were functionally anergic upon BCR engagement 80. Smith et al. reported on the loss of high‐affinity insulin‐binding functionally anergic B cells with the same naive IgD+, IgM– phenotype in prediabetic and new‐onset type 1 diabetic patients 81, while an expanded population of CD21‐/lo B cells was identified in individuals prone to autoimmune disease 82. These B cells were mainly autoreactive and unable to induce calcium flux, become activated or proliferate in response to BCR and/or CD40 triggering, but were able to respond to TLR engagement. Transcriptional analysis revealed that these anergic B cells down‐regulated the genes encoding activation and survival, including complement binding molecules, CD40, OX40L and B cell maturation antigen (BCMA), IL‐4R and IL‐13R while up‐regulating receptors with the immunoreceptor tyrosine‐based inhibition motif (ITIM) that inhibit B cell activation and proliferation, such as CD72, Fc receptors CD32/Fc fragment of Ig receptor IIb (FCGR2B) and FCRL5/Ig superfamily receptor translocation‐associated 2 (IRTA2), leucocyte Ig‐like receptor subfamily B member 1 (LILRB), sialic acid binding Ig‐like lectins (SIGLEC). Collectively, these data suggest that autoreactive B cells that are hyporesponsive to BCR engagement circulate in healthy individuals, but when this anergy is overcome by TLR engagement the B cells differentiate into plasma cells that secrete autoreactive antibodies.
Whether transplantation tolerance also induces a state of hyporesponsiveness on alloreactive B cells has not been well characterized. In models of central tolerance achieved through mixed bone marrow chimerism 83, 84, B cell tolerance was shown to be dependent upon early anergy followed by the deletion. In a model of peripheral allograft tolerance induced by co‐stimulation blockade to allogeneic heart grafts in BCR‐transgenic recipients, Li et al. 85 reported that alloreactive B cells were deleted, whereas in a non‐BCR‐transgenic model of co‐stimulation‐induced tolerance, restoration of alloantibody responses required both the deletion of CD25+ cells and the reconstitution of alloreactive B cells 86. Whether the alloreactive B cells were deleted or dysfunctional in the non‐BCR‐transgenic model were not ascertained because of the inability to track the alloreactive B cells. Overall, compared to B cells with regulatory activity, the investigation into the basis for cell intrinsic B cell tolerance in transplantation is more limited. Nevertheless, evidence for anergy as a major mechanism controlling peripheral autoreactive B cells, either through the down‐modulation of BCR signalling or suppression by Tfr cells, suggests that similar mechanisms may also be at play in allograft tolerance.
Conclusion
This review summarizes literature investigating B cells in transplantation tolerance, their limitations and future direction (Table 2). Investigations into biomarkers of tolerance in the peripheral blood of tolerant renal transplant recipients suggested a key finding of the preservation of a B cell compartment with IL‐10 production capacity and a deficiency in antibody‐producing plasma cells. While these findings have been tempered by recent reports that immunosuppressive agents have profound effects on B cell subsets, B cells producing suppressive cytokines, including IL‐10 and IL‐35, to control T cell responses may nevertheless be essential contributors to transplantation tolerance. Indeed, there is considerable literature supporting this notion, although details of Breg provenance remain controversial. Furthermore, the control of T cell help may be over‐ridden by inflammatory cues, so additional B cell‐intrinsic mechanisms may be necessary for robust tolerance. The mechanistic insights into Bregs and B cell dysfunction may lead to new therapeutic approaches and guide ongoing efforts to identify reliable biomarkers for transplantation tolerance.
Table 2.
Summary of major points of discussion and future directions.
| Major findings | Future directions |
|---|---|
| Enriched B cell signature of tolerance in renal transplantation tolerance compared to stable immunosuppressed patients is influenced strongly by the effects of immunosuppressive drugs | Comparison of signatures of PBMC and B cells from tolerant compared to healthy controls |
| B cells producing immunosuppressive cytokines such as IL‐10 and IL‐35 may modulate alloimmune responses and promote allograft tolerance | Better understanding of how immunomodulatory B cells function to inhibit T cell responses, signals that lead to their differentiation and/or expansion and identification of markers to allow their easy quantification |
| Control of B cell and antibody responses are critical for sustained tolerance and maintenance of allograft function. This can be achieved by the control of Tfh cells or B cell intrinsic hyporesponsiveness/anergy | Mechanistic studies on how Tfh cell differentiation is prevented in tolerance, and biomarker/mechanistic studies on allospecific B cell hyporesponsiveness/anergy |
IL = interleukin; PBMC = peripheral blood mononuclear cells; Tfh = T follicular helper.
Disclosure
The authors do not have any disclosures.
Acknowledgements
This work was supported in part by grants (R01AI110513, P01AI097113) from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health.
References
- 1. Loupy A, Hill GS, Jordan SC. The impact of donor‐specific anti‐HLA antibodies on late kidney allograft failure. Nat Rev Nephrol 2012; 8:348–57. [DOI] [PubMed] [Google Scholar]
- 2. Puttarajappa C, Shapiro R, Tan HP. Antibody‐mediated rejection in kidney transplantation: a review. J Transplant 2012; 2012:193724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hidalgo LG, Sellares J, Sis B, Mengel M, Chang J, Halloran PF. Interpreting NK cell transcripts versus T cell transcripts in renal transplant biopsies. Am J Transplant 2012; 12:1180–91. [DOI] [PubMed] [Google Scholar]
- 4. Thomas KA, Valenzuela NM, Reed EF. The perfect storm: HLA antibodies, complement, FcgammaRs, and endothelium in transplant rejection. Trends Mol Med 2015; 21:319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meehan SM, Kremer J, Ali FN, et al Thrombotic microangiopathy and peritubular capillary C4d expression in renal allograft biopsies. Clin J Am Soc Nephrol 2011; 6:395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Djamali A, Kaufman DB, Ellis TM, Zhong W, Mates A, Samaniego M. Diagnosis and management of antibody‐mediated rejection: current status and novel approaches. Am J Transplant 2014; 14:255–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Newell KA, Asare A, Kirk AD, et al Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest 2010; 120:1836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sagoo P, Perucha E, Sawitzki B, et al Development of a cross‐platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest 2010; 120:1848–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Massart A, Pallier A, Pascual J, et al The DESCARTES‐Nantes survey of kidney transplant recipients displaying clinical operational tolerance identifies 35 new tolerant patients and 34 almost tolerant patients. Nephrol Dial Transplant 2016; 31:1002–13. [DOI] [PubMed] [Google Scholar]
- 10. Hernandez‐Fuentes MP, Lechler RI. A ‘biomarker signature’ for tolerance in transplantation. Nat Rev Nephrol 2010; 6:606–13. [DOI] [PubMed] [Google Scholar]
- 11. Pallier A, Hillion S, Danger R, et al Patients with drug‐free long‐term graft function display increased numbers of peripheral B cells with a memory and inhibitory phenotype. Kidney Int 2010; 78:503–13. [DOI] [PubMed] [Google Scholar]
- 12. Tebbe B, Wilde B, Ye Z, et al Renal transplant recipients treated with calcineurin‐inhibitors lack circulating immature transitional CD19+CD24hiCD38hi regulatory B‐lymphocytes. PLOS ONE 2016; 11:e0153170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rebollo‐Mesa I, Nova‐Lamperti E, Mobilio P, et al Biomarkers of tolerance in kidney transplantation: are we predicting tolerance or response to immunosuppressive treatment? Am J Transplant 2016; 16(12):3443–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bottomley MJ, Chen M, Fuggle S, Harden PN, Wood KJ. Application of operational tolerance signatures are limited by variability and type of immunosuppression in renal transplant recipients: a cross‐sectional study. Transplant Direct 2017; 3:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leibler C, Matignon M, Pilon C, et al Kidney transplant recipients treated with belatacept exhibit increased naive and transitional B cells. Am J Transplant 2014; 14:1173–82. [DOI] [PubMed] [Google Scholar]
- 16. Evans JG, Chavez‐Rueda KA , Eddaoudi A, et al Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol 2007; 178:7868–78. [DOI] [PubMed] [Google Scholar]
- 17. Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10‐producing B cells. J Exp Med 2003; 197:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schioppa T, Moore R, Thompson RG, et al B regulatory cells and the tumor‐promoting actions of TNF‐alpha during squamous carcinogenesis. Proc Natl Acad Sci USA 2011; 108:10662–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ding Q, Yeung M, Camirand G, et al Regulatory B cells are identified by expression of TIM‐1 and can be induced through TIM‐1 ligation to promote tolerance in mice. J Clin Invest 2011; 121:3645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das A, Ellis G, Pallant C, et al IL‐10‐producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J Immunol 2012; 189:3925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL‐10. Nat Immunol 2002; 3:944–50. [DOI] [PubMed] [Google Scholar]
- 22. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity 2015; 42:607–12. [DOI] [PubMed] [Google Scholar]
- 23. Shen P, Roch T, Lampropoulou V, et al IL‐35‐producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014; 507:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van de Veen W, Stanic B, Yaman G, et al IgG4 production is confined to human IL‐10‐producing regulatory B cells that suppress antigen‐specific immune responses. J Allergy Clin Immunol 2013; 131:1204–12. [DOI] [PubMed] [Google Scholar]
- 25. Tedder TF. B10 cells: a functionally defined regulatory B cell subset. J Immunol 2015; 194:1395–401. [DOI] [PubMed] [Google Scholar]
- 26. Lenert P, Brummel R, Field EH, Ashman RF. TLR‐9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL‐10 production. J Clin Immunol 2005; 25:29–40. [DOI] [PubMed] [Google Scholar]
- 27. Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc Natl Acad Sci USA 2007; 104:14080–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell‐dependent inflammatory responses. Immunity 2008; 28:639–50. [DOI] [PubMed] [Google Scholar]
- 29. Stolp J, Turka LA, Wood KJ. B cells with immune‐regulating function in transplantation. Nat Rev Nephrol 2014; 10:389–97. [DOI] [PubMed] [Google Scholar]
- 30. Matsumoto M, Baba A, Yokota T, et al Interleukin‐10‐producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity 2014; 41:1040–51. [DOI] [PubMed] [Google Scholar]
- 31. Neves P, Lampropoulou V, Calderon‐Gomez E, et al Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during Salmonella typhimurium infection. Immunity 2010; 33:777–90. [DOI] [PubMed] [Google Scholar]
- 32. Ray A, Basu S. Regulatory B cells in experimental autoimmune encephalomyelitis (EAE). Methods Mol Biol 2014; 1190:243–55. [DOI] [PubMed] [Google Scholar]
- 33. Maseda D, Candando KM, Smith SH, et al Peritoneal cavity regulatory B cells (B10 cells) modulate IFN‐gamma+CD4+ T cell numbers during colitis development in mice. J Immunol 2013; 191:2780–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chesneau M, Michel L, Degauque N, Brouard S. Regulatory B cells and tolerance in transplantation: from animal models to human. Front Immunol 2013; 4:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haas KM, Watanabe R, Matsushita T, et al Protective and pathogenic roles for B cells during systemic autoimmunity in NZB/W F1 mice. J Immunol 2010; 184:4789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol 2015; 27:479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen P, Fillatreau S. Antibody‐independent functions of B cells: a focus on cytokines. Nat Rev Immunol 2015; 15:441–51. [DOI] [PubMed] [Google Scholar]
- 38. Fillatreau S. Regulatory plasma cells. Curr Opin Pharmacol 2015; 23:1–5. [DOI] [PubMed] [Google Scholar]
- 39. Dang VD, Hilgenberg E, Ries S, Shen P, Fillatreau S. From the regulatory functions of B cells to the identification of cytokine‐producing plasma cell subsets. Curr Opin Immunol 2014; 28:77–83. [DOI] [PubMed] [Google Scholar]
- 40. Yeung MY, Ding Q, Brooks CR, et al TIM‐1 signaling is required for maintenance and induction of regulatory B cells. Am J Transplant 2015; 15:942–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deng S, Moore DJ, Huang X, et al Cutting edge: transplant tolerance induced by anti‐CD45RB requires B lymphocytes. J Immunol 2007; 178:6028–32. [DOI] [PubMed] [Google Scholar]
- 42. Zhao G, Moore DJ, Lee KM, et al An unexpected counter‐regulatory role of IL‐10 in B‐lymphocyte‐mediated transplantation tolerance. Am J Transplant 2010; 10:796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee KM, Kim JI, Stott R, et al Anti‐CD45RB/anti‐TIM‐1‐induced tolerance requires regulatory B cells. Am J Transplant 2012; 12:2072–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee KM, Stott RT, Zhao G, et al TGF‐beta‐producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol 2014; 44:1728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lal G, Nakayama Y, Sethi A, et al Interleukin‐10 from marginal zone precursor B‐cell subset is required for costimulatory blockade‐induced transplantation tolerance. Transplantation 2015; 99:1817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lal G, Kulkarni N, Nakayama Y, et al IL‐10 from marginal zone precursor B cells controls the differentiation of Th17, Tfh and Tfr cells in transplantation tolerance. Immunol Lett 2016; 170:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Durand J, Huchet V, Merieau E, et al Regulatory B cells with a partial defect in CD40 signaling and overexpressing granzyme B transfer allograft tolerance in rodents. J Immunol 2015; 195:5035–44. [DOI] [PubMed] [Google Scholar]
- 48. Blair PA, Norena LY, Flores‐Borja F, et al CD19(+)CD24(hi) CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity 2010; 32:129–40. [DOI] [PubMed] [Google Scholar]
- 49. Flores‐Borja F, Bosma A, Ng D, et al CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med 2013; 5:173ra23. [DOI] [PubMed] [Google Scholar]
- 50. Khoder A, Sarvaria A, Alsuliman A, et al Regulatory B cells are enriched within the IgM memory and transitional subsets in healthy donors but are deficient in chronic GVHD. Blood 2014; 124:2034–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bigot J, Pilon C, Matignon M, et al Transcriptomic signature of the CD24hi CD38hi transitional B cells associated with an immunoregulatory phenotype in renal transplant recipients. Am J Transplant 2016; 16(12):3430–42. [DOI] [PubMed] [Google Scholar]
- 52. Nova‐Lamperti E, Fanelli G, Becker PD, et al IL‐10‐produced by human transitional B‐cells down‐regulates CD86 expression on B‐cells leading to inhibition of CD4+T‐cell responses. Sci Rep 2016; 6:20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nova‐Lamperti E, Chana P, Mobillo P, et al Increased CD40 ligation and reduced BCR signalling leads to higher IL‐10 production in B cells from tolerant kidney transplant patients. Transplantation 2016. doi: 10.1097/TP.0000000000001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cherukuri A, Rothstein DM, Clark B, et al Immunologic human renal allograft injury associates with an altered IL‐10/TNF‐alpha expression ratio in regulatory B cells. J Am Soc Nephrol 2014; 25:1575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Valenzuela NM, Reed EF. Antibodies in transplantation: the effects of HLA and non‐HLA antibody binding and mechanisms of injury. Methods Mol Biol 2013; 1034:41–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mauiyyedi S, Colvin RB. Humoral rejection in kidney transplantation: new concepts in diagnosis and treatment. Curr Opin Nephrol Hypertens 2002; 11:609–18. [DOI] [PubMed] [Google Scholar]
- 57. Lederer SR, Kluth‐Pepper B, Schneeberger H, Albert E, Land W, Feucht HE. Impact of humoral alloreactivity early after transplantation on the long‐term survival of renal allografts. Kidney Int 2001; 59:334–41. [DOI] [PubMed] [Google Scholar]
- 58. Dorner T, Giesecke C, Lipsky PE. Mechanisms of B cell autoimmunity in SLE. Arthritis Res Ther 2011; 13:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science 2003; 301:1374–7. [DOI] [PubMed] [Google Scholar]
- 60. Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B‐cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol 2007; 7:633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yarkoni Y, Getahun A, Cambier JC. Molecular underpinning of B‐cell anergy. Immunol Rev 2010; 237:249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eris JM, Basten A, Brink R, Doherty K, Kehry MR, Hodgkin PD. Anergic self‐reactive B cells present self antigen and respond normally to CD40‐dependent T‐cell signals but are defective in antigen‐receptor‐mediated functions. Proc Natl Acad Sci USA 1994; 91:4392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goodnow CC, Crosbie J, Adelstein S, et al Altered immunoglobulin expression and functional silencing of self‐reactive B lymphocytes in transgenic mice. Nature 1988; 334:676–82. [DOI] [PubMed] [Google Scholar]
- 64. Benschop RJ, Aviszus K, Zhang X, Manser T, Cambier JC, Wysocki LJ. Activation and anergy in bone marrow B cells of a novel immunoglobulin transgenic mouse that is both hapten specific and autoreactive. Immunity 2001; 14:33–43. [DOI] [PubMed] [Google Scholar]
- 65. Cooke MP, Heath AW, Shokat KM, et al Immunoglobulin signal transduction guides the specificity of B cell–T cell interactions and is blocked in tolerant self‐reactive B cells. J Exp Med 1994; 179:425–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Healy JI, Dolmetsch RE, Timmerman LA, et al Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity 1997; 6:419–28. [DOI] [PubMed] [Google Scholar]
- 67. Getahun A, Beavers NA, Larson SR, Shlomchik MJ, Cambier JC. Continuous inhibitory signaling by both SHP‐1 and SHIP‐1 pathways is required to maintain unresponsiveness of anergic B cells. J Exp Med 2016; 213:751–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Taylor JJ, Martinez RJ, Titcombe PJ, et al Deletion and anergy of polyclonal B cells specific for ubiquitous membrane‐bound self‐antigen. J Exp Med 2012; 209:2065–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014; 41:529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sage PT, Sharpe AH. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol 2015; 36:410–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chung Y, Tanaka S, Chu F, et al Follicular regulatory T cells expressing Foxp3 and Bcl‐6 suppress germinal center reactions. Nat Med 2011; 17:983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Linterman MA, Pierson W, Lee SK, et al Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 2011; 17:975–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wollenberg I, Agua‐Doce A, Hernandez A, et al Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol 2011; 187:4553–60. [DOI] [PubMed] [Google Scholar]
- 74. Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD‐1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol 2013; 14:152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev 2016; 271:246–59. [DOI] [PubMed] [Google Scholar]
- 76. Sage PT, Alvarez D, Godec J, von Andrian UH, Sharpe AH. Circulating T follicular regulatory and helper cells have memory‐like properties. J Clin Invest 2014; 124:5191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen‐specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA‐4. Immunity 2014; 41:1013–25. [DOI] [PubMed] [Google Scholar]
- 78. Sage PT, Ron‐Harel N, Juneja VR, et al Suppression by TFR cells leads to durable and selective inhibition of B cell effector function. Nat Immunol 2016; 17:1436–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chenouard A, Chesneau M, Bui Nguyen L, et al Renal operational tolerance is associated with a defect of blood Tfh cells that exhibit impaired B cell help. Am J Transplant 2016; doi: 10.1111/ajt.14142. [DOI] [PubMed] [Google Scholar]
- 80. Duty JA, Szodoray P, Zheng NY, et al Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J Exp Med 2009; 206:139–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Smith MJ, Packard TA, O'Neill SK, et al Loss of anergic B cells in prediabetic and new‐onset type 1 diabetic patients. Diabetes 2015; 64:1703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Isnardi I, Ng YS, Menard L, et al Complement receptor 2/CD21‐ human naive B cells contain mostly autoreactive unresponsive clones. Blood 2010; 115:5026–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ohdan H, Yang YG, Shimizu A, Swenson KG, Sykes M. Mixed chimerism induced without lethal conditioning prevents T cell‐ and anti‐Gal alpha 1,3Gal‐mediated graft rejection. J Clin Invest 1999; 104:281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kawahara T, Shimizu I, Ohdan H, Zhao G, Sykes M. Differing mechanisms of early and late B cell hyporesponsiveness induced by mixed chimerism. Am J Transplant 2005; 5:2821–9. [DOI] [PubMed] [Google Scholar]
- 85. Li Y, Ma L, Shen J, Chong AS. Peripheral deletion of mature alloreactive B cells induced by costimulation blockade. Proc Natl Acad Sci USA 2007; 104:12093–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li Y, Ma L, Yin D, Shen J, Chong AS. Long‐term control of alloreactive B cell responses by the suppression of T cell help. J Immunol 2008; 180:6077–84. [DOI] [PMC free article] [PubMed] [Google Scholar]