

Abstract
Failure to translate successful neuroprotective preclinical data to a clinical setting in Alzheimer's disease (AD) indicates that amyloidopathy and tauopathy alone provide an incomplete view of disease. We have tested here the relevance of additional homeostatic deviations that result from loss of activity of transcription factor NRF2, a crucial regulator of multiple stress responses whose activity declines with ageing. A transcriptomic analysis demonstrated that NRF2-KO mouse brains reproduce 7 and 10 of the most dysregulated pathways of human ageing and AD brains, respectively. Then, we generated a mouse that combines amyloidopathy and tauopathy with either wild type (AT-NRF2-WT) or NRF2-deficiency (AT-NRF2-KO). AT-NRF2-KO brains presented increased markers of oxidative stress and neuroinflammation as well as higher levels of insoluble phosphorylated-TAU and Aβ*56 compared to AT-NRF2-WT mice. Young adult AT-NRF2-KO mice exhibited deficits in spatial learning and memory and reduced long term potentiation in the perforant pathway. This study demonstrates the relevance of normal homeostatic responses that decline with ageing, such as NRF2 activity, in the protection against proteotoxic, inflammatory and oxidative stress and provide a new strategy to fight AD.
Keywords: NRF2, Alzheimer's disease, Transcriptomics, Proteotoxicity, Neuroinflammation, Oxidative stress
Graphical abstract
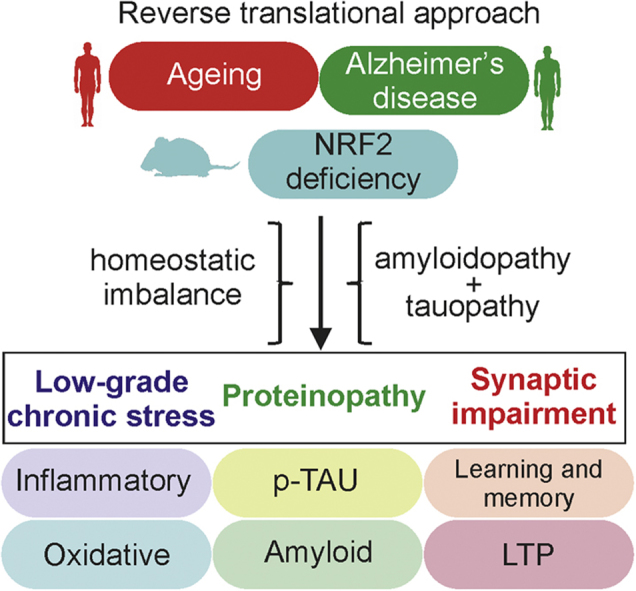
Highlights
-
•
NRF2-lack mimics many transcriptomic alterations of elderly and AD patients.
-
•
APP/TAU-induced oxidative and inflammatory stress is exacerbated by NRF2-deficiency.
-
•
APP/TAU-induced proteinopathy is exacerbated by NRF2-deficiency.
-
•
APP/TAU-induced deficits in cognition are aggravated by NRF2-deficiency.
1. Introduction
In spite of the efforts made over the last decades, the etiology of Alzheimer's disease (AD) remains largely unknown. This is in part caused by incomplete reproduction of the human pathology in animal models merely exhibiting the proteinopathy associated to deposition of amyloid-beta (Aβ) and hyperphosphorylation of TAU. Current AD-models do not reflect molecular mechanisms that correlate with the general decline of homeostatic capacity during ageing and may contribute significantly to the disease process. These mechanisms include oxidative, inflammatory and metabolic stress, which may precede the proteinopathy in prodromal and early phases of sporadic AD [1].
In this study, we have addressed this problem in the context of deficiency in the Nuclear factor (erythroid-derived 2)-like 2 (NRF2), the master regulator of homeostatic responses [2], [3], [4], [5], [6]. The NRF2 transcriptional signature provides an armamentarium to adapt reactive oxygen species signaling, inflammation and metabolism to normal physiological needs [7], [8]. Its transcriptional acivity declines with ageing [9], [10]. Also, in Hutchinson-Gilford progeria syndrome, characterized by premature ageing, NRF2 is mislocated in the nuclear periphery, resulting in impaired NRF2 activity and consequently increased chronic oxidative stress while NRF2-activators restore viability in an animal model of this syndrome [11]. Moreover, male mice pharmacologically treated with a NRF2-activator exhibit longer lifespan [12].
Ageing is the main risk factor for AD and some scattered studies have noted the correlation between deficits in NRF2 and AD. In humans, one haplotype allele in the NFE2L2 gene promoter, coding NRF2, was associated with an earlier onset of AD, implying that common variants of the NFE2L2 gene might affect AD progression [13]. In animal models, the relevance of NRF2 has been analyzed in transgenic mice with amyloidopathy [14], [15] or tauopathy [16], [17], but not combined, and the main focus was on the role of NRF2 in proteostasis.
Here, we have performed a transcriptomics study comparing pathways altered in elderly and AD brains with those of NRF2-knockout mouse brains. From this reverse translational approach, we have further combined expression of the human APPV717I and TAUP301L transgenes [18] with lack of NRF2 expression. The absence of NRF2 led to earlier onset of the disease, with more severe amyloidopathy and tauopathy and exacerbated cognitive defects. This study demonstrates for the first time the importance of homeostatic functions regulated by NRF2 in the brain pathophysiology of AD and represents a new tool for interventional studies.
2. Material and methods
2.1. Transgenic mice
Colonies of NRF2-KO mice and NRF2-WT littermates were described previously [19]. Heterozygous APPV717I mice (FVB/N), expressing the hAPP695 isoform with the V717I mutation under the control of the mouse Thy1 gene promoter, were crossed with C57/BL6j-NRF2-WT (APP-NRF2-WT) or C57/BL6j-NRF2-KO (APP-NRF2-KO). Similarly, TAUP301L mice (FVB/N), expressing homozygously the longest isoform of protein TAU with the P301L mutation (TAU 4 R/2 N P301L) under control of the same mouse Thy1 gene promoter, were crossed with C57/BL6j-NRF2-WT (TAU-NRF2-WT) or C57/BL6j-NRF2-KO (TAU-NRF2-KO). APP/TAU-NRF2-WT (AT-NRF2-WT) and APP/TAU-NRF2-KO (AT-NRF2-KO) in C57/BL6j background were obtained by crossing the above described genotypes for more than eight generations. Genotypic characterization of the APPV717I and TAUP301L transgenic mice was described previously [18], [20], [21].
2.2. RNA microarray expression analysis
Total mouse RNA was obtained from brain tissue by Trizol extraction (Invitrogen, California, USA) according to manufacturer's instruction and as previously described [22]. RNA quality was determined using a bioanalyzer (Agilent, California, USA). RIN values of all samples were greater than 9.5. For expression analysis 50 ng of each total RNA sample was converted and amplified to cDNA using the Ovation® Pico WTA System V2 according to manufacturer's instructions. Products were then hybridized on Mouse Gene 2.0 ST arrays containing probe sets for >28,000 coding and >7000 non-coding transcripts. The functional clustering tool DAVID (Database for Annotation, Visualization and Integrated Discovery) v6.7 [23] was used to look for functional enrichment of genes with more than 3-fold of change and adjusted p-value (FDR/BH) > 0.05 (644 genes). The number of genes analyzed in AD brain cohorts was: i) 214 from [24], ii) 860 from [25], iii) 156 from [26], iv) 743 from [27], v) 1785 from [28] and vi) 2027 from [29]. The number of genes analyzed in ageing brain cohorts were: i) 399 genes from [30], ii) 801 from [31], iii) 247 from [32]. Pathway analysis was performed with the Gene Ontology Biological Process (GOTERM_BP_FAT).
2.3. Analysis of mRNA levels
Total RNA extraction, reverse transcription and quantitative PCR were done as detailed elsewhere [33]. Primer sequences are shown in Suppl. Table S1. To ensure that equal amounts of cDNA were added to the PCR, the housekeeping genes ActB, Gapdh and Tbp were amplified. Data analysis was based on the ΔΔCT method with normalization of the average data of housekeeping genes. All PCRs were performed in triplicate.
2.4. Immunofluorescence, immunohistochemistry and immunoblotting
For immunofluorescence and immunohistochemistry, 30 µm-thick and 5 µm-thick sections from fixed brains and frozen spinal cords, respectively, were immunostained as described [33], [34]. Immunoblots were performed as described in [35]. Primary antibodies are annotated in Suppl. Table S2.
2.5. TAU extraction and oligomeric Aβ detection
Hippocampus, brainstem and spinal cord were homogenized as described in [34]. Sarkosyl soluble and insoluble TAU was run on 7.5% SDS-PAGE gels and transferred to PVDF membranes in low methanol transfer buffer (6.6% methanol, 25 mM Tris and 192 mM Glycine). Hippocampal samples were fractionated by a four step extraction method previously described [36]. One hundred micrograms of protein per sample was electrophoresed on 10–20% SDS-polyacrylamide Tris-tricine gels (Bio-Rad). Proteins were transferred to PVDF and membranes were boiled in PBS for 5 min after immunoblotting.
2.6. Morris water maze test
After a 1-day habituation trial (day 0) in which preferences between quadrants in the different experimental groups were ruled out, the animals learned to find a hidden platform over the following 5 days, through 4 trials/day, 60 s each trial, plus 20 s in the platform. If an animal failed to reach the platform, it was placed on it by the experimenter. Subsequently, the animals were subjected to two trials on the 7th day, the first without the platform to assess possible differences in swimming speed and preference for the platform quadrant. And in the second, a cued version protocol using a visible platform was conducted to determine sensorimotor and motivational status of the animals. The animal behavior was recorded through an automated tracking system (Smart video tracking system, version 2.0.14, Panlab; Harvard Apparatus).
2.7. Electrophysiological recordings
Data were obtained from six urethane-anaesthetized (1.6 g/kg i.p.) adult mice per group as described in [37]. Briefly, field potentials were recorded through tungsten macroelectrodes (1 MΩ; World Precision Instruments) stereotaxically implanted in the dentate gyrus (A: −2,3; L: 2; H: 3.5 mm from bregma) according to the Paxinos and Watson Atlas. Twisted bipolar electrodes for electrical stimulation were aimed at the perforant path (A: −2.5; L: 0.5; H: 3.5 mm from bregma). Baseline recordings were taken with test stimuli (10–50 µA, 0.3 ms, 0.5 Hz) during 15 min before tetanic stimulation consisting of three pulse trains of 10–50 µA, lasting each pulse 0.3 ms and at 50 Hz. Trains lasted 500 ms and the inter-train interval was 2 s. Recording was maintained for 30 min after tetanic stimulation. Field potentials were 0.1 Hz–1 kHz band-pass filtered, amplified (P15 Amplifier, Grass Co., USA), and digitized at 10 kHz (CED 1401; Cambridge Electronic Design). Signals analysis was carried out with Spike 2 software (Cambridge Electronic Design, Cambridge, UK). Field potential segments of 5 min were analyzed to obtain the response average. The mean average response during the 15 min period before the tetanic stimulation was considered as 100%. Recordings were accepted for analysis when baseline variability was less than 10%.
2.8. Determination of reduced and oxidized glutathione, protein carbonyl and malondialdehyde content
Brain samples were homogenized in 1 ml phosphate buffer and submitted to the protocol described in [38].
2.9. Image analysis and statistics
Calculations of the false discovery rates (FDR) were done accordingly with Benjamini Hochberg's FDR [39] employing IBM SPSS statistics 22 software. Adjusted p-value (FDR/BH) of ≤0.05 was considered significant. Band intensities corresponding to immunoblot detection of protein samples were quantified using Image J 1.48 software. Calculation of p-values from ANOVA one-way followed by Newman-Keulspost-hoc test, ANOVA two-way followed by Bonferroni post-hoc test and Student's t-test were assessed by GraphPad Prism 5 software among groups. A p value of ≤0.05 was considered significant. Unless otherwise indicated, all experiments were performed at least three times with similar results. Values presented in the graphs are means of at least three samples. Results are expressed as mean ± SEM.
3. Results
3.1. NRF2-deficient mouse brain replicates pathway alterations found in human elderly and AD brains
A microarray analysis of 25,293 genes was performed with brain samples from NRF2-knockout (NRF2-KO) vs. wild type (NRF2-WT) mice. We found significant up-regulation of 510 genes and down-regulation of 134 genes as assessed by more than 3-fold change expression and adjusted p≤0.05 (FDR/BH) (Suppl. Table S3). As shown in Table 1, we confirmed that the Nfe2l2 gene, coding NRF2, was not expressed in NRF2-KO animals (effect change = −3.23 and adjusted p = 0.035 (FDR/BH)). Among other altered genes, we validated the down-regulation of Gstm1, regulated by NRF2 [40] and Kras [41] and Wdfy1 [42] involved in cell signaling and inflammation. We also found up-regulation of Zbtb21/ZNF295, which has been implicated in Down syndrome and cognitive deficit [43]. These genes were further classified in functional clusters of biological processes by employing GOTERM_BP_FAT category (Suppl. Table S4) and compared with 3 cohorts of old human brain and 6 of AD brain (Fig. 1). All altered pathways in NRF2-KO brain mice were found to be also altered in either human ageing or AD brains, although some others were only altered in elderly subjects or AD patients but not in mice (Fig. 1C). Among the 20 gene clusters with the highest changes, aging and NRF2-KO groups share seven, AD and NRF2-KO groups share ten and the three groups combined share six (Figs. 1A and 1B). Altogether, these results show overlap in many dysregulated processes in the brain of NRF2-deficient mice, old people and AD patients and suggest the participation of NRF2 in the pathological process of AD.
Table 1.
Microarray validation by qRT-PCR. The expression data obtained using microarrays for selected genes was validated using qRT-PCR analysis. Fold of change in mRNA of the indicated genes was calculated relative to NRF2-WT brains and normalized to the average expression of ActB, Tbp and Gapdh house keeping genes. Data are mean ± SEM (n=4).
| Microarray |
qRT-PCR |
|||||
|---|---|---|---|---|---|---|
| Gene Entrez ID | Gene name | Effect change |
p-value (FDR/BH) |
NRF2-WT | NRF2-KO | p-value (t-student) |
| 14862 | Gstm1 | −3,87 | 0.0082 | 1.00±0.11 | 0.23±0.08 | 0.001 |
| 16653 | Kras | −5.10 | 0.0022 | 1.00±0.13 | 0.64±0.07 | 0.042 |
| 69368 | Wdfy1 | −6.97 | 0.00043 | 1.00±0.12 | 0.60±0.09 | 0.023 |
| 114565 | Zbtb21 | 11.36 | 0.000078 | 1.00±0.44 | 4.00±0.40 | 0.002 |
Fig. 1.

Comparison of the functional pathways altered in brain of NRF2-KO mice vs. human ageing and AD cohorts. Altered gene expression in NRF2-KO mice was analyzed in brain samples from 11-months-old NRF2-WT (n = 4 males) vs. NRF2-KO mice (n = 4 males). Comparison with human ageing and AD brain cohorts was done as indicated in Material and Methods. A, Venn diagram showing the overlapping distribution of functional pathways in AD, ageing and NRF2-null brains. Cluster numbers correspond to: 1, regulation of cell signaling; 2, phosphorous and phosphate metabolism; 3, cellular protein metabolic process; 4, neurodevelopment; 5, regulation of cell death; 6, protein complex assembly; 7, intracellular transport and protein location; 8, nucleoside, purine and ATP metabolism; 9, regulation of cell cycle; 10, acetylcholine metabolism; 11, transmembrane transport of ions; 12, synaptic function; 13, cognition, learning and memory; 14, host response to infection; 15, lipid and alcohol metabolism; 16, interspecies and multi-organ modulation; 17, secretory and exocytic pathways; 18, regulation of membrane and vesicles; 19, regulation of cytoskeleton; 20, regulation of angiogenesis; 21; protein modification by conjugation; 22, regulation of cell-environment interaction; 23, glucose metabolism; 24, cell morphogenesis; 25, immune response; 26, ROS metabolic process; 27, cellular motility; 28, regulation of transmembrane transport; 29, development of peripheral tissues. B and C, summary of selected categories as calculated by DAVID. The categories are ranked according to -log10 P-values.
3.2. Characterization of APPV717I and TAUP301L expression in mice with presence or absence of NRF2
Based on the overlapping alterations indicated above, and to determine if NRF2 deficiency might participate in AD pathology, we generated transgenic mice with combined neuronal expression of human mutant hAPPV717I and hTAUP301L proteins (for simplicity from now on denoted AT) under the neuronal Thy1 promoter in the C57/bl6 genetic background, with either wild type (AT-NRF2-WT) or NRF2 absence (AT-NRF2-KO). The combined expression of these transgenes has been described previously in wild type mice [18]. Both AT-NRF2-WT and AT-NRF2-KO mice exhibited short lifespan as expected from severe tauopathy [18], [21], [44]. Thus, the mean lifespan of AT-NRF2-WT and AT-NRF2-KO mice was 15 and 12 months, respectively (to be described elsewhere). Although the pathway analysis by biological process demonstrated significant alterations of ROS management and metabolism, as expected from previous reports [15], [34], many targets of NRF2 did not exhibit significant changes in the microarray analysis (see Discussion). Therefore, to more closely determine the effects of NRF2-deficiency on brain homeostasis, we explored markers of oxidative and inflammatory stress as these factors are tightly linked to NRF2 homeostatic adaptations [7], [33], [45], [46]. As shown in Fig. 2A-C, AT-NRF2-KO brains exhibited lower levels of reduced glutathione and higher levels of carbonylated proteins (adducts of 2,4.drinitrophenylhydrazzine, DNPH) and lipid peroxides (malondialdheyde, MDA) than AT-NRF2-WT mice. Regarding neuroinflammation, the levels of some pro-inflammatory markers, IL6, COX2 and iNOS, were modestly but significantly increased in AT-NRF2-KO vs. age-matched AT-NRF2-WT mice (Fig. 2D-G). These results are consistent with a preventive effect of NRF2 against low-grade chronic oxidative and inflammatory stress in the brain, which are two hallmarks of ageing and AD onset and progression.
Fig. 2.

Low-grade oxidative and inflammatory stress in AT-NRF2-KO mice. A, levels of reduced glutathione (GSH) normalized with total oxidized glutathione (GSSG). B, protein carbonyl content as determined by DNPH levels. C, thiobarbituric acid-reactive substances (TBARs) representing mostly MDA. Data are mean ± SEM (NRF2-WT= 4 males; NRF2-KO=4 males; AT-NRF2-WT = 2 males and 2 females; AT-NRF2-KO= 2 males and 2 females). Statistical analysis was performed with a one-way ANOVA followed by Neiman Keuls post-hoc test. *p ≤ 0.05, ***p ≤ 0.001 vs. NRF2-WT group. D, immunoblot analysis of IL6, COX2 and NOS2 in brain homogenates from mice of the indicated genotypes. Protein levels of GAPDH were analyzed to ensure similar load per lane. E, F and G, densitometric quantification of representative blots from B. Data are mean ± SEM (NRF2-WT= 2 males and 1 female; NRF2-KO= 3 males; AT-NRF2-WT = 2 males and 1 female; AT-NRF2-KO= 2 males and 1 female). Statistical analysis was performed with a one-way ANOVA followed by Neiman Keuls post-hoc test. *p ≤ 0.05, **p≤0.01 vs. NRF2-WT group.
3.3. Phosphorylated TAU and Aβ*56 oligomers are increased in hippocampus of AT-NRF2-KO mice
We next compared the phosphorylation status of TAU in hippocampus, brainstem and spinal cord by immunohistochemistry with the AT8 antibody, specific for phosphorylated hTAU at residues Ser202 and Thr205 (p-TAU) (Fig. 3E). As shown in Fig. 3A, no major differences in p-TAU-AT8 were observed in hippocampi of AT-NRF2-WT and AT-NRF2-KO mice. Neurons of the pontine reticular formation (Fig. 3B) contain p-TAU-AT8 in both genotypes, exhibiting intracellular p-TAU aggregates and thick dystrophic neurites. While most p-TAU-positive neurites were traceable in AT-NRF2-WT mice, these fibers were disorganized in AT-NRF2-KO mice (Fig. 3C). In the spinal cord (Fig. 3D), the motor neurons of the anterior horn also exhibited disorganized patterns, with a characteristic ballooned morphology of dystrophic fibers [47]. As shown in Figs. 3E and 3G, three forms of p-TAUP301L in the sarkosyl-insoluble fraction (SI) were separated by SDS-PAGE and termed hP1, hP2 and hP3, according to their lesser mobility caused by augmented phosphorylation [48], [49]. Densitometric quantification revealed that the less phosphorylated faster migrating hP1 form was most abundant in the sarkosyl-soluble fraction (SS) while the most phosphorylated slower migrating hP3 form was mainly found in the SI fraction. AT-NRF2-KO mice exhibited higher levels of hP2 and hP3 in the SI fraction than AT-NRF2-WT mice. These biochemical results confirmed the tauopathy observed immunohistochemically in the brainstem and spinal cord.
Fig. 3.

Lack of NRF2 leads to increased levels of insoluble TAUP301Lin brain and spinal cord. Immunostaining with antibody against p-TAU (AT8) in hippocampal CA1 and subiculum zones (A), pontine reticular formation (B and C) and spinal cord (D) from 11-month old AT-NRF2-WT and AT-NRF2-KO mice. The spinal cord sections were counterstained with hematoxylin. For A, B and C, the plates show representative preparations from NRF2-WT (2 males and 1 female) and NRF2-KO (2 males and 1 female) mice. E, scheme of protein TAU-4R showing the phosphorylated residues recognized by the indicated antibodies. F, immunoblot analysis of sarkosyl-insoluble (SI) and sarkosyl-soluble (SS) pTAU levels in hippocampus, brainstem and spinal cord homogenates respectively from AT-NRF2-WT and AT-NRF2-KO mice employing anti-PHF1 antibody. G, densitometric quantification of representative blots from F. Data are mean ± SEM (AT-NRF2-WT = 5 males and 1 female; AT-NRF2-KO = 3 males and 3 females). Statistical analysis was performed with Student's t-test. *p≤0.05, comparing AT-NRF2-WT and AT-NRF2-KO groups.
Hippocampal sections stained for APP/Aβ (4G8 antibody) revealed more and larger intracellular vesicles in neurons of AT-NRF2-KO compared with AT-NRF2-WT mice, particularly in the subiculum (Fig. 4A-B) as expected [34]. We analyzed the levels of Aβ-oligomeric species that have been proposed to correlate with cognitive impairment in humans. Hippocampi from AT-NRF2-WT and AT-NRF2-KO mice were separated in soluble (SF) (Figs. 4A, 4C) and membrane bound (MF) (Figs. 4A, 4D) fractions as described [36]. We did not detect significant differences in the levels of APP, sAPPα, sAPPβ, CTF/Aβ (3-mer), Aβ (6-mer) or Aβ (2-mer). Interestingly, NRF2 deficiency increased the levels of the presumed toxic Aβ*56 oligomer in both SF and MF fractions (Fig. 4E). The levels of mRNA encoding enzymes involved in APP processing, i.e. ADAM10, ADAM17, BACE1 and PS1, were not significantly altered by the lack of NRF2 (data not shown). Therefore, increased Aβ*56 levels should be related to either oxidative alterations of the amyloidogenic pathway or impaired clearance of the oligomers. In any case, NRF2-deficiency increased the levels of the potentially toxic proteins p-TAU and Aβ*56 and suggest a cell autonomous mechanism of neuronal damage in the absence of NRF2 defense.
Fig. 4.

NRF2 deficiency leads to Aβ*56 accumulation. A, scheme showing APP processing by α-, β- and γ-secretase and the location of the epitopes recognized by the antibodies employed in western blot analysis. (SF, soluble fraction and MF, membrane fraction). B, immunohistochemical analysis of APP/Aβ expression in hippocampus of 11-month old AT-NRF2-WT and AT-NRF2-KO mice stained with anti 4G8 antibodies in CA1 and subiculum. C and D, immunoblots with the anti-Aβ antibody as indicated SF and MF fractions from hippocampal homogenates of AT-NRF2-WT and AT-NRF2-KO mice. The right two lanes show samples from APP/PS1 and wild type mice as positive and negative controls, respectively. The asterisk in D indicates a non-specific band present in all samples including the negative control. β-actin detection demonstrates similar loading across lanes. E, densitometric quantification of blots depicted in C and D. Data are mean ± SEM (AT-NRF2-WT = 5 males; AT-NRF2-KO = 4 males and 1 female). Statistical analysis was performed with a Student's t-test. *p≤0.05, comparing AT-NRF2-WT and AT- NRF2-KO groups.
3.4. NRF2 deficiency aggravates learning and memory deficits induced by amyloidopathy and tauopathy
We analyzed the spatial acquisition and retention memory in 6-months-old NRF2-WT, NRF2-KO, AT-NRF2-WT and AT-NRF2-KO mice using the Morris water maze test. Swim speed was similar at this age for all genotypes, indicating uncompromised motor competency (data not shown). After five days of training, mice landed on the platform in less than 30 s except AT-NRF2-KO mice, who took significantly longer (Fig. 5A). On the fifth day, the platform was removed and the mice were also tested for their memory of the platform location (Fig. 5B). All mice, again with exception of the AT-NRF2-KO, exhibited a tendency to swim in the target sector where the platform used to be located. AT-NRF2-KO mice, however, displayed circling and tumbling swimming-tracks, indicating their incapacity to remember the original location of the platform. These results demonstrated impaired cognition already at 6 months of age in the AT-NRF2-KO mice despite lacking obvious amyloid load or TAU pathology (data not shown and [18]).
Fig. 5.

NRF2 deficiency impairs long term potentiation, and spatial learning and memory. A, acquisition time to reach the hidden platform in five trials during Morris water maze test of 6-months old-mice of the indicated genotypes. Data are mean ± SEM (NRF2-WT = 5 males and 5 females; NRF2-KO = 5 males and 5 females; AT-NRF2-WT = 6 males and 4 females; AT-NRF2-KO = 5 males and 5 females). Statistical analysis was performed with two-way ANOVA followed by Bonferroni post-hoc test. *p≤0.05 and **p≤0.01 versus NRF2-WT group. B. Upper circles, representative swimming tracks of the indicated genotypes during the probe phase in which the platform had been removed. Lower panel, quantification of the frequency to cross to the platform quadrant. Statistical analysis was performed with a one-way ANOVA followed by Neiman Keuls post-hoc test. *p<0.05 vs. NRF2-WT group. C, Upper registries correspond to representative responses recorded before (thin line) and after (thick line) high-frequency trains of tetanic stimulation. Calibration: 2 mV, 20 ms. The graph shows LTP of 6-months-old animals from the indicated genotypes. Data are mean ± SEM (n = 6). Statistical analysis was performed with two-way ANOVA followed by Bonferroni post-hoc test. **p < 0.01, and ***p < 0.001 comparing AT-NRF2-KO versus NRF2-WT group. ###p< 0.001 comparing NRF2-KO vs NRF2-WT group. $$$ p< 0.001 comparing AT-NRF2-WT versus NRF2-WT group.
Further exploration of cognitive function was performed electrophysiologically for hippocampal long term potentiation (LTP) in 6-months-old mice. The synaptic transmission in the neurons of the dentate gyrus was recorded in response to performant path stimulation (Fig. 5C). High frequency stimulation increased the field excitatory post-synaptic potential (fEPSP) in control NRF2-WT mice and to a lesser extent in AT-NRF2-WT and AT-NRF2-KO mice. This was expected, given the combined proteinopathy, but surprisingly there was lower LTP in the parental NRF2-KO mice, for the first time indicating that NRF2-deficiency in itself reduced the LTP in these hippocampal regions. We concluded that combined expression of human mutant APP and TAU with NRF2 depletion substantially compromised the electrophysiological capacity of neurons in the hippocampus.
4. Discussion
The use of network techniques to study alteration of brain functional pathways in the context of human ageing and AD allowed us to recreate some of the links shared in common with NRF2-deficiency, a master regulator of cellular homeostasis. In line with previous reports [15], [34], the microarray data did not show significant changes in expression of well-known NRF2 targets. This is most likely related to the low basal expression of NRF2 in brain. However, this study illustrates that subtle changes in NRF2-regulated genes may have a strong amplification in other pathophysiological pathways and uncovers crucial mechanisms that might have passed unnoticed in AD. In this regard, it is interesting to note that NRF2-encoding NFE2L2 haplotypes influence AD progression [13].
Based on the transcriptomics data, in a reverse translational approach, from man to mice, we introduced NRF2-deficiency as a novel variable. Previous studies have addressed the relevance of NRF2 in AD by combining NRF2 deficiency with either amyloidopathy [14], [17] or tauopathy [17] alone, but not in combination as presented here. These studies suggested that depletion of NRF2 activity might aggravate AD-related phenotypes but were unable to address the interactions between NRF2 expression and both pathologies when present, as they are in human disease, together. We emphasize this point because in humans, AD is by definition a combination of both hallmarks and it is still not clear what their mutual relation is, or what the contribution of each is in the development of idiopathic, sporadic AD.
In fact, we observed that the hippocampus of AT-NRF2-KO mice exhibited increased levels of oxidative and pro-inflammatory markers compared to AT-NRF2-WT mice, suggesting that the proteotoxic challenge, brought about by human APP and TAU, rendered them more sensitive to oxidative and inflammatory stress. Low-grade persistent oxidative stress is a characteristic of mild cognitive impairment indicating that low grade oxidative stress and probably inflammatory stress are early events in AD [50], [51], [52].
Memory and learning were severely impaired in the AT-NRF2-KO mice as early as 6 months, and preceding the presence of amyloid plaques and TAU fibrillar aggregates. These findings suggest that lack of NRF2 replicates a prodromal condition of human AD that is aggravated by loss of NRF2-related homeostasis and is in line with other findings from animal models of disease related phenotypes before neurodegeneration [53], [54].
An additional argument for the contribution of NRF2 to AD is its decline in activity with ageing, which remains the main risk factor for sporadic AD [55], [56]. The well-established genes with high impact in familial AD have a small incidence at the population level (APP, PS1, PS2). We envision NRF2 as a gene with a relatively small contribution to AD penetrance but with a large impact on the general population, particularly in the elderly where NRF2 activity is low because of genetic and as yet unknown environmental reasons. A consequence of this study is the generation of a mouse model that combines impaired homeostatic responses with proteinopathy and paves the way to future new pharmacological strategies aimed at NRF2 as the molecular target for treatment of the cognitive impairment in MCI and early AD.
Author contributions
Study design: AR and AC. Data acquisition and analysis: AR, AN, MP, PR, ER and AN. Drafting the manuscript: SL, RK, FL, MY, AR and AC.
Acknowledgemts
We thank Dr. Angel García-Yagüe for his help with animal handling and Dr. Mar Perez for providing the anti-PHF-1 antibody. This work was funded by SAF2016-76520-R of the Spanish Ministry of Economy and Competitiveness, a Pathfinder grant of the Centres of Excellence in Neurodegeneration (COEN) of the EU-Joint Program for Neurodegenerative diseases and European Regional Development Fund, Competitiveness Operational Program 2014–2020, through the grant P_37_732/2016 REDBRAIN.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.07.006.
Contributor Information
Ana I. Rojo, Email: airojo@iib.uam.es.
Antonio Cuadrado, Email: antonio.cuadrado@uam.es.
Appendix A. Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
References
- 1.Galimberti D., Scarpini E. Vol. 3. 2011. Inflammation and oxidative damage in Alzheimer's disease: friend or foe? pp. 252–266. (Front Biosci). [DOI] [PubMed] [Google Scholar]
- 2.Suzuki T., Motohashi H., Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013;34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Holmstrom K.M., Baird L., Zhang Y., Hargreaves I., Chalasani A., Land J.M., Stanyer L., Yamamoto M., Dinkova-Kostova A.T., Abramov A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open. 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirotsu Y., Katsuoka F., Funayama R., Nagashima T., Nishida Y., Nakayama K., Engel J.D., Yamamoto M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012;40:10228–10239. doi: 10.1093/nar/gks827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Hayes J.D., Dinkova-Kostova A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Rojo A.I., McBean G., Cindric M., Egea J., Lopez M.G., Rada P., Zarkovic N., Cuadrado A. Redox control of microglial function: molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014;21:1766–1801. doi: 10.1089/ars.2013.5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt H.H., Stocker R., Vollbracht C., Paulsen G., Riley D., Daiber A., Cuadrado A. Antioxidants in translational medicine. Antioxid. Redox Signal. 2015;23:1130–1143. doi: 10.1089/ars.2015.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman M.M., Sykiotis G.P., Nishimura M., Bodmer R., Bohmann D. Declining signal dependence of Nrf2-MafS-regulated gene expression correlates with aging phenotypes. Aging Cell. 2013;12:554–562. doi: 10.1111/acel.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suh J.H., Shenvi S.V., Dixon B.M., Liu H., Jaiswal A.K., Liu R.M., Hagen T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubben N., Zhang W., Wang L., Voss T.C., Yang J., Qu J., Liu G.H., Misteli T. Repression of the antioxidant NRF2 pathway in premature. Aging Cell. 2016;165:1361–1374. doi: 10.1016/j.cell.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strong R., Miller R.A., Antebi A., Astle C.M., Bogue M., Denzel M.S., Fernandez E., Flurkey K., Hamilton K.L., Lamming D.W., Javors M.A., de Magalhaes J.P., Martinez P.A., McCord J.M., Miller B.F., Muller M., Nelson J.F., Ndukum J., Rainger G.E., Richardson A., Sabatini D.M., Salmon A.B., Simpkins J.W., Steegenga W.T., Nadon N.L., Harrison D.E. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an alpha-glucosidase inhibitor or a Nrf2-inducer. Aging Cell. 2016 doi: 10.1111/acel.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Otter M., Landgren S., Nilsson S., Zetterberg M., Celojevic D., Bergstrom P., Minthon L., Bogdanovic N., Andreasen N., Gustafson D.R., Skoog I., Wallin A., Tasa G., Blennow K., Nilsson M., Hammarsten O., Zetterberg H. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer's disease and age-related cataract. Mech. Ageing Dev. 2010;131:105–110. doi: 10.1016/j.mad.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Kanninen K., Heikkinen R., Malm T., Rolova T., Kuhmonen S., Leinonen H., Yla-Herttuala S., Tanila H., Levonen A.L., Koistinaho M., Koistinaho J. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2009;106:16505–16510. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joshi G., Gan K.A., Johnson D.A., Johnson J.A. Increased Alzheimer's disease-like pathology in the APP/ PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol. Aging. 2015;36:664–679. doi: 10.1016/j.neurobiolaging.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lastres-Becker I., Innamorato N.G., Jaworski T., Rabano A., Kugler S., Van Leuven F., Cuadrado A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain. 2014;137:78–91. doi: 10.1093/brain/awt323. [DOI] [PubMed] [Google Scholar]
- 17.Jo C., Gundemir S., Pritchard S., Jin Y.N., Rahman I., Johnson G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014;5:3496. doi: 10.1038/ncomms4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terwel D., Muyllaert D., Dewachter I., Borghgraef P., Croes S., Devijver H., Van Leuven F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008;172:786–798. doi: 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys. Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 20.Moechars D., Dewachter I., Lorent K., Reverse D., Baekelandt V., Naidu A., Tesseur I., Spittaels K., Haute C.V., Checler F., Godaux E., Cordell B., Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 21.Terwel D., Lasrado R., Snauwaert J., Vandeweert E., Van Haesendonck C., Borghgraef P., Van Leuven F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J. Biol. Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- 22.Killick R., Ribe E.M., Al-Shawi R., Malik B., Hooper C., Fernandes C., Dobson R., Nolan P.M., Lourdusamy A., Furney S., Lin K., Breen G., Wroe R., To A.W., Leroy K., Causevic M., Usardi A., Robinson M., Noble W., Williamson R., Lunnon K., Kellie S., Reynolds C.H., Bazenet C., Hodges A., Brion J.P., Stephenson J., Simons J.P., Lovestone S. Clusterin regulates beta-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol. Psychiatry. 2014;19:88–98. doi: 10.1038/mp.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 24.Chandrasekaran S., Bonchev D. Network topology analysis of post-mortem brain microarrays identifies more alzheimer's related genes and MicroRNAs and points to novel routes for fighting with the disease. PLoS One. 2016;11:e0144052. doi: 10.1371/journal.pone.0144052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Twine N.A., Janitz K., Wilkins M.R., Janitz M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer's disease. PLoS One. 2011;6:e16266. doi: 10.1371/journal.pone.0016266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills J.D., Nalpathamkalam T., Jacobs H.I., Janitz C., Merico D., Hu P., Janitz M. RNA-Seq analysis of the parietal cortex in Alzheimer's disease reveals alternatively spliced isoforms related to lipid metabolism. Neurosci. Lett. 2013;536:90–95. doi: 10.1016/j.neulet.2012.12.042. [DOI] [PubMed] [Google Scholar]
- 27.Liang W.S., Dunckley T., Beach T.G., Grover A., Mastroeni D., Ramsey K., Caselli R.J., Kukull W.A., McKeel D., Morris J.C., Hulette C.M., Schmechel D., Reiman E.M., Rogers J., Stephan D.A. Neuronal gene expression in non-demented individuals with intermediate Alzheimer's Disease neuropathology. Neurobiol. Aging. 2010;31:549–566. doi: 10.1016/j.neurobiolaging.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blalock E.M., Geddes J.W., Chen K.C., Porter N.M., Markesbery W.R., Landfield P.W. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. USA. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang M., Roussos P., McKenzie A., Zhou X., Kajiwara Y., Brennand K.J., De Luca G.C., Crary J.F., Casaccia P., Buxbaum J.D., Ehrlich M., Gandy S., Goate A., Katsel P., Schadt E., Haroutunian V., Zhang B. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer's disease. Genome Med. 2016;8:104. doi: 10.1186/s13073-016-0355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu T., Pan Y., Kao S.Y., Li C., Kohane I., Chan J., Yankner B.A. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 31.Berchtold N.C., Cribbs D.H., Coleman P.D., Rogers J., Head E., Kim R., Beach T., Miller C., Troncoso J., Trojanowski J.Q., Zielke H.R., Cotman C.W. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. USA. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loerch P.M., Lu T., Dakin K.A., Vann J.M., Isaacs A., Geula C., Wang J., Pan Y., Gabuzda D.H., Li C., Prolla T.A., Yankner B.A. Evolution of the aging brain transcriptome and synaptic regulation. PLoS One. 2008;3:e3329. doi: 10.1371/journal.pone.0003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rojo A.I., Innamorato N.G., Martin-Moreno A.M., De Ceballos M.L., Yamamoto M., Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia. 2010;58:588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 34.Pajares M., Jimenez-Moreno N., Garcia-Yague A.J., Escoll M., de Ceballos M.L., Van Leuven F., Rabano A., Yamamoto M., Rojo A.I., Cuadrado A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy. 2016:1–15. doi: 10.1080/15548627.2016.1208889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rojo A.I., Medina-Campos O.N., Rada P., Zuniga-Toala A., Lopez-Gazcon A., Espada S., Pedraza-Chaverri J., Cuadrado A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: role of glycogen synthase kinase-3. Free Radic. Biol. Med. 2012;52:473–487. doi: 10.1016/j.freeradbiomed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Sherman M.A., Lesne S.E. Detecting abeta*56 oligomers in brain tissues. Methods Mol. Biol. 2011;670:45–56. doi: 10.1007/978-1-60761-744-0_4. [DOI] [PubMed] [Google Scholar]
- 37.Navarrete M., Perea G., Fernandez de Sevilla D., Gomez-Gonzalo M., Nunez A., Martin E.D., Araque A. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 2012;10:e1001259. doi: 10.1371/journal.pbio.1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rada P., Rojo A.I., Evrard-Todeschi N., Innamorato N.G., Cotte A., Jaworski T., Tobon-Velasco J.C., Devijver H., Garcia-Mayoral M.F., Van Leuven F., Hayes J.D., Bertho G., Cuadrado A. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell Biol. 2012;32:3486–3499. doi: 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hochberg Y., Benjamini Y. More powerful procedures for multiple significance testing. Stat. Med. 1990;9:811–818. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 40.Chanas S.A., Jiang Q., McMahon M., McWalter G.K., McLellan L.I., Elcombe C.R., Henderson C.J., Wolf C.R., Moffat G.J., Itoh K., Yamamoto M., Hayes J.D. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J. 2002;365:405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satoh H., Moriguchi T., Takai J., Ebina M., Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158–4168. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 42.Dutta S., Roy S., Polavaram N.S., Baretton G.B., Muders M.H., Batra S., Datta K. NRP2 transcriptionally regulates its downstream effector WDFY1. Sci. Rep. 2016;6:23588. doi: 10.1038/srep23588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spellman C., Ahmed M.M., Dubach D., Gardiner K.J. Expression of trisomic proteins in Down syndrome model systems. Gene. 2013;512:219–225. doi: 10.1016/j.gene.2012.10.051. [DOI] [PubMed] [Google Scholar]
- 44.Dutschmann M., Menuet C., Stettner G.M., Gestreau C., Borghgraef P., Devijver H., Gielis L., Hilaire G., Van Leuven F. Upper airway dysfunction of Tau-P301L mice correlates with tauopathy in midbrain and ponto-medullary brainstem nuclei. J. Neurosci. 2010;30:1810–1821. doi: 10.1523/JNEUROSCI.5261-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Innamorato N.G., Rojo A.I., Garcia-Yague A.J., Yamamoto M., de Ceballos M.L., Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- 46.Lastres-Becker I., Ulusoy A., Innamorato N.G., Sahin G., Rabano A., Kirik D., Cuadrado A. alpha-synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson'sdisease. Hum. Mol. Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- 47.Lenders M.B., Peers M.C., Tramu G., Delacourte A., Defossez A., Petit H., Mazzuca M. Dystrophic peptidergic neurites in senile plaques of Alzheimer's disease hippocampus precede formation of paired helical filaments. Brain Res. 1989;481:344–349. doi: 10.1016/0006-8993(89)90812-3. [DOI] [PubMed] [Google Scholar]
- 48.Theunis C., Crespo-Biel N., Gafner V., Pihlgren M., Lopez-Deber M.P., Reis P., Hickman D.T., Adolfsson O., Chuard N., Ndao D.M., Borghgraef P., Devijver H., Van Leuven F., Pfeifer A., Muhs A. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS One. 2013;8:e72301. doi: 10.1371/journal.pone.0072301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crespo-Biel N., Theunis C., Borghgraef P., Lechat B., Devijver H., Maurin H., Van Leuven F. Phosphorylation of protein Tau by GSK3beta prolongs survival of bigenic Tau.P301LxGSK3beta mice by delaying brainstem tauopathy. Neurobiol. Dis. 2014;67:119–132. doi: 10.1016/j.nbd.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 50.Pratico D. Evidence of oxidative stress in Alzheimer's disease brain and antioxidant therapy: lights and shadows. Ann. N. Y Acad. Sci. 2008;1147:70–78. doi: 10.1196/annals.1427.010. [DOI] [PubMed] [Google Scholar]
- 51.Nunomura A., Perry G., Pappolla M.A., Wade R., Hirai K., Chiba S., Smith M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J. Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gabbita S.P., Lovell M.A., Markesbery W.R. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J. Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 53.Hooper C., Markevich V., Plattner F., Killick R., Schofield E., Engel T., Hernandez F., Anderton B., Rosenblum K., Bliss T., Cooke S.F., Avila J., Lucas J.J., Giese K.P., Stephenson J., Lovestone S. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 2007;25:81–86. doi: 10.1111/j.1460-9568.2006.05245.x. [DOI] [PubMed] [Google Scholar]
- 54.Mudher A., Shepherd D., Newman T.A., Mildren P., Jukes J.P., Squire A., Mears A., Drummond J.A., Berg S., MacKay D., Asuni A.A., Bhat R., Lovestone S. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- 55.Ungvari Z., Bailey-Downs L., Gautam T., Sosnowska D., Wang M., Monticone R.E., Telljohann R., Pinto J.T., de Cabo R., Sonntag W.E., Lakatta E.G., Csiszar A. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J. Gerontol. A Biol. Sci. Med Sci. 2011;66:866–875. doi: 10.1093/gerona/glr092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomobe K., Shinozuka T., Kuroiwa M., Nomura Y. Age-related changes of Nrf2 and phosphorylated GSK-3beta in a mouse model of accelerated aging (SAMP8) Arch. Gerontol. Geriatr. 2012;54:e1–e7. doi: 10.1016/j.archger.2011.06.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material
Supplementary material
Supplementary material