

Summary
Complement C5 deficiency (C5D) is a rare primary immunodeficiency associated with recurrent infections, particularly meningitis, by Neisseria species. To date, studies to elucidate the molecular basis of hereditary C5D have included fewer than 40 families, and most C5 mutations (13 of 17) have been found in single families. However, the recently described C5 p.A252T mutation is reported to be associated with approximately 7% of meningococcal disease cases in South Africa. This finding raises the question of whether the mutation may be prevalent in other parts of Africa or other continental regions. The aim of this study was to investigate the prevalence of C5 p.A252T in Africa and other regions and discuss the implications for prophylaxis against meningococcal disease. In total, 2710 samples from healthy donors within various populations worldwide were analysed by quantitative polymerase chain reaction (qPCR) assay to detect the C5 p.A252T mutation. Eleven samples were found to be heterozygous for p.A252T, and nine of these samples were from sub‐Saharan African populations (allele frequency 0·94%). Interestingly, two other heterozygous samples were from individuals in populations outside Africa (Israel and Pakistan). These findings, together with data from genomic variation databases, indicate a 0·5–2% prevalence of the C5 p.A252T mutation in heterozygosity in sub‐Saharan Africa. Therefore, this mutation may have a relevant role in meningococcal disease susceptibility in this geographical area.
Keywords: Africa, C5 deficiency, complement, infection, meningococcal disease, primary immunodeficiency
Introduction
The complement system has a major role in the innate immune system's defence against pathogens. Complement activation upon recognition of certain molecular patterns by any of the three pathways leads to activation of a common terminal complement pathway 1, 2. C5 is the initial component of this pathway, and activation through the C5 convertases releases the potent anaphylatoxin peptide C5a and generates C5b. After C5 cleavage, the terminal components C6, C7, C8 and C9 bind to produce the membrane attack complex (MAC), which forms a hydrophilic channel through the membrane of target cells, causing cell death 3. The pores that the MAC generates on a pathogen's surface are key to host defence against Gram‐negative bacteria, particularly species from the Neisseria genus 4.
During recent decades, deficiencies of the terminal complement components that form the MAC (C5‐C9) have been described extensively 5. The risk of meningococcal disease in patients with these deficiencies is 7000–10 000‐fold higher than that of healthy individuals, and they also have a higher risk of recurrent invasive meningococcal infection 6, 7. In general, terminal complement deficiencies are rare, but the incidence varies considerably between different populations. A well‐recognized example is C9 deficiency, caused mainly by the non‐sense mutation, R95X. In Japan, it is one of the most common genetic disorders, with a population prevalence of one in 1000, whereas only a few patients with C9 deficiency have been identified in European countries 8. C7 deficiency caused by the G357R mutation is reported as having a high prevalence (1·1%) in the Israeli Moroccan Jewish population 9, and C6 deficiency has a high prevalence in Western Cape South Africans and in African Americans 10, 11, 12, 13.
Focusing upon C5 deficiency, approximately 50 cases have been published from around the world 14, 15, 16, 17, 18, 19, 20, 21, 22 and most of the causal mutations have been described in only one family. A striking exception is the p.A252T mutation, reported recently to be responsible for C5 deficiency and found in approximately 7% of meningococcal disease cases in black Africans from the Western Cape (South Africa) 21. This mutation has an allele frequency of 3% in the black African population and 0·66% in the Cape Coloured population of the Western Cape area 21.
Meningococcal meningitis cases occur throughout the world. However, large, recurrent epidemics affect an extensive region of sub‐Saharan Africa known as the ‘meningitis belt’, which covers 26 countries from Senegal in the West to Ethiopia in the East 23. Considering the previously observed allelic distribution of p.A252T in South Africa, the question arises as to whether the mutation may be prevalent in other parts of Africa or other continental regions. The aim of this study was to evaluate the presence of the C5 p.A252T mutation in Africa and other continental regions and discuss the implications for prophylaxis against meningococcal disease.
Materials and methods
Samples
In total, 2710 gDNA samples were tested. Almost half these samples (n = 1064) were a part of the Human Genome Diversity Panel (HGDP), a widely used resource for studies of human genetic variation. Genomic DNA samples from these fully consenting individuals were collected by the Human Genome Diversity Project in collaboration with the Centre Etude Polymorphism Humain (CEPH) in Paris. Fifty‐one different populations from Africa, Europe, the Middle East, South and Central Asia, East Asia, Oceania and the Americas are represented in the HGDP 24, 25.
To increase the number of samples from Africa, a collection of 352 samples from Gabon and 768 samples from North African countries (Morocco, Algeria, Tunisia and Libya) were also included. The presence of the mutated allele was also evaluated in 526 local samples (Barcelona, Spain) obtained from blood donors.
Although all the samples included in the study were from healthy individuals, a history of previous meningococcal disease was not recorded specifically. All samples were collected with the informed consent of the volunteers participating in the study.
Screening for the C5 p.A252T mutation
Quantitative polymerase chain reaction (qPCR) assay design
The properly tagged consensus sequence containing the C5 c.754G > A (p.A252T) variant and all other single nucleotide polymorphisms (SNPs) in the vicinity was submitted to the design tool at the Applied Biosystems website for custom TaqMan assay design. The TaqMan genotyping assay mix contained a forward and reverse primer for the submitted sequence, one probe that matched perfectly the wild‐type sequence variant (c.754G) labelled with VIC, and a second probe that matched the mutant (c.754A) variant, labelled with FAM.
Allele discrimination assay
To obtain a final PCR volume of 10 μl, a working master mix was prepared containing 0·25 μl of TaqMan genotyping assay mix (×40), 5 μl TaqMan UNG Master Mix ×2 (Applied Biosystems, Carlsbad, CA, USA) and 2·75 μl of water for each reaction. Samples were placed directly into each well of a 96‐well plate. Two controls for each condition (wild‐type, heterozygous for the mutation and homozygous for the mutation) were used in each assay. Plates were loaded onto an Applied Biosystems 7500 fast real‐time PCR system, and the 7500 (version 2.0.5) software was used to run the assay, following the default standard allelic discrimination genotyping assay protocol. Briefly, this consisted of an end‐point PCR in which fluorescence was read prior to PCR and after completion of the last PCR cycle. The software calculated the normalized dye fluorescence (ΔRn) as a function of cycle number for allele 1 (wild‐type) and allele 2 (mutant). The results were displayed on a plot, where each axis represented the fluorescence level of each fluorochrome, thus grouping the samples on the three different genotypes (Fig. 1a).
Figure 1.
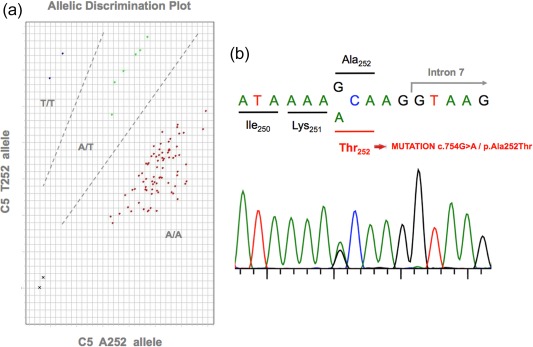
C5 p.A252T mutation screening. (a) Cluster plot of 96 DNA samples that underwent C5 p.A252T mutation genotyping by TaqMan assay using the Applied Biosystems 7500 real‐time polymerase chain reaction (RT–PCR) system. Red indicates wild‐type homozygous samples (A252). Green indicates heterozygous samples. Blue indicates mutated homozygous samples (T252). Black indicates the negative controls. Two controls for each genotype were included in all plates. (b) Sanger sequencing of one heterozygous carrier of the C5 p.A252T mutation. All heterozygous samples detected by quantitative PCR were sequenced to confirm the results. [Colour figure can be viewed at wileyonlinelibrary.com]
Sanger sequencing
Samples identified as heterozygous for the c.754G > A/p.A252T mutation by the qPCR genotyping assay were then confirmed by Sanger sequencing of C5 exon 7 (Fig. 1b).
Results
The C5 p.A252T mutation is prevalent in, but not restricted to, sub‐Saharan Africa
Screening for the C5 c.754G > A/p.A252T mutation was carried out in 2710 samples from different populations worldwide. Initially, to provide a general view of p.A252T distribution, we grouped the samples into seven continental regions and calculated the frequencies of the mutated allele in each one (Table 1). Results for individual populations are shown in the Supporting information, Table S1. In total, 11 samples heterozygous for the p.A252T mutation were found, resulting in an overall mutated allele frequency of 0·2% in our samples. However, the distribution of the mutation was not random: sub‐Saharan Africa populations showed the highest number of mutated alleles, with nine heterozygous samples among the 958 tested (allele frequency 0·94%). These included seven heterozygous samples among 352 individuals tested in Gabon (allele frequency 1%), one of 25 in Nigeria (allele frequency 2%) and one of eight in South Africa (allele frequency 6·25%) (Table 2). In this last case, and to lesser extent in Nigeria, the allele frequency was probably over‐estimated because of the small number of samples tested.
Table 1.
Allelic frequencies of the C5 c.754G > A/p.A252T mutation
| Continental region | Alleles tested | Mutated alleles | Mutated allele frequency (%) |
|---|---|---|---|
| Sub‐Saharan Africa | 958 | 9 | 0·94 |
| America | 216 | 0 | 0 |
| East Asia | 502 | 0 | 0 |
| Central and South Asia | 400 | 1* | 0·25 |
| Middle East and North Africa | 1892 | 1 † | 0·05 |
| Oceania | 78 | 0 | 0 |
| Europe | 1374 | 0 | 0 |
All mutated alleles detected correspond to heterozygous individuals. *Individual from Pakistan; †individual from Israel (Negev).
Table 2.
Allelic frequencies of c.754G > A/p.A252T in sub‐Saharan African countries
| Country | Alleles tested | Mutated alleles | Mutated allele frequency (%) |
|---|---|---|---|
| South Africa | 16 | 1 | 6·25 |
| Kenya | 24 | 0 | 0 |
| Namibia | 14 | 0 | 0 |
| Central African Republic | 72 | 0 | 0 |
| Nigeria | 50 | 1 | 2 |
| Democratic Republic of Congo | 30 | 0 | 0 |
| Gabon | 704 | 7 | 1 |
| Senegal | 48 | 0 | 0 |
Interestingly, we found two heterozygous samples from other continental regions: one from Israel (Middle East and North Africa region) and one from Pakistan (Central and South Asia region). The mutation was absent in the remaining continental regions studied.
Overall analysis of the C5 p.A252T mutation worldwide distribution
The C5 c.754G > A/p.A252T allelic variation is reported in the SNP database (dbSNP) (code rs112959008), and has an overall minor allele frequency (MAF) of 0·0012, as reported in the 1000 Genomes Project (http://www.1000genomes.org) 26. However, as our results indicate, there are huge differences in this frequency depending on the continental region. Within the 1000 Genomes Project (1000G), the mutated allele was reported in individuals from Barbados (of African origin), Nigeria and Kenya (Table 3), with an overall estimated heterozygosity of 1–2% in these Central African countries. This data is concordant with our reported heterozygosity in Gabon (1%). The p.A252T mutation was not found in any of the other populations examined in 1000G, which unfortunately does not include samples from South Africa or countries in the Middle East/North Africa.
Table 3.
Review of the reported populations with presence of C5 p.A252T mutation
| Population | Heterozygosity frequency (%) | Count (total) | Source |
|---|---|---|---|
| Africa | |||
| Barbados (African Caribbean) | 2·1 | 2 (96) | 1000G |
| Nigeria (Esan) | 2 | 2 (98) | 1000G |
| Kenya (Luhya in Webuye) | 1 | 1 (99) | 1000G |
| Nigeria (Yoruba in Ibadan) | 0·93 | 1 (108) | 1000G |
| African American | 0·72 | 15 (2090) | NHLBI Exome Sequencing Project |
| South Africa (black Africans) | 6 | 45 (750) | Owen et al. |
| South Africa (Cape Coloured) | 1·33 | 10 (750) | Owen et al. |
| South Africa (Bantu S.U. Zulu) | 12·5 | 1 (8) | This study |
| Nigeria (Yoruba) | 4 | 1 (25) | This study |
| Gabon | 1·99 | 7 (352) | This study |
| South Asia | |||
| Pakistan | 0·5 | 1 (200) | This study |
| Middle East | |||
| Israel (Negev) | 2 | 1 (49) | This study |
1000G = 1000 Genomes Project; NHLBI = National Heart, Lung and Blood Institute.
Further evidence of the higher prevalence of the p.A252T mutation in Africa relies upon a report from the National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/). In the ESP, the mutated allele was identified in in 15 of 2090 samples from African American individuals (allele frequency, 0·36%) (Table 3). In contrast, it was not found in any of the 4044 samples tested from Americans of European ancestry.
Recently, Owen et al. investigated the incidence of the mutated allele in 1500 samples from South African neonates. The allele was present in both black Africans and Cape Coloured individuals, with allelic frequencies of 3 and 0.66%, respectively 21. The populations in which the C5 p.A252T mutation has been detected, together with their heterozygosity frequencies, are summarized in Table 3.
Discussion
The MAC has a major role in defending the host from Neisseria infection, but it cannot form if any of the five final complement components are absent. Consequently, C5 deficiency (C5D, OMIM #609536) is associated with recurrent episodes of infection by Neisseria species, particularly meningitis and extragenital gonorrhoea. To date, studies to elucidate the molecular basis of hereditary C5D have included more than 30 families, with descriptions of 17 different mutations 22 distributed randomly along the C5 gene, and including missense, non‐sense, insertion–deletion and splicing mutations. Most C5 mutations (13 of the 17) have been described in only one family 22. The most relevant exception to this general observation is the p.A252T mutation, reported by Owen et al. 21.
The results of our study, together with data recorded in public databases, demonstrate the high prevalence of p.A252T in sub‐Saharan Africa, and show for the first time the presence of this mutation in regions outside Africa (Middle East and South Asia).
Although it is difficult to establish the true frequency of the mutation, our results may be helpful for outlining an estimate. As shown in Table 3, various unrelated data have proved the presence of the A252T variant in heterozygosity in healthy individuals from several parts of Africa, and our results broaden the distribution of this mutation to other regions, specifically Israel and Pakistan.
However, when interpreting the frequency of p.A252T, several factors must be considered, particularly the size and origin of the population studied. The specific populations included in the 1000G and HGDP used in this study are well defined, but relatively small in size (around 100 individuals or less). To increase the representation from Africa, we collected 352 samples from Gabon (Central Africa) and 768 from North African countries (Morocco, Algeria, Tunisia and Libya). Other relevant data were provided by the 2090 African American individuals within the NHLBI Exome Sequencing Project (although their specific origin is not reported) and the 1500 individuals from South Africa described by Owen et al. 21. Based on the data from the present study and the above‐mentioned available information, we estimated the heterozygosity frequency of the C5 p.A252T mutation in sub‐Saharan Africa to be 0·5–2%. The true accuracy of this frequency and whether it substantially varies between the different populations in the region are questions that remain to be elucidated in studies with larger sample sizes. What seems clear, however, is that the high prevalence of this mutation in Africa is restricted to the sub‐Saharan region, as no carriers were found among the 768 samples from North African countries. Nevertheless, we cannot exclude the possibility that the mutation is present at a low frequency in North Africa.
Based on these data, it seems likely that the C5 p.A252T mutation could have a relevant role in meningococcal disease susceptibility in sub‐Saharan Africa. In all individuals homozygous for p.A252T, C5 protein levels are < 4% of the mean control level measured by enzyme‐linked immunosorbent assay (ELISA) and are undetectable by Western blot 21, providing proof of the functional importance of this mutation. The amino acid A252 is highly conserved among the species (Supporting information, Fig. S1), and estimation of the putative pathogenicity of A252T using two widely used prediction software packages (SIFT and PolyPhen‐2) indicated that pathogenicity was probable. Therefore, p.A252T homozygosity would lead to de‐facto C5 deficiency (with extremely low serum C5 protein levels) and a high susceptibility to meningococcal disease.
Why this mutation has survived evolutionary pressure is a question still to be answered. It is reported that individuals with deficiencies of late complement components have a lower incidence of septic shock after Neisseria meningitides infection, which results in lower mortality from this disease 7. Following this rationale, it is possible that carriers of the mutation in the meningitis belt countries of sub‐Saharan Africa would have some protection from septic shock during infection by this pathogen, thus increasing their chances of survival. Nonetheless, meningococcal disease is often associated with serious sequelae, such as a loss of digits or limbs, or persistent neurological damage. Hence, emphasis should be placed upon prevention of the disease through vaccination.
Based on the lower range of the estimated frequency of C5 p.A252T heterozygosity in this study (0·5%) and considering a population of 1 billion individuals living in sub‐Saharan Africa (source: http://www.worldbank.org), a few million heterozygous and thousands of homozygous individuals with C5 deficiency would be living in this area. These estimated values should be confirmed in further studies including a significantly larger sample size from countries within the meningitis belt. It would also be interesting to evaluate the prevalence of p.A252T in meningococcal disease patients in sub‐Saharan African countries, which could be carried out with the simple, low‐cost method used in the present study. Moreover, if the prevalence of the mutation is as high as suspected, a routine test could be used to detect it and facilitate a prompt diagnosis of complement deficiency 27.
Many countries outside Africa have been a common destination for African immigrants over the years; consequently, the mutation can be spread to other countries. An example of this occurred in the present study. We collected 527 local samples, and after rigorous selection to ensure their European origin, one of them was excluded from the study because it came from an individual from The Gambia. The sample was tested and was found to carry the mutated allele in heterozygosis.
In summary, the C5 p.A252T mutation is indeed prevalent in sub‐Saharan Africa. Testing for this mutation in meningococcal disease patients would be of value to identify C5‐deficient individuals. A further step would be newborn screening, which could enable more effective prevention of the primary disease through vaccination.
Disclosure
None of the authors has any potential financial conflict of interest related to this paper.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Allelic frequencies of C5 c.754G > A/p.A252T mutation
Fig. S1. Phylogenetic comparison of human C5 protein sequence. C5 multiple sequence alignment focusing on A252 is shown. Amino acid sequences from different organisms have been obtained from the UniProt database (http://www.uniprot.org).
Acknowledgements
This study was funded by Instituto de Salud Carlos III, grant PI14/00405, co‐financed by the European Regional Development Fund (ERDF), and by MINECO grant CGL2013‐44351‐P. We thank Jean‐Marie Hombert and Lolke van der Veen (Dynamique du Langage, Institut des Sciences De l'Homme, Lyon, France), as well as Patrick Mouguiama Daouda (University Omar Bongo, Libreville, Gabon) and the Centre International des Recherches Médicales de Franceville (CIRMF, Gabon) for sharing the Gabon samples. We thank Karima Fadhlaoui‐Zid for providing some North African DNA samples. We would like to thank Felicity Leisegang for providing the samples that were used as controls for the technique. Ann Orren acknowledges the support of the SA MRC, and the Department of Medicine and the Division of Chemical Pathology at the University of Cape Town. We also acknowledge Celine Cavallo for English language assistance.
References
- 1. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 2010; 11:785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walport MJ. Complement. First of two parts. N Engl J Med 2001; 344:1058–66. [DOI] [PubMed] [Google Scholar]
- 3. Tegla CA, Cudrici C, Patel S et al Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res 2011; 51:45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lewis LA, Ram S. Meningococcal disease and the complement system. Virulence 2014; 5:98–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Petersen BH, Lee TJ, Snyderman R, Brooks GF. Neisseria meningitidis and Neisseria gonorrhoeae bacteremia associated with C6, C7, or C8 deficiency. Ann Intern Med 1979; 90:917–20. [DOI] [PubMed] [Google Scholar]
- 6. Tedesco F, Nürnberger W, Perissutti S. Inherited deficiencies of the terminal complement components. Int Rev Immunol 1993; 10:51–64. [DOI] [PubMed] [Google Scholar]
- 7. Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev 1991; 4:359–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kira R, Ihara K, Watanabe K et al Molecular epidemiology of C9 deficiency heterozygotes with an arg95stop mutation of the C9 gene in Japan. J Hum Genet 1999; 44:109–11. [DOI] [PubMed] [Google Scholar]
- 9. Halle D, Elstein D, Geudalia D et al High prevalence of complement C7 deficiency among healthy blood donors of Moroccan Jewish ancestry. Am J Med Genet 2001; 99:325–7. [DOI] [PubMed] [Google Scholar]
- 10. Hobart MJ, Fernie BA, Fijen KA, Orren A. The molecular basis of C6 deficiency in the Western Cape, South Africa. Hum Genet 1998; 103:506–12. [DOI] [PubMed] [Google Scholar]
- 11. Orren A, Owen EP, Henderson HE et al Complete deficiency of the sixth complement component (C6Q0), susceptibility to Neisseria meningitidis infections and analysis of the frequencies of C6Q0 gene defects in South Africans. Clin Exp Immunol 2012; 167:459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parham KL, Roberts A, Thomas A et al Prevalence of mutations leading to complete C6 deficiency (C6Q0) in the Western Cape, South Africa and detection of novel mutations leading to C6Q0 in an Irish family. Mol Immunol 2007; 44:2756–60. [DOI] [PubMed] [Google Scholar]
- 13. Zhu Z, Atkinson TP, Hovanky KT et al High prevalence of complement component C6 deficiency among African Americans in the south‐eastern USA. Clin Exp Immunol 2000; 119:305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X, Fleischer DT, Whitehead WT et al Inherited human complement C5 deficiency. Nonsense mutations in exons 1 (gln1 to stop) and 36 (arg1458 to stop) and compound heterozygosity in three African‐American families. J Immunol 1995; 154:5464–71. [PubMed] [Google Scholar]
- 15. Delgado‐Cerviño E, Fontán G, López‐Trascasa M. C5 complement deficiency in a Spanish family. Molecular characterization of the double mutation responsible for the defect. Mol Immunol 2005; 42:105–11. [DOI] [PubMed] [Google Scholar]
- 16. Pfarr N, Prawitt D, Kirschfink M et al Linking C5 deficiency to an exonic splicing enhancer mutation. J Immunol 2005; 174:4172–7. [DOI] [PubMed] [Google Scholar]
- 17. Aguilar‐Ramirez P, Reis ES, Florido MP et al Skipping of exon 30 in C5 gene results in complete human C5 deficiency and demonstrates the importance of c5d and CUB domains for stability. Mol Immunol 2009; 46:2116–23. [DOI] [PubMed] [Google Scholar]
- 18. López‐Lera A, Garrido S, de la Cruz RM, Fontán G, López‐Trascasa M. Molecular characterization of three new mutations causing C5 deficiency in two non‐related families. Mol Immunol 2009; 46:2340–7. [DOI] [PubMed] [Google Scholar]
- 19. Arnaout R, Al Shorbaghi S, Al Dhekri H et al C5 complement deficiency in a Saudi family, molecular characterization of mutation and literature review. J Clin Immunol 2013; 33:871–5. [DOI] [PubMed] [Google Scholar]
- 20. Schejbel L, Fadnes D, Permin H, Lappegård KT, Garred P, Mollnes TE. Primary complement C5 deficiencies – molecular characterization and clinical review of two families. Immunobiology 2013; 218:1304–10. [DOI] [PubMed] [Google Scholar]
- 21. Owen EP, Würzner R, Leisegang F et al A complement C5 gene mutation, c.754G>A:P.A252T, is common in the Western Cape, South Africa and found to be homozygous in seven percent of Black African meningococcal disease cases. Mol Immunol 2015; 64:170–6. [DOI] [PubMed] [Google Scholar]
- 22. Colobran R, Franco‐Jarava C, Martín‐Nalda A et al Novel mutations causing C5 deficiency in three North‐African families. J Clin Immunol 2016; 36:388–96. [DOI] [PubMed] [Google Scholar]
- 23. Trotter CL, Greenwood BM. Meningococcal carriage in the African meningitis belt. Lancet Infect Dis 2007; 7:797–803. [DOI] [PubMed] [Google Scholar]
- 24. Cann HM, de Toma C, Cazes L et al A human genome diversity cell line panel. Science 2002; 296:261–2. [DOI] [PubMed] [Google Scholar]
- 25. Rosenberg NA, Pritchard JK, Weber JL et al Genetic structure of human populations. Science 2002; 298:2381–5. [DOI] [PubMed] [Google Scholar]
- 26. Auton A, Brooks LD, Durbin RM et al A global reference for human genetic variation. Nature 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Owen EP, Leisegang F, Whitelaw A et al Complement component C5 and C6 mutation screening indicated in meningococcal disease in South Africa. S Afr Med J 2012; 102:525–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Allelic frequencies of C5 c.754G > A/p.A252T mutation
Fig. S1. Phylogenetic comparison of human C5 protein sequence. C5 multiple sequence alignment focusing on A252 is shown. Amino acid sequences from different organisms have been obtained from the UniProt database (http://www.uniprot.org).