

Summary
Neutrophils play a crucial role in the pathophysiology of rheumatoid arthritis (RA) via the release of reactive oxygen species (ROS), proteases and cytokines. Orally active Janus kinase (JAK) inhibitors (JAKi), e.g. baricitinib and tofacitinib, have high clinical efficacy in RA but are linked with neutropenia and increased infections. Our aim was to determine the effect of JAK inhibition with baricitinib and tofacitinib on healthy control and RA neutrophil lifespan and function. RA (n = 7) and healthy control (n = 7) neutrophils were treated with baricitinib or tofacitinib for 30 min, prior to incubation in the absence or presence of granulocyte–macrophage colony‐stimulating factor (GM‐CSF) or interferon (IFN)‐γ. JAKi prevented GM‐CSF‐ and IFN‐γ‐induced apoptosis delay in RA and healthy control neutrophils in a dose‐dependent manner. Baricitinib decreased the rate of chemotaxis towards interleukin (IL)‐8, but not f‐Met‐Leu‐Phe (fMLP) in RA neutrophils. While healthy control neutrophils incubated with GM‐CSF became primed to produce ROS in response to stimulation with fMLP and phorbol‐12‐myristate‐12‐acetate (PMA), RA neutrophils produced increased levels of ROS without the need for priming. JAKi prevented ROS release from primed healthy control neutrophils in response to fMLP, but had no effect on ROS production by RA neutrophils. Baricitinib reversed GM‐CSF priming of ROS production in response to fMLP in healthy control, but not RA, neutrophils. We conclude that incubation with JAKi prevents chemotaxis of RA neutrophils towards IL‐8, but does not prevent the production of ROS or increase the level of apoptosis. This may be due to the in‐vivo exposure of RA neutrophils to priming agents other than those that activate JAK/signal transducer and activator of transcription (STAT) signalling.
Keywords: apoptosis, baricitinib, JAK inhibitors, neutrophils, rheumatoid arthritis, ROS, tofacitinib
Introduction
Orally active JAK inhibitors (JAKi), including tofacitinib (JAK3/JAK1 inhibitor) and baricitinib (JAK1/JAK2 inhibitor), show efficacy in treating rheumatoid arthritis (RA) patients who are refractory to disease‐modifying anti‐rheumatic drugs (DMARDs) and biological therapies, including tumour necrosis factor inhibitors (TNFi) 1, 2, 3, 4, 5. The molecular mechanisms of JAK inhibitors in decreasing disease activity in RA are still emerging. For example, tofacitinib decreases interleukin (IL)‐17/interferon (IFN)‐γ production and CD4+ T cell proliferation 6, and suppresses synovial production of matrix metalloproteinases (MMPs) and chemokines via decreased signal transducer and activator of transcription (STAT)‐1 and STAT‐3 activation 7. However, while baricitinib is clinically effective in the treatment of RA 4, to date there are no published studies on the molecular effects of baricitinib at the cellular level in RA or any other disease.
Neutrophils are implicated in the pathophysiology of RA via secretion of degradative enzymes, such as elastase and collagenase, and reactive oxygen species (ROS) 8, 9, 10. Neutrophils also contribute to the cytokine/chemokine cascades that accompany inflammation and regulate immune responses 11. Production of neutrophil extracellular traps (NETs), containing citrullinated peptide residues, is implicated in the production of anti‐citrullinated peptide antibodies (ACPA) and development of RA 12, 13, 14, 15, and neutrophils are one of the few cells expressing high levels of active protein arginine deiminase 4 (PAD4), the enzyme principally responsible for the citrullination of peptides 8, 15, 16. However, neutrophils play a key role in immunity and host protection against micro‐organisms via phagocytosis and production of ROS 10, and therefore drug‐induced neutropenia and/or impairment of normal neutrophil function has serious implications for host defence in immunosuppressed patients. Clinical trials of JAKi have reported transient drops in neutrophil counts during therapy 1, 4, 5, 17 and increased rates of infection in patients receiving JAKi [compared to tumour necrosis factor inhibitor (TNFi) or placebo]. More specifically, clinical trials have reported increased cases of bronchitis, herpes zoster, influenza and urinary and upper respiratory tract infections in patients receiving JAKi 2, 3, 5, 18, suggesting drug‐induced neutropenia and/or decreased neutrophil function.
The aim of this study was to investigate the effect of JAK inhibition with baricitinib (JAK1/2 inhibitor) and tofacitinib (JAK3 inhibitor) on the normal neutrophil response to in‐vitro cytokine priming in healthy controls, and on the ex‐vivo‐activated phenotype of neutrophils isolated from the peripheral blood of patients with RA. We present data showing the effect of baricitinib and tofacitinib on three key aspects of neutrophil function which are important in the response to infection, but are also implicated in unwanted activation during inflammatory disease: apoptosis, chemotaxis and ROS production.
Methods
Materials
Ficoll‐Paque, horseradish peroxidase (HRP)‐linked anti‐rabbit immunoglobulin (Ig)G antibody, photographic film (GE Healthcare, Chalfont St Giles, UK); RPMI‐1640 media, annexin V–fluorescein isothiocyanate (FITC) (Life Technologies, Paisley, UK); 3 μm porous membrane hanging cell inserts, phosphatase inhibitor cocktail II, polyvinylidene difluoride (PVDF) membrane, enhanced chemiluminescence (ECL) reagent (Merck, Watford, UK); propidium iodide (PI), luminol, phorbol 12‐myristate 13‐acetate (PMA), human AB serum, f‐Met‐Leu‐Phe (fMLP), IL‐8, HRP‐linked anti‐mouse IgG antibody, poly‐HEMA (Sigma, Gillingham, UK); actin antibody (Abcam, Cambridge, UK); phosphorylated‐STAT‐1, phosphorylated‐STAT‐3 antibodies (New England Biosciences, Hitchin, UK); tofacitinib, baricitinib (Stratech, Newmarket, UK); and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) and IFN‐γ (Roche Applied Sciences, Burgess Hill, UK).
Patients and controls
This study was approved by the University of Liverpool Committee on Research Ethics for healthy controls and NRES Committee North West (Greater Manchester West, UK) for RA patients. All participants gave written, informed consent. Patients with active RA were recruited from University Hospital Aintree. Clinical demographics are shown in Table 1. All patients were receiving DMARD therapy with methotrexate (n = 7), along with concomitant hydroxychloriquine (n = 4), sulphasalazine (n = 3) and/or leflunamide (n = 1).
Table 1.
Clinical demographics of rheumatoid arthritis (RA) patients included in the study
| n | 7 |
| Age (years)*† | 58 (42–84) |
| Sex: male, female | 4, 3 |
| Disease duration (years)*† | 5 (0–11) |
| RF+ | 5 |
| ACPA+ | 3 |
| CRP (g/l)*† | 11 (2–28) |
| ESR (mm/h)*† | 23 (12–40) |
| DAS28*† | 4·67 (3·1–6·6) |
| Current therapy | |
| Methotrexate | 7 |
| Hydroxychloriquine | 4 |
| Sulfasalazine | 3 |
| Leflunamide | 1 |
*Mean; †range.
Isolation of neutrophils
Neutrophils (purity typically > 97%) were isolated from heparinized whole blood using Ficoll‐Paque, as described previously 9, 19, and resuspended in RPMI‐1640 media plus 25 mM Hepes. Neutrophils were incubated with therapeutically and experimentally relevant concentrations 6, 20 of tofacitinib (200 ng/ml) and baricitinib (200 ng/ml) for 30 min prior to assay. Dimethylsulphoxide (DMSO) was used as a vehicle control in all incubations at the same concentration as JAKi (v/v).
Measurement of apoptosis
Neutrophils (106/ml) were incubated at 37°C in 5% CO2 in RPMI‐1640 (+Hepes, +L‐glutamine, +10% AB serum) for up to 20 h in the absence/presence of GM‐CSF (5 ng/ml) or IFN‐γ (10 ng/ml). Apoptosis was measured using annexin V–FITC/PI (1 μg/ml) staining 19 before analysis by flow cytometry (> 5000 events analysed) using a Dako CyAn flow cytometer. Apoptotic cells were defined as those being positive for annexin V.
Chemotaxis assay
The chemotaxis assay was carried out in 24‐well tissue culture plates (coated with 12 mg/ml poly‐HEMA to prevent cell adhesion) using hanging chamber inserts with a 3 μM porous membrane to separate media in the top and bottom chambers, as described previously 21. fMLP (10−8 M) or IL‐8 (100 ng/ml) was added to RPMI media in the bottom chamber. Neutrophils (106/ml) were added to the top chamber and incubated for 90 min at 37°C and 5% CO2. The number of migrated cells after 90 min incubation was measured using a Coulter Counter Multisizer‐3 (Beckman Coulter, High Wycombe, UK).
Measurement of the respiratory burst
Neutrophils (5 × 106/ml) were incubated with JAK inhibitors for 30 min prior to GM‐CSF (5 ng/ml) priming for 45 min. ROS production was stimulated with fMLP (1 μM) or PMA (100 ng/ml). Luminol‐ECL (luminol, 10 μM) was measured continuously for 30 min using a Tecan plate reader at 37°C. For reverse‐priming experiments, neutrophils were incubated with GM‐CSF (5 ng/ml) prior to the addition of JAKi. ROS production was measured in response to fMLP and PMA at 30‐min intervals for up to 120 min after addition of JAKi.
Protein analysis
Neutrophils were incubated as detailed in the Results, with or without the addition of JAKi (0–200 ng/ml), GM‐CSF (5 ng/ml), IFN‐γ (10 ng/ml), fMLP (10−8 M) or IL‐8 (100 ng/ml) for up to 60 min. Neutrophils were lysed in boiling, reducing Laemmli buffer containing phosphatase inhibitor cocktail II, and proteins separated by 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) prior to transfer onto PVDF membrane. Primary antibodies were: anti‐phosphorylated STAT‐1 (1 : 1000), anti‐phosphorylated STAT‐3 (1 : 1000) and actin (1 : 10 000). Secondary antibodies were HRP‐linked anti‐rabbit IgG (1 : 20 000) and HRP‐linked anti‐mouse IgG (1 : 10 000). Bound antibodies were detected using the ECL system on carefully exposed film to avoid saturation. Protein expression was measured as relative abundance to actin loading control.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 5.
Results
Effect of JAK inhibition on neutrophil apoptosis
Delayed neutrophil apoptosis is an important response to infection, but an unwanted phenomenon in inflammatory diseases such as RA 22. We determined the dose‐dependent effect of JAKi on neutrophil apoptosis using healthy control neutrophils incubated with baricitinib and tofacitinib for 20 h over a range of clinically relevant concentrations, with and without the addition of GM‐CSF or IFN‐γ, cytokines known to signal via the JAK/STAT pathway. GM‐CSF and IFN‐γ delayed neutrophil apoptosis significantly compared to unstimulated (constitutive) levels (Fig. 1a, n = 4, §P < 0·05). Neither JAKi exerted any effect on the constitutive rate of neutrophil apoptosis. GM‐CSF‐delayed apoptosis was abrogated by both baricitinib and tofacitinib over a range of concentrations (10–200 ng/ml) (Fig. 1a, n = 4, *P < 0·05). While baricitinib inhibited IFN‐γ‐delayed apoptosis at higher concentrations (100–200 ng/ml), tofacitinib had a significant effect only at the highest concentration (200 ng/ml). The specificity of the inhibition of GM‐CSF‐ and IFN‐γ‐induced JAK signalling was confirmed by Western blotting for STAT proteins known to be unphosphorylated in healthy control neutrophils 23, 24, but activated by both cytokines (STAT‐3 by GM‐CSF; STAT‐1 by IFN‐γ). Neutrophils were incubated with increasing concentrations of baricitinib or tofacitinib (0–200 ng/ml) for 30 min prior to the addition of GM‐CSF (5 ng/ml) or IFN‐γ (10 ng/ml). Baricitinib and tofacitinib both blocked the phosphorylation of STAT‐3 by GM‐CSF at concentrations > 50 ng/ml (Fig. 1b, n = 3, *P < 0·05). However, significant inhibition of STAT‐1 phosphorylation by IFN‐γ was obtained only when incubating with baricitinib at 100 and 200 ng/ml and tofacitinib at the highest concentration (200 ng/ml, Fig. 1b, n = 3, *P < 0·05). A concentration of 200 ng/ml was therefore used for both drugs for further experiments.
Figure 1.
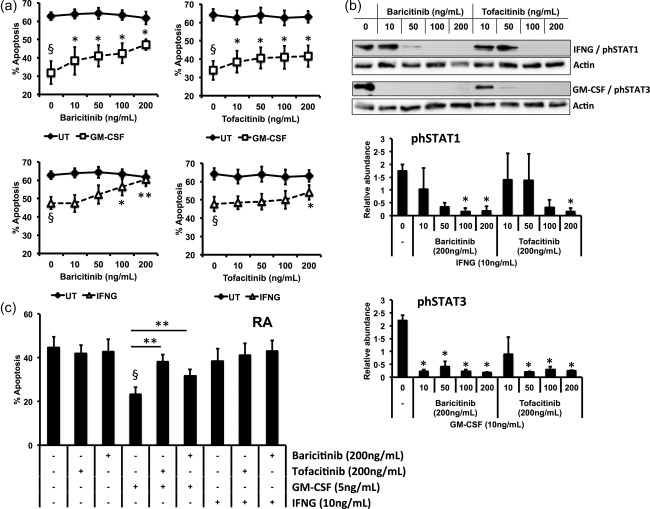
Effect of Janus kinase (JAK) inhibition on neutrophil apoptosis. Neutrophils from healthy controls (a,b) and (c) rheumatoid arthritis (RA) patients were incubated with baricitinib or tofacitinib over a range of concentrations (50–200 ng/ml) for 30 min prior to addition of granulocyte–macrophage‐colony stimulating factor (GM‐CSF) (5 ng/ml) or interferon (IFN)‐γ, 10 ng/ml) for 20 h. Dimethylsulphoxide (DMSO) was used as a vehicle control. (a) GM‐CSF‐ and IFN‐γ‐delayed apoptosis (§P < 0·01) was abrogated by baricitinib and tofacitinib in a dose‐dependent manner (*P < 0·05, **P < 0·01). (b) Phosphorylation of signal transducer and activator of transcription (STAT)‐3 by GM‐CSF and STAT‐1 by IFN‐γ occurred after 15 min incubation, and was inhibited by high concentrations of baricitinib and tofacitinib (n = 3, *P < 0·05). (c) GM‐CSF‐delayed apoptosis was abrogated by baricitinib and tofacitinib (200 ng/ml) in RA neutrophils (*P < 0·05; **P < 0·01).
We next measured the effect of JAK inhibition by baricitinib and tofacitinib at the highest concentration (200 ng/ml) on apoptosis in neutrophils from patients with RA, with and without stimulation with GM‐CSF and IFN‐γ. After 2 h incubation the JAK inhibitors had no significant effect on constitutive or cytokine‐delayed neutrophil apoptosis (data not shown). By 20 h, both baricitinib and tofacitinib inhibited the anti‐apoptotic effect of GM‐CSF (Fig. 1c, n = 7; *P < 0·05, **P < 0·01). IFN‐γ did not delay RA neutrophil apoptosis significantly.
Effect of JAK inhibitors on neutrophil chemotaxis
Neutrophil migration from the bloodstream to sites of inflammation is an essential process in the innate immune response to infection. However, in inflammatory diseases such as RA, migration of neutrophils into synovial joints, and their subsequent activation within the joint, contributes to joint damage and persistent inflammation 8. In order to determine the effect of baricitinib and tofacitinib on neutrophil chemotaxis we measured the effect of both drugs on migration towards fMLP (a bacterial peptide) and IL‐8 (a chemokine found at high levels in RA joints) 25. Increased numbers of neutrophils from healthy controls and patients with RA migrated towards fMLP and IL‐8. While this was significant in healthy controls (Fig. 2a, n = 6, *P < 0·05, **P < 0·01), this did not reach significance across the entire population of RA patients (n = 7, P = 0·06). Similarly, comparison of the number of neutrophils migrating towards fMLP between healthy controls and patients with RA was not statistically significant (P = 0·06). Both inhibitors decreased significantly the level of random migration in RA neutrophils (Fig. 2b, n = 7, *P < 0·05), and baricitinib decreased significantly the number of RA neutrophils that migrated towards IL‐8 (Fig. 2c, n = 7, *P < 0·05). Neither inhibitor affected the rate of chemotaxis towards fMLP in healthy control or RA patients. Western blotting of neutrophils incubated for up to 60 min with fMLP or IL‐8 showed that neither STAT‐1 nor STAT‐3 was phosphorylated by either chemoattractant at the concentrations used in the migration experiments (Fig. 2e, n = 3).
Figure 2.
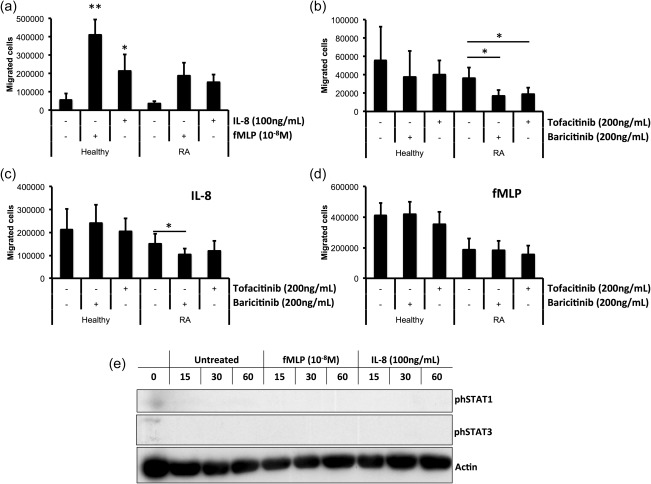
Effect of Janus kinase (JAK) inhibition on neutrophil chemotaxis. (a) Neutrophil migration towards f‐Met‐Leu‐Phe (fMLP) (10−8 M) and interleukin (IL)‐8 (100 ng/ml) after 90 min was increased compared to random migration (*P < 0·05, **P < 0·01). (b) Preincubation with baricitinib and tofacitinib (200 ng/ml) for 30 min decreased the rate of random migration in RA neutrophils (*P < 0·05; **P < 0·01). Chemotaxis towards IL‐8 (c) but not fMLP (d) was inhibited by baricitinib in RA neutrophils (*P < 0·05). (e) fMLP and IL‐8 did not phosphorylate signal transducer and activator of transcription (STAT)‐1 or STAT‐3 in healthy neutrophils during a period of 15–60 min (representative Western blot of n = 3 experiments).
Effect of JAK inhibitors on ROS production
Neutrophils produce ROS as part of the respiratory burst, a vital process in bacterial killing 10. However, ROS production within RA synovial joints contributes to inflammation and is associated with the release of proteases that can damage the cartilage and joint tissues 8, 9. We measured production of ROS stimulated by fMLP (receptor‐dependent) and PMA (receptor‐independent) in neutrophils incubated with baricitinib and tofacitinib for 30 min, and in JAKi‐treated neutrophils incubated with and without GM‐CSF priming for 45 min. Neither inhibitor affected the level of ROS production in unprimed control or RA neutrophils stimulated with fMLP (Fig. 3a, n = 7). In healthy controls, the level of ROS production in response to fMLP stimulation was increased significantly by GM‐CSF priming, and this effect was blocked completely by baricitinib and tofacitinib (Fig. 3b, n = 7, *P < 0·05). The level of ROS produced by unprimed RA neutrophils was higher than in healthy controls, and was not increased following incubation with GM‐CSF (Fig. 3b). Incubation with baricitinib and tofacitinib did not inhibit the production of ROS by RA neutrophils in response to fMLP (Fig. 3b). We also measured ROS production in response to PMA in inhibitor‐treated neutrophils, before and after priming with GM‐CSF. Tofacitinib increased the amount of ROS produced by unprimed RA neutrophils significantly in response to PMA (Fig. 3c, n = 7, *P < 0·05). Following priming with GM‐CSF, the amount of ROS produced by control neutrophils treated with baricitinib and tofacitinib was increased significantly (Fig. 3d, n = 7, *P < 0·05).
Figure 3.
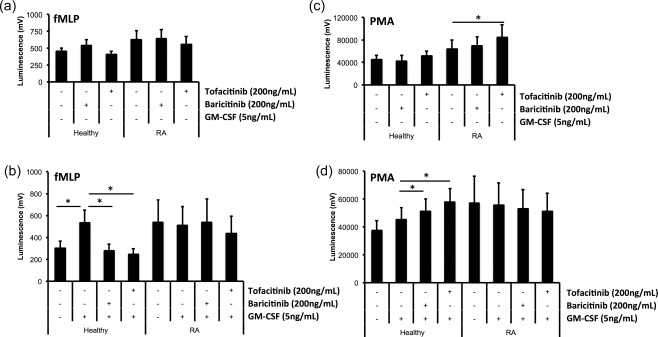
Effect of Janus kinase (JAK) inhibition on neutrophil reactive oxygen species (ROS) production. (a) Preincubation with baricitinib and tofacitinib for 30 min did not affect the production of ROS by unprimed neutrophils in response to f‐Met‐Leu‐Phe (fMLP) (1 μM), but inhibited ROS production in healthy control neutrophils primed with granulocyte–macrophage‐colony stimulating factor (GM‐CSF) for 45 min (b, *P < 0·05). GM‐CSF priming did not increase ROS production in RA neutrophils, and JAK inhibitors had no effect on ROS production. (c) ROS production was increased by tofacitinib in response to phorbol‐12‐myristate‐12‐acetate (PMA) (100 ng/ml) in RA neutrophils (*P < 0·05). (d) Both baricitinib and tofacitinib increased ROS production in GM‐CSF‐primed neutrophils from healthy controls (*P < 0·05).
We next measured whether JAK inhibitors could ‘reverse‐prime’ neutrophils that had been primed with GM‐CSF. Neutrophils from healthy controls and RA patients were primed with GM‐CSF (5 ng/ml) for 45 min prior to the addition of baricitinib and tofacitinib (200 ng/ml). ROS production was measured every 30 min for a further 120 min. Baricitinib decreased ROS production significantly in response to fMLP, but not PMA, in GM‐CSF‐primed control neutrophils after 30 and 60 min (Fig. 4a,b *P < 0·05, **P < 0·01, n = 3). This effect was not observed in RA neutrophils (Fig. 4c,d, n = 3). Tofacitinib had no reverse‐priming effect in either control or RA neutrophils.
Figure 4.
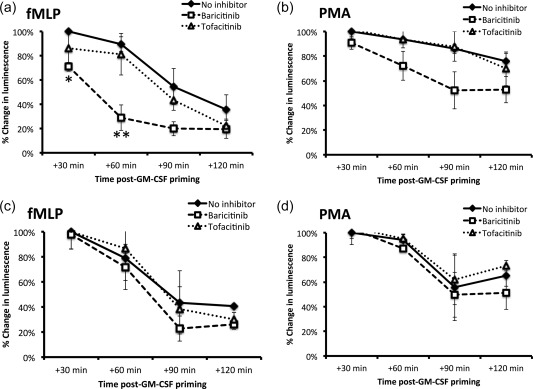
Effect of Janus kinase (JAK) inhibition on reverse priming of reactive oxygen species (ROS) production. The priming effect of granulocyte–macrophage‐colony stimulating factor (GM‐CSF) (5 ng/ml) on neutrophils from healthy controls (a) and RA patients (c) stimulated with f‐Met‐Leu‐Phe (fMLP) (10−8 M) decreased to ∼40% of the levels observed at 45 min, following a further 120 min in culture. Baricitinib (200 ng/ml) enhanced significantly the depriming of healthy control neutrophils after 30 and 60 min incubation (a, *P < 0·05; **P < 0·01, n = 3), but was not able to reverse GM‐CSF priming of rheumatoid arthritis (RA) neutrophils (c). Tofacitinib was not able to reverse prime GM‐CSF‐treated neutrophils from controls (a,b) or RA patients (c,d). No significant reverse priming effect was seen with either JAK inhibitor in phorbol‐12‐myristate‐12‐acetate (PMA)‐treated (100 ng/ml) neutrophils from healthy controls (b) or RA patients (d).
Effect of JAK inhibitors on STAT activation in RA neutrophils
We have shown previously that STAT‐1 and STAT‐3 are activated in neutrophils isolated freshly from the peripheral blood of RA patients 23. In order to investigate the effect of JAK inhibitors on STAT activation in RA neutrophils, we incubated freshly isolated neutrophils for up to 60 min in the absence and presence of baricitinib and tofacitinib (200 ng/ml). Western blotting for phosphorylated STAT‐1 and STAT‐3 showed that in untreated RA neutrophils, STAT‐1 and STAT‐3 activation decreased by approximately 50% during 60‐min incubation (Fig. 5, n = 4). The addition of baricitinib and tofacitinib increased the loss of phosphorylated STAT‐1 and STAT‐3 to approximately 50% by 30 min, although this did not reach statistical significance.
Figure 5.
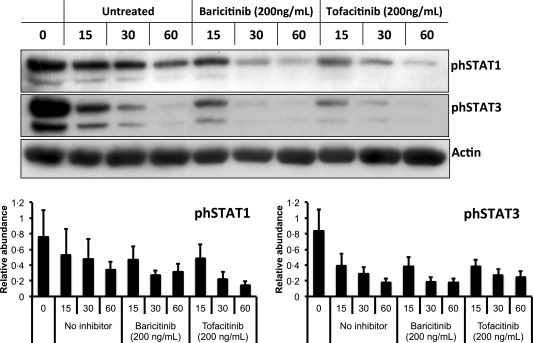
Effect of Janus kinase (JAK) inhibition on phosphorylated signal transducer and activator of transcription (STAT) phosphorylation in RA neutrophils. Freshly isolated (0 h) RA neutrophils exhibited elevated levels of STAT‐1 and STAT‐3, which decreased during 60 min incubation in untreated conditions. Addition of baricitinib and tofacitinib (200 ng/ml) at 0 h induced a loss of phosphorylated STAT‐1 and STAT‐3 compared to untreated cells at 30 and 60 min.
Discussion
In this study we determined the effect of JAK inhibition by baricitinib (JAK1/2 inhibitor) and tofacitinib (JAK3 inhibitor) on three key aspects of neutrophil function which are important in the response to infection, but implicated in unwanted activation during inflammatory disease: apoptosis, chemotaxis and ROS production. In line with other published work, we found that GM‐CSF decreased the level of apoptosis in healthy control and RA neutrophils 22. IFN‐γ delayed apoptosis in healthy control, but not RA, neutrophils. Neutrophils from RA patients have a gene and protein expression profile indicating activation by interferons in vivo 23. Therefore, one explanation of our observation is that RA neutrophils are unresponsive to further stimulation by IFN‐γ in vitro because this signalling pathway has already been activated in vivo 23. Baricitinib and tofacitinib abrogated significantly the anti‐apoptotic effect of GM‐CSF and IFN‐γ on healthy control neutrophils in a dose‐dependent manner, although the effect of tofacitinib was less evident. The GM‐CSF receptor signals via a JAK2 homodimer, while the IFN‐γ receptor signals via a JAK1/JAK2 heterodimer 1; therefore, both cytokine–receptor signalling complexes should be direct targets for baricitinib (JAK1/2 inhibitor). The main inhibitory target of tofacitinib is JAK3. However, data from cell‐free kinase assays demonstrate that tofacitinib also inhibits JAK2 and JAK1, albeit with 20‐ and 112‐fold less potency 26. While tofacitinib had a less potent effect on GM‐CSF‐delayed neutrophil apoptosis in vitro than baricitinib, we did not investigate higher concentrations of tofacitinib, as this would not have reflected serum concentrations of the drug 6, 20. Clinical studies have also shown that tofacitinib inhibits signalling via JAK1 and prevents phosphorylation of STAT‐1 in RA synovial tissue 7. Indeed, our experiments show that incubation of RA neutrophils with tofacitinib (and baricitinib) enhanced the loss of phosphorylated STAT‐1. These published data, in concert with our own findings, would indicate that tofacitinib has a greater effect on the JAK1/JAK2 heterodimer than previously thought.
Both healthy control and RA neutrophils migrated towards fMLP and IL‐8. The number of migrated cells was lower in RA patients, in line with other studies 21, and may be due to previous exposure to DMARDs in vivo. All patients in the study were receiving methotrexate, which has been shown previously to decrease neutrophil migration both in vivo and in vitro 27. Baricitinib decreased RA neutrophil migration towards IL‐8 consistently and significantly. IL‐8 has been described previously as activating JAK2 in hepatocellular carcinoma cell lines 28, which could explain our observation. However, we did not see phosphorylation of STAT‐1 or STAT‐3 in neutrophils in response to IL‐8 in this study, and therefore this phenomenon remains unexplained and warrants further investigation. The effect of tofacitinib on migration towards IL‐8 was not significant in our study.
Priming of healthy control neutrophils with GM‐CSF prior to fMLP activation resulted in significantly greater ROS production compared to unstimulated neutrophils, in line with previous publications 24. Priming was inhibited by pretreatment with both baricitinib and tofacitinib in healthy control neutrophils. Unprimed RA neutrophils demonstrated higher levels of ROS production than healthy controls after being activated by fMLP. This was prior to the addition of the priming agent GM‐CSF, suggesting that RA neutrophils were already primed for ROS production in vivo 29. GM‐CSF priming did not increase ROS production by RA neutrophils, suggesting that once RA neutrophils have been primed in vivo they do not have the ability to ‘re‐prime’. Healthy control and RA neutrophils lost the GM‐CSF‐induced priming response over time in vitro, so that after 165 min in culture the levels of ROS produced in response to fMLP were only ∼40% of the levels produced after 45 min priming. This is in line with observations of de‐priming following exposure of neutrophils to other agents, such as platelet activating factor 30. Addition of baricitinib to healthy neutrophils primed with GM‐CSF reversed the priming response (i.e. enhanced de‐priming) and generation of ROS successfully in response to fMLP after 30 min. However, neither JAK inhibitor reversed the priming effect in RA neutrophils either before or after the addition of GM‐CSF, suggesting that RA neutrophils had been primed in vivo by an inflammatory agonist that did not activate the JAK/STAT pathway. These data are supported further by our observation that JAK inhibitors were able to increase the rate of STAT de‐phosphorylation in RA neutrophils.
All RA patients in our study were receiving DMARDs, including methotrexate. The effect of existing medications such as methotrexate and TNFi on neutrophils has been described extensively in the case of methotrexate, which has been shown to abrogate delayed apoptosis of neutrophils, and decrease both chemotaxis and ROS production 27, 31. To reduce the potential effect of this confounder, further studies analysing the effect of JAK inhibitors should recruit newly diagnosed RA patients prior to commencing DMARD therapy, as this may provide a clearer picture as to how JAK inhibitors affect RA neutrophil function. Further work should now focus upon the ability of JAK inhibitors to modulate other key aspects of neutrophil activation in RA (e.g. NET production) 8, 9, with particular attention to the risk of impaired host defence and the ability of JAK inhibitor‐treated neutrophils to carry out key protective functions such as bacterial killing.
In conclusion, incubation of healthy control and RA neutrophils with baricitinib and tofacitinib inhibited the anti‐apoptotic effect of GM‐CSF and IFN‐γ in a dose‐dependent manner. JAKi increased the turnover of active, phosphorylated STAT‐1 and STAT‐3 in RA neutrophils, and prevented ex‐vivo chemotaxis of RA neutrophils towards IL‐8. However, JAKi did not prevent the production of ROS or increase the level of constitutive apoptosis in RA neutrophils. This may be due to the prior exposure of RA neutrophils to priming agents other than those which activate JAK/STAT signalling in vivo.
Disclosure
None.
Acknowledgements
T. S. M. was funded by the University of Liverpool MRes Clinical Sciences Research Support Fund. H. L. W. was funded by the University of Liverpool Faculty of Health and Life Sciences. We would like to thank the rheumatology nurses and consultants at University Hospital Aintree for their assistance in recruiting patients.
References
- 1. Hodge JA, Kawabata TT, Krishnaswami S et al The mechanism of action of tofacitinib – an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 2016; 34:318–28. [PubMed] [Google Scholar]
- 2. Burmester GR, Blanco R, Charles‐Schoeman C et al Tofacitinib (CP‐690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 2013; 381:451–60. [DOI] [PubMed] [Google Scholar]
- 3. Fleischmann R, Kremer J, Cush J et al Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012; 367:495–507. [DOI] [PubMed] [Google Scholar]
- 4. Dougados M, van der Heijde D, Chen YC et al Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA‐BUILD study. Ann Rheum Dis 2017; 76:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Vollenhoven RF, Fleischmann R, Cohen S et al Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 2012; 367:508–19. [DOI] [PubMed] [Google Scholar]
- 6. Maeshima K, Yamaoka K, Kubo S et al The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon‐gamma and interleukin‐17 production by human CD4+ T cells. Arthritis Rheum 2012; 64:1790–8. [DOI] [PubMed] [Google Scholar]
- 7. Boyle DL, Soma K, Hodge J et al The JAK inhibitor tofacitinib suppresses synovial JAK1‐STAT signalling in rheumatoid arthritis. Ann Rheum Dis 2015; 74:1311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol 2014; 10:593–601. [DOI] [PubMed] [Google Scholar]
- 9. Thieblemont N, Wright HL, Edwards SW, Witko‐Sarsat V. Human neutrophils in auto‐immunity. Semin Immunol 2016; 28:159–73. [DOI] [PubMed] [Google Scholar]
- 10. Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 2010; 49:1618–31. [DOI] [PubMed] [Google Scholar]
- 11. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011; 11:519–31. [DOI] [PubMed] [Google Scholar]
- 12. Scally SW, Petersen J, Law SC et al A molecular basis for the association of the HLA‐DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med 2013; 210:2569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khandpur R, Carmona‐Rivera C, Vivekanandan‐Giri A et al NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 2013; 5:178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pratesi F, Dioni I, Tommasi C et al Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann Rheum Dis 2014; 73:1414–22. [DOI] [PubMed] [Google Scholar]
- 15. Spengler J, Lugonja B, Ytterberg AJ et al Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol 2015; 67:3135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y, Li M, Stadler S et al Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009; 184:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi JG, Chen X, Lee F et al The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol 2014; 54:1354–61. [DOI] [PubMed] [Google Scholar]
- 18. Cohen S, Radominski SC, Gomez‐Reino JJ et al Analysis of infections and all‐cause mortality in phase II, phase III, and long‐term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol 2014; 66:2924–37. [DOI] [PubMed] [Google Scholar]
- 19. Wright HL, Makki F, Moots RJ, Edwards SW. Low‐density granulocytes: functionally distinct, immature neutrophils in rheumatoid arthritis with altered properties and defective TNF signalling. J Leukoc Biol 2017; 101:599–611. [DOI] [PubMed] [Google Scholar]
- 20. Dowty ME, Lin J, Ryder TF et al The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab Dispos 2014; 42:759–73. [DOI] [PubMed] [Google Scholar]
- 21. Wright HL, Cross AL, Edwards SW, Moots RJ. Effects of IL‐6 and IL‐6 blockade on neutrophil function in vitro and in vivo . Rheumatology (Oxford) 2014; 53:1321–31. [DOI] [PubMed] [Google Scholar]
- 22. Wright HL, Chikura B, Bucknall RC, Moots RJ, Edwards SW. Changes in expression of membrane TNF, NF‐{kappa}B activation and neutrophil apoptosis during active and resolved inflammation. Ann Rheum Dis 2011; 70:537–43. [DOI] [PubMed] [Google Scholar]
- 23. Wright HL, Thomas HB, Moots RJ, Edwards SW. Interferon gene expression signature in rheumatoid arthritis neutrophils correlates with a good response to TNFi therapy. Rheumatology (Oxford) 2015; 54:188–93. [DOI] [PubMed] [Google Scholar]
- 24. Wright HL, Thomas HB, Moots RJ, Edwards SW. RNA‐seq reveals activation of both common and cytokine‐specific pathways following neutrophil priming. PLOS ONE 2013; 8:e58598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wright HL, Bucknall RC, Moots RJ, Edwards SW. Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatology (Oxford) 2012; 51:451–9. [DOI] [PubMed] [Google Scholar]
- 26. Changelian PS, Flanagan ME, Ball DJ et al Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003; 302:875–8. [DOI] [PubMed] [Google Scholar]
- 27. Kraan MC, de Koster BM, Elferink JG, Post WJ, Breedveld FC, Tak PP. Inhibition of neutrophil migration soon after initiation of treatment with leflunomide or methotrexate in patients with rheumatoid arthritis: findings in a prospective, randomized, double‐blind clinical trial in fifteen patients. Arthritis Rheum 2000; 43:1488–95. [DOI] [PubMed] [Google Scholar]
- 28. Fu XT, Dai Z, Song K et al Macrophage‐secreted IL‐8 induces epithelial–mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol 2015; 46:587–96. [DOI] [PubMed] [Google Scholar]
- 29. El Benna J, Hayem G, Dang PM et al NADPH oxidase priming and p47phox phosphorylation in neutrophils from synovial fluid of patients with rheumatoid arthritis and spondylarthropathy. Inflammation 2002; 26:273–8. [DOI] [PubMed] [Google Scholar]
- 30. Kitchen E, Rossi AG, Condliffe AM, Haslett C, Chilvers ER. Demonstration of reversible priming of human neutrophils using platelet‐activating factor. Blood 1996; 88:4330–7. [PubMed] [Google Scholar]
- 31. Weinmann P, Moura RA, Caetano‐Lopes JR et al Delayed neutrophil apoptosis in very early rheumatoid arthritis patients is abrogated by methotrexate therapy. Clin Exp Rheumatol 2007; 25:885–7. [PubMed] [Google Scholar]