

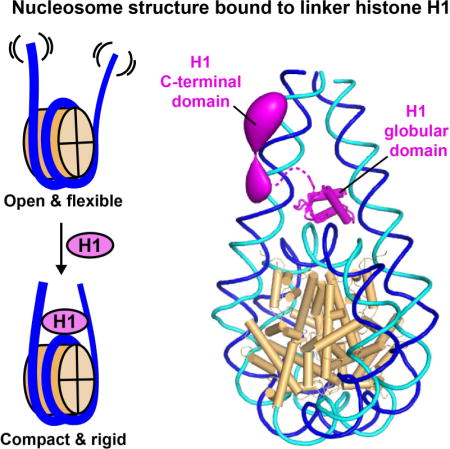
SUMMARY
Linker histones associate with nucleosomes to promote the formation of higher-order chromatin structure, but the underlying molecular details are unclear. We investigated the structure of a 197 base-pair nucleosome bearing symmetric 25 base-pair linker DNA arms in complex with vertebrate linker histone H1. We determined electron cryo-microscopy (cryo-EM) and crystal structures of unbound and H1-bound nucleosomes and validated these structures by site-directed protein cross-linking and hydroxyl radical footprinting experiments. Histone H1 shifts the conformational landscape of the nucleosome by drawing the two linkers together and reducing their flexibility. The H1 C-terminal domain (CTD) localizes primarily to a single linker, while the H1 globular domain contacts the nucleosome dyad and both linkers, associating more closely with the CTD-distal linker. These findings reveal that H1 imparts a strong degree of asymmetry to the nucleosome, which is likely to influence the assembly and architecture of higher-order structures.
Graphical Abstract
INTRODUCTION
The basic repeat unit of eukaryotic chromatin, the nucleosome, comprises a nucleosome core particle (NCP), linker DNA and a linker histone (H1) (van Holde, 1988). Each NCP contains 147 DNA base pairs (bp) wrapped around a histone core octamer (Luger et al., 1997) and connects to neighbouring NCPs by a variable length of linker DNA. Linker histones induce the formation of an apposed linker DNA stem motif (Bednar et al., 1998; Hamiche et al., 1996) and extend the amount of DNA protected from micrococcal nuclease digestion by ~20 bp beyond the 147 bp protected by the core octamer. Linker histones are a key determinant of nucleosome repeat length (Woodcock et al., 2006) and are critical for the assembly and maintenance of the 30-nm chromatin fibre (Bednar et al., 1995; Makarov et al., 1983; Thoma et al., 1979; Tremethick, 2007) and of higher-order chromatin structures (Maresca et al., 2005). Multiple isoforms with distinct species, tissue and developmental specificity have been identified, including eleven mammalian H1 sub-types and the avian erythrocyte variant H5 (Izzo et al., 2008).
Histone H1/H5 family members share a tripartite structure consisting of a conserved globular domain of ~75 residues, an N-terminal tail of 20–35 residues and a highly basic C-terminal domain (CTD) of ~100 residues (Allan et al., 1980). Whereas the N-terminal tail has little effect on chromatin binding (Allan et al., 1980; Hendzel et al., 2004; Syed et al., 2010), the globular domain is sufficient for structure-specific nucleosome recognition and for protecting additional DNA from nuclease digestion (Allan et al., 1980; Simpson, 1978). The CTD is required for linker stem formation (Syed et al., 2010) and for stabilizing secondary chromatin structures (Allan et al., 1980; Allan et al., 1986). The CTD is intrinsically disordered but becomes more structured and compact upon nucleosome binding (Fang et al., 2012). However, the localization of the CTD on the nucleosome remains a major open question.
The linker histone globular domain adopts a winged-helix DNA-binding fold (Ramakrishnan et al., 1993). Studies support distinct models for how this domain interacts with nucleosomes. One model positions the domain on the nucleosome dyad in contact with core DNA and with both DNA linkers (Allan et al., 1980; Simpson, 1978; Staynov and Crane-Robinson, 1988; Syed et al., 2010), whereas in alternative models the domain is displaced from the dyad and contacts only a single linker as well as core DNA (Brown et al., 2006; Pruss et al., 1996; Zhou et al., 1998). This debate has been significantly clarified by recent structural work, which demonstrated distinct binding modes for different linker histone-nucleosome complexes. Specifically, the crystal structure of the globular domain of chicken H5 (GH5) bound to a 167-bp particle exhibits an on-dyad binding mode (Zhou et al., 2015). In contrast, an NMR study reported an off-dyad binding mode for the Drosophila H1 globular domain (GH1) (Zhou et al., 2013). The different binding modes observed for these two histone isoforms have been ascribed to differences in a few DNA-contacting residues (Zhou et al., 2016). A distinct off-dyad binding mode was reported for human histone H1.4 in the cryo-EM structure of a condensed 12-nucleosome array (Song et al., 2014). Taken together, these studies suggest that the orientation of the linker histone globular domain may vary with H1 isoform and with the overall structural context.
The GH5-bound nucleosome structure was determined using the isolated GH5 domain bound to a 167-bp nucleosome bearing short (10 bp) linker DNA arms (Zhou et al., 2015). The present study reports the structure of a larger particle, a 197-bp nucleosome containing two 25-bp DNA linkers and full-length linker histone H1, determined by cryo-EM and X-ray crystallography and validated by biochemical analysis. Our results reveal that the H1 globular domain adopts an on-dyad binding mode while the CTD associates primarily with a single linker, strongly disrupting the two-fold symmetry of the nucleosome. These findings advance our understanding of how linker histones associate with nucleosomes and provide an enhanced framework for investigating the assembly of higher-order chromatin structures.
RESULTS
H1 stabilises a compact and rigid nucleosome conformation
We reconstituted nucleosomes using recombinant Xenopus laevis or human core histones and a 197-bp DNA duplex comprising the Widom 601 positioning sequence (Lowary and Widom, 1998) or a palindromic derivative (601L) of this sequence (Chua et al., 2012), respectively. The 601 and 601L nucleosomes were complexed with X. laevis histone H1.0b or with a truncation mutant of human H1.5 lacking 50 C-terminal residues (Syed et al., 2010), hereafter called H1.0 and H1.5ΔC50, respectively. (We use “H1” below to refer generically to linker histones without specifying the precise isoform or species). Nucleosomes were then analysed by single particle cryo-EM (Figures 1A–C and S1A–F). We first determined the structure of 601 nucleosomes lacking H1 and used three-dimensional (3D) classification to sort conformational variants. The cryo-EM reconstruction at 11.4 Å resolution (Figure S1D) agreed well with the NCP crystal structure (Luger et al., 1997) and allowed fitting of the linker DNA. We observed a range of linker DNA configurations (Figures 1A and 1D, representative conformations 1–3), which we characterized by measuring the angle between each linker and the dyad axis in the planes parallel (angle α) and perpendicular (angle β) to the nucleosomal disc plane (Figure 1E). Both angles varied by 25–30°, revealing the highly dynamic character of the linkers, likely due to “breathing” of the histone-DNA interactions near NCP exit. Although the linker arms appear convergent when viewed from the “front” (mean value for α=17.5° ± 7.5°), side views show that they diverge from the nucleosomal disc plane (mean β=18.3° ± 8.9°), placing the two linker DNA ends far apart (mean separation of 10.1 ± 0.9 nm).
Figure 1. H1 stabilises a compact nucleosome conformation.
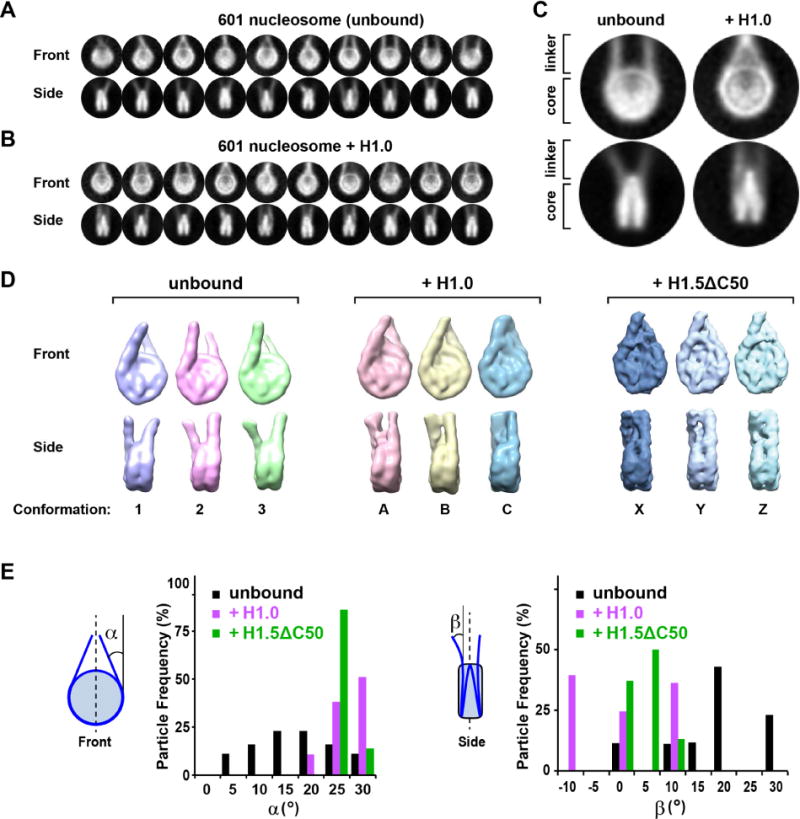
(A,B) Gallery of class averages of 197-bp 601 nucleosomes in the (A) absence and (B) presence of histone H1.0. (C) Close-up views. (D) Representative 3D classes showing different linker DNA orientations in the unbound state (3 of 8 conformational classes are shown) or bound to H1.0 or H1.5ΔC50 (all 3 classes are shown). (E) Distribution of linker DNA exit angles in the unbound state (black) or bound to H1.0 (magenta) or H1.5ΔC50 (green). See also Figure S1.
We next analysed H1.0- and H1.5ΔC50-bound nucleosomes. Cryo-EM reconstructions at 11.5 and 6.2 Å resolution, respectively (Figures S1E and S1F) (the resolution difference primarily reflects data collection on different electron microscopes and detectors) revealed a significant change in linker DNA orientation upon H1 binding, giving particles a more compact appearance (Figures 1B–D). 3D classification revealed that the linker orientation was less variable compared to unbound nucleosomes, with narrower distributions for both angles α and β (Figure 1E). The α angle shifted towards higher values (mean α: 27.0° ± 3.4° for H1.0; 25.7° ± 1.7° for H1.5ΔC50), while that for β shifted to lower values (mean β: −0.3° ± 8.7° for H1.0; 3.8° ± 3.3° for H1.5ΔC50), indicating stabilization of the most convergent linker DNA conformations. Thus, H1.0 and H1.5ΔC50 shift the conformational landscape of the nucleosome to a more compact, rigid state.
H1 confers polarity to the nucleosome
We further analysed the cryo-EM reconstructions obtained for compact H1.0- and H1.5ΔC50-bound nucleosomes (conformations C and Y in Figure 1D and shown in Figure 2A after high-pass filtering or B-factor sharpening, respectively). To localize H1, we compared the cryo-EM density with that calculated from the fitted atomic structures of the NCP and linker DNA. Difference maps revealed additional density on the NCP dyad between the two linker arms attributable to the H1 globular domains (GH1.0 and GH1.5; collectively referred to as GH1; Figure 2B). The local map resolution for these domains (18 Å and 8 Å for GH1.0 and GH1.5, respectively; Figure 2C) is lower than the overall resolution, suggesting minor variability in the GH1 domain orientation. Fitting the GH5-bound 167-bp nucleosome structure (Zhou et al., 2015) into our cryo-EM maps showed a strong overlap between the GH5 domain and the GH1 densities (Figures S1G and S1H), indicating an on-dyad binding mode for the GH1 domain (confirmed below). Interestingly, the GH1 domain density is unevenly centered (“lopsided”) relative to the nucleosome dyad and appears more intimately associated with one linker than with the other. Indeed, for the more open conformational classes, the GH1-distal linker appears completely detached from the GH1 domain density (Figure 2E), revealing at least one of the two GH1-linker interfaces to be unstable.
Figure 2. Localization of H1 on the nucleosome.
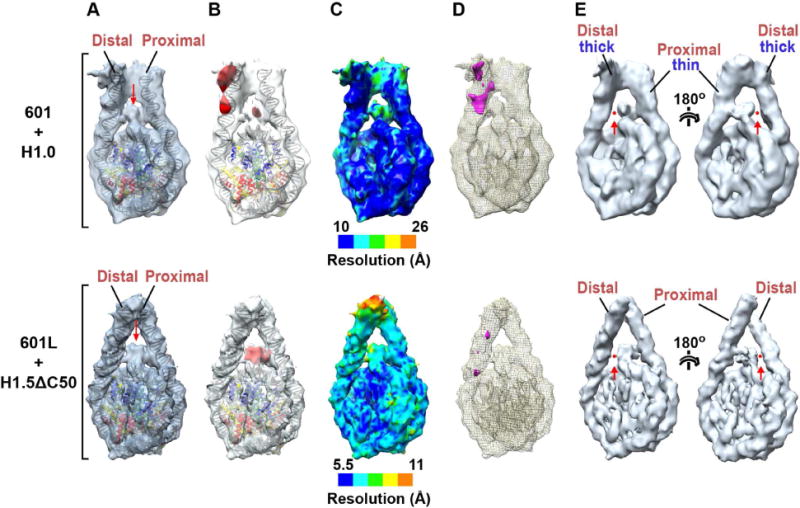
(A) Atomic models of the NCP and linker DNA fitted into 3D reconstructions of the H1.0-bound 601 nucleosome (top) and H1.5ΔC50-bound 601L nucleosome (bottom). (Structures are of conformations C and Y in Fig. 1D). To highlight H1-occupied density, the H1.0-bound 601 nucleosome map was bandpass filtered to keep spatial frequencies between 10 and 40 Å while that of the H1.5ΔC50-bound 601L nucleosome was sharpened by applying a negative B-factor. The red arrow indicates density attributed to the GH1 domain. (B) Density difference maps (red) calculated between the cryo-EM reconstruction and fitted atomic structures of the NCP and linker DNA. (C) Local resolution maps. (D) Difference map between the two linker arms. The proximal linker density was excised and aligned with the distal linker density. Alignment was performed at a high density threshold to favor the contribution of DNA in linker alignment. A difference map between the aligned linker arms is shown in magenta (threshold: 3 sigma). (E) Views of the H1.0-bound 601 nucleosome (top; bandpass filtered between 10 and 40 Å) and H1.5ΔC50-bound 601L nucleosome (bottom; with B-factor sharpened). Maps are displayed at a higher threshold than in panels A–D. The red arrow and dot show the loss of contact between the GH1 domain density and one of the linker arms (the thicker distal arm in the case of the 601/H1.0 complex).
Strikingly, the difference map calculated for H1.0-bound 601 nucleosomes revealed a second region of additional density on the distal linker (Figure 2B, top panel), consistent with the thicker appearance of this linker in the original map. The increased thickness is apparently not due to greater linker flexibility, which would have been detected by 3D classification or local resolution measurements (Figure 2C, top). Indeed, aligning the two linker arm densities and calculating a difference map between them revealed strong positive density indicative of additional mass on the distal linker (Figure 2D, top). We attribute the extra density to the H1 CTD. This hypothesis is confirmed by the absence of such density in H1.5ΔC50-bound nucleosomes, consistent with the loss of 50 C-terminal residues in this mutant (Figures 2B and 2D, bottom panels). This localization of the CTD strongly differentiates the two linker arms, breaking the two-fold symmetry of the nucleosome. Moreover, the highly basic CTD would largely neutralize the negative charge on the CTD-bound linker. Thus, the binding of H1 transforms the nucleosome from a two-fold symmetric particle to one that is strongly polarized both in mass and electrostatic charge distribution.
The GH1 domain displays an on-dyad mode of nucleosome recognition
We further investigated the H1-bound nucleosome by X-ray crystallography. Crystals diffracting at 5.5 Å resolution (Table 1) were obtained with a 197-bp palindromic (601L) nucleosome bound to histone H1.0. The structure contains one and a half H1-bound nucleosomes in the asymmetric unit (Figures S2A–C) and was solved by molecular replacement using the NCP as a search model. This allowed us to trace the linker DNA and to position the GH1 domain (a homology model based on the chicken GH5 structure) within density (Figure S2D and Movies S1 and S2). Density for the H1 N- and C-terminal domains was poor and therefore not interpreted. The GH1.0 domain localizes to the dyad axis and interacts with nucleosomal core DNA and with both linkers, exhibiting an on-dyad binding mode similar to that reported for chicken GH5 (Zhou et al., 2015) (Figure 3A), consistent with the 80% sequence identity between these two linker histone globular domains. Our crystal structure also agrees with the cryo-EM reconstruction of the H1.5ΔC50-bound nucleosome (Figure 3B and Movie S3), confirming that the H1.0 and H1.5 globular domains adopt the same binding mode.
Table 1.
Crystallographic data collection and refinement statistics
| Data Collection1 | |
|---|---|
| Synchrot r on beamline | ESRF ID – 29 |
| Wavelength (Å) | 0.99987 |
| Space group | C222 1 |
| Unit cell dimensions | a= 61.7 Å, b= 405.7 Å, c= 348.2 Å |
| Resolution range (Å) | 49.1 – 5.50 |
| (outer shell) | (5.59 – 5.50) |
| Number of measured reflections | 66,144 (3,419) |
| Number of unique reflections | 14,807 (748) |
| Multiplicity | 4.5 (4.6) |
| Completeness (%) | 99.3 (99.5) |
| Mean I/sigma(I) | 9.3 (2.0) |
| Rmerge | 0.098 (0.778) |
| Rmeas | 0.113 (0.887) |
| Rpim | 0.053 (0.417) |
| CC 1/2 | 0.995 (0.671) |
|
| |
| Refinement | |
| Resolution used for refinement | 49.1 – 5.5 |
| Reflections used (total/Rfree) | 13,308/1,479 |
| Rwork/Rfree | 0.2370/0.2625 |
| Number of atoms/Mean B – factor | |
| All | 21,493/308.0 |
| Core Histones | 9,065/240.7 |
| GH1 domain | 559/399.8 |
| Core DNA | 9,040/335.0 |
| Linker DNA | 2829/419.5 |
| RMS deviations: | |
| Bond distances (Å) | 0.008 |
| Bond angles (°) | 1.33 |
| Ramachandran analysis (%) | |
| Favored/outliers | 95.2/0.3 |
| Molprobity analysis | |
| Clash Score/Overall score | 5.43/1.94 |
Values in parenthesis are for the highest-resolution shell.
Figure 3. Orientation of the GH1 domain.
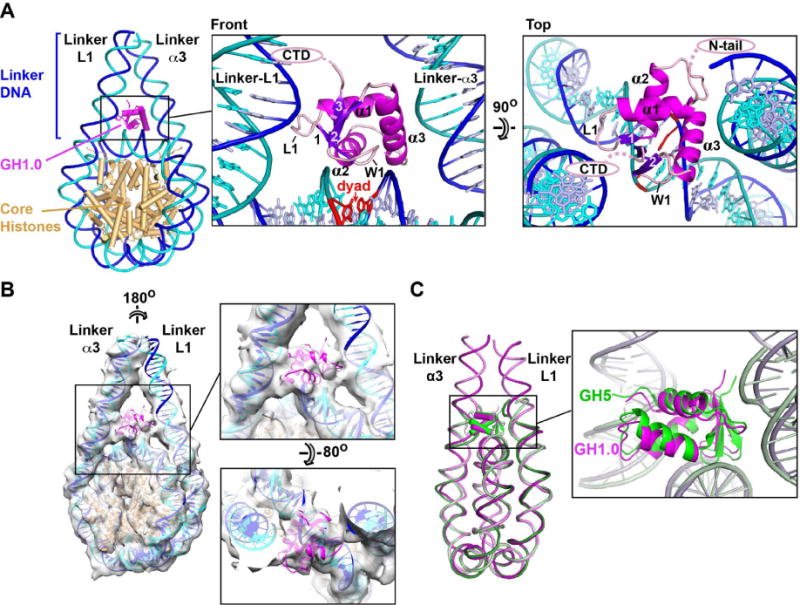
(A) Crystal structure showing the GH1.0 domain orientation. The winged-helix fold of GH1 includes a helix-turn-helix (HTH) motif formed by helices α2 and α3 and a “wing” (W1) defined by the β2-β3 loop. The base pair on the dyad axis is in red. (B) H1.0-bound 601L nucleosome crystal structure fitted into the cryo-EM map of the H1.5ΔC50-bound 601L nucleosome. (C) Alignment of the H1.0-bound nucleosome with that of chicken GH5 bound to a 167-bp nucleosome (PDB entry 4QLC)(Zhou et al., 2015). The GH1.0 and GH5 domains are related by a 10.5° rotation and by 0.5 Å shift in centre of mass. See also Figure S2 and Movies S1–S4.
The linker arms in our H1-bound crystal and cryo-EM structures are farther apart (by ≥5 Å measured half a DNA helical turn from NCP exit) than in the GH5-bound nucleosome structure and the GH1.0 domain is slightly rotated relative to GH5 (Figure 3C). The more open linker conformation results in a considerably reduced GH1-linker DNA contact surface (620 Å2 of buried surface area for GH1.0 versus 1320 Å2 for GH5), rationalizing the disrupted GH1-linker interface observed by cryo-EM (Figure 2E). The centre-of-mass of the GH1.0 domain lies midway between the dyad and linker-α3 [linkers are named as in (Zhou et al., 2015)], consistent with the lopsided density observed in the cryo-EM maps (Figure 2A). The C-terminal residue of the GH1.0 domain is next to linker-L1 (Figure 3A, front view), suggesting that the H1 CTD associates with this linker. This attribution is confirmed by fitting our crystal structure into the cryo-EM map of the H1.0-bound nucleosome, which identifies the thicker DNA arm as linker-L1 (Movie S4). Interestingly, the GH1 domain positions its N-terminal residue next to linker-α3 (Figure 3A, top view), suggesting that the H1 N-terminal tail may preferentially bind this linker.
The GH1 domain recognizes the nucleosome mainly through core DNA
As in the GH5-bound nucleosome (Zhou et al., 2015), the GH1.0 domain positions its DNA-proximal residues on the minor groove side of the phosphate backbone, with helix α2 and the W1 “wing” next to the core DNA, helix α3 next to one linker, and loop L1 next to the other (Figures 4A and 4B). Helices α1 and α2 point their N-termini towards the linker-α3 and core DNA, respectively, stabilizing these GH1-DNA interfaces via the positive charge of the helix dipole (Figures 3A and 4B), reminiscent of the effect observed for helices in the core histones (Luger et al., 1997). Nucleotides within contact distance of the GH1.0 domain are disposed nearly symmetrically about the dyad, and include seven nucleotides within the core DNA and three on each linker (Figure 4B, red circles). This contrasts with the GH5-nucleosome complex, where the more “closed” linker arms allow GH5 to contact a total of twelve linker nucleotides (Zhou et al., 2015). Indeed, of the total surface area (1460 Å2) buried between the GH1.0 domain and the nucleosome, over half (58%) is in the interface with the core DNA, compared to only 24% and 18% for the α3 and L1 linkers, respectively, revealing the core DNA to form the primary binding surface recognized by the GH1.0 domain. Consistent with this observation, plotting the sequence conservation derived from an alignment of H1/H5 orthologs (Figure S3) onto the GH1.0 domain surface shows that the best-conserved residues localize next to the core DNA, whereas residues next to the linkers are more variable (Figure 4C). Also consistent is a study in which the residence time of H1 on chromatin was measured in vivo using FRAP (Brown et al., 2006). H1 point mutations that reduced residence time localize to DNA-proximal residues, whereas those that had little effect map to DNA-distal residues (Figure 4D). Strikingly, the four mutations with the strongest effect (R47, K69, K73, K85) involve residues located next to the nucleosomal core close to the dyad, whereas mutation of residues adjacent to linker DNA had milder effects. This indicates that, in vivo, GH1-linker interactions are weaker than those with the core DNA, consistent with the sizes of the corresponding interfaces in our crystal structure.
Figure 4. Nucleosome recognition by GH1.0.
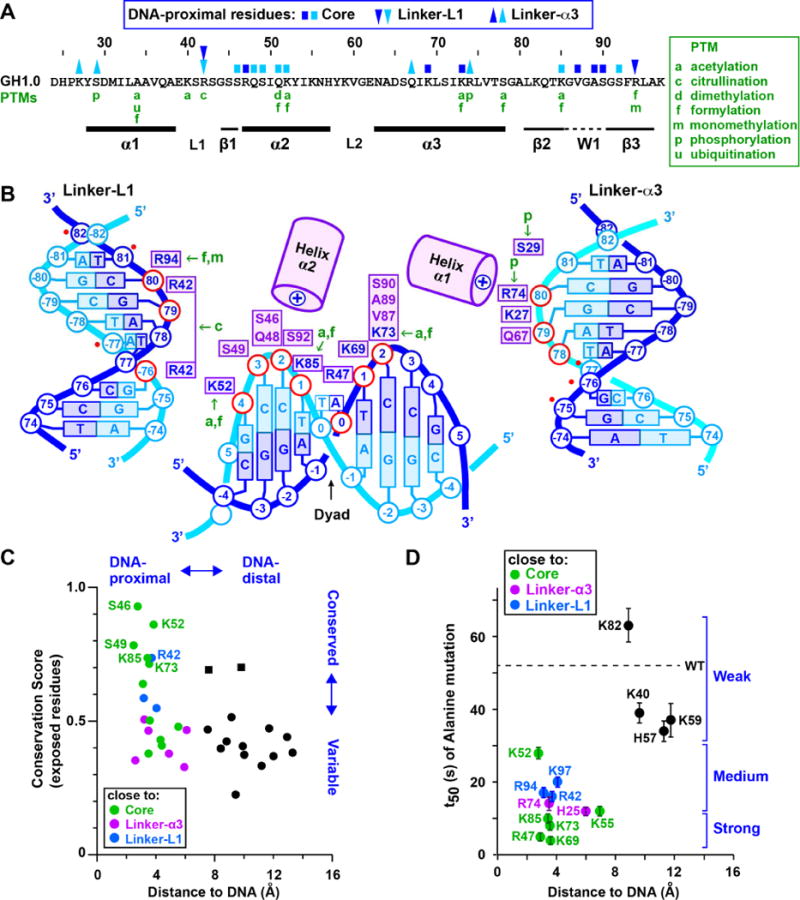
(A) Primary structure of the X. laevis GH1.0 domain. Residues close to core or linker DNA are marked by blue (sense) or cyan (anti-sense) squares and triangles, respectively, coloured as in panel B. Post-translational modifications (PTMs) in mammalian histones H1.1-H1.5 (Christophorou et al., 2014; Wisniewski et al., 2007; Wisniewski et al., 2008) are in green. (B) Summary of DNA-proximal residues. GH1.0 residues are shown next to the DNA phosphate group (in red) to which they are most proximal. Residues shown are within ~4 Å of the DNA, except for Ser29 which is ~5 Å away. Basic residues are in blue, other residues in violet. The six additional linker nucleotide positions contacted by the GH5 domain in the structure of (Zhou et al., 2015) are indicated by a red dot. (C) Plot of sequence conservation versus distance from DNA for surface-exposed residues in the GH1.0 domain. For each residue, the distance from each stereochemically allowed rotamer to the closest DNA phosphate atom was measured and the shortest distance was plotted. Residues close to the core DNA or to the α3 and L1 linkers are shown in green, magenta, and blue respectively. DNA-distal residues are in black. The best-conserved residues localize close to nucleosomal DNA, while most DNA-distal residues are poorly conserved. Exceptions (conserved and DNA-distal; black squares) are Lys40, consistent with an alanine substitution of Lys40 having little effect on stability of the H1-nucleosome complex (Brown et al., 2006) (see panel D) and Ser41, which corresponds to an acidic residue in most H1 orthologs (Figure S3). (D) Effect of alanine mutations on half-time of FRAP recovery (t50) plotted versus distance from DNA. FRAP data (mean ± S.D.) are those of (Brown et al., 2006). Brackets indicate mutations with a strong, medium or weak effect on t50. See also Figure S3.
H1 adopts an on-dyad binding mode in solution
To validate our crystal structure, we performed site-specific cross-linking and DNA footprinting experiments to confirm specific H1-nucleosome interactions. (Histone H1.0 was used for all experiments unless otherwise specified). GH1 domain residues Arg42 and Ser66 (located next to linkers-L1 and −α3, respectively) were mutated to cysteine (absent from wildtype H1.0) and reacted with 4-azido phenacylbromide (APB), which forms a specific covalent adduct with the cysteine thiol group. Both H1 mutants retain the ability to bind nucleosomes efficiently (e.g., Figure 5A, top). Radiolabeled nucleosomes incubated with APB-derivatized H1 were exposed to UV radiation, which causes the APB nitrene group to react with nearby nucleotides, generating an H1-DNA cross-link (Figures 5A, bottom and 5B). A base elimination reaction and sequencing gel analysis revealed that Cys42 formed cross-links with the half-turn of linker DNA preceding NCP entry (nucleotide positions −80 and −77; Figure 5C, orange arrowheads), while Cys66 formed cross-links with the same linker (positions −77 to −74) and with the opposite linker (at approximate positions +75 and +80) (Figure 5C, magenta arrowheads). The results are consistent with the GH1 domain adopting two dyad-related orientations corresponding to our crystal structure (Figure 5D).
Figure 5. Mapping of H1-nucleosomal DNA interactions.
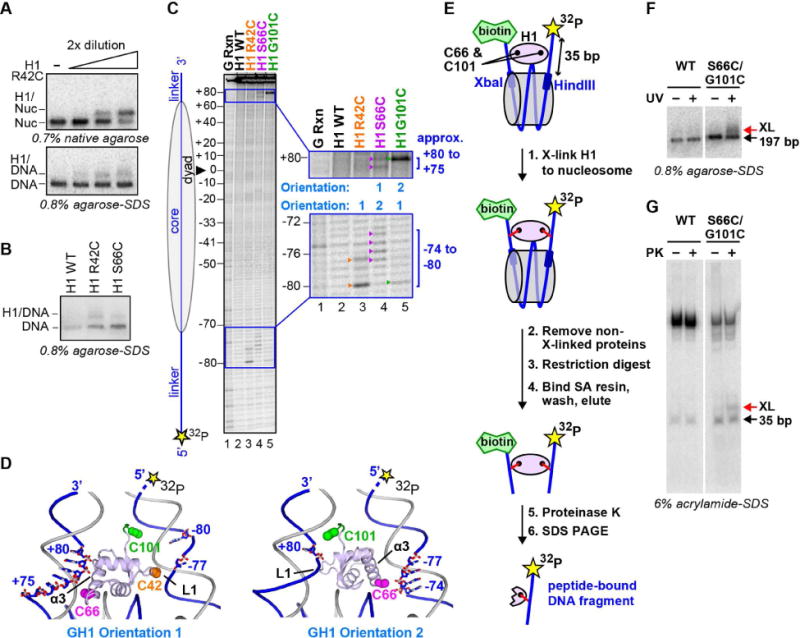
(A to D) Site-specific cross-linking of GH1 to nucleosomal DNA. (A) Top. Native gel showing the binding of APB-derivatized H1 mutant R42C (R42C-APB) to the nucleosome. Bottom. Denaturing gel showing cross-linking of H1 R42C-APB to nucleosomal DNA after UV irradiation. (B) Denaturing gel showing cross-linking of H1 R42C-APB and H1 S66C-APB to nucleosomal DNA upon UV irradiation. (C) Mapping of cross-linked nucleotides by piperidine base elimination cleavage of the DNA and subsequent sequencing gel analysis. Nucleotides cross-linked to R42C-APB, S66C-APB and G101C-APB are indicated by orange, magenta and green arrowheads, respectively. (D) Crystal structure (orientation 1) and dyad-related orientation of GH1 (orientation 2) showing the proximity of GH1 residues to specific linker nucleotides on the radiolabeled strand. Residues 98–101 (green; absent from the crystal structure) were modeled in an extended conformation. (E to G) Simultaneous cross-linking of H1 residues to both DNA linkers. (E) Summary of the cross-linking experiment. Nucleosomes were reconstituted using 5′ biotinylated and 5′ radiolabeled 197-bp DNA containing a specific restriction endonuclease (Xba I and Hind III) site next to each linker arm. (F) APB-derivatized H1 S66C/G101C mutant binds and cross-links in a UV-dependent manner to the 197-bp nucleosome. (G) Elutions with or without proteinase K (PK) were analyzed on 6% acrylamide-SDS gel revealing a distinct band (XL) consistent with double cross-link dependent retention of the radiolabeled linker arm.
To verify that the GH1 domain contacts both linker arms in solution, we sought to cross-link this domain to both linkers simultaneously. Because attempts using the H1 double mutant R42C/S66C yielded inefficient double cross-link formation (due to the low efficiency of the individual reactions), we exploited an alternate double mutant, S66C/G101C. Residue Gly101 is located immediately C-terminal to the GH1 domain next to the same linker as Arg42 (Figure 5D). The corresponding Cys mutant yields a highly efficient cross-link (with nucleotide +80, approximately; Figure 5C). We reconstituted 197-bp nucleosomes containing a radiolabel on one linker and a biotin tag on the other, each flanked by core DNA bearing a specific restriction site (Figure 5E). We cross-linked the full-length H1 S66C/G101C mutant to the nucleosome (Figure 5F), cleaved the linkers from the core DNA, and affinity purified the biotinylated linker and cross-linked adducts. Proteinase K treatment of the eluted fraction followed by denaturing gel analysis revealed a specific radiolabeled band consistent with H1-mediated tethering of the two linkers (Figure 5G). This demonstrates that H1 residues 66 and 101 can simultaneously cross-link to opposite linkers, corroborating our crystal structure.
We next performed hydroxyl-radical footprinting to verify the position of the GH1 domain on the nucleosome core. Both full-length H1 and the isolated GH1 domain make a symmetric footprint on the core DNA, protecting the central base pair plus 3–4 flanking nucleotides on each strand (Figures 6A and 6B; compare lanes 1 and 2 at magenta asterisks). In addition, both H1 and the isolated GH1 domain protect nucleotides within the first turn of linker DNA (Figures 6A and 6B; red and black asterisks) and enhance the protection of core nucleotides in the DNA turn preceding the linkers (Figures 6A and 6B; green asterisks), indicating that H1 induces tighter DNA wrapping around the histone core octamer. These findings recapitulate the footprinting pattern observed for the binding of H1.5 to di- and tri-nucleosomes (Syed et al., 2010). We observed the identical protection pattern on dinucleosomes with H1 histones isolated from HeLa cells, as well as with the X. laevis oocyte histone B4, an isoform present in early embryonic chromatin which diverges significantly from H1.0 (26% sequence identity overall, 25% in the globular domain) (Figure S4A). In all cases the observed protection agrees well with the specific protein-DNA interfaces in our crystal structure and with the effect of H1 on linker conformation seen by cryo-EM (Figures 1D and 1E).
Fig. 6. DNA footprinting and cross-linking analysis of H1 binding to symmetric and asymmetric nucleosomes.
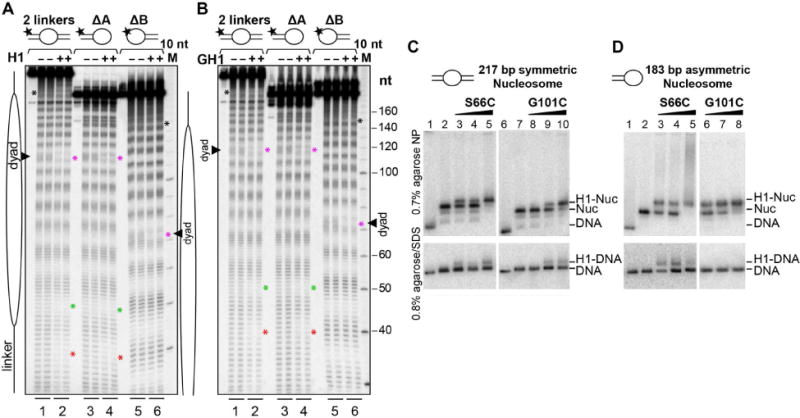
(A,B) Hydroxyl radical footprinting of centrally positioned nucleosomes bearing two linkers (lanes 1–2) compared to nucleosomes with only one linker (lanes 3–6). Reactions were performed in the absence (lanes 1, 3 and 5) or presence (lanes 2, 4 and 6) of (A) H1 or (B) the isolated GH1 domain. Cleavage patterns are shown in duplicate. Nucleotide regions protected by H1 or GH1 are indicated by asterisks as described in the text. (C,D) APB-derivatized H1 binding and cross-linking to (C) symmetric, 2-linker nucleosomes or (D) asymmetric, single-linker nucleosomes. Top. Native gels showing the binding of H1 S66C-APB and H1 G101C-APB to both (C) symmetric and (D) asymmetric nucleosomes. Bottom. Denaturing gels showing cross-linking of H1 S66C-APB and G101C-APB to nucleosomal DNA following UV irradiation. See also Figures S4 and S5.
While the above footprinting results are consistent with an on-dyad binding mode, they do not formally exclude off-dyad binding, since two dyad-related orientations of an asymmetrically positioned GH1 domain can combine to yield a symmetric footprint (see Figure S4B, experiment 1). To address this issue we prepared nucleosomes lacking either one or the other linker (designated Δlinker-A and Δlinker-B) and confirmed their ability to bind GH1 for linker lengths ranging from 10 to 25 bp (Figures 6B and S4C). In the off-dyad, single-linker binding scenario, the GH1 domain should bind mono-linker nucleosomes with a preferred orientation and therefore yield distinct patterns of nucleotide protection on the nucleosome core for Δlinker-A, Δlinker-B and the two-linker nucleosome (Figure S4B, left panels). In fact, incubating either H1 or the isolated GH1 domain with mono-linker nucleosomes yields a footprint on the dyad resembling that observed with two-linker nucleosomes (Figures 6A, 6B and S4D, magenta asterisks), consistent with the on-dyad binding mode observed in our crystal structure (Figure S4B, right panels). These data strongly argue against H1 adopting an off-dyad binding mode in solution.
Our crystal and cryo-EM structures show the GH1 domain to be more closely associated with linker-α3 than with linker-L1. Accordingly, H1 should associate with a mono-linker nucleosome by preferentially orienting the linker-α3 binding surface of the GH1 domain towards the single linker. To verify this, we assessed the ability of H1 point mutants S66C and G101C to be covalently cross-linked with mono-linker nucleosomes. Strikingly, whereas both mutants formed cross-links to symmetric two-linker nucleosomes (Figure 6C, bottom), only S66C was efficiently cross-linked to the mono-linker nucleosome (Figure 6D, bottom), confirming the greater stability of the GH1/linker-α3 interface. This is consistent with previous observations that mutations on the linker-α3 binding surface of GH5 more significantly reduced nucleosome binding affinity than those on the linker-L1 binding surface (Zhou et al., 2015).
As further validation of our crystal structure, we performed molecular docking analyses to identify the most probable GH1.0 domain orientation in solution compatible with the above biochemical data. We docked the GH1.0 domain to the 197-bp nucleosome by two approaches. In a data-driven approach using the program HADDOCK (de Vries et al., 2010), we used the above cross-linking and footprinting results as interaction restraints to guide the docking procedure (Table S1). In certain docking experiments we also included previous data reporting close proximity of specific residues (His25 and Lys85) to DNA (Buckle et al., 1992; Mirzabekov et al., 1990; Thomas and Wilson, 1986) as additional restraints. Using either the full set of restraints or various partial subsets, the best-scoring solutions consistently displayed an on-dyad binding orientation resembling the GH1.0 domain orientation in our crystal structure (Figures S5A and S5B and Table S1). In a separate, unbiased docking approach, we used the program Autodock Vina (Trott and Olson, 2010) to generate energetically favoured GH1.0-nucleosome configurations which were subsequently screened for consistency with the biochemical data. This approach identified a single solution similar to the configuration observed in our crystal structure (Figure S5B). Thus, both molecular docking approaches support an on-dyad binding mode for H1 in solution.
DISCUSSION
In this study, we used structural and biochemical techniques to investigate an intact 197 bp nucleosome containing full-length histone H1. Our cryo-EM analysis shows that H1 binding induces the nucleosome to adopt a more compact conformation with reduced linker arm flexibility. This is significant because a more homogeneous nucleosome conformation would likely facilitate assembly into a regular helical structure and promote condensed fibre formation. The binding of full-length and C-terminally truncated H1 constructs yielded similar effects on linker conformation and dynamics, suggesting that much of the CTD is dispensible for inducing a more compact and rigid nucleosome structure. The linker arms in our H1-bound crystal and cryo-EM structures are farther apart than in that of the chicken GH5-bound nucleosome (Zhou et al., 2015), resulting in relatively small GH1-linker interfaces. Our cryo-EM data show that dynamic flexibility of the linker DNA can lead to increased linker separation and to the disruption of one of the GH1-linker interfaces, yielding a “two-contact” binding mode in which only a single linker and the core DNA interact with the GH1 domain (Figure 2E). This interdependence between linker separation and the size of the H1-nucleosome interaction surface suggests how factors affecting the exit/entry angle of linker DNA could modulate the stability of H1 binding. For example, the binding of linker histones is abrogated by the defective docking domain in the H2A histone variant H2A. Bbd, which causes the unwrapping of ∼10–15 bp at each end of the NCP (Shukla et al., 2011). Dynamic linker flexibility may also contribute to the ability of transcription factors to compete with H1 to bind cognate sites located within the linker DNA (Lone et al., 2013).
A striking result of our study is the observation that the H1 CTD associates primarily with a single linker. [We cannot exclude that part of the CTD associates at least transiently with the opposite linker, such as the C-terminal seven residues previously implicated in linker stem formation (Syed et al., 2010)]. The observed localization is consistent with a recent study which reported that deletion of the CTD yielded a similar drop in affinity for a nucleosome bearing two linkers as for a nucleosome bearing only one (White et al., 2016). The observed CTD localization confers a notable asymmetry to the nucleosome, both in mass distribution and electrostatic character, as the highly basic CTD would neutralize the negative charge of the associated DNA linker. The lopsided positioning of the GH1 domain relative to the dyad and to the two linkers also contributes to the particle’s asymmetry. Such asymmetry in H1-bound nucleosomes is likely to have significant consequences for the formation of higher-order chromatin structures. For example, in a nucleosomal array with a two-start helical configuration, H1 proteins bound to adjacent nucleosomes with the same (head-to-tail) polarity would yield a different spatial arrangement of CTD-bound linkers than proteins bound with opposite (head-to-head) polarity, resulting in distinct mass and electrostatic charge distributions (Figure 7A). These two configurations are characterized by different repeating structural units (dinucleosome versus tetranucleosome) and could conceivably stabilize different higher-order chromatin conformations.
Figure 7. Implications for higher-order chromatin structures.
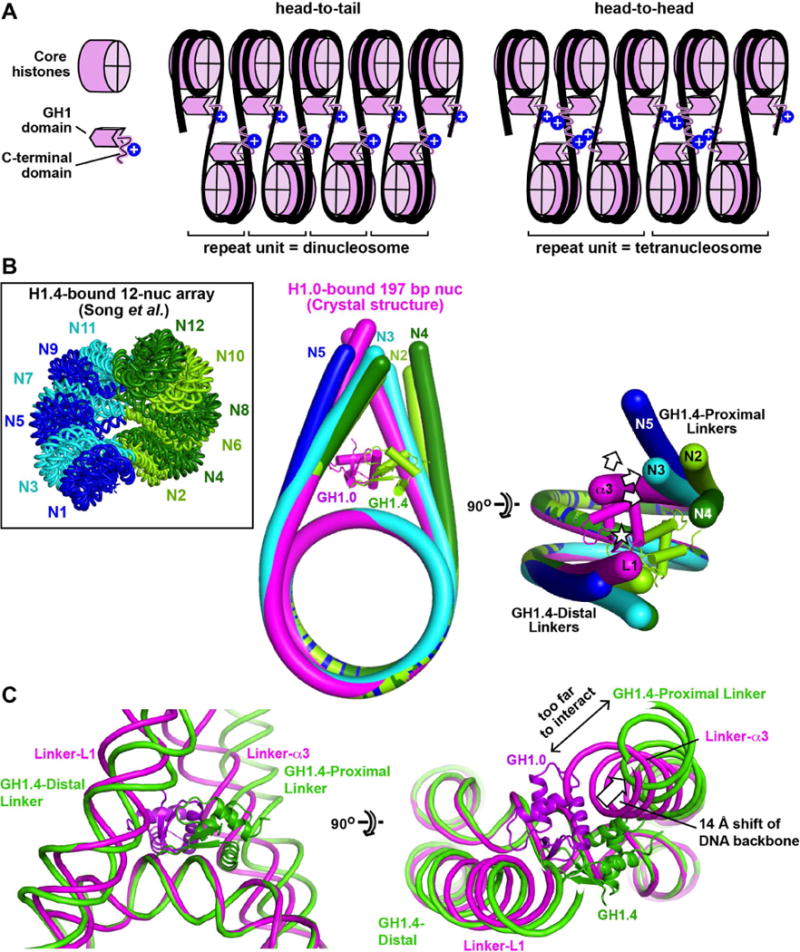
(A) The asymmetric localization of the CTD may influence the assembly and properties of higher-order structures. Two hypothetical arrangements shown for H1-bound nucleosomes within a two-start helical array give rise to distinct mass and electrostatic charge distributions. (B) Comparison of linker arm geometry with that observed in the condensed 12-nucleosome array of (Song et al., 2014). Nucleosomes N2-N5 of the 12-nucleosome array were aligned onto the H1.0-bound 601L nucleosome crystal structure (complex A) by superimposing the nucleosomal cores. The DNA from the crystal structure is in magenta, while that for N2-N5 are in lime, cyan, dark green and blue, respectively. (Only 4 nucleosomes of the array are shown because the 3 tetranucleosomal units have similar conformations. N5 is shown instead of N1 because the latter lacks the first linker arm). The GH1.0 domain from the crystal structure and the GH1.4 domain bound to N2 are also shown. The asterisk indicates the pseudodyad axis. The arrows show the displacement of GH1.4-proximal linkers relative to Linker-α3 of our crystal structure. The mean displacement of the DNA backbone measured one helical turn from NCP exit is 14.5 ± 6.3 Å between the GH1.4-proximal linkers and Linker-α3, and 4.0 ± 1.6 Å between the GH1.4-distal linkers and Linker-L1. (C) Comparison of linker arm geometry between the H1.0-bound crystal structure and nucleosome N2 of the 12-nucleosome array. See also Figure S6.
Our crystal structure reveals the GH1 domain to interact with both linkers and with the nucleosome dyad, similar to the on-dyad configuration reported for chicken GH5 (Zhou et al., 2015). Cryo-EM, crosslinking, and footprinting analyses confirm that this binding mode also occurs in solution. We observe the same on-dyad binding mode for the globular domains of both Xenopus H1.0 and human H1.5. These two domains share 47% sequence identity and differ at numerous (23 out of 35) solvent-exposed residues (Figures S6A and S6B), indicating that even considerably divergent H1 isoforms can adopt the same binding configuration. By contrast, the Drosophila GH1 domain (43–46% identical to chicken GH5, Xenopus GH1.0 and human GH1.5; Figure S6A) has been observed to bind off the dyad (Zhou et al., 2013). Moreover, a chicken GH5 mutant could be engineered to adopt an off-dyad binding mode by replacing five surface-exposed residues with the corresponding Drosophila GH1 residue, confirming that GH1 sequence variation can modulate binding mode (Zhou et al., 2016). Interestingly, human GH1.5 matches Drosophila GH1 at two of these mutated positions (Figure S6B), raising the possibility that the H1.5 on-dyad configuration may be less stable than that of the H1.0 isoform.
On a related note, the on-dyad binding configuration observed for the globular domains of chicken H5 (Zhou et al., 2015), Xenopus H1.0 and human H1.5 (this work) bound to a mononucleosome differs markedly from the off-dyad binding reported for the human H1.4 globular domain in condensed 12-nucleosome arrays (Song et al., 2014) (Figures S6C and S6D). This is striking because H1.4 and H1.5 are close paralogs (95% sequence identity in the GH1 domain) and the few divergent residues are unlikely to account for the different binding configurations (Figures S6A and S6B). Conceivably, the discrepancy may be due to differences in sample preparation (e.g., the use of a cross-linking reagent in the study by Song et al.). Alternatively, the different binding modes might reflect different local stereochemical constraints. Indeed, aligning the individual nucleosomes of the 12-nucleosome array with our H1-bound crystal or cryo-EM structures reveals substantial differences in linker arm conformation: whereas the linkers in our structures are essentially symmetrical relative to the nucleosome dyad, those of the condensed array show much greater asymmetry (Figure 7B). This is due to the twisted fibre geometry of the array, which requires the two linkers of each nucleosome to follow non-superimposable trajectories as they connect to the preceding and subsequent nucleosome. Consequently, whereas the GH1.4-distal linkers of the array superimpose reasonably well with either of the two linkers in our crystal structure, the GH1.4-proximal linkers do not (see Figure 7B legend). The latter linkers are displaced away from the pseudodyad axis, too far to interact with a GH1 domain bound on the dyad (Figure 7C). Thus, the DNA conformation in the condensed array would significantly destabilize the on-dyad configuration, as a GH1 domain adopting such a binding mode could at best interact with only one linker, not two. Indeed, the observed GH1.4 domain in the array adopts a completely different orientation (rotated by 85°) and is substantially shifted (by ~20 Å) relative to the on-dyad GH1 orientation, presumably so as to optimize interactions with linker and core DNA. Because our crystal and cryo-EM structures likely represent nucleosomes in the uncondensed state, the above findings suggest that (at least for histones H1.4 and H1.5) chromatin condensation is associated with a switch from on-dyad to off-dyad binding. More generally, these findings suggest that the linker conformation may vary with GH1 binding mode, and hence that different GH1 binding configurations might be associated with distinct higher-order chromatin structures.
A number of post-translational modifications (PTMs) have been reported for the globular domain of mammalian somatic linker histones (Christophorou et al., 2014; Wisniewski et al., 2007; Wisniewski et al., 2008). Many of these occur on DNA-proximal residues and are predicted to destabilize the H1-nucleosome complex (Figures 4A and 4B). For example, phosphorylation has been observed on Ser29 (H1.0 numbering) in histones H1.1-H1.4 from multiple murine tissues, and on a serine corresponding to H1.0 residue Arg74 in mouse kidney histones H1.2-H1.4 (Wisniewski et al., 2007). Phosphorylation at these sites would cause electrostatic repulsion with the linker-α3 phosphate backbone. Citrullination of Arg42 (H1.0 numbering) in histones H1.2-H1.5 in mouse pluripotent stem cells has been linked to chromatin decondensation and to the enhanced expression of genes involved in stem cell development and maintenance (Christophorou et al., 2014). The loss of positive charge induced by citrullination would weaken the interaction of Arg42 with linker-L1 and promote H1 dissociation. The interaction between GH1 and linker-L1 would similarly be destabilized by the formylation of Lys106 in histone H1.2 (corresponding to Arg94 in H1.0) observed in murine seminal vesicles (Wisniewski et al., 2007). Likewise, the acetylation or formylation of three lysines (K52, K73 and K85 in H1.0) located next to nucleosomal core DNA in histones H1.1-H1.4 in human cell lines and in H1.2-H1.5 in various murine tissues (Wisniewski et al., 2007; Wisniewski et al., 2008) would favour the eviction of H1 from the nucleosome. Our structural data thus provide insights into how the post-translational regulation of histone H1 may affect chromatin structure.
In conclusion, our structural and biochemical results paint a coherent picture of how histone H1 interacts with a ~200-bp nucleosome. These results advance our understanding of nucleosome recognition by linker histones and will inform future efforts to elucidate the mechanism of chromatin condensation and the architecture of higher-order chromatin structures.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stefan Dimitrov (stefan.dimitrov@univ-grenoble-alpes.fr).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Histones and NAP-1 were expressed using BL21(DE3) competent Escherichia coli (Thermo Fisher Scientific and NEB) as previously described (Syed et al., 2010; Caterino et al., 2011; Tachiwana et al., 2011).
METHOD DETAILS
Human core histone purification
Human histones H2A, H2B and H3 were expressed from a pHCE vector in E. coli BL21(DE3) cells. Human histone H4 was expressed from a pET15b vector in E. coli JM109(DE3) cells. Core histones were expressed as N-terminal His-tagged proteins in the absence of T7 RNA polymerase by omitting the addition of isopropyl-P-D-thiogalactopyranoside (IPTG), which induces T7 RNA polymerase production in BL21(DE3) and JM109(DE3) cells. For each histone, 200 ng of plasmid were used to transform the relevant E. coli strain. 10 colonies were inoculated into 2 L LB medium (containing 50 μg/ml ampicillin) in a 5 L flask and incubated overnight at 37°C with shaking at 200 rpm. Each liter of bacteria was pelleted at 5000 g for 20 min at 4°C. Cells were collected and disrupted by sonication in 50 ml buffer A [50 mM Tris-HC1 pH 8.0, 500 mM NaC1, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 5% glycerol]. After centrifugation (27,000 g for 20 min at 4°C), the pellet containing His-tagged histones as insoluble forms was resuspended in 50 ml buffer A containing 7 M guanidine hydrochloride. After centrifugation (27,000 g for 20 min at 4°C), the supernatants containing His-tagged histones were incubated with NiNTA resin (Complete His-Tag purification Resin, Roche) (1mL of resin per 1 L of bacterial culture) for 1 hr at 4 °C. The resin was packed into an Econo-column (Bio-Rad), and then washed with 100 ml buffer B (50 mM Tris-HC1 pH 8.0, 500 mM NaC1, 6 M urea, 5 mM imidazole, and 5% glycerol). His-tagged histones were eluted by a 100 ml linear gradient of imidazole from 5 to 500 mM in buffer B, and the samples were dialyzed against buffer C (5 mM Tris-HC1 pH 7.5 and 2 mM β-mercaptoethanol). The N-terminal His tag was removed by thrombin protease (GE Healthcare) treatment using 1 unit per mg of protein for 3–5 h at 4°C. The untagged histone was then subjected to Resource S cation exchange column chromatography (GE Healthcare). The column was washed with buffer D (20 mM sodium acetate pH 5.2, 200 mM NaC1, 5 mM β-mercaptoethanol, 1 mM EDTA, and 6 M urea), and each histone was eluted by a linear gradient of NaCl from 200 to 900 mM in buffer D. Fractions containing the pure histone were pooled and stored at −80°C.
Preparation of histone tetramers and dimers
To prepare histone tetramers and dimers, human H3 & H4 and human H2A & H2B were mixed in an equimolar ratio and dialyzed overnight against HFB buffer (2 M NaCl, 10 mM Tris pH7.4, 1 mM EDTA pH 8 and 10 mM β-mercaptoethanol). After dialysis the supernatant containing folded tetramers and dimers was subjected to Superose 6 prep grade XK 16/70 size exclusion column (GE Healthcare) chromatography using HFB buffer. The major fractions containing purified tetramers and dimers were mixed together. For long-term storage, tetramers and dimers were mixed with NaCl-saturated glycerol to achieve a final glycerol concentration of 15–20% and stored at −20°C.
NAP-1 purification
N-terminally His-tagged mouse NAP-1 was expressed from a pET15b vector in E. coli BL21(DE3) cells, as previously described (Syed et al., 2010). Briefly, transformed cells were grown in LB medium containing ampicillin (50 μg/mL) and chloramphenicol (25 μg/ml) at 37°C until reaching an OD600 of 0.5–0.6, induced with 0.2 mM IPTG and further incubated at 37°C for 3–4 h. Cells were pelleted at 5000 g for 20 min at 4°C. NAP-1 was purified from the supernatant of the bacterial lysate using a Ni-NTA resin (Complete His-Tag purification Resin, Roche), followed by Resource Q anion exchange column chromatography (GE Healthcare).
Purification of Linker histones
Untagged linker histones X. laevis H1.0b, human H1.5 C50 (residues 1–177) and human GH1.5 (residues 40–112) were expressed from a pET15b vector in E. coli BL21(DE3) cells, as described above for NAP-1. These linker histones were purified from the supernatant of the bacterial lysate using a Bio-Rex 70 resin 50–100 mesh (Bio-rad) followed by Resource S cation exchange column chromatography (GE Healthcare). N-terminally GST-tagged X. laevis histone B4 was expressed from a pGEX6P-3 vector in E. coli BL21(DE3) cells. Transformed cells were grown at 37°C until reaching an OD600 of 0.4–0.6, induced by the addition of 0.2 mM IPTG and further incubated at 16°C for 16 hours. Collected cells were resuspended in 40 ml of lysis buffer (1x PBS, 0.5 M NaCl, 0.2% Triton X-100, 1 mM EDTA, 2 mM dithiothreitol (DTT), 1 mM PMSF, and 25 g/ml lysozyme), incubated on ice for 30 min, lysed by sonication and centrifuged at 27,000 g for 20 min at 4°C. Supernatants containing GST-tagged histone B4 were incubated with 2 ml of Glutathione Sepharose 4 Fast Flow (GE Healthcare) for 1 h at 4 °C. Beads were packed into an Econo-column (Bio-Rad), washed with 100 ml of wash buffer (1x PBS, 0.25 M NaCl, 0.1% Triton X-100, and 1 mM DTT) followed by 10 ml of cleavage buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.0). GST-histone B4 bound beads were incubated with PreScission Protease at 4°C for 16 hours. After removal of the GST tag, cleaved proteins were eluted with cleavage buffer.
Preparation of 197 bp Widom 601 DNA
The 197-bp Widom 601 DNA was prepared and purified as described in (Syed et al., 2010). Briefly, multiple repeats of the 197-bp 601 sequence were inserted into the EcoRV site of a pGEMT Easy vector and expressed in E.Coli DH5α cells. The fragments were excised by digestion with EcoRV (New England Biolabs), followed by phenol chloroform extraction and ethanol precipitation. The excised 197 bp nucleosome positioning DNA fragments were separated from linearized plasmid by preparative electrophoresis on a 5% polyacrylamide gel using a Prep Cell (BioRad). The nucleotide sequence is as follows:
ATCGATGGACCCTATACGCGGCCGCCCTGGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCTGTGCATGTATTGAACAGCGACCTGAT
Preparation of 197 bp 601L DNA
The 197-bp palindromic 601L DNA was prepared and purified as described in (Chua et al., 2012). Briefly, multiple repeats of one half of a 601L palindromic DNA fragments were generated in a pGEMT easy vector. The half 601L fragments were excised from the vector by digestion with ScaI (New England Biolabs). The excised blunt end fragments were separated from the linearized plasmid by PEG precipitation using (0.154 ml 5M NaCl + 0.346 ml 40% PEG-6000)/1 ml of DNA solution. Purified DNA fragments were dephosphorylated by Calf Intestinal Alkaline Phosphatase (CIAP; Thermo Fisher Scientific) followed by phenol chloroform extraction and ethanol precipitation. Further blunt end DNA fragments were digested by HinfI (New England Biolabs) to create cohesive ends and purified by preparative electrophoresis on a 5% polyacrylamide gel using a Prep Cell (BioRad). To generate palindromic 197 bp DNA, purified DNA fragments were self-ligated using T4 DNA ligase (New England Biolabs) and further purified through TSK-DEAE ion exchange chromatography to separate them from unligated DNA fragments. The nucleotide sequence is as follows:
ACTACGTAATATTGGCCAGCTAGGATATCACAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGGAATCCGTACGTGCGTTTAAGCGGTGCTAGAGCTGTCTACGACCAATTGAGCGGCCTCGGCACCGGGATTGTGATATCCTAGCTGGCCAATATTACGTAGT
Preparation of radiolabeled Widom 601 sequence
Widom 601 sequences harboring different linker DNA lengths (total length of 240 bp for centrally positioned nucleosomes; 197 bp, 157 bp and 162 bp for single-linker nucleosomes) were subcloned in a pGEMT vector between the EcoRI and AflII sites and expressed in E.Coli DH5α cells. Inserts were excised from the vector using restriction enzymes EcoRI and AflII, purified through a 1% crystal violet agarose gel and klenow filled with α32P-dTTPs.
Nucleosome reconstitution
Centrally positioned nucleosomes were reconstituted by the salt-dialysis method, as previously described (Syed et al., 2009; Syed et al., 2010). Specifically, for hydroxyl radical footprinting experiments, approximately 500 ng of 32P-labeled Widom 601 DNA and 4.5 μg of unlabeled 601 DNA were mixed with human histone tetramers and dimers approximately in a 1:0.5:0.5 ratio in HFB buffer (2M NaCl, 10 mM Tris pH7.4, 1 mM EDTA pH 8 and 10 mM β-mercaptoethanol), respectively. For Cryo-EM and X-ray crystallography studies, approximately 100–500 μg of 197-bp 601 DNA or 197-bp 601L DNA were mixed with human histone tetramers and dimers approximately in a 1:0.5:0.5 ratio in HFB buffer. The mixtures were transferred to dialysis tubing and the reconstitution was performed by dialysis against a slowly decreasing salt buffer: the NaCl concentration starts at 2 M and decreases slowly to 500 mM NaCl. Using a peristaltic pump, low salt buffer is added to the high salt buffer beaker at a rate of 1.5 ml/min for 18 h. Once finished, the dialysis bags were transferred to 300 mM NaCl, 10 mM Tris pH7.4, 1 mM EDTA pH 8 and 10 mM β-mercaptoethanol for 2 h, followed by a final dialysis overnight in 10 mM NaCl, 10 mM Tris pH7.4, 0.25 mM EDTA pH 8 without β-mercaptoethanol.
NAP-1 mediated H1 deposition
NAP-1-mediated H1 deposition was performed as in (Syed et al., 2010). Briefly, the H1 linker histone [Xenopus laevis H1.0b, human H1.5 C50 (residues 1–177) or GH1.5 (residues 40–112)] was mixed with histone chaperone NAP-1 in a 1:2 molar ratio and incubated at 30°C for 15 min in 20 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 100 mM NaCl, 1 mM DTT, 10% glycerol, 0.1 mM PMSF. After incubation an equimolar amount of nucleosomes and linker histone/NAP-1 complexes were mixed together and further incubated at 30°C for 30 min. His-tagged NAP-1 was removed by using a Ni-affinity column and nucleosomes were further purified by preparative electrophoresis using a Prep Cell (BioRad). The final samples were estimated to be >95% free of NAP-1.
Cryo-electron microscopy and image processing
Data collection
Reconstituted nucleosomes in a low ionic strength buffer (10 mM Tris, 1 mM EDTA, 10 mM NaCl) were diluted in the same buffer to a concentration of 150 μg/ml of DNA. Three microliters of the specimen were deposited on a holey carbon film (C-flat 2/2-2, EMS), rendered hydrophilic by a 20 s glow discharge in air, and flash frozen in liquid ethane using an automated plunger (Vitrobot2, FEI) with controlled blotting time (1 s), blotting force (5), humidity (100%) and temperature (4°C). Unbound (1412 frames) and H1.0-bound (555 frames) 601 nucleosomes were imaged using a cryo-transmission electron microscope (Polara, FEI) equipped with a field emission gun operating at 100 kV. Images were recorded under low-dose condition (total dose of 18 e−/Å2) on a direct electron detector (Falcon I, FEI) at a nominal magnification of 59,000 x, resulting in a pixel size on the specimen of 0.178 nm. 8911 images of H1.5ΔC50-bound 601L nucleosomes were recorded on a Titan Krios electron microscope (FEI) with a field emission gun operating at 300 kV, using a Falcon II direct electron detector (FEI) operating in dose-fractionation mode at a nominal magnification of 47,000 x, resulting in a pixel size on the specimen of 0.11 nm. Nine movie frames were recorded at a dose of 2.9 electrons per Å2 per frame corresponding to a total dose of 26.1 electrons per Å2 but only the 7 last frames were kept for further processing.
Image processing
Movie frames were aligned using MotionCorr (Li et al., 2013) and Xmipp Optical Flow (Abrishami et al., 2015) to correct for specimen charging and motion. To obtain initial references for automatic particle picking, approximately 2000 nucleosomal particles were selected manually using the Boxer application in the EMAN2 software package (Ludtke et al., 1999). The Contrast Transfer Function (CTF) of the microscope was determined for each micrograph using the CTFFIND3 program (Mindell and Grigorieff, 2003). The initial small dataset was subjected to reference-free classification in RELION (Scheres, 2012). Four class average images showing nucleosomes from different orientations were used as reference to automatically pick particles using Gautomatch (http://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch/) or RELION, yielding datasets of 94,264, 84,530 and 262,489 molecular images for unbound 601, H1.0-bound 601 and H1.5ΔC50-bound 601L nucleosomes, respectively. The images were subjected to reference-free 2D classification to remove images containing contamination or bad particles. At this stage about 75% of the selected particles were rejected. The molecular images were then re-extracted from the micrographs using centered coordinates calculated from the reference-free 2D classification. The structures were refined using a starting model obtained for the NCP by the reference-free angular reconstitution method (Van Heel, 1987) implemented in the IMAGIC software (van Heel et al., 1996). Maximum-likelihood based image sorting was found necessary to select the most homogeneous particles and another 30% of the particles were rejected. At the end of the selection procedure 29,711, 27,279 and 42,292 molecular images were retained for unbound 601, H1.0-bound 601 and H1.5ΔC50-bound 601L nucleosomes, respectively. Illustrations were prepared using the Chimera visualization software (Pettersen et al., 2004). The B-factor was determined in an automatic manner in RELION for the sharpening of the structure of H1.5ΔC50-bound 601L nucleosome. Local resolution estimations were performed using ResMap (Kucukelbir et al., 2014). To calculate the density difference map between the cryo-EM maps of the H1-containing nucleosomes and the modeled density of an H1-free nucleosome (Figure 2B), the crystal structure of the NCP was used as a starting point, the DNA linkers were extended as B-DNA stretches and were fitted into the cryo-EM map using program SITUS (Wriggers, 2012). The model was converted into a density map in Chimera and imported into the IMAGIC software package. For the H1-bound 601 nucleosome both the experimental and model maps were low pass filtered at 1/25 Å−1 and high pass filtered at 1/50 Å−1. In the case of the H1.5ΔC50-bound 601L nucleosome both maps were low pass filtered at 1/8Å−1. All maps were normalized to the same average density and standard deviation, before positive and negative density difference maps were calculated. The maps were thresholded at 3 sigma values and showed significant additional density only in the positive difference map.
X-ray crystallography
Preparation of crystals
Nucleosomes reconstituted from human core histones and 197-bp palindromic 601L DNA in complex with X. laevis histone H1.0b were crystallized using the hanging drop vapour diffusion method at 20°C by mixing equal volumes of the nucleosome/H1 complex (25–30 μM) and a crystallization solution composed of MPD (6% v/v), 50 mM NaCl, and 50 mM sodium potassium phosphate pH 6.4. Crystals were transferred to crystallization solution supplemented with MPD and ethylene glycol to a final concentration of 20% and 30% (v/v), respectively, and flash-cooled in liquid nitrogen.
Crystal structure determination
Diffraction data were collected at ESRF beamline ID29 on a Pilatus 6M-F detector. Data collection and refinement statistics are summarized in Table 1. Data were integrated with XDS (Kabsch, 2010) and scaled with AIMLESS (Evans, 2006). Crystals belong to space group C2221. The asymmetric unit contains a complete H1/nucleosome complex (complex A) plus half of a second complex (complex B) whose nucleosome dyad coincides with a crystallographic dyad (Figure S2A). The nucleosome cores of both complexes were positioned by molecular replacement using the X. laevis NCP-601L crystal structure (Chua et al., 2012) (PDB 3UT9) as a search model in Phaser (McCoy et al., 2007). Rigid-body and TLS refinement in Phenix (Adams et al., 2010) yielded Rcryst and Rfree values of 0.2928 and 0.3266, respectively. Linker DNA was extended from the nucleosome core as B-form DNA, adjusted into density using Coot (Emsley et al., 2010) and subjected to rigid body and TLS refinement in Phenix, resulting in improved R-values (Rcryst =0.2634, Rfree=0.3045). The 2Fo-Fc and Fo-Fc maps calculated using phases from the NCP and linker DNA revealed additional density corresponding to the GH1 domain in both complexes A and B (Figure S2D and Movies S1 and S2).
The structure of X. laevis GH1.0b was modeled by homology with the crystal structure of chicken GH5 (Ramakrishnan et al., 1993) (PDB 1HST), which shares 80% amino acid sequence identity with X. laevis GH1.0b. Modeling was performed with the SWISS-MODEL server (Biasini et al., 2014), using both the extended and compact conformations of GH5 (1HST chains A and B) as modeling templates. Whereas the GH1 density observed for nucleosome complex B in our crystals showed 2-fold disorder, that for complex A was interpretable: tubular density corresponding to the domain’s three α helices allowed manual positioning of GH1 into the map (Figure S2D and Movie S1). This interpretation was independently verified by performing a 6-dimensional search using the Colores program in SITUS (Wriggers, 2012), which allows the docking of atomic structures into low-resolution density maps. The highest-scoring fit obtained with Colores (correlation coefficient of 0.722 versus 0.653 for the next best fit) matched the position and orientation of the manually fitted GH1 domain. The extended GH1 conformation (based on 1HST chain A) gave a steric clash with the core DNA and was therefore discarded, whereas the compact conformation (based on 1HST chain B) exhibited no such clash. Two-fold averaging of the GH1 orientation determined for complex A accounts for the density observed in complex B (Movie S2), further confirming the orientation of this domain. Whereas the two linkers in complex B are perfectly symmetric, the two linkers in complex A deviate from symmetry, with linker-α3 displaced away from the pseudodyad and linker-L1 displaced towards it (Figure S2B). Linker-L1 sterically hinders GH1 from adopting both dyad-related orientations (Figure S2C), explaining why only one GH1 orientation is observed in complex A. Side chain conformations were kept as those in PDB 1HST, except for a few surface-exposed side chains which were adjusted to avoid a steric clash or make a favourable contact with the DNA, in which case either the most preferred rotamer or the rotamer present in GH5-bound nucleosome structure (PDB 4QLC) (Zhou et al., 2015) was used.
The final model was refined in Phenix using grouped isotropic B factor refinement (2 groups per residue) and deformable elastic network (DEN) restraints (Schroder et al., 2010), using the local structural information present in a reference model comprising chicken GH5 [PDB 1HST, chain B, rectified for a minor error in geometry (residue 41 was in the forbidden region of the Ramachandran plot) by replacing residues 41–43 by those of GH5 from PDB 4QLC (Zhou et al., 2015)] and X. laevis NCP-601L (PDB 3UT9) extended with ideal B-form DNA. The mean B-factor for the GH1 domain (400 Å2) is comparable to that of the linker DNA (420 Å2) and significantly higher than that of the core histones (241 Å2). This likely reflects the higher mobility of GH1 on the nucleosome due to the smaller interaction surfaces and the lack of crystal packing interactions constraining the orientation of this domain. Software used for crystallographic analysis was compiled by SBGrid (Morin et al., 2013).
Hydroxyl radical footprinting
Hydroxyl radical footprinting was carried out in 15 μl reaction mixture containing 150 ng of radiolabeled 601 nucleosomes either in the unbound state or bound to a linker histone [X. laevis H1.0b, GH1.5 (residues 40–112), X. laevis histone B4, or total H1 from HeLa cells)] in nucleosomal buffer. 240 bp 601 DNA was used for centrally positioned nucleosomes; 197 bp, 157 bp and 162 bp 601 DNA was used for single-linker nucleosomes. The hydroxyl radicals were generated by mixing 2.5 μl each of 2 mM FeAmSO4/4 mM EDTA, 0.1 M ascorbate, and 0.12% H2O2 together in a drop on the side of the reaction tube before mixing rapidly with the reaction solution. The reaction was terminated after 2 min by the addition of 100 μL stop solution (0.1% SDS, 25 mM EDTA, 1% glycerol, and 100 mM Tris, pH 7.4), and the DNA was purified by phenol/chloroform extraction and ethanol/glycogen precipitation. The DNA was resuspended in formamide loading buffer, heated for 3 minutes at 80°C and run on an 8% denaturing polyacrylamide gel in 1X TBE buffer. The gels were dried and exposed overnight and imaged on a phosphorimager (Fuji-FLA5100). Gel scans were analyzed by Multi-Gauge (Fuji) software.
Protein-DNA crosslinking
H1 modification
Site directed H1 cysteine substitutions were sequence confirmed, and H1 proteins expressed and purified as described previously (Caterino et al., 2011). Briefly, H1 mutant expression vectors were transformed into E. coli BL21 (DE3) and grown at 37°C to an OD600 of 0.6, induced with 0.4 mM IPTG and harvested 3–4 hours post induction by centrifugation at 6000 g for 15 min at 4°C. The cell pellet was resuspended in TE buffer (10 mM Tris, 1 mM EDTA pH 8.0) and treated with final concentrations of 0.72 mg/mL lysozyme, 0.4% Triton X-100, 4 mM PMSF and 50 mM DTT for 1 h at room temperature. Following incubation, cell lysates were diluted with TE buffer to 1M NaCl final and samples sonicated on ice. Cell debris was removed by centrifugation at 26,000 g for 30 min at 4°C. The supernatant was removed by decanting and diluted with TE buffer to a final concentration of 0.6 M NaCl before adding 3 mL 50% suspension of Bio-rex 70 cation exchange resin 50–100 mesh (Bio-Rad) prepared in 0.6 M NaCl. After incubating at 4°C for 2 h, the samples were added to a poly-prep chromatography column, the column was washed with increasing concentrations of NaCl, and H1 was eluted in 1M NaCl TE. An additional round of purification using Bio-rex 70 cation exchange resin 100–200 mesh was performed as above. Following purification, H1 was treated with 50 mM DTT for 1 h on ice. After removing the DTT, the protein was modified with the cross-linking reagent 4-azidophenacyl bromide (APB) by incubation with 3–10 fold molar excess APB for 1 h at room temperature under reduced lighting conditions. Excess APB was removed as described previously (Lee et al., 1999), using the cation exchange column strategy for protein purification above.
Nucleosome reconstitution
Nucleosomes were reconstituted by salt dialysis with chicken erythrocyte histone proteins as described in (Caterino et al., 2011) and empirically optimized by independent adjustment of histone amounts to maximize mononucleosome formation. Typically, reconstitutions included 5 μg H3/H4, 5.2 μg H2A/H2B, 10 μg BamHI-digested pBS plasmid and 1 × 106 cpm radiolabeled 217 bp (or 183 bp for single linker nucleosomes) 601 DNA fragment in reconstitution buffer (10 mM Tris, pH 8.0, 1 mM EDTA, 5 mM DTT, 2 M NaCl). Reconstitutions were then dialyzed against decreasing concentrations of NaCl (1.2, 1.0, 0.8, and 0.6 M) in TE buffer for 2 hours each at 4° followed by dialysis against TE alone overnight. Nucleosomes were purified by sedimentation through 7% to 20% sucrose gradients, (34,000 g for 18 h in a Beckman SW41 rotor at 4°C) and nucleosome-containing fractions collected in 0.6 ml tubes pretreated with 0.3 mg/ml bovine serum albumin (BSA) overnight at 4°C. Fractions were analyzed by electrophoresis on a 0.7% native nucleoprotein agarose gel to ensure that carrier pBS DNA was separated from radiolabeled nucleosomes.
Linker histone binding and cross-linking
APB-modified H1 proteins were mixed with a 2-fold excess of NAP-1 then incubated with radiolabeled 217 bp (or 183 bp for single linker nucleosomes) 601 nucleosomes in binding buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 50 mM NaCl, 150 ng/l BSA, and 5% v/v glycerol) at 4°C for 30 min. One-half of each reaction was analysed by electrophoresis on a 0.7% native nucleoprotein agarose gel followed by autoradiography. Cross-linking was induced by placing the rest of the reaction in a pyrex tube and exposing to UV irradiation at 365 nm for 90 s as described (Lee et al., 1999). The yield of cross-links was determined by electrophoresis on 0.8% agarose gels containing 0.02% SDS, followed by autoradiography of the dried gels.
Cross-link mapping
Cross-linking reactions containing 50,000 cpm were performed as above using the ratio of H1/NAP-1:nucleosome that yielded maximal cross-linking. A portion of the reaction was analysed by electrophoresis on agarose/SDS gels as described above to ensure cross-linking. DNA from the remaining cross-link reaction was precipitated by adding 200 ng calf thymus DNA, SDS (0.02% final), 0.3 M sodium acetate, and 2 volumes 95% ethanol. Centrifugation at 13,000 g for 30 min was followed by washing the pellet with 70% ethanol and centrifugation at 13,000 g for 10 min. Ethanol was removed, and H1-DNA cross-linked species resuspended in water. Cleavage of DNA at sites of cross-linking was induced by incubation in 1 M piperidine at 90°C for 30 min; the sample was subsequently dried under vacuum, resuspended in pure water, and dried again. The latter step was repeated once and the final pellet resuspended in water and the DNA precipitated, resuspended in formamide and loaded onto pre-run 6% acrylamide/8M urea sequencing gels. Gels were run at 55 W for 1 h, dried and the gels autoradiographed. Sites of H1-DNA cross-linking were mapped by analysis relative to Maxam-Gilbert sequencing reactions with the 217 bp 601 DNA fragment.
Simultaneous cross-link assay
Nucleosomes were reconstituted as above using 5′ biotinylated and 5′ radiolabeled 197-bp 601 DNA in which we introduced a specific restriction endonuclease, Xba I and Hind III, site next to each linker arm. H1 cysteine substitutions, APB modification and subsequent binding and cross-linking verification were performed as above. H1 cross-link reactions were performed as above, followed by incubation in 2 M NaCl and precipitation to remove proteins not cross-linked. Precipitated cross-link material was digested with 10 U each Xba I and Hind III in supplied reaction buffer (NEB) at 37°C for 1 h, and the reaction was stopped with 10 mM EDTA. Streptavidin agarose resin was prepared by successive washing of resin in TE buffer and blocked for 1 h in blocking buffer (50 mM NaCl, 800 ng/mL sheared calf thymus DNA). The resin was washed with TE and prepared as a 50% slurry in TE. Digested cross-link reactions were bound to 45 μl prepared bead slurry in 100 mM NaCl final at 4°C overnight. Resin binding reaction was loaded onto 0.22 μM filter columns, and the flow-through collected. Resin was washed twice with 2 M NaCl in TE followed by two TE washes to remove non-specific and unbound material; retained material was eluted by incubation with elution buffer (0.5% SDS, 50 mM NaCl, 50 μM biotin) at 37°C overnight. Eluted material was treated with or without 2.5 μg/mL final Proteinase K at 50°C for 2 h, and run on 6% acrylamide, 0.02% SDS, 0.25x TBE gels in 0.5x TBE running buffer, followed by autoradiography of the dried gels.
Molecular Docking
Docking with HADDOCK
The GH1.0 structure (simply called GH1 below) was modeled on the crystal structure of chicken GH5, as described above (see ‘Crystal structure determination’). Nucleosomal and linker DNA coordinates were taken from our crystal structure of the H1.0-bound nucleosome. GH1 was docked onto the DNA using the HADDOCK webserver (de Vries et al., 2010). HADDOCK uses biochemical information on interacting residues as ambiguous interaction restraints (AIRs) to drive the docking. Residues directly implicated or potentially involved in mediating the interaction are defined as “active” and “passive”, respectively. Each AIR is defined between an active residue of one molecule and specific active or passive residues of the other. Passive residues for GH1 were defined as all surface exposed residues with over 50% solvent accessibility [as calculated by Naccess (Hubbard et al., 1991)], while DNA passive residues were defined as all nucleotides within the central helical turn of nucleosomal core DNA (spanning the dyad) and the first two turns of each linker (Table S1). The set of AIRs used to dock GH1 are summarized in Table S1. The default target distance of 2 Å was used for all AIRs, except for the cross-linking restraints, where a distance of 12 Å was used.
The HADDOCK docking protocol consists of randomization of orientations and rigid body energy minimization, semi-rigid simulated annealing in torsion angle space, and final refinement in Cartesian space with explicit solvent. Ten independent docking jobs were performed using different subsets of AIRs, as indicated in Table S1. For each job, a total of 1000 complex configurations were calculated in the randomization stage using an ensemble model of GH1 that comprised both the extended and compact conformations (corresponding to 1HST chains A and B, respectively). The best 200 solutions were then used in the semi-rigid and final refinement stages, in which the side chain and backbone atoms of GH1 were allowed to move while the DNA was held fixed. The final structures were ranked according to HADDOCK score and clustered by pairwise backbone RMSD. Clusters were then ranked according to the average of the four best-scoring structures within each cluster.
For the majority of docking runs listed in Table S1, the best cluster scored substantially better than the next best cluster and was frequently the largest in size. The best-scoring solutions in each run consistently exhibited the compact (B) conformation of GH1. For all 10 runs, the best cluster closely resembled the configuration observed in our crystal structure. Compared to the crystal structure, clusters showed a difference in GH1 orientation that ranged between 14.7° and 35.0° (mean of 23.4° ± 6.2° over all runs) and a difference in interface RMSD (calculated on DNA atoms and main chain GH1 atoms within 10 Å of the protein-DNA interface) between 1.81 and 3.98 Å (mean of 2.43 ± 0.65 Å over all runs).
Docking with Autodock Vina
An alternative approach to predicting binding positions of GH1 used the docking program Autodock Vina (Trott and Olson, 2010). Vina docking calculations involve independent optimization runs, starting from a set of random positions of the ligand (GH1) with respect to the receptor (nucleosomal DNA). Vina uses a stochastic global optimization algorithm to improve the docking score. The scoring function includes both the ligand-receptor interaction energy and the deformation energy of both interacting species. Vina was used to dock GH1 within a defined search space without any imposed interaction restraints. During docking the DNA was treated as a rigid body while GH1 was either held rigid or its side chains were allowed to be flexible. Both the A (extended) and B (compact) conformations of GH1 were tested. The dimensions of the search space were larger than GH1 by 3 Å along each Cartesian axis. The centre of the search space was chosen so that the minimal distance of the GH1 centre of mass from the nucleosomal DNA and two linker arms was 11 Å. Subsequent optimization runs involved shifting the centre of the search space by ± 3 Å in orthogonal directions. This allowed the centre of GH1 to move up to 9 Å away from its initial position. Multiple docking simulations (17 with rigid H1 and 6 with flexible H1 side chains), using different random seeds for the search procedure, resulted in a total 70 proposed complexes (with rigid GH1), and 30 proposed complexes (with flexible GH1 side chains). The Vina scores for these optimized structures ranged from −16.0 to −11.0 kcal.mol−1. These structures were subsequently screened according to their ability to satisfy the following experimentally defined interaction restraints: (1) Arg42 cross-links to nucleotides −77 and −80 on chain I; (2) Ser66 cross-links to nucleotides −75 to −77 on chain J; (3) His25 is close to DNA; (4) Lys85 is close to DNA; (5) 6–8 nucleotide pairs centred around the nucleosome dyad are protected by GH1, implying GH1 proximity to nucleotides −3 to +3 on both DNA strands; (6) the C-terminus of GH1 points away from the nucleosome core towards the stem formed by the DNA linker arms. During the screening, restraints 1–4 were counted as satisfied if the Cα atom of Arg42 (restraint 1) or Ser66 (restraint 2) was located within 13.0 Å of any atom of the corresponding DNA nucleotides, or if any atom of His25 (restraint 3) or Lys85 (restraint 4) was within 5.0 Å of any DNA atom.
Structures satisfying at least four of the six above restraints were retained and re-optimized in Vina and subsequently energy minimized in AMBER 12 (Case et al., 2012) without any conformational restraints on the system to remove residual steric hindrance (having embedded the complex in an explicit solvent/salt environment using a TIP3P water model (Jorgensen, 1981) and Na+ and Cl- ions with Dang parameters (Dang, 1995) corresponding to a 0.15 M concentration). Several structures from the initial docking runs satisfied four of the six experimentally identified restraints with Vina scores of −13.0 kcal.mol−1. After re-optimization in Vina and energy minimization in AMBER, we obtained a single structure that satisfied all restraints. This structure had an improved Vina score of −13.8 kcal.mol−1 and closely resembled the structure determined from the X-ray data as well as the HADDOCK models.
QUANTIFICATION AND STATISTICAL ANALYSIS
Protein-DNA crosslinking and hydroxyl radical footprinting experiments were each performed at least twice and gave reproducible results.
DATA AND SOFTWARE AVAILABILITY
The cryo-EM maps of the nucleosome in the unbound state, bound to X. laevis H1.0b and bound to human H1.5ΔC50 have been deposited in the Electron Microscopy Data Bank with accession codes EMD-3660, EMD-3659 and EMD-3657, respectively. The crystallographic model coordinates of the nucleosome bound to X. laevis histone H1.0b have been deposited in the Protein Data Bank with the accession number 3NL0. Original gel scans for hydroxyl radical footprinting and protein-DNA crosslinking experiments have been deposited in the Mendeley Data repository (http://dx.doi.org/10.17632/x9y79wwswc.1).
Supplementary Material
Movie S1. Related to Figure 3. Fo-Fc omit map showing electron density for the GH1 domain in complex A. Compared to omit maps for the core histones, lower contour levels are required to visualize the omitted GH1 helices. This likely reflects the higher mobility of the GH1 domain on the nucleosome due to the smaller interaction surfaces and the lack of crystal packing interactions constraining this domain.
Movie S2. Related to Figure 3. Fo-Fc omit map showing the two-fold averaged electron density for the GH1 domain in complex B. The two copies of the GH1 domain related by a crystallographic dyad are shown in magenta and light orange. Each copy is at half occupancy, and so the density is considerably weaker than in complex A.
Movie S3. Related to Figure 3. Cryo-EM map of the H1.5ΔC50-bound 601L nucleosome and agreement with the crystal structure of the H1.0-bound 601L nucleosome. The cryo-EM structure corresponds to conformation Y in Fig. 1D.
Movie S4. Related to Figure 3. Cryo-EM map of the H1.0-bound 601 nucleosome and agreement with the crystal structure of the H1.0-bound 601L nucleosome. The cryo-EM structure corresponds to conformation C in Fig. 1D.
Acknowledgments
We acknowledge the ESRF for provision of synchrotron radiation facilities. We thank EMBL and ESRF staff for providing assistance in using beamline ID29, ID23–1 and ID30B, Jean-François Ménétret for dedicated help with microscopes, and Ping Zhu for kindly providing the atomic coordinates of the 12-nucleosome array. This work was supported by the Fondation pour la Recherche Médicale (Programme “Epigénétique et Stabilité du Genome”), the Institut National de la Santé et de la Recherche Médicale, the Centre National pour la Recherche Scientifique, Strasbourg University, University Grenoble Alpes, the Association pour la Recherche sur le Cancer and the Agence Nationale pour la Recherche (Chromcomp and Chrome grants), the Labex INRT, the French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INSB-05-01] and Strasbourg Instruct Center platforms, JSPS KAKENHI grant number JP25116002 (to H.K), and National Institutes of Health grant GM052426 (to J.J.H). A.J.B. was supported by NIH Training Grant T32GM068411. This work used the platforms of the Grenoble Instruct Center (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.D. conceived the project and coordinated partner contributions. R.B., A.R.C, S.H.S., I.N.L., O.T., H.M., C.Pa., H.K., A.H., J.J.H. and D.A. performed the molecular biology and biochemistry work. J.B., G.B., C.C., and P.S. performed the cryo-EM work. I.G.S., D.A.S and C.Pe. performed crystallography. A.R., R.L. and C.Pe performed in silico modeling. P.S., S.D. and C.Pe. wrote the manuscript with input from all authors.
References
- Abrishami V, Vargas J, Li X, Cheng Y, Marabini R, Sorzano CO, Carazo JM. Alignment of direct detection device micrographs using a robust Optical Flow approach. J Struct Biol. 2015;189:163–176. doi: 10.1016/j.jsb.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D. Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan J, Hartman PG, Crane-Robinson C, Aviles FX. The structure of histone H1 and its location in chromatin. Nature. 1980;288:675–679. doi: 10.1038/288675a0. [DOI] [PubMed] [Google Scholar]
- Allan J, Mitchell T, Harborne N, Bohm L, Crane-Robinson C. Roles of H1 domains in determining higher order chromatin structure and H1 location. J Mol Biol. 1986;187:591–601. doi: 10.1016/0022-2836(86)90337-2. [DOI] [PubMed] [Google Scholar]
- Bednar J, Horowitz RA, Dubochet J, Woodcock CL. Chromatin conformation and salt-induced compaction: three-dimensional structural information from cryoelectron microscopy. J Cell Biol. 1995;131:1365–1376. doi: 10.1083/jcb.131.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Natl Acad Sci U S A. 1998;95:14173–14178. doi: 10.1073/pnas.95.24.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DT, Izard T, Misteli T. Mapping the interaction surface of linker histone H1(0) with the nucleosome of native chromatin in vivo. Nat Struct Mol Biol. 2006;13:250–255. doi: 10.1038/nsmb1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckle RS, Maman JD, Allan J. Site-directed mutagenesis studies on the binding of the globular domain of linker histone H5 to the nucleosome. J Mol Biol. 1992;223:651–659. doi: 10.1016/0022-2836(92)90981-o. [DOI] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheathman TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, et al. AMBER 12. 2012 San Francisco. [Google Scholar]
- Caterino TL, Fang H, Hayes JJ. Nucleosome linker DNA contacts and induces specific folding of the intrinsically disordered H1 carboxyl-terminal domain. Mol Cell Biol. 2011;31:2341–2348. doi: 10.1128/MCB.05145-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, Mowen KA, Bertone P, Silva JC, Zernicka-Goetz M, et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature. 2014;507:104–108. doi: 10.1038/nature12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua EY, Vasudevan D, Davey GE, Wu B, Davey CA. The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 2012;40:6338–6352. doi: 10.1093/nar/gks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang LX. Mechanism and thermodynamics of ion selectivity in aqueous-solutions of 18-crown-6 ether – A molecular dynamics study. J Am Chem Soc. 1995;117:6954–6960. [Google Scholar]
- de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica. Section D. Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta crystallographica. Section D. Biological crystallography. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Fang H, Clark DJ, Hayes JJ. DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic Acids Res. 2012;40:1475–1484. doi: 10.1093/nar/gkr866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamiche A, Schultz P, Ramakrishnan V, Oudet P, Prunell A. Linker histone-dependent DNA structure in linear mononucleosomes. J Mol Biol. 1996;257:30–42. doi: 10.1006/jmbi.1996.0144. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Lever MA, Crawford E, Th’ng JP. The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo. J Biol Chem. 2004;279:20028–20034. doi: 10.1074/jbc.M400070200. [DOI] [PubMed] [Google Scholar]
- Hubbard SJ, Campbell SF, Thornton JM. Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J Mol Biol. 1991;220:507–530. doi: 10.1016/0022-2836(91)90027-4. [DOI] [PubMed] [Google Scholar]
- Izzo A, Kamieniarz K, Schneider R. The histone H1 family: specific members, specific functions? Biol Chem. 2008;389:333–343. doi: 10.1515/BC.2008.037. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL. Transferable intermolecular potential functions for water, alcohols and ethers. Application to liquid water. J Am Chem Soc. 1981;103:335–340. [Google Scholar]
- Kabsch W. Xds. Acta crystallographica. Section D. Biological crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KM, Chafin DR, Hayes JJ. Targeted cross-linking and DNA cleavage within model chromatin complexes. Methods Enzymol. 1999;304:231–251. doi: 10.1016/s0076-6879(99)04014-8. [DOI] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Peterson C, Hayes JJ. SWI/SNF and RSC Catalyzed Nucleosome Mobilization Requires Internal DNA Loop Translocation Within Nucleosomes. Mol Cell Biol. 2011 doi: 10.1128/MCB.05605-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lone IN, Shukla MS, Charles Richard JL, Peshev ZY, Dimitrov S, Angelov D. Binding of NF-kappaB to Nucleosomes: Effect of Translational Positioning, Nucleosome Remodeling and Linker Histone H1. PLoS Genet. 2013;9:e1003830. doi: 10.1371/journal.pgen.1003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Makarov VL, Dimitrov SI, Petrov PT. Salt-induced conformational transitions in chromatin. A flow linear dichroism study. Eur J Biochem. 1983;133:491–497. doi: 10.1111/j.1432-1033.1983.tb07491.x. [DOI] [PubMed] [Google Scholar]
- Maresca TJ, Freedman BS, Heald R. Histone H1 is essential for mitotic chromosome architecture and segregation in Xenopus laevis egg extracts. J Cell Biol. 2005;169:859–869. doi: 10.1083/jcb.200503031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins. 2003;52:51–67. doi: 10.1002/prot.10393. [DOI] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Mirzabekov AD, Pruss DV, Ebralidse KK. Chromatin superstructure-dependent crosslinking with DNA of the histone H5 residues Thr1, His25 and His62. J Mol Biol. 1990;211:479–491. doi: 10.1016/0022-2836(90)90366-T. [DOI] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P. Collaboration gets the most out of software. eLife. 2013;2:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pruss D, Bartholomew B, Persinger J, Hayes J, Arents G, Moudrianakis EN, Wolffe AP. An asymmetric model for the nucleosome: a binding site for linker histones inside the DNA gyres. Science. 1996;274:614–617. doi: 10.1126/science.274.5287.614. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V, Finch JT, Graziano V, Lee PL, Sweet RM. Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature. 1993;362:219–223. doi: 10.1038/362219a0. [DOI] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder GF, Levitt M, Brunger AT. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464:1218–1222. doi: 10.1038/nature08892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla MS, Syed SH, Goutte-Gattat D, Richard JL, Montel F, Hamiche A, Travers A, Faivre-Moskalenko C, Bednar J, Hayes JJ, et al. The docking domain of histone H2A is required for H1 binding and RSC-mediated nucleosome remodeling. Nucleic Acids Res. 2011;39:2559–2570. doi: 10.1093/nar/gkq1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RT. Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry. 1978;17:5524–5531. doi: 10.1021/bi00618a030. [DOI] [PubMed] [Google Scholar]
- Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science. 2014;344:376–380. doi: 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- Staynov DZ, Crane-Robinson C. Footprinting of linker histones H5 and H1 on the nucleosome. EMBO J. 1988;7:3685–3691. doi: 10.1002/j.1460-2075.1988.tb03250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed SH, Boulard M, Shukla MS, Gautier T, Travers A, Bednar J, Faivre-Moskalenko C, Dimitrov S, Angelov D. The incorporation of the novel histone variant H2AL2 confers unusual structural and functional properties of the nucleosome. Nucleic Acids Res. 2009;37:4684–4695. doi: 10.1093/nar/gkp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, Everaers R, Angelov D, Bednar J, Dimitrov S. Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc Natl Acad Sci U S A. 2010;107:9620–9625. doi: 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachiwana H, Kagawa W, Shiga T, Osakabe A, Miya Y, Saito K, Hayashi-Takanaka Y, Oda T, Sato M, Park SY, et al. Crystal structure of the human centromeric nucleosome containing CENP-A. Nature. 2011;476:232–235. doi: 10.1038/nature10258. [DOI] [PubMed] [Google Scholar]
- Thoma F, Koller T, Klug A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J Cell Biol. 1979;83:403–427. doi: 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JO, Wilson CM. Selective radiolabelling and identification of a strong nucleosome binding site on the globular domain of histone H5. EMBO J. 1986;5:3531–3537. doi: 10.1002/j.1460-2075.1986.tb04679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremethick DJ. Higher-order structures of chromatin: the elusive 30 nm fiber. Cell. 2007;128:651–654. doi: 10.1016/j.cell.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar WS. Scoring residue conservation. Proteins. 2002;48:227–241. doi: 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- Van Heel M. Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy. 1987;21:111–123. doi: 10.1016/0304-3991(87)90078-7. [DOI] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- van Holde K. Chromatin. Springer-Verlag KG; Berlin, Germany: 1988. [Google Scholar]
- Vasudevan D, Chua EY, Davey CA. Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. J Mol Biol. 2010;403:1–10. doi: 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
- Wang X, Hayes JJ. Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol Cell Biol. 2008;28:227–236. doi: 10.1128/MCB.01245-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AE, Hieb AR, Luger K. A quantitative investigation of linker histone interactions with nucleosomes and chromatin. Sci Rep. 2016;6:19122. doi: 10.1038/srep19122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Kruger S, Mann M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol Cell Proteomics. 2007;6:72–87. doi: 10.1074/mcp.M600255-MCP200. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Mann M. Nepsilon-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 2008;36:570–577. doi: 10.1093/nar/gkm1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock CL, Skoultchi AI, Fan Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 2006;14:17–25. doi: 10.1007/s10577-005-1024-3. [DOI] [PubMed] [Google Scholar]
- Wriggers W. Conventions and workflows for using Situs. Acta crystallographica. Section D. Biological crystallography. 2012;68:344–351. doi: 10.1107/S0907444911049791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BR, Feng H, Ghirlando R, Li S, Schwieters CD, Bai Y. A small number of residues can determine if linker histones are bound on or off dyad in the chromatosome. J Mol Biol. 2016 doi: 10.1016/j.jmb.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BR, Feng H, Kato H, Dai L, Yang Y, Zhou Y, Bai Y. Structural insights into the histone H1-nucleosome complex. Proc Natl Acad Sci U S A. 2013;110:19390–19395. doi: 10.1073/pnas.1314905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BR, Jiang J, Feng H, Ghirlando R, Xiao TS, Bai Y. Structural Mechanisms of Nucleosome Recognition by Linker Histones. Mol Cell. 2015;59:628–638. doi: 10.1016/j.molcel.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YB, Gerchman SE, Ramakrishnan V, Travers A, Muyldermans S. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature. 1998;395:402–405. doi: 10.1038/26521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Related to Figure 3. Fo-Fc omit map showing electron density for the GH1 domain in complex A. Compared to omit maps for the core histones, lower contour levels are required to visualize the omitted GH1 helices. This likely reflects the higher mobility of the GH1 domain on the nucleosome due to the smaller interaction surfaces and the lack of crystal packing interactions constraining this domain.
Movie S2. Related to Figure 3. Fo-Fc omit map showing the two-fold averaged electron density for the GH1 domain in complex B. The two copies of the GH1 domain related by a crystallographic dyad are shown in magenta and light orange. Each copy is at half occupancy, and so the density is considerably weaker than in complex A.
Movie S3. Related to Figure 3. Cryo-EM map of the H1.5ΔC50-bound 601L nucleosome and agreement with the crystal structure of the H1.0-bound 601L nucleosome. The cryo-EM structure corresponds to conformation Y in Fig. 1D.
Movie S4. Related to Figure 3. Cryo-EM map of the H1.0-bound 601 nucleosome and agreement with the crystal structure of the H1.0-bound 601L nucleosome. The cryo-EM structure corresponds to conformation C in Fig. 1D.