

Abstract
Background
Scottish Terriers have a high incidence of juvenile onset hereditary ataxia primarily affecting the Purkinje neuron of the cerebellar cortex and causing slowly progressive cerebellar dysfunction.
Objective
To identify chromosomal regions associated with hereditary ataxia in Scottish Terriers.
Animals
One hundred and fifty‐three Scottish Terriers were recruited through the Scottish Terrier Club of America.
Materials and Methods
Prospective study. Dogs were classified as affected if they had slowly progressive cerebellar signs. When possible, magnetic resonance imaging and histopathological evaluation of the brain were completed as diagnostic aids. To identify genomic regions connected with the disease, genome‐wide mapping was performed using both linkage‐ and association‐based approaches. Pedigree evaluation and homozygosity mapping were also performed to examine mode of inheritance and to investigate the region of interest, respectively.
Results
Linkage and genome‐wide association studies in a cohort of Scottish Terriers both identified a region on CFA X strongly associated with the disease trait. Homozygosity mapping revealed a 4 Mb region of interest. Pedigree evaluation failed to identify the possible mode of inheritance due to the lack of complete litter information.
Conclusion and Clinical Importance
This finding suggests that further genetic investigation of the potential region of interest on CFA X should be considered in order to identify the causal mutation as well as develop a genetic test to eliminate the disease from this breed.
Keywords: Canine, Cerebellar abiotrophy, Cerebellar ataxia, Neurodegeneration
Abbreviations
- AKC
American Kennel Club
- CFA
Canis familiaris
- CMH
Cochran‐Mantel‐Haenszel
- GC
genomic control
- GWAS
genome‐wide association study
- IBS
identical‐by‐state
- MAF
minor allele frequency
- Mb
mega base
- MRI
magnetic resonance images
- MSS‐2
minimal screening set 2
- NCSU
North Carolina State University
- PARs
pseudoautosomal regions
- SD
standard deviation
- SNP
single nucleotide polymorphism
- Xa
active X chromosome
- XCI
X‐chromosome inactivation
- Xic
X‐inactivation center
- Xi
inactive X chromosome
- Xist
X‐inactivation‐specific transcript
Hereditary ataxias are a heterogeneous group of neurodegenerative diseases that cause cerebellar ataxia with a wide range of clinical and pathological manifestations, constituting an important problem affecting purebred dogs.1 To date, mutations in 8 different genes have been associated with hereditary ataxias in more than 10 breeds of dog.1 , 1, 2, 3, 4, 5, 6, 7, 8, 9 These genes encode proteins that have a variety of different functions including protein degradation and autophagy (RAB24 in Old English Sheep dogs and Gordon Setters,2 SEL1L in Finnish hounds,3 and CAPN1 in Parson Russell Terriers4), CNS cytoskeleton integrity (SPTBN2 in Beagles5), cation trafficking (ITPR1 in Italian Spinones,6 GRM1 in Coton de Tulears,7 and KCNJ10 in Russell Terrier Group and smooth‐haired Fox Terrier8, 9), and mitochondrial function and cholesterol trafficking (SERAC1 in Kerry Blue and Chinese Crested1). Mutations include exonic nonsynonymous nucleotide substitution, retrotransposon insertion in the exon and intronic GAA repeat expansion.1 , 2, 3, 4, 5, 6, 7, 8 An autosomal recessive mode of inheritance has been identified in these breeds.1 , 2, 3, 4, 5, 6, 7, 8 An X‐linked mode of inheritance has been reported in the English pointer dogs,10 but the mutation has not been identified.
Hereditary ataxia or hereditary cerebellar degeneration has been recognized in Scottish Terriers for more than a decade.11 The cerebellar cortex is the primary site of neurodegeneration, which is characterized by severe loss of Purkinje cells, depletion of granule cells and atrophy of the molecular layer as well as polyglucosan body accumulation.12 Clinical signs reflect cerebellar dysfunction including a wide‐based stance, dysmetria producing a hypermetric gait, difficulty negotiating stairs and intention tremors. Onset of signs usually occurs during the first year of life and in many dogs, progression is slow with signs ultimately stabilizing, resulting in a lifelong relatively mild phenotype.13 The objective of this study was to investigate the genetic basis of hereditary ataxia in Scottish Terriers. We performed genome‐wide microsatellite and single nucleotide polymorphism (SNP) genotyping in affected Scottish Terriers and their extended families to identify a chromosomal locus associated with the disorder using linkage and genome‐wide association analyses.
Materials and Methods
This was a prospective study aimed at mapping the locus of cerebellar degeneration in Scottish Terriers following genotyping with microsatellites using linkage analysis in related families. To confirm findings of the linkage study, a broader population of dogs was then genotyped on single nucleotide polymorphisms and a genome‐wide association study was performed.
Study Population
Affected (case) and normal (control) Scottish Terriers were recruited through the Scottish Terrier Club of America. All participating dogs were privately owned pets. Dogs were classified as affected if they had slowly progressive cerebellar signs as described previously13 (established by direct evaluation of the dog by a veterinary neurologist, or by evaluating video of gait when walking, trotting, and negotiating stairs). When possible, cerebellar atrophy was identified on MRI, or a definitive diagnosis was established by necropsy. As the age of onset of clinical signs ranges from 2 months to 6 years,13 control dogs were required to be older than 7 years with no evidence of gait abnormalities as reported by owners. Dogs reported by owners as affected but not exhibiting typical signs of cerebellar degeneration as determined by review of video‐taped gait by the investigators or by evaluation of the patient by a veterinary neurologist were classified as “undetermined.”13 DNA samples were obtained either from whole blood or from saliva. DNA was extracted from whole blood with the QIAamp® DNA Blood Midi Kit2 and from saliva with Oragene Animal kit.3 DNA concentrations were measured with a ND‐1000 NanoDrop spectrophotometer.4 Pedigrees of all dogs were requested to construct the family pedigree and examine their relationships. All protocols were performed with approval from North Carolina State University's Institutional Animal Care and Use Committee.
Genome‐Wide Microsatellite Genotyping and Linkage Analysis
Related dogs were genotyped with a genome‐wide panel of 296 autosomal fluorescently labeled (FAM, VIC, NED, and PET) canine microsatellite markers from the minimal screening set 2 (MSS‐2)14 (representing approximately 10 cM resolution across the entire canine genome), organized into multiplex PCR groups. The PCR conditions used have been described elsewhere.14 PCR fragments were visualized by an ABI 3730xl DNA Analyzer5, and genotypes were assigned by GeneMapper v3.7 software.5 Homozygous (uninformative) markers were excluded from further analysis. Initially, 48 Scottish Terriers (10 cases, 37 controls and 1 undetermined) were genotyped on the all autosomes. Then, an additional 47 Scottish Terriers (11 cases, 34 controls, and 2 undetermined) were genotyped on chromosomes of interest. Finally, chromosome (Canis familiaris, CFA) X was genotyped with 7 microsatellite markers from MSS‐2 in 96 dogs (22 cases, 71 controls, and 3 undetermined, one of which was a new case that was not genotyped on the autosomes).
Multipoint linkage analysis on autosomal chromosomes was computed with the MORGAN (v.2.8.1) software package.15 The penetrance was initially set at 98%. The trait frequency was set to default at 61% for the common allele and 39% for the risk allele based on previous data suggesting an approximately 40% prevalence of risk allele in canine highly penetrant autosomal recessive disease.16 Non‐parametric linkage analysis was performed on X chromosomal loci by the X‐linked version of Genehunter (xgh), GENEHUNTER‐IMPRINTING version 1.3 software.17 Due to limitations with the computation time of this program, the 96 dogs were divided into 10 pedigrees in which linkage analysis was performed separately and the LOD score results from each family were summed to represent the total LOD score of CFA X from 96 dogs. Genotypes that were not consistent with Mendelian inheritance were re‐evaluated, and individuals with multiple Mendelian errors were removed from the analysis. LOD scores greater than 3 were considered significant. However, autosomes that had a LOD score greater than 1 were examined further by genotyping the additional 47 dogs. The location of microsatellites in CanFam 2.0 was determined from the online canine genetic linkage map at the University of California, Davis Veterinary Genomics Laboratory (http://www.vgl.ucdavis.edu/dogmap/). Regions of interest were compared between case and control dogs for evidence of segregation of homozygosity with phenotype. Pedigrees were re‐examined later to evaluate whether an X‐linked mode of inheritance was possible.
Genome‐Wide Association Study
Genome‐wide SNP genotyping in the case and control dogs was performed with the Illumina® CanineSNP20 BeadChip6 (22,362 SNPs, n = 70 dogs) and Illumina® CanineHD genotyping BeadChip6 (173,662 SNPs, n = 59 dogs, 13 of which were also genotyped on the 22 K array). The assays were performed at the National Institutes of Health's Laboratory of Neurogenetics (Bethesda, MD) following the manufacturer's instructions. The amplified DNA products were imaged with a BeadArray Reader.6 The assay intensity data were then loaded into Illumina® BeadStudio software6 to score and generate the SNP genotypes.
The genome‐wide association study (GWAS) and data pruning as well as the adjustment for multiple testing were carried out by PLINK v1.07 software package18 (http://pngu.mgh.harvard.edu/purcell/plink/). A case‐control GWAS was performed on the SNPs common to both the 22 K and 173 K platforms. These SNPs had to have identical alleles in the 13 duplicate dogs genotyped on both arrays. Data were pruned by removing individuals with a call rate below 96%, and SNPs with minor allele frequency (MAF) less than 1%, or with more than 10% missing genotypes. Individuals were clustered on the basis of genetic identity by Identical‐by‐state (IBS) clustering, and the Cochran‐Mantel‐Haenszel (CMH) association analysis was implemented to test SNP‐disease association conditional on the IBS cluster provided. Bonferroni adjustment was used to correct multiple comparisons with adjusted P‐values less than .05 considered significant. JMP® Genomics software (SAS; Cary, NC) was used to create QQ‐plots from the genomic control (GC) data to assess the adequacy of correction of population structure.
Disease‐segregating homozygous regions were identified based on the SNP genotypes by visual inspection of the data. The UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway) was used to search for candidate genes in regions of interest (CanFam2.0).
Results
A total of 153 dogs (69 males, 84 females) were genotyped in this study. There are 46 cases (27 males, 19 females), 97 controls (36 males and 61 females), and 10 undetermined (6 males and 4 females). Hereditary ataxia was confirmed by histopathology in 11 cases, 3 of which had an MRI of the brain. An additional 3 cases underwent an MRI of the brain. The rest of the cases were diagnosed based on clinical history, and signs of cerebellar disease on videotapes of gait.
Genome‐Wide Microsatellite Genotyping and Linkage Analysis
A total of 96 dogs from one pedigree were selected to be genotyped (Fig S1). There were 71 controls (25 males and 46 females), 22 cases (14 males and 8 females), and 3 undetermined status dogs (one 3‐year‐old male, and two 4‐ and 9‐year‐old females). The diagnosis was confirmed in 5 of the cases by histopathology, one of which was also examined by MRI. The rest of the cases were diagnosed based on clinical history, and signs of cerebellar disease on videotapes of gait.
The maximum LOD score from performing linkage analysis of the first 48 Scottish Terriers was 1.16 located on CFA 11 between markers C11.873 (66 Mb) and DGN13 (73 Mb). When the genotypes of an additional 47 dogs from the same pedigree were added, the LOD score in the same region decreased to 0.99. There were no LOD scores greater than 0.50 on any other autosomes. In contrast, LOD scores greater than 3 were found on CFA X between markers FH3027 (41.0 Mb) and REN75A05 (117.2 Mb). There was only 1 more informative marker in this region (FH2584 at 103.9 Mb), and the highest LOD score of 5.6 was located at this marker (Fig 1). Evaluation of the haplotypes in the region failed to identify clear segregation with disease using either a dominant or recessive model of inheritance. There were 9 genes within this 76.2 Mb disease‐linked region (Table 1) that have been associated with X‐linked syndromes in humans in which cerebellar degeneration or dysfunction was a component. However, none of these syndromes cause a pure cerebellar ataxia as seen in the Scottish Terrier.19, 20, 21
Figure 1.
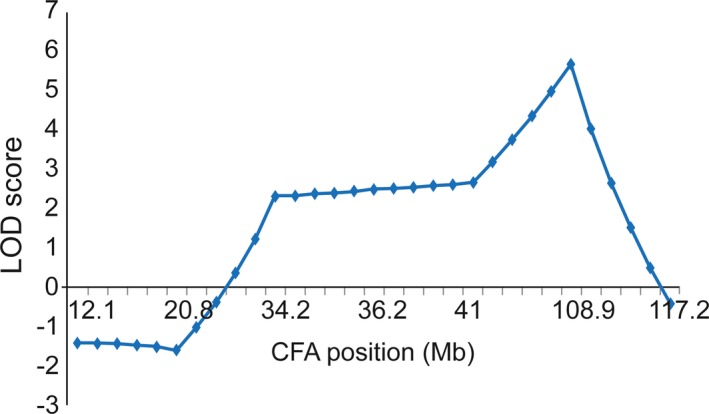
A plot of LOD score on CFA X. Y‐axis is the LOD score, and x‐axis is the position on CFA X in Mb. The significant LOD score (>3) region is located between 41 and 117.2 Mb with the highest LOD score of 5.6 at 103.9 Mb.
Table 1.
| Human X‐linked syndrome with cerebellar ataxia | Human chromosomal locus | Dog chromosomal locus | Gene | Gene product | Mutation |
|---|---|---|---|---|---|
| Oligophrenin‐1 syndrome | chrX:67262186‐67653299 | chrX:55377327‐55908921 | OPHN1 | Oligophrenin‐1 | Point mutation |
| X‐link sideroblastic anemia with ataxia (XLSA/A) | chrX:74273105‐74376132 | chrX:61359945‐61523487 | ABCB7 | ATP‐binding cassette subfamily B member 7 | Point mutation |
| X‐linked Opitz/GBBB syndrome | chrX:10413350‐10851809 | chrX:7041201‐7198729 | MID1 | Midline‐1 | Point mutation |
| X‐linked lissencephaly type I | chrX:110537007‐110655460 | chrX:87469155‐87586087 | DCX | Neuronal migration protein doublecortin | Point mutation, duplication |
| Oral‐facial‐digital type I/X‐linked Joubert syndrome | chrX:13752832‐13787480 | chrX:10098404‐10151431 | OFD1 | Oral‐facial‐digital syndrome 1 protein | Point mutation |
| Candidate gene for X‐linked mental retardation in 12 families | chrX:128580478‐128657460 | chrX:103732870‐103805637 | SMARCA1/SNF2L | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 1 | Unknown |
| Lesch‐Nyhan Syndrome (LNS) | chrX:133594175‐133634698 | chrX:108177629‐108214917 | HPRT1 | Hypoxanthine‐guanine phosphoribosyltransferase | Point mutation |
| X‐linked Angelman‐like syndrome | chrX:135067586‐135129428 | chrX:109487347‐109534427 | SLC9A6 | Sodium/hydrogen exchanger 6 isoform a precursor | Point mutation |
| X‐linked visceral heterotaxy | chrX:136648346‐136654259 | chrX:110890225‐110896083 | ZIC3 | Zinc finger protein ZIC 3 | Point mutation |
To determine whether the disease could be inherited as an X‐linked dominant or recessive trait, dogs’ relationships in the pedigrees genotyped were re‐examined. Full litter information was not available for most of the families; therefore, it was not possible to make a statistical comparison of actual and expected findings given different modes of inheritance. Overall, there were approximately twice as many females (46) as males (25) in the control dogs, while there were nearly twice as many males (14) as females (8) in the case dogs. If the control population was taken to be the baseline ratio of females to males, the ratio of male to female cases becomes more substantial. If the mode of inheritance is X‐linked recessive trait, affected females would have affected male offspring. There was only 1 litter for which we had data on offspring from an affected female (Fig 2), and in this case, the male offspring was affected. Notably, in the litter with several male and female dogs (Fig 2), all the males were affected and all the females had a normal phenotype. These litters came from normal parents implying a recessive mode of inheritance, or incomplete penetration of a dominant mode. In one instance, the family structure is not consistent with an X‐linked recessive mode of inheritance (Fig 2). In this family, normal parents (8‐year‐old dam and sire reported to be normal by the owners) gave rise to an affected female. There was only 1 subfamily in which an affected male and normal female produced normal male offspring. To investigate the results of the linkage study further a genome‐wide association study was performed using SNP genotypes.
Figure 2.
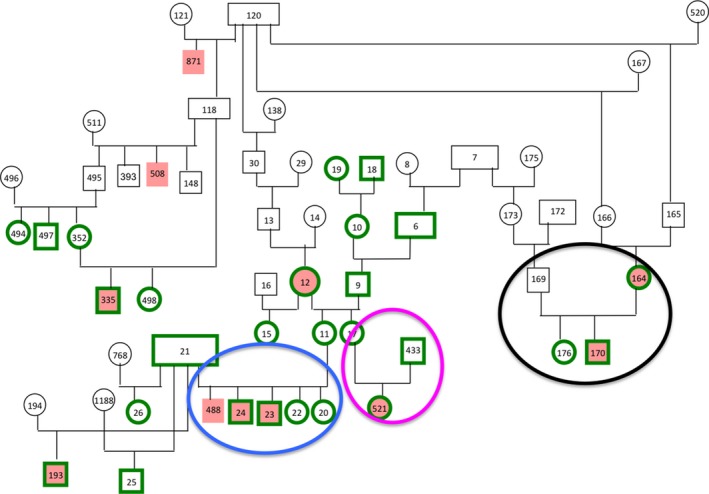
A Scottish Terrier pedigree illustrating 27 dogs that were genotyped (green border). Females are ovals, and males are rectangles. Affected (case) dogs are solid red and normal (control) dogs are white. The black oval highlights the subfamily in which the affected dam has an affected male and a normal female offspring. The blue oval highlights a litter containing affected males and normal females. The pink oval highlights a female affected dog from apparently normal parents.
Genome‐Wide Association Study
A total of 116 dogs (46 cases, 60 controls, and 10 undetermined) were genotyped on SNP chips, 59 (22 cases, 34 controls, and 3 undetermined) of which were included in the linkage analysis study. Thirty‐four cases (15 females and 19 males), 34 controls (19 females and 15 males), and 2 undetermined dogs (1 female and 1 male) were genotyped on the 22 K array, whereas 24 cases (10 females and 14 males), 27 controls (16 females and 11 males), and 8 undetermined dogs (3 females, 5 males) were genotyped on the 173 K array. Thirteen dogs (6 female cases, 6 male cases, and 1 male control) were genotyped on both arrays for quality control. Before pruning, there were 19,215 SNPs common to both 22 K and 173 K platforms, all of which had identical alleles in the 13 duplicate dogs genotyped on both arrays. SNPs with MAF <1% and missing genotype calls >10% were removed from the analysis, resulting in a final data set of 14,365 SNPs that underwent case‐control association analysis. The genotyping call rate in all individuals was more than 99%. As most of the dogs were related and some dogs were closely related, population stratification was expected. GWAS with CMH testing for SNP‐disease association conditional on IBS was therefore performed. A significantly associated region on CFA X was identified. It extended from 103 to 109 Mb (CanFam2.0) with the strongest Praw equal to 5.249 × 10−9 (Pgenome = 3.076 × 10−5 and Bonferroni adjusted multiple testing significance value = 7.497 × 10−5). The Q‐Q plot of the observed p‐value (genomic control) against the expected p‐value also demonstrated an appropriate adjustment for population stratification (Fig 3A and B). This finding confirmed the results of the linkage analysis study. In addition, there were weaker, but still significant signals on CFA 2, 4, and 17 (Fig 3A).
Figure 3.
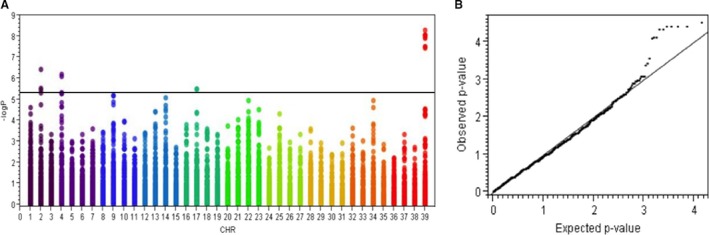
Genome‐wide association mapping of hereditary ataxia in 116 Scottish Terriers. (A) A plot of –log10 (P) on the y‐axis and chromosome (CHR) on the x‐axis, where 39 is CFA X. The strongest association is demonstrated by –log10 (P) on CFA X (Praw = 5.249 × 10−9, Pgenome = 3.076 × 10−5, and Bonferroni adjusted multiple testing = 7.497 × 10−5). The horizontal black line represents the cutoff of significant signals. (B) A Q–Q plot of Pgenome demonstrated where the significant signals on CFA X (observed: y‐axis) deviated from the expected value (x‐axis).
The regions of interest on CFA X, 2, 4, and 17 were subjected to further investigation using homozygosity mapping (Fig 4). The X‐chromosome SNP genotypes of all male dogs were displayed as homozygous, although they are hemizygous because they only possess 1 X chromosome. Genotype data were visually inspected for regions of homozygosity shared only in the affected dogs. There are 2 candidate regions from 103,456,993 bp to 103,530,501 bp and 104,813,634 bp to 106,189,685 bp. While 93.47% of the cases in these 2 blocks are identical, 35% (103 Mb) and 31.67% (104–106 Mb) of the controls also shared the same haplotype. Three female cases did not share the haplotype of most cases (Fig 4 and Table 2). When control dogs had the case genotype, this was true across both regions of interest on the X chromosome. The SNP genotypes on CFA 2, 4, and 17 associated with a weaker GWAS signal were also evaluated, but there was no segregation of genotype with phenotype.
Figure 4.
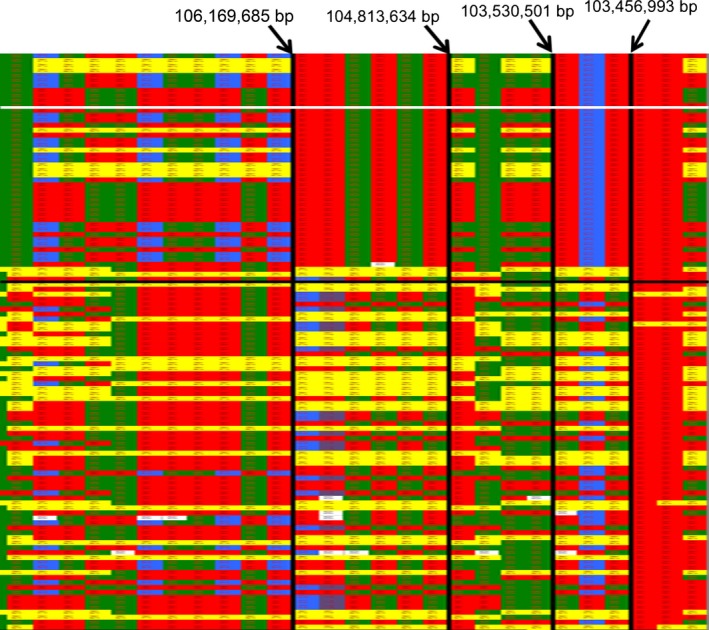
SNP genotypes of 116 dogs on CFA X from 100,095,625 bp to 110,167,038 bp. Each row is a different individual, and each column is a different SNP. Solid green, red, purple, and blue boxes indicate homozygosity whereas yellow boxes indicate heterozygosity. The genotype of necropsy confirmed cases is displayed in the top segment of the chart above the horizontal white line, other cases are shown above the horizontal black line, and the remaining phenotypically normal dogs are in the bottom segment of the chart below the horizontal black line. The large and small regions of homozygosity in most of the affected dogs extend from 104,813,634 bp to 106,189,685 bp and from 103,456,993 bp to 103,530,501 bp, respectively.
Table 2.
Homozygosity mapping by examining dogs’ genotypes on the common 14,365 SNPs (116 dogs: 46 cases (19 females and 27 males); 60 controls (35 females and 25 males); and 10 undetermined dogs (4 females and 6 males)). F is female and M is male
| Affected status | First haplotype | Second haplotype | Heterozygous |
|---|---|---|---|
| 103.4–103.6 Mb (116 dogs: 58 F, 58 M) | |||
| Cases | 43 (18 F, 25 M) | 1 (F) | 2 (F) |
| Controls | 21 (6 F, 15 M) | 15 (6 F, 9 M) | 24 (F) |
| Undetermined | 3 (1 F, 2 M) | 4 (M) | 3 (F) |
| 104.8–106.2 Mb (116 dogs: 58 F, 58 M) | |||
| Cases | 43 (18 F, 25 M) | 1 (F) | 2 (F) |
| Controls | 19 (4 F, 15 M) | 15 (6 F, 9 M) | 26 (F) |
| Undetermined | 3 (1 F, 2 M) | 4 (M) | 3 (F) |
Discussion
Whole genome mapping using both linkage analysis and GWAS identified a region on CFA X that is strongly associated with hereditary ataxia in Scottish Terriers. Linkage analysis revealed a region extending from 41.0 to 117.2 Mb and GWAS confirmed the association on CFA X and narrowed the region down to 6 Mb (from 103 to 109 Mb). This finding appears to be robust as it was duplicated with 2 separate mapping techniques, but was unexpected given the apparent autosomal recessive mode of inheritance described previously.13 While the pedigrees were evaluated, the lack of complete litter information prevented a segregation analysis from being performed. Several explanations could be made to support an X‐linked mode of inheritance for this disease in Scottish Terriers. In X‐linked recessive traits, males have a higher risk of being affected than females because males carry only 1 copy of the X chromosome. In our population of cases, the number of female cases was lower than males with an overall ratio of 0.7 females to 1 male, but still higher than expected for a recessive X‐linked trait. The pedigree contains 3 litters with complete information (Figs 2 and S1, blue ovals). In these litters, both parents were reported to be normal, and only male offspring were affected, and the females were normal. In another family, the female offspring from an affected dam was normal, whereas the male offspring was affected (Fig 2, black oval). These families suggest the disease could potentially be transmitted in an X‐linked recessive manner. However, there is 1 affected female (Fig 2, pink oval) that has normal parents (as reported by the owners of the parents), which is inconsistent with an X‐linked recessive mode of inheritance, so an X‐linked dominant trait with incomplete penetrance was considered.
In female mammals, 1 of the X chromosomes is silenced in order to avoid overexpression of the genes compared to males.22 The process of X‐chromosome inactivation (XCI) begins randomly in each cell of the embryo resulting in a genetic mosaic of cells from expression of either paternal or maternal X‐chromosome origin. XCI is regulated initially by the long noncoding RNA which is transcribed from the X‐inactivation‐specific transcript (Xist) gene located in the X‐inactivation center (Xic) locus on the X chromosome.23, 24 However, about 15% of human X‐linked genes can escape silencing by XCI and are expressed in both the active (Xa) and inactive (Xi) X chromosome in females.25 These escaped XCI genes are located mostly in the pseudoautosomal regions (PARs) on the X chromosome. PARs contain homologous nucleotide sequences between mammalian X and Y chromosomes, and they are inherited like autosomes. Nevertheless, in humans and mice, some of the escaped genes can be found distributed throughout the X chromosome.25, 26, 27 It is possible that the causal mutation of hereditary ataxia in Scottish Terriers is in an escaped gene on the X chromosome reflecting the apparent segregation of the disease in the population as an autosomal recessive trait. However, the identified region cannot be located in the PAR as the canine PAR spans approximately 6.6 Mb from the telomere of the Xp arm.28 Nonrandom XCI (skewed X‐chromosome inactivation) in the female may occur by paternally or maternally biased inactivation of the X chromosome and can also result in tissue‐specific skewing.29, 30 This could also explain the affected female with normal parents. Unfortunately, the pattern of inheritance of the disease cannot be determined by examining the available pedigree. In order to accurately determine the mode of inheritance, a breeding colony of Scottish Terriers with hereditary ataxia would need to be established to perform selective breedings.
The possibility that more than one form of hereditary ataxia is segregating within the Scottish Terrier breed as was found in Parson/Jack Russell Terriers4, 8 has also been considered. In addition, the lack of a definitive diagnosis made by MRI or histopathology in every dog means that some could have suffered from an acquired cause of cerebellar ataxia. However, the presence of phenocopies typically reduces the study power, making associated chromosomal loci impossible to detect.
Homozygosity mapping by visual inspection of the genotypes identified 2 nonoverlapping regions, within the 103 Mb and 104 to 106 Mb, in which the overwhelming majority of cases were homozygous. However, in both regions, approximately 30% of the controls were also homozygous for the same allele. This could be explained in 2 ways: Firstly, it was possible that the dogs had such a mild phenotype that it was unrecognized by their owners. Owners of control dogs were reluctant to send videotapes so it was difficult to confirm the reported phenotype of the control group. Secondly, as most of the controls that had the same haplotypes as cases were from the same family, the haplotype segregation could be explained by the presence of a common haplotype in this region with the mutation located within the haplotype.
In conclusion, linkage analysis, GWAS, and homozygosity mapping have identified a candidate region on CFA X associated with the hereditary ataxia in Scottish Terriers. Although, there was no perfect disease‐segregating haplotype between cases and controls and no strong candidate gene within the region, it is worth investigating this region further using the new build of canine genome (CanFam3) and performing deep sequencing for this region to identify the potential mutations.
Supporting information
Fig S1. A Scottish Terrier pedigree. Ninety‐six dogs were genotyped (green border). Females are ovals, and males are rectangles. Affected (case) dogs are solid red, normal (control) dogs are white, and undetermined status dogs are solid yellow. The vertical black oval highlights the sub‐family in which the affected dam has an affected male and a normal female offspring. The horizontal blue ovals highlight litters containing affected males and normal females. The pink circle highlights a female affected dog from normal parents.
Acknowledgments
This work could not have been completed without the help of veterinarians and Scottish Terrier breeders and owners. The work was supported by the Health Trust Fund of the Scottish Terrier Club of America and the American Kennel Club Canine Health Foundation.
Conflict of Interest Declaration: Authors declare no conflict of interest.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
The work was completed at North Carolina State University, Raleigh, NC and the Laboratory of Neurogenetics, NIA, Bethesda, MD
This study was funded by the American Kennel Club Canine Health Foundation (AKC‐CHF) with support from the Health Trust Fund of the Scottish Terrier Club of America.
Footnotes
O'Brien DP, Zeng R, Schnabel RD, Taylor JF, Johnson GS. Identification of Two Breed‐Specific Mutations Associated with Canine Multiple System Degeneration Using Whole Genome Resequencing. Proc ACVIM Forum 2013
QIAamp® DNA Blood Midi Kit: Qiagen, Valencia, CA
Oragene Animal kit: DNA Genotek, Kanata, Ontario
ND‐1000 NanoDrop spectrophotometer: Thermo Scientific, Wilmington, DE
ABI 3730xl DNA Analyzer and GeneMapper v3.7 software: Applied Biosystems, Carlsbad, CA
Illumina® CanineSNP20 BeadChip, Illumina® CanineHD genotyping BeadChip, BeadArray Reader, and Illumina® BeadStudio software: Illumina Inc., San Diego, CA
References
- 1. Urkasemsin G, Olby NJ. Canine hereditary ataxia. Vet Clin North Am Small Anim Pract 2014;44:1075–1089. [DOI] [PubMed] [Google Scholar]
- 2. Agler C, Nielsen DM, Urkasemsin G, et al. Canine hereditary Ataxia in old English sheepdogs and Gordon setters is associated with a defect in the Autophagy gene encoding RAB24. PLoS Genet 2014;10:e1003991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kyöstilä K, Cizinauskas S, Seppälä EH, et al. A SEL1L mutation links a canine progressive early‐onset cerebellar ataxia to the Endoplasmic Reticulum‐Associated Protein Degradation (ERAD) machinery. PLoS Genet 2012;8:e1002759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Forman OP, De Risio L, Mellersh CS. Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the parson Russell terrier dog breed. PLoS ONE 2013;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forman OP, De Risio L, Stewart J, et al. Genome‐Wide mRNA sequencing of a single canine cerebellar cortical degeneration case leads to the identification of a disease associated SPTBN2 mutation. BMC Genet 2012;13:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forman OP, De Risio L, Matiasek K, et al. Spinocerebellar ataxia in the Italian spinone dog is associated with an intronic GAA repeat expansion in ITPR1. Mamm Genome 2015;26:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeng R, Farias FH, Johnson GS, et al. A truncated retrotransposon disrupts the GRM1 coding sequence in Coton de Tulear dogs with Bandera's neonatal Ataxia. J Vet Intern Med 2011;25:267–272. [DOI] [PubMed] [Google Scholar]
- 8. Gilliam D, O'Brien DP, Coates JR, et al. A homozygous KCNJ10 mutation in jack Russell terriers and related breeds with spinocerebellar ataxia with Myokymia, seizures, Or both. J Vet Intern Med 2014;28:871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rohdin C, Gilliam D, O'Leary CA, et al. A KCNJ10 mutation previously identified in the Russell group of terriers also occurs in smooth‐haired fox terriers with hereditary ataxia and in related breeds. Acta Vet Scand 2015;57:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Brien DP, Shibuya H, Zhou T, et al. X‐linked cerebellar ataxia in English pointer dogs: Phenotype and linkage data. Canine Pract 1998;23:46. [Google Scholar]
- 11. van der Merwe LL, Lane E. Diagnosis of cerebellar cortical degeneration in a Scottish terrier using magnetic resonance imaging. J Small Anim Pract 2001;42:409–412. [DOI] [PubMed] [Google Scholar]
- 12. Urkasemsin G, Linder KE, Bell JS, et al. Mapping of Purkinje neuron loss and Polyglucosan body accumulation in hereditary cerebellar degeneration in Scottish Terriers. Vet Pathol 2012;49:852–859. [DOI] [PubMed] [Google Scholar]
- 13. Urkasemsin G, Linder KE, Bell JS, et al. Hereditary cerebellar degeneration in Scottish Terriers. J Vet Intern Med 2010;24:565–570. [DOI] [PubMed] [Google Scholar]
- 14. Clark LA, Tsai KL, Steiner JM, et al. Chromosome‐specific microsatellite multiplex sets for linkage studies in the domestic Dog. Genomics 2004;84:550–554. [DOI] [PubMed] [Google Scholar]
- 15. Thompson EA. MCMC estimation of multi‐locus genome sharing and multipoint gene location scores. Int Stat Rev 2000;68:53–73. [Google Scholar]
- 16. Olby N, Blot S, Thibaud JL, et al. Cerebellar cortical degeneration in adult American Staffordshire Terriers. J Vet Intern Med 2004;18:201–208. [DOI] [PubMed] [Google Scholar]
- 17. Strauch K, Fimmers R, Kurz T, et al. Parametric and nonparametric multipoint linkage analysis with imprinting and two‐locus‐trait models: Application to mite sensitization. Am J Hum Genet 2000;66:1945–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Finsterer J. Ataxias with autosomal, X‐chromosomal or maternal inheritance. Can J Neurol Sci 2009;36:409–428. [DOI] [PubMed] [Google Scholar]
- 20. Zanni G, Bertini ES. X‐linked disorders with cerebellar dysgenesis. Orphanet J Rare Dis 2011;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Del Bigio MR, Halliday WC. Multifocal atrophy of cerebellar internal granular neurons in Lesch‐Nyhan disease: Case reports and review. J Neuropathol Exp Neurol 2007;66:346–353. [DOI] [PubMed] [Google Scholar]
- 22. Lyon MF. Sex chromatin and gene action in the mammalian X‐chromosome. Am J Hum Genet 1962;14:135–148. [PMC free article] [PubMed] [Google Scholar]
- 23. Arthold S, Kurowski A, Wutz A. Mechanistic insights into chromosome‐wide silencing in X inactivation. Hum Genet 2011;130:295–305. [DOI] [PubMed] [Google Scholar]
- 24. Pontier DB, Gribnau J. Xist regulation and function explored. Hum Genet 2011;130:223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carrel L, Willard HF. X‐inactivation profile reveals extensive variability in X‐linked gene expression in females. Nature 2005;434:400–404. [DOI] [PubMed] [Google Scholar]
- 26. Berletch JB, Yang F, Disteche CM. Escape from X inactivation in mice and humans. Genome Biol 2010;11:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang F, Babak T, Shendure J, Disteche CM. Global survey of escape from X inactivation by RNA‐sequencing in mouse. Genome Res 2010;20:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Young AC, Kirkness EF, Breen M. Tackling the characterization of canine chromosomal breakpoints with an integrated in‐situ/in‐silico approach: The canine PAR and PAB. Chromosome Res 2008;16:1193–1202. [DOI] [PubMed] [Google Scholar]
- 29. Desai V, Donsante A, Swoboda KJ, et al. Favorably skewed X‐inactivation accounts for neurological sparing in female carriers of Menkes disease. Clin Genet 2011;79:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Orstavik KH. X chromosome inactivation in clinical practice. Hum Genet 2009;126:363–373. [DOI] [PubMed] [Google Scholar]
- 31. Lazzaro MA, Todd MAM, Lavigne P, et al. Characterization of novel Isoforms and evaluation of SNF2L/SMARCA1 as a Candidate gene for X‐linked mental retardation in 12 families linked to Xq25‐26. BMC Med Genet 2008;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. A Scottish Terrier pedigree. Ninety‐six dogs were genotyped (green border). Females are ovals, and males are rectangles. Affected (case) dogs are solid red, normal (control) dogs are white, and undetermined status dogs are solid yellow. The vertical black oval highlights the sub‐family in which the affected dam has an affected male and a normal female offspring. The horizontal blue ovals highlight litters containing affected males and normal females. The pink circle highlights a female affected dog from normal parents.