

Abstract
The host defence against infection is an adaptive response in which several mechanisms are deployed to decrease the pathogen load, limit tissue injury and restore homeostasis. In the past few years new evidence has suggested that the ability of the immune system to limit the microbial burden — termed resistance — might not be the only defence mechanism. In fact, the capacity of the host to decrease its own susceptibility to inflammation- induced tissue damage — termed tolerance — might be as important as resistance in determining the outcome of the infection. Metabolic adaptations are central to the function of the cellular immune response. Coordinated reprogramming of metabolic signalling enables cells to execute resistance and tolerance pathways, withstand injury, steer tissue repair and promote organ recovery. During sepsis-induced acute kidney injury, early reprogramming of metabolism can determine the extent of organ dysfunction, progression to fibrosis, and the development of chronic kidney disease. Here we discuss the mechanisms of tolerance that act in the kidney during sepsis, with particular attention to the role of metabolic responses in coordinating these adaptive strategies. We suggest a novel conceptual model of the cellular and organic response to sepsis that might lead to new avenues for targeted, organ-protective therapies.
Host defence against infection involves several mechanisms that decrease pathogen load, limit tissue injury and restore homeostasis. Most efforts to understand the infection response have focused on defining the mechanisms by which the immune system recognizes, attacks, eliminates or blocks the invasion of pathogens. This capacity to reduce pathogen load is known collectively as resistance1, and is considered the cornerstone of defence against infection. Adjuvants to boost the resistance capacity, such as antibiotics, are effective at reducing mortality resulting from all sorts of infections, including sepsis, which has been defined as “life-threatening organ dysfunction caused by a dysregulated host response to infection” (REF. 2). However, tissue injury occurs not only as a consequence of pathogenic factors, but also as collateral damage resulting from the proinflammatory immune response (collectively known as immunopathology). Thus, despite efficiently reducing the pathogen load, resistance can result in substantial tissue injury.
Conversely, failure of resistance mechanisms secondary to apoptotic immune cell death, T-cell exhaustion, monocyte deactivation and defective immune cell metabolism3 (that is, acquired immunosuppression), is also a key driver of mortality during sepsis, presumably because this failure makes the host susceptible to secondary infections4. These realizations have fuelled interest in testing immune-modulating agents targeted at either dampening the immune response to limit immunopathology, or boosting the immune response to overcome acquired immunosuppression. Despite these advances the mechanisms of sepsis-induced organ dysfunction remain incompletely understood, and effective interventions to limit tissue injury and organ dysfunction or to prevent progression to chronic organ dysfunction remain largely unavailable5.
In the past few years, two fundamental discoveries have expanded understanding of the natural response to infection and its relationship with tissue injury. The first is that resistance is not the only defence mechanism and that decreasing susceptibility to tissue damage, the protective strategy known as tolerance, is at least as important. This discovery is crucial because enhancing tolerance can be a complementary or even an alternative strategy to improve survival as such an approach might decrease mortality regardless of pathogen load6. The second discovery is that the early metabolic responses of immune and nonimmune cells have fundamental roles in both resistance and tolerance. Metabolic adaptations that occur in response to inflammation not only participate in the triggering and execution of key resistance pathways (such as the Warburg effect in immune cells), but also in reprogramming metabolic signalling at the tissue level to withstand the acute inflammatory insult, determine the fate of tissue repair and promote recovery of organ function as manifestations of tolerance7. In the setting of sepsis-induced acute kidney injury (AKI) for example, early reprogramming of metabolic pathways can not only protect the kidney from further injury, but also determine the fate of tissue repair and the progression to fibrosis and chronic organ dysfunction7. This point is relevant because AKI induced by critical illness increases mortality 6–8-fold8, can predispose to infection9, and is a risk factor for progression to chronic kidney disease (CKD)10.
In this Review we explore the characteristics of potential tolerance mechanisms in the kidney during sepsis and the role of metabolic responses in coordinating these adaptive strategies. We discuss factors that currently limit evaluation of the relationship between resistance and tolerance mechanisms in sepsis-induced AKI, and propose furthering the translational research agenda to overcome these barriers. Finally, we introduce the hypothetical concept of a metabolic vaccine as a potential strategy to boost tolerance and protect organs during the acute phases of sepsis.
Resistance and tolerance
Cross-pollination from the fields of evolutionary biology, plant ecology and immunology has provided fundamental concepts to better understand the mechanisms by which invertebrates and vertebrates defend themselves from infection. Based on the relationship between the health status of plants (measured using outcomes such as seed production) and pathogen load, plant ecologists described two defence mechanisms: the capacity of the plant to decrease the pathogen load, termed resistance, and the ability of the plant to limit the health impact of a given pathogen load, termed tolerance1. By graphing the relationship between pathogen load and health status (known as reaction norms), ecologists have been able to describe the roles of these two mechanisms in response to different environmental conditions (FIG. 1). Importantly, controlling the pathogen load carries a health cost to the host resulting in a decline in health status, and tolerance capacity might differ between genotypes. These novel concepts provide a starting point for an entirely unexplored, mechanism-targeted approach to the prevention and treatment of organ failure in the context of sepsis.
Figure 1. The relationship between health fitness and pathogen load.
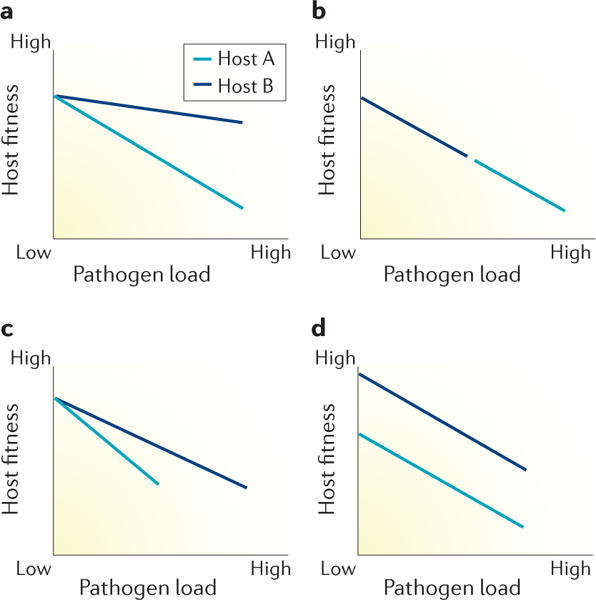
The slope of the lines represent the health cost associated with controlling a given pathogen burden for different groups of hosts in different situations. a | Both host genotypes have the same resistance capacity but host A has a worse tolerance capacity (steeper slope) so a greater health cost. b | The hosts have equivalent tolerance but host B has higher resistance because the pathogen load is lower. c | The hosts have different resistance and tolerance capacities. d | The hosts have equivalent resistance (same pathogen load) and tolerance capacity (same slope) but differ in fitness even in the absence of infection. The difference in fitness across pathogen load is constant indicating that this difference is the result of traits other than defence against infection. Modified with permission from Macmillan Publishers Limited © Schneider, D. S. & Ayres, J. S. Nat. Rev. Immunol. 8, 889–895 (2008).
Resistance
Resistance is the mechanism by which the immune system recognizes and targets foreign microbial agents to attenuate and control the pathogen load1,11. The innate immune system is an evolutionarily conserved machinery that directs this early response12,13. Innate immune cells (for example monocytes and neutrophils) express pattern recognition receptors (PRRs)12, which bind to pathogen-derived and host-derived inflammatory mediators (pathogen-associated molecular patterns (PAMPS) and damage-associated molecular patterns (DAMPs)) and trigger signal transduction cascades that result in activation of the immune response12. Importantly a subfamily of PRRs, the Toll-like receptors (TLRs), are also expressed on various types of epithelial cells, including renal tubular epithelial cells (TECs)14.
As the kidney filters plasma at a rate of ~150 ml/min, the renal tubular epithelium is exposed to circulating PAMPs and DAMPs, many of which are readily filtered through the glomerulus15. Once in the tubule, TECs can recognize PAMPs and DAMPs via their constitu-tively expressed TLRs14,16. Stimulation of TECs with lipopolysaccharide (LPS) increases the recruitment of RNA polymerase II to cytokine-specific genes, which promote the tubular secretion of tumour necrosis factor (TNF) and monocyte chemoattractant protein-1 (MCP-1)17. Experimental sepsis also induces increased renal expression of TLR4, resulting in an increase in receptor density in the entire nephron14. These findings are clinically relevant because higher cytokine concentrations have been associated with slower renal recovery and mortality among critically ill patients with AKI18. Importantly, in mouse models, prior episodes of AKI or pretreatment with LPS have been shown to repro-gram TECs, resulting in a state of hyper-responsiveness to inflammatory stimuli (PAMPs or DAMPs)17. This finding suggests that similar to immune cells, TECs can develop immunologic memory7. Rather than being passive bystanders that are damaged by the inflammatory response in sepsis, these cells might be active participants that contribute to the resistance mechanisms of the host.
Tolerance
Tissue injury depends not only on direct damage caused by a pathogenic agent or by immunopathology, but also on the intrinsic susceptibility of the host and tissue to the specific type of injury. This concept of tolerance has long been accepted in plant immunology19, but was only described in animals in the past decade1. Importantly this ecological definition of tolerance must not be confused with the concept of immune tolerance, which is the ability of the immune system to avoid autoimmune injury by not recognizing or reacting to self-antigens12. Several laboratories have now provided evidence that tolerance might be an important protective mechanism that not only acts during the acute phase of sepsis, but also has a role in the repair processes that occur after organ failure, and in the transition to chronic organ dysfunction in survivors6,20.
Rather than being a specific protective pathway, tolerance encompasses multiple mechanisms because pathogens and immunopathology can affect virtually any physiologic process11. Many mechanisms that maintain homeostasis at multiple levels during infection potentially contribute to the tolerance capacity of the host. Råberg et al. were the first to characterize differences in tolerance capacity in mammals21. They exposed five strains of mice to different intensities of infection (using different strains of Plasmodium chabaudi, a model of malaria), and measured parasite density to assess resistance, and red blood cell (RBC) density and weight as a surrogate for host fitness. They found that the slopes of the reaction norms between RBC density or weight, and parasite density differed between mouse strains, demonstrating variation in tolerance capacity21. Subsequently, Ferreira et al. demonstrated that expression of haem oxygenase 1 (HO-1) during malaria is a key mechanism that protects against the severe adverse effects of cell-free haemoglobin released during haemolysis22. The protective effects of HO-1 did not correlate with parasite burden, suggesting that HO-1 expression is a tolerance mechanism.
The harmful effect of free haem has also been documented in a model of polymicrobial sepsis after cecal ligation and puncture (CLP). In this model, administration of the haem-binding protein haemopexin prevented organ dysfunction and mortality without altering the pathogen load, suggesting that haemopexin might enhance tolerance20. This mechanism seems to be clinically relevant because reduced haemopexin levels were associated with mortality in patients with septic shock. Moreover, the murine kidney was more sensitive to damage by free haem than was heart or liver from the same host, suggesting that tolerance capacity varies between organs20.
Sepsis and inflammation at the tissue and cellular level are associated with decreased levels of intracellular adenosine triphosphate (ATP) and with mitochondrial injury in various organs, including the kidney23,24. Despite uncontrolled energy imbalance, mitochondrial injury and damage resulting from mitochondria-derived reactive oxygen species (ROS) and reactive nitrogen species, the levels of necrotic and apoptotic cells in the hearts and kidneys of patients with sepsis are surprisingly limited25 and certainly fail to explain early organ dysfunction. We found that stimulation of energy regulatory pathways, for example via activation of AMP-activated protein kinase (AMPK)26, can protect against sepsis-induced AKI in rodents, and might confer a survival advantage27. Furthermore, enhancing intracellular quality control processes such as autophagy protects against AKI in the setting of experimental sepsis28. Whether these mechanisms exert their protective effects by enhancing resistance, tolerance or both is unclear, but these findings indicate that the metabolic response to inflammation is a key protective mechanism during sepsis and a potential target for therapy.
The key role of metabolic regulation
Metabolic regulation has a central role in determining the stage and phenotype of the immune response to infection. Moreover, this response seems to be biphasic, with an early, anabolic phase fuelled by glucose and thus supported by a switch in metabolism towards aerobic glycolysis, similar to the Warburg effect in cancer cells; and an adaptive, catabolic phase, fuelled by fatty acid oxidation (FAO) and supported by a shift in metabolism to oxidative phosphorylation (OXPHOS). Similar to immune cells, TECs undergo metabolic reprogramming in response to sepsis.
The Warburg effect
The capacity of cells to induce Warburg metabolism or aerobic glycolysis is not exclusive to cancer cells. In 2002, activated T cells were shown to switch their metabolism to glycolysis; instead of feeding pyruvate into the tricarboxylic acid cycle to generate ATP through OXPHOS, they convert pyruvate to lactate, which is then excreted29. This discovery is profoundly important because it suggests that proliferative cells are capable of reprogramming their metabolism to adjust their energy and substrate requirements in response to changing environmental conditions. In this context, the advantage of Warburg metabolism over OXPHOS is that despite being energetically less efficient, aerobic glyco-lysis can provide sufficient energy for cell survival as well as essential structural components such as fatty acids, amino acids and nucleotides, which are necessary for cell functions such as mitosis30.
Although the evidence for metabolic reprogramming is substantially less robust for TECs than for immune cells, data suggest that TECs undergo similar metabolic changes in response to sepsis. We and others have shown that experimental sepsis induces an early anabolic response in renal tissue that is characterized by a shift of metabolism towards aerobic glycolysis31,32, followed by a decline in total renal ATP levels33. Smith et al. showed a significant increase in renal cortical glycolytic metabolism in rodents early after administration of LPS, which was associated with a decline in renal function31. In a rodent model of CLP-induced sepsis, we used metabolomic analysis to demonstrate an increase in the levels of glycolytic intermediates concomitant with a decrease in flux through the tricarboxylic acid cycle32. In a model of LPS-induced sepsis, oxidative phosphorylation genes were selectively suppressed in the acute phase of sepsis-induced AKI, and importantly, reactivated in those animals that recovered renal function34. In agreement with this finding, our preliminary data suggest that this initial phase might be followed by activation of AMPK35, a known inhibitor of Warburg metabolism36, suggesting that the response of TECs might also be biphasic.
In actively proliferating tumour cells37,38, naive macrophages and dendritic cells39–42, the metabolic shift towards aerobic glycolysis is driven by mTOR complex 1 (mTORC1)-induced stabilization of hypoxia inducible factor-1α (HIF-1α) through the Akt/phosphatidylin-ositol 3-kinase/mTORC1 pathway (FIG. 2). Activation of HIF-1α in proliferating cells increases glucose uptake by inducing expression of glucose transporter 1, promotes the transformation of pyruvate into lactate by inducing the expression of LDH, and inhibits the transformation of pyruvate into acetyl-coenzyme A (acetyl-CoA) by inducing the expression of pyruvate dehydrogenase kinase (PHDK), thus blocking entry into the Krebs cycle3,39,40. Importantly, HIF-1α induces the expression of the M2 isoform of pyruvate kinase (PKM2), a key enzyme that catalyses the last step of the glycolytic pathway (that is the conversion of phosphoenolpyruvate (PEP) to pyruvate)39. In contrast to the more commonly expressed M1 isoform (PKM1), PKM2 slows down the conversion of PEP to pyruvate, resulting in a bottleneck at the end of the pathway43. This slowing of glycolysis is advantageous because intermediary metabolites that accumulate behind the bottleneck can be used as precursors for other anabolic pathways in the proliferating cell. PKM2 can also act as a transcriptional co-activator of HIF-1α, thus forming a positive feedback loop for expression of HIF-1α44 and PDHK and contributing to activation of the glycolytic pathway.
Figure 2. Putative pathways that regulate the metabolic response of naive macrophages to sepsis.
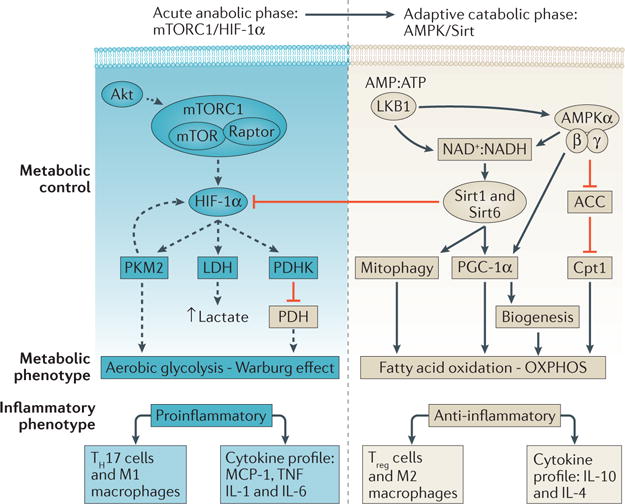
During the early phase response, the Akt/mTOR complex 1 (mTORC1)/hypoxia inducible factor-1 (HIF-1α) pathway drives the induction of aerobic glycolysis by increasing the expression of glycolytic enzymes including lactate dehydrogenase (LDH), the M2 isoform of pyruvate kinase (PKM2) and pyruvate dehydrogenase kinase (PDHK). PKM2 slows the conversion of PEP to pyruvate, so forces entry of glycolysis intermediates into the pentose phosphate pathway, whereas PHDK inhibits conversion of pyruvate to acetyl-coenzyme A so blocks entry into the Krebs cycle and decreases oxidative phosphorylation (OXPHOS). Excess pyruvate is converted to lactate by LDH. During the adaptive catabolic phase, liver kinase B1 (LKB1), AMP activated protein kinase (AMPK), sirtuin 1 (Sirt1) and sirtuin 6 (Sirt6) are likely the key regulators that switch metabolism from aerobic glycolysis back to OXPHOS. Activation of AMPK leads to activation of Sirt1, which in turn activates Sirt6. Sirt6 blocks the effects of HIF-1α, thus ‘turning off’ aerobic glycolysis, whereas sirt1 and AMPK activate peroxisome proliferator-activated receptor γ co-activator 1-α (PGC-1α), which together with carnitin palmitoyltransferase 1 (Cpt1), stimulates fatty acid oxidation to reconstitute mitochondrial oxidative metabolism. PGC-1α and the activation of mitophagy by AMPK and sirt1 further promote the coordination of cellular and mitochondrial DNA transcription to trigger biogenesis. ACC, acetyl co-enzyme A carboxylase; AMP, adenosine monophosphate; ATP, adenosine triphosphate; MCP-1, monocyte chemoattractant molecule 1; NAD+, nicotinamide adenine dinucleotide (oxidized); NADH, nicotinamide adenine dinucleotide (reduced); TH17, type 17 T helper; TNF, tumour necrosis factor; Treg, regulatory T cell.
Based on the above evidence, we speculate that TECs might use similar mechanisms to reprogramme metabolism in response to sepsis. Namely, these cells might undergo a metabolic switch from the Akt/mTORC1/ HIF-1α pathway to aerobic glycolysis, resulting in an early anabolic, proinflammatory stage, followed by a switch back to OXPHOS and entry into a late catabolic, anti-inflammatory stage (FIG. 3). Although these mechanisms are yet to be confirmed in TECs and other epithelial cells, several independent studies suggest that the induction of aerobic glycolysis or OXPHOS can affect the development of organ injury and outcome, and that the direction of the effect might be closely related to the timing of induction45,46.
Figure 3. Model of metabolic reprogramming in tubular epithelial cells during sepsis.
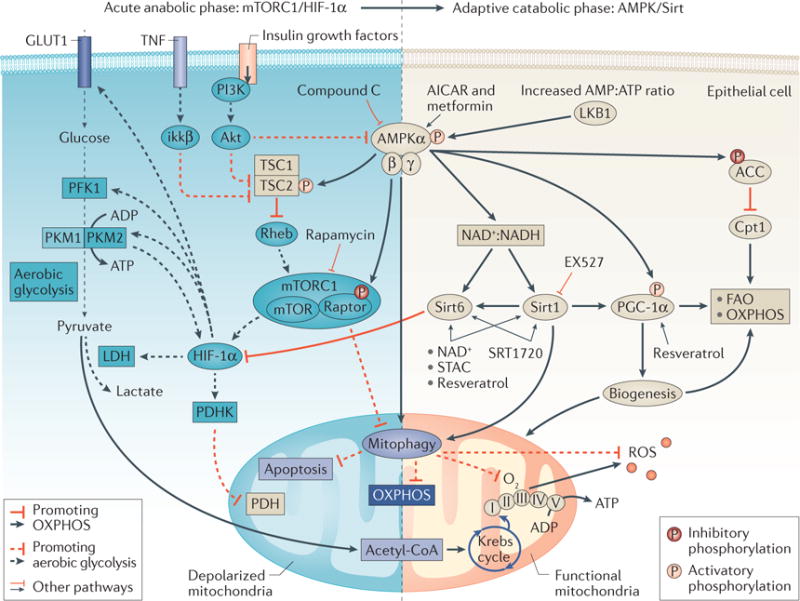
During the early metabolic response to sepsis, tubular epithelial cells might undergo a metabolic switch from oxidative phospharylation (OXPHOS) to aerobic glycolysis via the Akt/mTOR complex 1 (mTORC1)/hypoxia inducible factor 1α (HIF-1α) pathway, resulting in an early anabolic, proinflammatory stage. This stage is followed by a switch back to OXPHOS and entry into a late catabolic, anti-inflammatory stage. Dashed lines represent pathways that lead to aerobic glycolysis. Continuous lines represent pathways that might coordinate the switch from aerobic glycolysis back to OXPHOS during the adaptive catabolic phase. Various agents including 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), metformin and resveratrol and rapamycin have been used to activate or inactivate key portions of these pathways in experimental studies. ACC, acetyl-coenzyme A carboxylase; acetyl-CoA, acetyl-coenzyme A; AMP, adenosine monophosphate; ATP, adenosine triphosphate; AMPKα, AMP activated protein kinase catalytic subunit α; Cpt1, carnitin palmitoyltransferase 1; FAO, fatty acid oxidation; GLUT1, glucose transporter 1; ikkβ, inhibitor of NF-κB kinase subunit β; LDH, lactate dehydrogenase; LKB1, liver kinase B1; MCP-1, monocyte chemoattractant molecule 1; NAD+, nicotinamide adenine dinucleotide (oxidized); NADH, nicotinamide adenine dinucleotide (reduced); PDH, pyruvate dehydrogenase; PDHK, pyruvate dehydrogenase kinase; PFK1, phopsphofructokinase 1; PKM1, M1 isoform of pyruvate kinase; PKM2, M2 isoform of pyruvate kinase; PGC-1α, peroxisome proliferator-activated receptor γ co-activator 1-α; PI3K, phosphatidylinositol 3-kinase; Rheb, GTP-binding protein Rheb; ROS, reactive oxygen species; Sirt1, sirtuin 1; Sirt6, sirtuin 6; STAC, sirtuin activating compounds; TNF, tumour necrosis factor; TSC1, tuberous sclerosis 1; TSC2, tuberous sclerosis 2.
Proximal TECs have been suggested to act as danger sensors in the nephron by responding to PAMPs and secreting cytokines into the tubule, which in turn leads to activation of TECs in neighbouring nephron segments16,47. The diversion of TEC metabolism towards aerobic gly-colysis might also enhance the capacity of the cell to cope with oxidative damage by decreasing OXPHOS and thus the production of mitochondrial ROS, and by activating the pentose phosphate pathway, a major source of NADPH48, which regenerates the reduced glutathione that is necessary to eliminate mitochondrial H2O2 (REF. 49). Consistent with this hypothesis, in a mouse model of sepsis-induced AKI, LPS stimulation increased the entrance of glucose 6 phosphate into the pentose phosphate pathway by increasing the activity of glucose 6 phosphate dehydrogenase in the renal cortex31. Overall, early induction of aerobic glycolysis might provide the cell with an opportunity to mount appropriate inflammatory responses and signals, and perhaps pre-empt other cells to prepare for inflammatory injury by limiting oxidative damage and rerouting substrate allocation while generating sufficient energy to avoid cell death.
Conversely, evidence that a switch to aerobic glyco-lysis might be harmful comes from the demonstration that stimulation of promoters of OXPHOS during the early course of experimental sepsis results in protection from organ damage and improved survival. In rodents, administration of a PKM2 inhibitor 30 min before as well as 12 h and 24 h after injection of LPS, or 24 h and 48 h after CLP significantly reduced mortality40. Similarly, pharmacologic activation of NAD-dependent protein deacetylase sirtuin-1 (Sirt1) at the time of CLP in rodents improved survival45, whereas inhibition of Sirt1 at the time of CLP increased mortality46. Importantly the timing of Sirt1 inhibition determined its effect on outcome; inhibition of Sirt1 24 h after CLP improved survival46. Our preliminary data suggest a similar time dependency of outcomes because activation of AMPK (a negative regulator of aerobic glycolysis36) 24 h before, but not 18 h after CLP improved survival (G.H., unpublished work). As AMPK indirectly regulates activation of Sirt1 by increasing NAD+ availability36, it is reasonable to postulate that AMPK/Sirt1-6 is a key axis in the regulation of the response to inflammation. After ischae-mia, mice deficient in peroxisome proliferator-activated receptor γ co-activator 1-α (PGC-1α, a downstream substrate of both Sirt1 and AMPK) develop local deficiency of the NAD+ precursor niacinamide or NAM, accumulate fat and fail to re-establish normal renal function50. Together these findings suggest that early, exaggerated inflammation driven by aerobic glycolysis might be harmful, perhaps as a result of increased immunopathol-ogy (that is, decreased tolerance), whereas a weak inflammatory response at later stages might result in increased mortality presumably because of poor resistance.
Renal mitochondrial injury occurs early in the course of sepsis23,24 and is associated with cell damage as a result of ROS generation51–55 and decreased ATP synthesis. Although such damage can trigger apoptosis56, the mitochondrial apoptotic response is limited in TECs during sepsis25. Removal of injured, ROS-producing mitochondria might be a complementary mechanism to glutathione regeneration to avoid oxidative injury during the early glycolytic phase. The TEC is capable of targeting, digesting and removing dysfunctional mitochondria from the cytosol through a specialized form of auto-phagy, called mitophagy23,28,57. Inhibition of autophagy during CLP is associated with increased apoptosis in the liver57, suggesting that an early autophagic response might be necessary to limit cell death. Furthermore, an impaired autophagic response, such as that seen in ageing rodents, is associated with impaired recovery from CLP-induced AKI, and pharmacologic induction of autophagy in this setting promotes renal recovery28. Treatment with antracyclines (anticancer medications), which act by inhibiting mTOR and thus activating auto-phagy, limited AKI and improved survival in a rodent model of CLP-induced sepsis6. Importantly, this effect was independent of the pathogen load, indicating that it was mediated through enhanced tolerance.
Finally, replenishment of a pool of functional mitochondria is necessary to survive sepsis and critical illness. Widespread renal mitochondrial damage triggers the induction of repair processes, including biogenesis24. This process is upregulated by carbon monoxide (CO) via activation of the redox-regulated NF-E2-related factor transcription factor, nuclear respiratory factor 1 (NRF1), NRF2 and PGC-1α58. CO-induced biogenesis conferred protection from hepatic injury in rodents during Staphylococcus aureus-induced sepsis55, whereas inhibition of autophagy or TLR9 signalling has been shown to prevent activation of CLP-induced biogen-esis, suggesting that these mechanisms are linked59. We postulate that activation of autophagy might be necessary during the early glycolytic phase to decrease ROS production, OXPHOS and oxygen consumption by decreasing mitochondrial mass, and to limit cell injury, whereas restitution of a functional mitochondrial pool through biogenesis might be required to maintain organ protection, and promote organ recovery and survival.
Early metabolic reprogramming
An important lesson from the field of immunometabolism is that early changes in cellular metabolism in response to infection can affect cell behaviour beyond the acute phases of disease. This concept is key because patients with sepsis are exposed to repeated injury beyond that caused by the source infection, from the effects of immunopathology, shock and co-infections, which exacerbate or even promote persistent organ dysfunction.
A switch towards aerobic glycolysis is necessary for the development of trained immunity, which is the ability of the innate immune system to develop memory39 and modify the response to future insults7. In monocytes, pre-stimulation of the Akt/mTORC1/ HIF1-α pathway with β-glucan (by exposure to sub-lethal concentrations of Candida albicans) resulted in increased secretion of TNF and IL-6 upon stimulation with S. aureus; in mice such prestimulation resulted in improved survival39. The protective trained immunity achieved by pretreatment with β-glucan was lost when inhibiting Akt, mTOR or HIF-1α, suggesting that the Akt/mTORC1/HIF1-α pathway, which is also involved in metabolic reprogramming and regulation during infection, has a key role in the development of trained immunity. Zager et al. postulated a similar mechanism of biologic memory in the kidney, whereby diverse insults that are usually associated with critical illness such as ischaemia or exposure to nephrotoxins, reprogramme the kidney to ‘hyper-respond’ to sepsis7. They showed that mice that were pre-exposed to ischaemia60, nephrotoxins, or urinary tract obstruction61, had a more robust inflammatory response (that is, a higher release of TNF and MCP-1) to LPS and lipoteichoic acid than mice that had not been primed by these other insults. Importantly, they demonstrated that this biologic memory was driven by the activation of chromatin-remodelling enzymes such as histone deacetylases, which reprogramme TECs to hyper-respond to the septic insult.
The AKI to CKD transition
A persistent proinflammatory state is associated with worse organ dysfunction and sepsis outcome40; therefore, the capacity to de-escalate the inflammatory response to sepsis is as important as the ability to mount a robust immune response. An inability to switch metabolism from aerobic glycolysis to OXPHOS, and thus ‘turn off’ inflammation, has been associated with chronic inflammation and impaired organ recovery62. In inflammatory cells, the switch from this early anabolic glycolytic phase to an adaptive catabolic phase in which metabolism is reprogrammed towards OXPHOS and regulated to conserve energy, has been attributed to the action of Sirt1 and Sirt6 (REF. 63). Sirt6 opposes HIF-1α by silencing genes that encode key glycolytic enzymes64, whereas Sirt1 deacetylates PGC-1α, a promoter of FAO, OXPHOS and biogenesis65,66 (FIG. 3). Mice that were deficient in upstream regulators of NAD+/NADPH or were exposed to FAO inhibitors had substantial energy deficiencies, increased apoptosis and profibrotic phenotypes, all of which could be reversed by treatment with upstream activators of PGC-1α such as fenofibrate62.
In sepsis, AKI is usually present at the time of presentation67,68. Identifying strategies to interrupt AKI progression is therefore paramount, especially as AKI can lead to CKD69–71. Maladaptive processes such as decreased capillary density (which exacerbates hypoxia), expansion of interstitial fibroblasts and myofibroblasts, and epithelial-to-mesenchymal transition (EMT), presumably drive the development of insterstitial fibrosis72, and thus CKD73.
Human and murine fibrotic kidneys had lower expression of FAO enzymes and regulators than did kidneys from humans and rodents without fibrotic changes, and inhibition of FAO in primary human cells resulted in phenotypic changes consistent with fibrosis74. Moreover, restoring FAO by activating key regulators such as PPAR-α and PGC-1α protected mice from tubulointerstitial fibrosis after exposure to folic acid. Mice that lacked LKB1, an upstream regulator of AMPK, also progressed to a fibrotic phenotype62, which was rescued and FAO restored using fenofibrate74. Activation of AMPK with A769662 resulted in the same effects, again suggesting that different mediators of this energy regulatory pathway have key roles in regulating progression to the mechanisms that lead to CKD.
Using a model of fibrosis after unilateral ureteral obstruction, Qui et al. demonstrated that animals deficient in AMPKα2 not only showed increased production of cytokines and chemokines, but also increased EMT and fibrosis. Conversely, activation of AMPKα2 protected against this profibrotic phenotype75. These data are reminiscent of those of Naito et al. who demonstrated that TECs display a proinflammatory phenotype with secretion of cytokines in the setting of enhanced aerobic glycolysis17. Together, these data might suggest that although the early switch to glycolysis seems to be necessary for a robust inflammatory response to sepsis, failure to restore FAO and OXPHOS might result in progression to fibrosis and CKD.
The feasibility of a metabolic vaccine
The understanding that metabolic reprogramming is used by host cells to drive an efficient immune response and enhance tolerance to noxious stimuli at different stages of infection provides an opportunity to explore interventions that could enhance these protective mechanisms and provide the foundations for a metabolic antipathology vaccine. Greater understanding is required, however, particularly with regard to tolerance mechanisms. Although many pathways could potentially exert protection through tolerance, only a few (such as expression of haemopexin and HO-1) have been shown to have protective effects in experimental models. Importantly, the degree of specificity of these pathways in terms of their protective effects during different types of infection is unclear. Different infectious agents might have very diverse pathogenic mechanisms, and thus a pathway that enhances tolerance during one type of infection, might have no effect or even be harmful during a different type of infection. Finally, the available data demonstrate that the timing of the intervention will determine the effect on tissue protection and outcome. With these caveats in mind, manipulation of the Akt/ mTORC/HIF-1α pathway, the AMPK/Sirt1-6 pathway, mitophagy and HO-1/CO-induced biogenesis might hold promise as potential therapeutic or vaccination targets in future experimental studies.
Future directions
Applying the concept of reaction norms to the study of experimental and human sepsis is desirable because it represents the first step in applying a tool to characterize the impact of tolerance mechanisms in this setting. Understanding of these mechanisms at the experimental level is still at an early stage, however, and far from clinical application. Thus, research should focus on understanding the biologic implications of these mechanisms in sepsis and in formulating translational avenues to bridge this knowledge to patient care.
The study of tolerance mechanisms is challenging for several reasons. First, the identification of surrogates of health status that are representative of the health cost of a particular infection is difficult. For example, weight loss is a suitable measure of health status in a patient with cholera because it is related to dehydration and reflects the impact of treatment (rehydration) on outcome, but is an insensitive and misleading measure in sepsis because rapid, profound weight loss is not typical and weight gain owing to fluid resuscitation does not necessarily translate into a better health status or outcome. Second, a translational barrier exists as illustrated by the fact that although misleading in human sepsis, weight loss might be a good measure of fitness in rodent models because fluid resuscitation and its influence on weight can be accurately measured and accounted for.
As organ dysfunction during sepsis is a direct measure of tissue damage and a determinant of survival, and different organs and tissues have different tolerance capacities, the identification of organ-specific markers of fitness has been suggested as an alternative strategy to identify surrogates of health status1,11. For example, the levels of urinary metalloproteinase inhibitor 2 (TIMP-2) and insulin-like growth factor-binding protein 7 have been shown to predict AKI in different settings76,77. These markers can be used to screen for tubular damage 12–24 h before a rise in serum creatinine level occurs, and thus provide an opportunity not only to assess fitness, but also to gauge the response of therapies to enhance fitness. Another organ-specific marker is renal functional reserve, which quantifies the capacity of the kidney to increase the glomerular filtration rate (GFR) in response to a protein load78. This test is interesting because usual measures of renal function such as GFR or serum creatinine remain normal until approximately 50% of nephrons are lost and the remainder can no longer compensate for their functional loss78–80.
Another translational challenge in tolerance research is measurement of pathogen load in the clinical setting. Such measurement is often unreliable owing to a lack of tissue samples and a dependency on cultures of indirect samples such as bronchoalveolar lavage, urine, blood or other bodily fluids (such as pleural fluid or drained abscesses). The absence of bacteria in any of these samples might lead to the false impression that the bacterial load has been controlled. This problem is not a barrier in experimental sepsis, and thus a need exists to find suitable means or surrogates to measure pathogen load at the bedside. Given that an important source of tissue damage and injury is immunopathology, an alternative, potentially more feasible approach to measurement of pathogen load might be measurement of the immune response. Such an approach might provide important insights, particularly in sepsis and in non-infectious inflammatory settings such as cardiac surgery or pancreatitis. Measuring organ fitness against inflammatory burden might provide a method to monitor tolerance as well as the health cost of a particular immune response to infection (FIG. 4). In the future, this approach could enable the evaluation of immune-targeted and tolerance-targeted strategies.
Figure 4. Potential strategies for the assessment of renal tolerance in patients with sepsis.
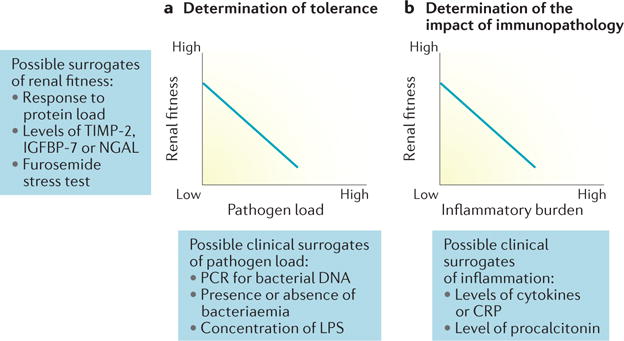
a | Determination of tolerance (that is, reaction norms) could potentially be achieved at the bedside using existing kidney-specific markers of renal fitness and clinical surrogates of pathogen load. b | An alternative, potentially more feasible approach might be to determine the impact of immunopathology on renal fitness using clinical surrogates of inflammatory burden. CRP, C-reactive protein; IGFBP-7, insulin-like growth factor-binding protein; LPS, lipopolysaccharide; NGAL, neutrophil gelatinase-associated lipocalin; TIMP-2, urinary metalloproteinase inhibitor 2.
Conclusions
Host defence against infection is governed at least in part by resistance and tolerance mechanisms that act to decrease the microbial load and the susceptibility of the host to damage inflicted by microorganisms or by immunopa-thology. Metabolic reprogramming is essential to protect the cell from energy imbalance, determine the phenotype of the immune response, dictate the phasic response in monocytes and possibly in TECs, and regulate repair programmes. Future research should consider tolerance to be an integral part of the natural response to infection and inflammation, and metabolic regulation to be the mechanism that steers these responses. Accordingly, forthcoming investigations should dedicate efforts to better understand the natural course of specific metabolic pathways that are associated with protection during different types of sepsis, the optimum timing of activation of these pathways, and importantly, the long-term effects on organ dysfunction, persistent inflammation and survival after the acute insult. In the future, this knowledge might provide sufficient basis to adequately phenotype patients with sepsis and to design mechanism-specific, phase-oriented treatments. Finally, this framework might open the possibility to use long lasting targeted antipathology vaccines to avoid further tissue damage, correct maladaptive repair processes and avoid chronic organ dysfunction and death.
Key points.
Resistance and tolerance mechanisms govern the infection response and determine host survival
A switch to Warburg metabolism seems to be an important survival strategy in immune cells and possibly in tubular epithelial cells in response to inflammation
Reprogramming of metabolic pathways is a key cellular strategy to execute resistance and tolerance protective programmes
Early metabolic reprogramming in the response to inflammation is crucial to determine the acute response of the cell, to avoid cell death and to determine the repair phenotype during recovery
Understanding of resistance and tolerance mechanisms in sepsis might provide a basis for the development of therapeutic strategies to prevent or reverse organ damage, promote recovery and decrease mortality
Acknowledgments
H.G. is supported by NIH grants 1K12HL109068-02 and 1K08GM117310-01A1.
Glossary
- Warburg effect
A coordinated change in cellular metabolism from oxidative phosphorylation to glycolysis to support the production of ATP and essential structural components required by the cell. This switch in metabolism can occur even in aerobic conditions
- Autophagy
An intracellular degradation pathway in which cellular components are taken up by autophagosomes and transported to lysosomes for degradation and recycling
- Oxidative phosphorylation
A metabolic process in which electrons derived from oxidation of carbon fuels (such as glucose and fats) are transported through electron acceptors and donors in the inner mitochondrial membrane. This process releases the energy required to generate adenosine triphosphate in the presence of oxygen
- Tricarboxylic acid cycle
A series of enzyme-catalysed reactions that through oxidation of acetyl-coenzyme A (derived from carbohydrate, fat and protein), yields reducing equivalents such as NADH, which are necessary for many processes including ATP generation
- Mitophagy
A specialized form of autophagy that targets and degrades dysfunctional mitochondria
- Biogenesis
Mitochondrial biogenesis is a coordinated process that involves both nuclear and mitochondrial DNA and results in the regeneration of new mitochondria
- Epithelial-to-mesenchymal transition
In the context of organ injury, epithelial-to-mesenchymal transition refers to the process by which epithelial cells undergo a transition to myofibroblasts, which produce collagenous matrix and promote tissue fibrosis
Footnotes
Competing interests statement
J.A.K. has received consulting fees and grant support from Astute Medical. H.G. and C.R. declare no competing interests.
Author contributions
All authors researched the data, discussed the content, wrote the article and reviewed or edited the manuscript before submission.
References
- 1.Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. 2008;8:889–895. doi: 10.1038/nri2432. This opinion article summarizes the evidence for specific tolerance mechanisms in animals and discusses the potential clinical implications of these findings. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng SC, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17:406–413. doi: 10.1038/ni.3398. [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen J, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15:581–614. doi: 10.1016/S1473-3099(15)70112-X. [DOI] [PubMed] [Google Scholar]
- 6.Figueiredo N, et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity. 2013;39:874–884. doi: 10.1016/j.immuni.2013.08.039. This study in a rodent model of sepsis demonstrates that low-dose anthracyclines confer robust protection, independent of bacterial burden, via a mechanism that inves DNA-damage-response-dependent activation of autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zager RA. ‘Biologic memory’ in response to acute kidney injury: cytoresistance, toll-like receptor hyper-responsiveness and the onset of progressive renal disease. Nephrol Dial Transplant. 2013;28:1985–1993. doi: 10.1093/ndt/gft101. This experimental study demonstrates that ischaemic or toxic renal injury triggers cellular adaptations that reprogramme the response of the kidney to subsequent injury. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchino S, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–818. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 9.Thakar CV, Yared JP, Worley S, Cotman K, Paganini EP. Renal dysfunction and serious infections after open-heart surgery. Kidney Int. 2003;64:239–246. doi: 10.1046/j.1523-1755.2003.00040.x. [DOI] [PubMed] [Google Scholar]
- 10.Murugan R, Kellum JA. Acute kidney injury: what’s the prognosis? Nat Rev Nephrol. 2011;7:209–217. doi: 10.1038/nrneph.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. This review summarizes the host defence against infection and discusses the integral role of tolerance in this response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 14.El-Achkar TM, et al. Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am J Physiol Renal Physiol. 2006;290:F1034–F1043. doi: 10.1152/ajprenal.00414.2005. [DOI] [PubMed] [Google Scholar]
- 15.El-Achkar TM, Hosein M, Dagher PC. Pathways of renal injury in systemic gram-negative sepsis. Eur J Clin Invest. 2008;38:39–44. doi: 10.1111/j.1365-2362.2008.02007.x. [DOI] [PubMed] [Google Scholar]
- 16.Kalakeche R, et al. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol. 2011;22:1505–1516. doi: 10.1681/ASN.2011020203. This study demonstrates that LPS can be filtered through the glomerulus, tubular epithelial cells can bind LPS through TLR4, and uptake of LPS by S1 tubular epithelial cells triggers a coordinated response characterized by oxidative stress in neighbouring cells in the S2 and S3 segments of the nephron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naito M, Bomsztyk K, Zager RA. Endotoxin mediates recruitment of RNA polymerase II to target genes in acute renal failure. J Am Soc Nephrol. 2008;19:1321–1330. doi: 10.1681/ASN.2007121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murugan R, et al. Plasma inflammatory and apoptosis markers are associated with dialysis dependence and death among critically ill patients receiving renal replacement therapy. Nephrol Dial Transplant. 2014;29:1854–1864. doi: 10.1093/ndt/gfu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schafer JF. Tolerance to plant disease. Annu Rev Phytopathol. 1971;9:235–252. This review describes the importance of tolerance mechanisms in plants. [Google Scholar]
- 20.Larsen R, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med. 2010;2:51ra71. doi: 10.1126/scitranslmed.3001118. This experimental study demonstrates that free haemoglobin induces substantial organ injury during murine sepsis, and that activation of haem-oxygenase-1 or administration of haemopexin confers protection against organ injury and a survival advantage by increasing tolerance. [DOI] [PubMed] [Google Scholar]
- 21.Råberg L, Sim D, Read AF. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science. 2007;318:812–814. doi: 10.1126/science.1148526. The first study to demonstrate tolerance mechanisms in mammals using a model of murine malaria and a statistical framework developed by plant ecologists. [DOI] [PubMed] [Google Scholar]
- 22.Ferreira A, Balla J, Jeney V, Balla G, Soares MP. A central role for free heme in the pathogenesis of severe malaria: the missing link? J Mol Med. 2008;86:1097–1111. doi: 10.1007/s00109-008-0368-5. [DOI] [PubMed] [Google Scholar]
- 23.Hsiao HW, et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37:289–296. doi: 10.1097/SHK.0b013e318240b52a. [DOI] [PubMed] [Google Scholar]
- 24.Bartz RR, et al. Staphylococcus aureus sepsis induces early renal mitochondrial DNA repair and mitochondrial biogenesis in mice. PLoS ONE. 2014;9:e100912. doi: 10.1371/journal.pone.0100912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takasu O, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187:509–517. doi: 10.1164/rccm.201211-1983OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hallows KR, Mount PF, Pastor-Soler NM, Power DA. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol. 2010;298:F1067–F1077. doi: 10.1152/ajprenal.00005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escobar DA, et al. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J Surg Res. 2015;194:262–272. doi: 10.1016/j.jss.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howell GM, et al. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS ONE. 2013;8:e69520. doi: 10.1371/journal.pone.0069520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frauwirth KA, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. Experimental study that demonstrates that the metabolic phenotype of activated T cells is aerobic glycolysis, and that CD28 co-stimulation through the PI3K//Akt pathway is required to increase glycolytic flux in this context. [DOI] [PubMed] [Google Scholar]
- 30.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith JA, Stallons LJ, Schnellmann RG. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am J Physiol Renal Physiol. 2014;307:F435–F444. doi: 10.1152/ajprenal.00271.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waltz P, Carchman E, Gomez H, Zuckerbraun B. Sepsis results in an altered renal metabolic and osmolyte profile. J Surg Res. 2016;202:8–12. doi: 10.1016/j.jss.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol. 2014;306:F734–F743. doi: 10.1152/ajprenal.00643.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran M, et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121:4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin K, et al. Is acute kidney injury in the early phase of sepsis a sign of metabolic downregulation in tubular epithelial cells? Crit Care. 2015;19(Suppl 1):P286. [Google Scholar]
- 36.Faubert B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth. in vivo Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. This experimental study demonstrates that AMPK negatively regulates aerobic glycolysis in cancer cells, and that inactivation of the AMPKα catalytic subunit promotes a metabolic shift to aerobic glycolysis, which requires HIF-1α stabilization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hudson CC, et al. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dodd KM, Yang J, Shen MH, Sampson JR, Tee AR. mTORC1 drives HIF-1α; and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene. 2014;34:2239–2250. doi: 10.1038/onc.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng SC, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. This study demonstrates that a shift of central glucose metabolism from oxidative phosphorylation to aerobic glycolysis is the metabolic basis for trained immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang L, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436. doi: 10.1038/ncomms5436. This experimental study demonstrates that pyruvate kinase M2 interacts with HIF-1α to induce aerobic glycolysis (Warburg effect) in macrophages, and that PKM2 is a key regulator of the release of high mobility group box 1 during sepsis and inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Neill LAJ, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 42.Krawczyk CM, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang L, DeBerardinis RJ. Cancer metabolism: when more is less. Nature. 2012;489:511–512. doi: 10.1038/489511a. [DOI] [PubMed] [Google Scholar]
- 44.Luo W, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Opal S, et al. Sirt1 activation markedly alters transcription profiles and improves outcome in experimental sepsis. Shock. 2015 doi: 10.1097/SHK.0000000000000528. http://dx.doi.org/10.1097/SHK.0000000000000528. [DOI] [PubMed]
- 46.Vachharajani VT, et al. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol. 2014;96:785–796. doi: 10.1189/jlb.3MA0114-034RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gomez H, et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41:3–11. doi: 10.1097/SHK.0000000000000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39:347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2013;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran MT, et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531:528–532. doi: 10.1038/nature17184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brealey D, et al. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol. 2004;286:R491–R497. doi: 10.1152/ajpregu.00432.2003. [DOI] [PubMed] [Google Scholar]
- 52.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 53.Sayeed MM, Zhu M, Maitra SR. Alterations in cellular calcium and magnesium during circulatory/ septic shock. Magnesium. 1989;8:179–189. [PubMed] [Google Scholar]
- 54.Duchen MR, Verkhratsky A, Muallem S. Mitochondria and calcium in health and disease. Cell Calcium. 2008;44:1–5. doi: 10.1016/j.ceca.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 55.Haden DW, et al. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med. 2007;176:768–777. doi: 10.1164/rccm.200701-161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053–2062. doi: 10.1002/hep.24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. 2011;1813:1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carchman EH, et al. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. FASEB J. 2013;27:4703–4711. doi: 10.1096/fj.13-229476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zager RA, Johnson ACM, Hanson SY, Lund S. Ischemic proximal tubular injury primes mice to endotoxin-induced TNF-alpha generation and systemic release. Am J Physiol Renal Physiol. 2005;289:F289–F297. doi: 10.1152/ajprenal.00023.2005. [DOI] [PubMed] [Google Scholar]
- 61.Zager RA, Johnson ACM, Hanson SY, Lund S. Acute nephrotoxic and obstructive injury primes the kidney to endotoxin-driven cytokine/ chemokine production. Kidney Int. 2006;69:1181–1188. doi: 10.1038/sj.ki.5000022. [DOI] [PubMed] [Google Scholar]
- 62.Han SH, et al. Deletion of Lkb1 in renal tubular epithelial cells leads to CKD by altering metabolism. J Am Soc Nephrol. 2016;27:439–453. doi: 10.1681/ASN.2014121181. This experimental study demonstrates a role of Lkb1 (a master regulator of metabolism) in the progression from AKI to CKD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+-dependent Sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. 2012;287:25758–25769. doi: 10.1074/jbc.M112.362343. This experimental study demonstrates the roles of Sirt1 and Sirt6 in the metabolic switch from aerobic glycolysis to oxidative phosphorylation in THP-1 promonocytes during the acute inflammatory response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhong L, Mostoslavsky R. SIRT6: a master epigenetic gatekeeper of glucose metabolism. Transcription. 2014;1:17–21. doi: 10.4161/trns.1.1.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lehman JJ, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kellum JA, et al. The effects of alternative resuscitation strategies on acute kidney injury in patients with septic shock. Am J Respir Crit Care Med. 2016;193:281–287. doi: 10.1164/rccm.201505-0995OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kellum JA, et al. Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch Intern Med. 2007;167:1655–1663. doi: 10.1001/archinte.167.15.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zager RA, Johnson ACM, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and ‘end-stage’ kidney disease. Am J Physiol Renal Physiol. 2011;301:F1334–F1345. doi: 10.1152/ajprenal.00431.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grgic I, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82:172–183. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heung M, Chawla LS. Predicting progression to chronic kidney disease after recovery from acute kidney injury. Curr Opin Nephrol Hypertens. 2012;21:628–634. doi: 10.1097/MNH.0b013e3283588f24. [DOI] [PubMed] [Google Scholar]
- 72.Basile DP, et al. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. 2016;27:687–697. doi: 10.1681/ASN.2015030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hodgkins KS, Schnaper HW. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr Nephrol. 2011;27:901–909. doi: 10.1007/s00467-011-1992-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kang HM, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. doi: 10.1038/nm.3762. This study demonstrates that human and murine kidneys with tubulointerstitial fibrosis have reduced expression of enzymes inved in fatty acid oxidation, and that inhibition of these enzymes in a murine model results in renal fibrosis, which can be reversed by restoring fatty acid metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qiu S, et al. AMPKα2 reduces renal epithelial transdifferentiation and inflammation after injury through interaction with CK2β. J Pathol. 2015;237:330–342. doi: 10.1002/path.4579. [DOI] [PubMed] [Google Scholar]
- 76.Bihorac A, et al. Validation of cell-cycle arrest biomarkers for acute kidney injury using clinical adjudication. Am J Respir Crit Care Med. 2014;189:932–939. doi: 10.1164/rccm.201401-0077OC. [DOI] [PubMed] [Google Scholar]
- 77.Kashani K, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care. 2013;17:R25. doi: 10.1186/cc12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sharma A, Mucino MJ, Ronco C. Renal functional reserve and renal recovery after acute kidney injury. Nephron Clin Pract. 2014;127:94–100. doi: 10.1159/000363721. [DOI] [PubMed] [Google Scholar]
- 79.Liu KD, Brakeman PR. Renal repair and recovery. Crit Care Med. 2008;36:S187–S192. doi: 10.1097/CCM.0b013e318168ca4a. [DOI] [PubMed] [Google Scholar]
- 80.Fliser D, Zeier M, Nowack R, Ritz E. Renal functional reserve in healthy elderly subjects. J Am Soc Nephrol. 1993;3:1371–1377. doi: 10.1681/ASN.V371371. [DOI] [PubMed] [Google Scholar]