

Abstract
Orbital extension is a major cause of death in children with retinoblastoma in the developing countries. Delayed detection and inappropriate management contribute to poor outcome. Conventional treatment including primary orbital exenteration or chemotherapy or radiotherapy alone result in mortality as high as 70%. The recent understanding on the role of sequential multimodal therapy with a combination of high-dose chemotherapy, followed by appropriate surgery, radiotherapy, and additional adjuvant chemotherapy has helped dramatically improve life salvage.
Keywords: Multimodal therapy, orbit, retinoblastoma
Survival of patients with retinoblastoma has dramatically improved in the past few decades, essentially due to early diagnosis and better management protocols.[1] At 5-year survival rates of 88%, 91%, and 93% have been reported from developed countries such as the United Kingdom,[2] Japan,[3] and the United States, respectively.[1,4] However, the mortality is still high in the developing countries.[5,6] Delayed medical attention due to compounding social and economic factors and the paucity of standard treatment protocols for the comprehensive management of advanced retinoblastoma are believed to be the main causes for poor survival.[3] One of the significant contributors to mortality is orbital retinoblastoma.[7,8,9] This review provides an update on the current concepts in the management of orbital retinoblastoma.
Definition
Clinically, radiologically, or histopathologically detected extension of retinoblastoma outside the confines of the eye is termed as orbital retinoblastoma.
Incidence
Orbital retinoblastoma is rare in developed countries. Ellsworth observed a steady decline in the incidence of orbital retinoblastoma in his large series of 1160 patients collected over 50 years.[10] Overall incidence was 8.2% in the period 1925–1959 and 7.6% between years 1959 and 1974.[10] Later, authors from the same center reported that 6.3% (11 of 175) patients presented with primary orbital retinoblastoma from 1980 to 1986.[11] The histopathologic evidence of scleral invasion, extrascleral extension, and optic nerve infiltration, although variable, is about 2%.[12]
Orbital retinoblastoma is relatively more common in the developing countries. In a recent large multi-center study from Mexico, 18% of 500 patients presented with an orbital retinoblastoma.[13] A Taiwanese group reported that 36% (42 of 116) of their patients manifested with orbital retinoblastoma.[14] The incidence is higher (40%, 19 of 43) in Nepal, with proptosis was the most common clinical manifestation of retinoblastoma.[15] However, the incidence of orbital retinoblastoma is much less in our series from India (80 of 1543, 0.5%).[16]
Classification
Orbital retinoblastoma is variably classified based on clinical, radiological, and histopathological characteristics. Classes 1–3 of Children's Cancer Group classification pertain to orbital retinoblastoma.[17] In the new international staging of retinoblastoma, Stages 2 and 3 refer to orbital retinoblastoma.[18] Clinical tumor, node, metastasis (TNM) classification stages T4a and T4b, and pathological TNM stages PT4a and PT4b classify orbital retinoblastoma.[19] Honavar has classified orbital retinoblastoma based on clinical, radiological, and histopathological manifestations.[20]
Clinical Manifestations
Age of presentation
Orbital retinoblastoma generally manifests much later than intraocular retinoblastoma, at around 3 years of age. Various series report the average age of presentation ranging from 30 to 38 months. Patients in our series presented at an average age of 39 (range 4.6–108) months.[20]
Presenting features
While leukocoria was the most common presenting feature (68%) in a series by Antoneli et al.,[21] proptosis predominated (83%) in Menon et al.'s series.[22] The predominant presenting symptoms in our series were proptosis (36%), pain, redness and swelling (30%), and leukocoria (23%), whereas proptosis (73%) was the most common presenting sign.[16] The presenting features vary based on the type of orbital retinoblastoma.
Primary orbital retinoblastoma
Primary orbital retinoblastoma refers to clinically or radiologically detected orbital [Fig. 1], optic nerve [Fig. 2], or full-thickness scleral or extrascleral extension [Fig. 3] of an intraocular retinoblastoma at the initial presentation, with or without proptosis or a fungating mass. Asymptomatic proptosis without orbital and periocular inflammation in a patient with manifest intraocular tumor is the characteristic presentation. Proptosis is often subtle and can be easily missed unless specifically looked for. It is necessary to carefully assess for proptosis in a patient with advanced retinoblastoma with conjunctival congestion or chemosis, dilated episcleral blood vessels, enlarged cornea, neovascularization of the iris, neovascular glaucoma, anterior segment infiltration, hyphema, vitreous hemorrhage, or tumor filling the entire vitreous cavity. Proptosis with inflammation generally indicates reactive sterile orbital cellulitis secondary to necrosis of an intraocular tumor [Fig. 4]. The other clinical manifestations include an obvious vascular, cherry red or pale pink fleshy conjunctival or subconjunctival nodule fixed to the episclera; “solid” chemosis anterior scleral, or an episcleral mass; full-thickness scleral infiltration simulating clinically evident anterior, ciliary or intercalary staphyloma; or palpable orbital mass. Exuberant fungating orbital mass, a classic dramatic manifestation of orbital retinoblastoma, is rarely seen now.
Figure 1.
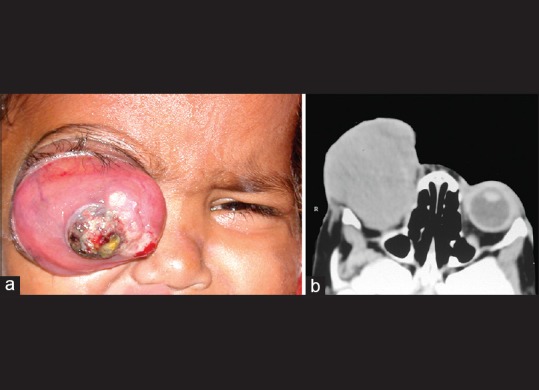
Primary orbital retinoblastoma. Orbital extension of an intraocular retinoblastoma at the initial clinical presentation, manifesting with massive proptosis of the right eye (a), computed tomography scan showing an orbital mass (b)
Figure 2.
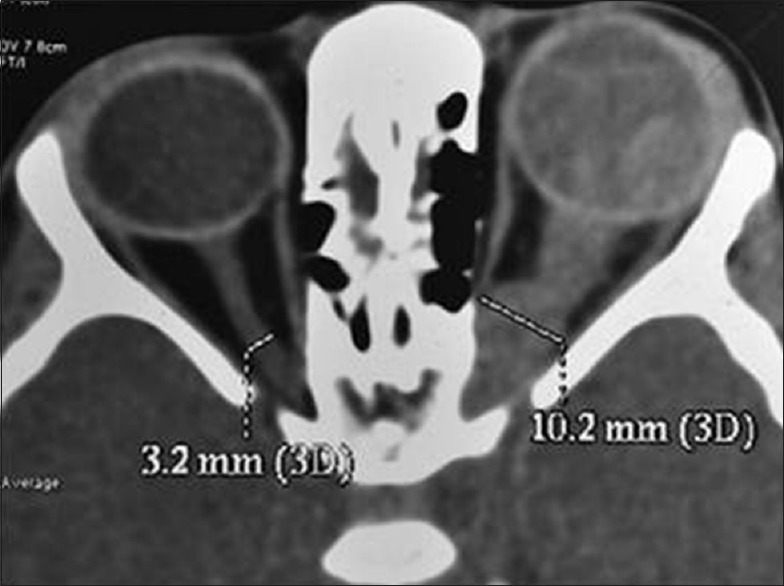
Primary orbital retinoblastoma, optic nerve extension. Axial computed tomography scan showing optic nerve extension of retinoblastoma in the left eye as indicated by gross thickening of the optic nerve up to the orbital apex
Figure 3.
Primary orbital retinoblastoma, episcleral nodule. Bilateral retinoblastoma in a 4-year-old child. The left eye has anterior orbital extension with a vascular pink episcleral nodule
Figure 4.
Sterile orbital inflammation. A 3-year-old child presenting with pain, redness and swelling in the left eye of 4-day duration in the background history of leukocoria for 6 months. Note mild proptosis, intense conjunctival congestion, corneal haze and dirty yellow papillary reflex indicating necrotic intraocular retinoblastoma with sterile orbital inflammation (a). Computed tomography scan shows calcified intraocular tumor in the left eye with diffuse soft tissue thickening around the eye with no evidence of an orbital mass, thus confirming the diagnosis (b)
Patients with suspected primary orbital retinoblastoma need an orbital imaging, computed tomography scan or magnetic resonance imaging. A phthisical eye with retinoblastoma but without evident enophthalmos is highly suspicious of an orbital extension and hence mandates imaging. Imaging may reveal a frank orbital mass contiguous with the eyeball or a thickened optic nerve. It is imperative to compare the optic nerve size with that in the other eye in axial, coronal and sagittal orientations before commenting about optic nerve infiltration. Subtle optic nerve extension may be seen as thickening of the base of the optic nerve. A posterior staphyloma evident on imaging or loss of posterior rounded contour of the eyeball are highly suspicious of extraocular extension.
Secondary orbital retinoblastoma
Orbital recurrence following enucleation for intraocular retinoblastoma is termed secondary orbital retinoblastoma [Fig. 5]. Orbital recurrence of retinoblastoma may present as an orbital mass several weeks to years after the primary surgery. Recurrent orbital tumor could arise either from:
Figure 5.
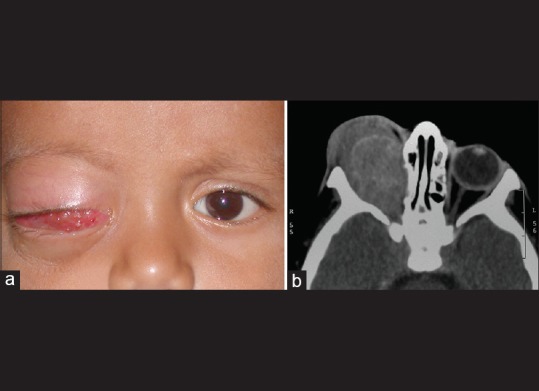
Secondary orbital retinoblastoma. Orbital recurrence of retinoblastoma 6 months following enucleation for intraocular retinoblastoma in the right eye (a). Computed tomography scan showing an orbital mass (b)
Viable residual tumor cells in the orbit in eyes with clinically undetected extraocular extension or unrecognized perforation of the eyeball or
Tumor residual at the optic nerve transection.
In both these situations, careful attention during surgery and histopathologic features following enucleation, followed by appropriate adjuvant therapy may help prevent orbital recurrence.
The recurrent tumor would be predominantly in the orbit if it were to arise from the residual viable tumor cells in orbit, while tumor arising from the residual at the optic nerve transection may present with an intracranial extension that may go clinically undetected until it is advanced. Unexplained displacement, bulge or extrusion of an orbital implant or a previously well-fitting conformer or a prosthesis is an ominous sign suggestive of an orbital recurrence. A vascular conjunctival or subconjunctival nodule may also be a feature of secondary orbital retinoblastoma.
It is considered a good clinical practice to examine for bulge or displacement of the implant and prosthesis and perform careful inspection and palpation of the socket on every periodic follow-up visit, specifically if enucleation has been performed elsewhere and detailed histopathology report is not accessible.
Accidental orbital retinoblastoma
Inadvertent perforation during enucleation, and fine-needle aspiration biopsy (FNAB) or intraocular surgery in an eye with unsuspected intraocular retinoblastoma should be considered as accidental orbital retinoblastoma [Fig. 6]. The implanted viable cells have a potential to develop into an orbital tumor. Patients with a prior history of intraocular surgery for unsuspected retinoblastoma should be specifically examined for a conjunctival, episcleral or orbital mass, nodules at the sites of surgical incisions and vitrectomy ports, and for the enlargement of regional lymph nodes.
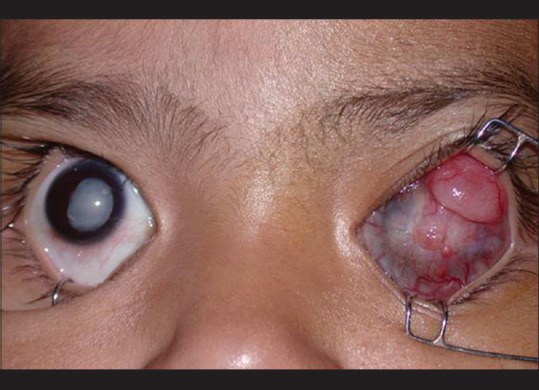
Figure 6.
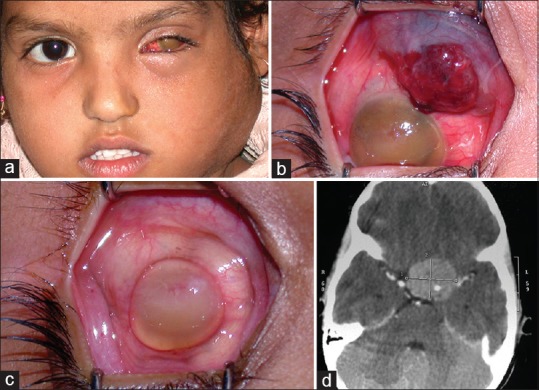
Accidental orbital retinoblastoma. Inadvertent hyphema drainage in an eye with unsuspected retinoblastoma in a 6-year-old girl. She was referred 6 months later with massive cervical lymphadenopathy (a), and a vascular conjunctival mass (b). Although the conjunctival mass resolved with high-dose chemotherapy (c), the child succumbed to intracranial metastasis (d)
Overt orbital retinoblastoma
Previously unrecognized extrascleral or optic nerve extension discovered during enucleation qualifies as overt orbital retinoblastoma [Fig. 7]. Pale pink to the cherry red episcleral nodule, generally juxtapapillary in location or at the site of vortex veins, may be visualized during enucleation. An enlarged and inelastic optic nerve with or without adherent and nodular optic nerve sheath are clinical indicators of optic nerve extension of retinoblastoma that should be recognized during enucleation.
Figure 7.
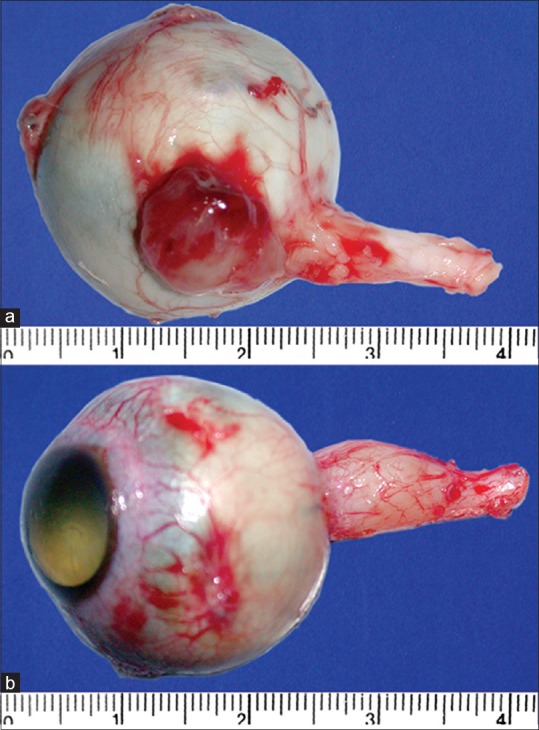
Overt orbital retinoblastoma. Previously unrecognized extrascleral mass (a) and optic nerve extension (b) discovered during enucleation
Microscopic orbital retinoblastoma
In several instances, an orbital extension of retinoblastoma may not be clinically evident and may only be microscopic. Detection of full-thickness scleral infiltration; extrascleral extension; invasion of the optic nerve transection in case of intraneuritic extension; optic nerve sheath infiltration, either contiguous or skip lesions at transection in perineuritic infiltration; or presence of loose tumor cells in the subarachnoid space between the optic nerve and its sheath at transection on histopathologic evaluation of an eye enucleated for intraocular retinoblastoma are unequivocal features of microscopic orbital retinoblastoma [Fig. 8]. Full-thickness choroidal invasion and tumor cells in the sclera, scleral emissaria and optic nerve sheath indicate possible orbital extension, mandating further serial sections and detailed histopathologic analysis to rule out orbital extension.
Figure 8.
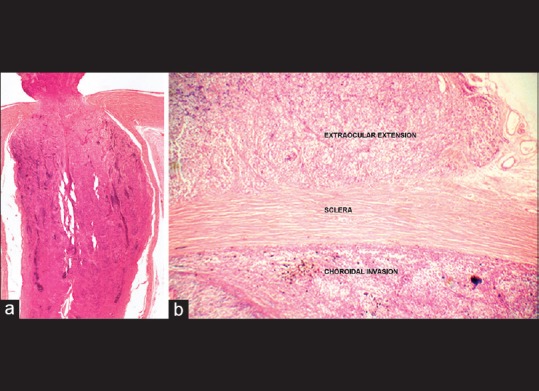
Microscopic orbital retinoblastoma. Detection of invasion of the optic nerve to the level of transection (a), scleral infiltration and extrascleral extension (b) on histopathologic evaluation of an eye enucleated for intraocular retinoblastoma
Diagnostic Evaluation
A thorough clinical evaluation paying attention to the subtle signs of orbital retinoblastoma is necessary. Magnetic resonance imaging preferably or computed tomography scan of the orbit and brain in axial and coronal orientation with 2-mm slice thickness helps confirm the presence of orbital retinoblastoma and determine its extent. Systemic evaluation, including a detailed physical examination, palpation of the regional lymph nodes and FNAB of the same if involved, imaging of orbit and brain, chest X-ray, ultrasonography of the abdomen, bone marrow biopsy, and cerebrospinal fluid cytology are necessary to stage the disease. Technetium-99 bone scan and positron emission tomography coupled with computed tomography may be useful modalities for the early detection of subclinical systemic metastasis.[23,24] Orbital biopsy is rarely required and should be considered specifically when a child presents with an orbital mass following enucleation or evisceration where the primary histopathology is unavailable.
Management
Primary orbital retinoblastoma
Primary orbital retinoblastoma has been managed in the past with orbital exenteration, chemotherapy or external beam radiotherapy in isolation or in combination, with variable results.[25,26,27,28,29] It is well known that local treatments have a limited effect on the course of this advanced disease[28,29] because:
Orbital exenteration alone is unlikely to achieve surgical clearance and preclude recurrence
Systemic chemotherapy alone is unlikely to eradicate residual orbital disease
External beam radiotherapy is unlikely to prevent systemic metastasis.
Therefore, a combination therapy is considered to be more effective. In a case series of five children, Goble and associates demonstrated long-term survival with local surgical excision, orbital radiotherapy, and systemic chemotherapy.[28] We have developed a treatment protocol [Table 1] comprising initial three drug (vincristine, etoposide, carboplatin) high dose chemotherapy (3–6 cycles), followed by surgery (enucleation, extended enucleation or orbital exenteration as appropriate) after complete resolution of the orbital component as detected by serial imaging followed by orbital radiotherapy, and extended chemotherapy for 12 cycles [Table 2].[30] In twenty carefully selected cases without intracranial extension and systemic metastasis, there was dramatic resolution of orbital involvement. The involved eyes became phthisical after 3–6 cycles of high-dose chemotherapy. No clinically apparent orbital tumor was found during enucleation. However, histopathology revealed viable tumor cells in 50%. All the patients completed the treatment protocol of orbital external beam radiotherapy and 12 cycles of chemotherapy. Eighteen (90%) patients were free of local recurrence or systemic metastasis at a mean follow-up of 36 months and achieved acceptable cosmetic outcome [Figs. 9 and 10].[30]
Table 1.
Suggested protocol for management of primary orbital retinoblastoma
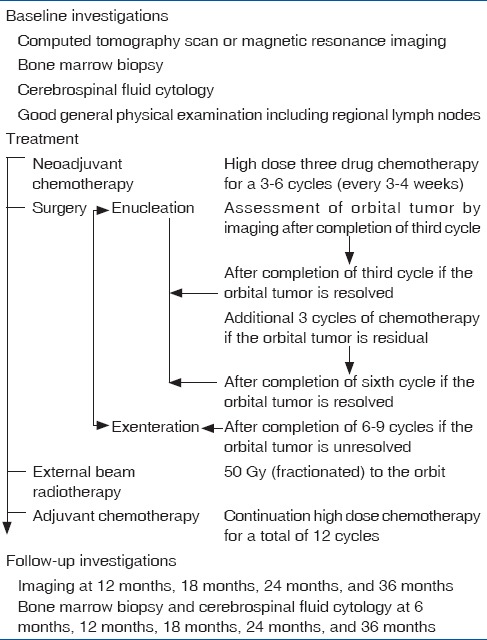
Table 2.
Chemotherapy drugs, dose (mg/kg body weight) and schedule for treatment of orbital retinoblastoma
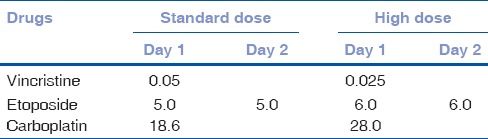
Figure 9.

Outcome of management of primary orbital retinoblastoma. A 3-year-old child with primary orbital retinoblastoma in the right eye with an anterior episcleral nodule (top left and middle), with axial computed tomography scan showing massive orbital extension (top right). Following 3 cycles of high-dose neoadjuvant chemotherapy, the right eye has undergone a quiet phthisis (bottom left and middle), with the computed tomography scan shows complete resolution of the orbital tumor (bottom right)
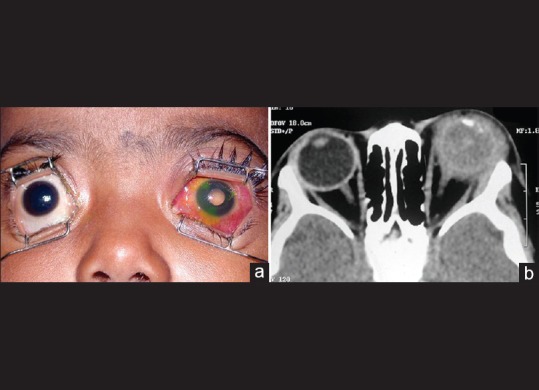
Figure 10.
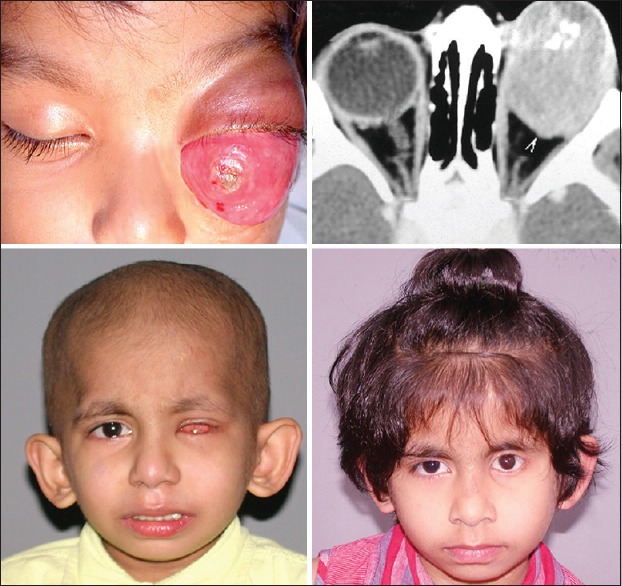
Outcome of management of primary orbital retinoblastoma. A 2-year-old child with primary orbital retinoblastoma in the left eye (top left). Computed tomography scan showing massive orbital tumor (top right). Following 3 cycles of neoadjuvant chemotherapy, enucleation, orbital external beam radiotherapy, and completion of 9 cycles of adjuvant chemotherapy, the orbital tumor is completely resolved (bottom left). Three years later, the child is free of local or systemic recurrence and has acceptable cosmetic outcome with a customized ocular prosthesis (bottom right)
Secondary orbital retinoblastoma
Our treatment protocol outlined for primary orbital retinoblastoma is currently under evaluation for secondary orbital retinoblastoma, and early results have been very encouraging. Surgical intervention in such cases may be limited to excision of the residual orbital mass or an orbital exenteration depending on the extent of the residual tumor after the initial 3–6 cycles of high-dose chemotherapy, followed by external beam radiation therapy and continued chemotherapy for 12 cycles. All the three cases managed by us with the multimodal treatment protocol achieved complete local tumor regression and were free of systemic metastasis.
Accidental orbital retinoblastoma
Accidental orbital retinoblastoma caused by intraocular surgery in an eye with unsuspected retinoblastoma
Patients with a prior history of intraocular surgery should undergo imaging and metastasis work-up. If there is a manifest orbital recurrence, they should be treated as for primary orbital retinoblastoma.
All the eyes that have undergone a recent intraocular surgery for unsuspected retinoblastoma should be promptly enucleated.[31] However, the choice of retaining the eye could be very judiciously exercised in consultation with the family only if the tumor is otherwise treatable, and if there is reasonable visual prognosis. If the eye is retained, no additional therapy (beyond what is planned specifically for the intraocular tumor) would be necessary in cases that have undergone an FNAB or cataract surgery by the clear corneal approach or cataract surgery by the scleral tunnel with an intact posterior capsule. In cases where cataract surgery has been performed by the sclera tunnel with posterior capsule breach or when pars plana vitrectomy has been performed, 6 cycles of high-dose chemotherapy and external beam radiotherapy are mandated.
If enucleation is performed, conjunctiva overlying the ports or surgical incisions with about 4 mm clear margin should be included en-bloc with enucleation. Random orbital biopsy may be also obtained, but there is no data to support its utility. If immediate enucleation is not logistically possible, then the vitrectomy ports or the surgical incision should be subjected to triple-freeze-thaw cryotherapy, sutured, and further sealed with cyanoacrylate glue, and enucleation should be performed at the earliest possible convenience. Histopathologic evaluation of such eyes may specifically involve analysis of the sites of sclerotomy ports or the cataract incision for tumor cells. Further to enucleation, adjuvant treatment depends on the nature and extent of intraocular surgery.
Patients who have undergone only an FNAB by the clear corneal approach without breach of conjunctiva may not need any further treatment. However, if FNAB has been performed by the pars plana route with breach of the conjunctiva, a 4-mm conjunctival margin around the site of FNAB should be included in en bloc enucleation. If the site of FNAB is clear for tumor cells on histopathology, an adjuvant chemotherapy for 6 cycles is provided. However if the site of FNAB shows needle track infiltration and tumor cells within the full-thickness sclera or extrascleral spread, then external beam radiotherapy to the orbit, followed by 6 cycles of adjuvant chemotherapy are mandated.
Patients who have undergone cataract surgery by the clear corneal approach without breach of the conjunctiva may not need any further treatment following enucleation. However, if the cataract surgery has been performed by the scleral tunnel, then an en bloc enucleation with excision of conjunctiva 4 mm around the cataract surgery incision is indicated. Histopathologic evidence of tumor cells in the incision or breach of the posterior capsule would mandate external beam radiotherapy to the orbit, and 6 cycles of adjuvant chemotherapy.
Nondrainage scleral buckle surgery would not mandate adjuvant treatment while scleral buckle with external drainage of subretinal fluid would need external beam radiotherapy and 6 cycles of adjuvant chemotherapy.
Patients who have undergone pars plana vitrectomy should have en bloc enucleation with excision of 4 mm conjunctiva around the ports, followed by external beam radiation and 6 cycles of adjuvant chemotherapy.
Accidental orbital retinoblastoma caused by inadvertent intraoperative perforation of an eye with retinoblastoma
The surgeon should be careful not to accidentally perforate the eye during enucleation for retinoblastoma. Many surgeons prefer to avoid traction sutures applied at the insertion of extraocular muscles to minimize the risk of accidental perforation. Instead, hemostat applied to a purposely kept longer medial or lateral rectus muscle stump or cryoprobe applied at the limbus can provide adequate traction. Eyes manifesting tumor necrosis with aseptic orbital cellulitis pose specific risk for accidental perforation. Surgery in such eyes is best performed when the inflammation is resolved with a brief course of systemic and topical steroids. Enucleation of phthisical eyes with conjunctival fibrosis following resolution of severe inflammation can be a challenge. Conjunctival fibrosis is also encountered in eyes that have been treated with periocular carboplatin injections. Careful conjunctival dissection under magnification in such situations can prevent perforation. Eyes with staphyloma and thin sclera are also prone to perforation and hence need delicate maneuvers during dissection.
All patients with accidental orbital retinoblastoma undergo baseline systemic evaluation for metastasis. Orbital external beam radiotherapy and 12 cycles of high dose chemotherapy are recommended.[31] None of the ten patients with accidental retinoblastoma managed by us developed systemic metastasis.
Overt orbital retinoblastoma
If an extraocular extension is macroscopically visualized during enucleation, special precaution is taken to excise it completely along with the eyeball, preferably along with the layer of Tenon's capsule intact in the involved area.[30] Moreover, steps should be taken to obtain about 18–20 mm long optic nerve stump in all cases of advanced retinoblastoma.[30] In case the optic nerve is thickened and inelastic and is suspected to be involved, and the optic nerve stump is small (<10 mm), it may be best to explore the orbit and attempt to obtain an additional length of the optic nerve. This seemingly difficult maneuver is made easier by hemostasis, suction, good magnification, and direct illumination. Placement of a biointegrated implant such as hydroxyapatite or porous polyethylene is generally avoided if orbital extension is suspected.[30] Although most implants structurally tolerate radiotherapy well, implant vascularization may be compromised by radiotherapy, thus increasing the risk of implant exposure. A polymethylmethacrylate or a silicone implant is recommended in such situations.
All patients with overt orbital retinoblastoma undergo baseline systemic evaluation to rule out metastasis. Orbital external beam radiotherapy and 12 cycles of high dose chemotherapy are recommended in patients who have an extraocular extension with a scleral or an episcleral nodule. In patients who have optic nerve extension, only 6 cycles of standard dose chemotherapy are recommended if the transection is free of tumor on histopathology. However, if the transection of the optic nerve is involved, then orbital external beam radiotherapy and 12 cycles of high dose chemotherapy are recommended. Both the patients with overt orbital retinoblastoma in our series are alive and well, 36 months following completion of treatment.
Microscopic orbital retinoblastoma
The management protocol for patients with microscopic extension of retinoblastoma up to the level of optic nerve transection, full-thickness scleral infiltration, and extrascleral extension detected on histopathologic evaluation of the enucleated eye includes orbital external beam radiotherapy (fractionated 45–50 Gy) and 12 cycles of high-dose chemotherapy.[12,31]
Prognosis
Orbital retinoblastoma has conventionally carried poor prognosis with mortality rates ranging from 25% to 100%.[26,27,28,32] The presence of orbital invasion was associated with a 10–27 times higher risk of systemic metastasis as compared to cases without orbital invasion.[5] In a group of 30 patients with orbital retinoblastoma managed by a multi-modal treatment protocol, only 2 (6.7%) developed systemic metastasis. In addition to our observations, several authors have reported improved survival when surgery (usually exenteration) was combined with chemotherapy.[10,25,29,32] Compared to the previously reported survival, our current protocol has provided better survival in a limited number of selected patients. However, longer follow-up of the larger numbers of treated patients is mandatory before a definite recommendation about the sequential combination of high-dose chemotherapy, followed by surgery, external beam radiotherapy, and extended chemotherapy can be made.[30]
Prognostic factors
The identification of frequency and significance of high-risk histopathologic factors that can reliably predict orbital recurrence of retinoblastoma and subsequent systemic metastasis is vital for patient selection for adjuvant therapy.[5,9,12] It is generally agreed that invasion of the optic nerve to transection, scleral infiltration and extrascleral extension are the risk factors that are predictive of orbital recurrence.[5,9] The role of adjuvant therapy in minimizing the risk of systemic metastasis and ultimate survival in patients with various histopathologic risk factors is now well-established.[30] The Children's Oncology Group trials (COG ARET0321, Trial of Intensive Multi-Modality Therapy for Extra-ocular Retinoblastoma; COG ARET 0332, Study of Unilateral Retinoblastoma With and Without Histopathologic High-Risk Features and the Role of Adjuvant Therapy) may provide further conclusive data.
Conclusions
Orbital retinoblastoma encompasses the spectrum of primary clinical manifestation (primary), orbital recurrence following enucleation (secondary), inadvertent perforation or intraocular surgery in an eye with unsuspected retinoblastoma (accidental), intraoperative discovery of extraocular or optic nerve extension (overt) and full-thickness scleral, extrascleral and optic nerve transection involvement on histopathology (microscopic). Although each clinical situation is unique with a gross variation in tumor load, the currently preferred management is multimodal with a combination of chemotherapy, surgery, and external beam radiotherapy, with an excellent prognosis for patient survival.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
Fig 1: Reprinted with permission from Honavar SG, Singh AD. Management of advanced retinoblastoma. Ophthalmol Clin North Am 2005;18:65-73. Copyright (2007), permission from Elsevier.
Fig 4-5, 7-8, 9-10: Reprinted with permission from from Honavar SG. Orbital retinoblastoma. In: Singh AD, Damato BE, Pe'er J, editors. Clinical Ophthalmic Oncology. PA: Saunders Elsevier; 2007. p. 477-83., Copyright (2007), permission from Elsevier.
Fig 6: Reprinted with permission from Murthy R, Honavar S, Vemuganti G, Naik M, Reddy V. Systemic Metastasis Following Hyphema Drainage in an Unsuspected Retinoblastoma. J Pediatr Ophthalmol Strabismus. 2007; 44: 120-123, permission from SLACK.
References
- 1.Young JL, Smith MA, Roffers SD, Liff JM, Bunin GR. Retinoblastoma. In: Ries LA, Smith MA, Gurney GJ, editors. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995. NIH Publication No. 99-4649. Bethesda, MD: National Cancer Institute, SEER Program; 1999. [Google Scholar]
- 2.Sanders BM, Draper GJ, Kingston JE. Retinoblastoma in great Britain 1969-80: Incidence, treatment, and survival. Br J Ophthalmol. 1988;72:576–83. doi: 10.1136/bjo.72.8.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anonymous Survival rate and risk factors for patients with retinoblastoma in Japan. The Committee for the National Registry of Retinoblastoma. Jpn J Ophthalmol. 1992;36:121–31. [PubMed] [Google Scholar]
- 4.Abramson DH, Niksarli K, Ellsworth RM, Servodidio CA. Changing trends in the management of retinoblastoma: 1951-1965 vs.1966-1980. J Pediatr Ophthalmol Strabismus. 1994;31:32–7. doi: 10.3928/0191-3913-19940101-07. [DOI] [PubMed] [Google Scholar]
- 5.Singh AD, Shields CL, Shields JA. Prognostic factors in retinoblastoma. J Pediatr Ophthalmol Strabismus. 2000;37:134–41. doi: 10.3928/0191-3913-20000501-04. [DOI] [PubMed] [Google Scholar]
- 6.Ajaiyeoba IA, Akang EE, Campbell OB, Olurin IO, Aghadiuno PU. Retinoblastomas in Ibadan: Treatment and prognosis. West Afr J Med. 1993;12:223–7. [PubMed] [Google Scholar]
- 7.Stannard C, Lipper S, Sealy R, Sevel D. Retinoblastoma: Correlation of invasion of the optic nerve and choroid with prognosis and metastases. Br J Ophthalmol. 1979;63:560–70. doi: 10.1136/bjo.63.8.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hungerford J. Factors influencing metastasis in retinoblastoma. Br J Ophthalmol. 1993;77:541. doi: 10.1136/bjo.77.9.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finger PT, Harbour JW, Karcioglu ZA. Risk factors for metastasis in retinoblastoma. Surv Ophthalmol. 2002;47:1–16. doi: 10.1016/s0039-6257(01)00279-x. [DOI] [PubMed] [Google Scholar]
- 10.Ellsworth RM. Orbital retinoblastoma. Trans Am Ophthalmol Soc. 1974;72:79–88. [PMC free article] [PubMed] [Google Scholar]
- 11.Grabowski EF, Abramson DH. Intraocular and extraocular retinoblastoma. Hematol Oncol Clin North Am. 1987;1:721–35. [PubMed] [Google Scholar]
- 12.Honavar SG, Singh AD, Shields CL, Meadows AT, Demirci H, Cater J, et al. Postenucleation adjuvant therapy in high-risk retinoblastoma. Arch Ophthalmol. 2002;120:923–31. doi: 10.1001/archopht.120.7.923. [DOI] [PubMed] [Google Scholar]
- 13.Leal-Leal C, Flores-Rojo M, Medina-Sansón A, Cerecedo-Díaz F, Sánchez-Félix S, González-Ramella O, et al. A multicentre report from the Mexican Retinoblastoma Group. Br J Ophthalmol. 2004;88:1074–7. doi: 10.1136/bjo.2003.035642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kao LY, Su WW, Lin YW. Retinoblastoma in Taiwan: Survival and clinical characteristics 1978-2000. Jpn J Ophthalmol. 2002;46:577–80. doi: 10.1016/s0021-5155(02)00546-4. [DOI] [PubMed] [Google Scholar]
- 15.Badhu B, Sah SP, Thakur SK, Dulal S, Kumar S, Sood A, et al. Clinical presentation of retinoblastoma in Eastern Nepal. Clin Exp Ophthalmol. 2005;33:386–9. doi: 10.1111/j.1442-9071.2005.01010.x. [DOI] [PubMed] [Google Scholar]
- 16.Honavar SG. Orbital Retinoblastoma, Meeting proceedings, International Society of Genetic Eye Diseases and Retinoblastoma, Bangalore, India. 2011 [Google Scholar]
- 17.Wolff JA, Boesel C, Ellsworth RM. Childrens Cancer Study Group Protocol CCSG 962. New York: Children Cancer Group; 1978. [Google Scholar]
- 18.Chantada G, Doz F, Antoneli CB, Grundy R, Clare Stannard FF, Dunkel IJ, et al. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer. 2006;47:801–5. doi: 10.1002/pbc.20606. [DOI] [PubMed] [Google Scholar]
- 19.Edge SD, Byrd DR, Carducci MA, Compton CC, editors. AJCC Cancer Staging Manual. 7th ed. Ch. 44. New York: Springer; 2009. Retinoblastoma. [Google Scholar]
- 20.Honavar SG. Orbital retinoblastoma. In: Singh AD, Damato BE, Pe’er J, editors. Clinical Ophthalmic Oncology. PA: Saunders Elsevier; 2007. pp. 477–83. [Google Scholar]
- 21.Antoneli CB, Steinhorst F, de Cássia Braga Ribeiro K, Novaes PE, Chojniak MM, Arias V, et al. Extraocular retinoblastoma: A 13-year experience. Cancer. 2003;98:1292–8. doi: 10.1002/cncr.11647. [DOI] [PubMed] [Google Scholar]
- 22.Menon BS, Reddy SC, Maziah WM, Ham A, Rosline H. Extraocular retinoblastoma. Med Pediatr Oncol. 2000;35:75–6. doi: 10.1002/1096-911x(200007)35:1<75::aid-mpo13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 23.Kiratli PO, Kiratli H, Ercan MT. Visualization of orbital retinoblastoma with technetium-99m (V) dimercaptosuccinic acid. Ann Nucl Med. 1998;12:157–9. doi: 10.1007/BF03164782. [DOI] [PubMed] [Google Scholar]
- 24.Moll AC, Hoekstra OS, Imhof SM, Comans EF, Schouten-van Meeteren AY, van der Valk P, et al. Fluorine-18 fluorodeoxyglucose positron emission tomography (PET) to detect vital retinoblastoma in the eye: Preliminary experience. Ophthalmic Genet. 2004;25:31–5. doi: 10.1076/opge.25.1.31.29001. [DOI] [PubMed] [Google Scholar]
- 25.Rootman J, Ellsworth RM, Hofbauer J, Kitchen D. Orbital extension of retinoblastoma: A clinicopathological study. Can J Ophthalmol. 1978;13:72–80. [PubMed] [Google Scholar]
- 26.Pratt CB, Crom DB, Howarth C. The use of chemotherapy for extraocular retinoblastoma. Med Pediatr Oncol. 1985;13:330–3. doi: 10.1002/mpo.2950130606. [DOI] [PubMed] [Google Scholar]
- 27.Kiratli H, Bilgiç S, Ozerdem U. Management of massive orbital involvement of intraocular retinoblastoma. Ophthalmology. 1998;105:322–6. doi: 10.1016/s0161-6420(98)93367-x. [DOI] [PubMed] [Google Scholar]
- 28.Goble RR, McKenzie J, Kingston JE, Plowman PN, Hungerford JL. Orbital recurrence of retinoblastoma successfully treated by combined therapy. Br J Ophthalmol. 1990;74:97–8. doi: 10.1136/bjo.74.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doz F, Khelfaoui F, Mosseri V, Validire P, Quintana E, Michon J, et al. The role of chemotherapy in orbital involvement of retinoblastoma. The experience of a single institution with 33 patients. Cancer. 1994;74:722–32. doi: 10.1002/1097-0142(19940715)74:2<722::aid-cncr2820740228>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 30.Honavar SG, Singh AD. Management of advanced retinoblastoma. Ophthalmol Clin North Am. 2005;18:65–73. doi: 10.1016/j.ohc.2004.09.001. viii. [DOI] [PubMed] [Google Scholar]
- 31.Shields CL, Honavar S, Shields JA, Demirci H, Meadows AT. Vitrectomy in eyes with unsuspected retinoblastoma. Ophthalmology. 2000;107:2250–5. doi: 10.1016/s0161-6420(00)00427-9. [DOI] [PubMed] [Google Scholar]
- 32.Hungerford J, Kingston J, Plowman N. Orbital recurrence of retinoblastoma. Ophthalmic Paediatr Genet. 1987;8:63–8. doi: 10.3109/13816818709028518. [DOI] [PubMed] [Google Scholar]