

Arrhythmias and sudden cardiac arrest are among the most important causes of morbidity and mortality in the developed world, but their underlying causes remain elusive. Arrhythmogenic substrates may be established by ischemia, infarction, heart failure, drugs and genetic mutations, all of which may cause inflammation, extracellular matrix remodeling, interstitial fibrosis, fibrofatty infiltration and changes in the 3-dimensional cellular composition and architecture of the myocardium.1 Although cardiomyocytes make up the bulk of the tissue in the normal heart, non-myocytes, specifically cardiac fibroblasts, can overwhelm the cardiac tissue during the initiation and progression of various cardiac diseases.2 Myocardial diseases can promote fibroblast differentiation into myofibroblasts, which are hypersecretory of extracellular matrix (ECM) components.3 Hence, myofibroblasts are the primary mediators of wound healing in the damaged ventricle.4 They contribute to cardiac fibrosis and scar formation through their relatively greater ability to produce fibrillar and non-fibrillar collagens.5 They also contribute to ECM remodeling through their production of focal adhesion-associated proteins.6
Cardiac myofibroblasts interact with cardiomyocytes through cell surface proteins of the β-integrin and cadherin families.7 N-cadherin in particular has been shown to be involved in cardiomyocyte-myofibroblast interactions.8 Electrotonic coupling of neonatal cardiomyocytes and non-myocytes in vitro was first demonstrated in the early 1970s.9 Subsequently, Rook et al10 showed that neonatal rat cardiac fibroblasts can connect with other fibroblasts and with cardiomyocytes through gap junctions with single channel conductance values of 21 pS and 32 pS, respectively. They also found in vitro evidence for Cx43 gap junctions between myocytes and fibroblasts.10 Gaudesius et al11 and Miragoli et al12 were first to demonstrate connexin43 (Cx43) and Cx45 expression both between fibroblasts and at fibroblast–to-cardiomyocyte junctions in cardiac strands.
A number of years ago, we investigated the effects of cardiomyocyte-myofibroblast interactions on wave propagation dynamics and reentry in co-cultured neonatal cell monolayers and computer simulations.13 By changing the myofibroblast-to-cardiomyocyte ratio and modifying junctional coupling systematically, we demonstrated that increasing the percentage of myofibroblasts, as seen in various cardiac diseases, reduced the conduction velocity, increased wave fragmentation and reduced action potential duration.13 The results indicated that, in addition to their association with increased fibrosis, myofibroblasts can directly or indirectly affect the electrical properties of the myocytes.
Cardiac myofibroblasts are usually thought to be electrically inactive cells but recent studies have demonstrated their ability to express an assortment of membrane ion channels, whose biophysical properties and density change under disease conditions.14 For example, increased differentiation of fibroblasts into myofibroblasts is associated with the expression of transient receptor potential channels, which are known to be increased in the atria of AF patients.15 In stem cells a change in the membrane potential correlates with increased fibroblast proliferation and differentiation which provides an association between channel biophysical properties and cell function.16
One of the major functions of cardiac fibroblasts is to produce and secrete soluble signaling molecules, including growth factors and cytokines that are known to affect cardiac myocyte morphology and function. Pro-fibrotic cytokines, such as transforming growth factor(TGF)-β1 are significantly upregulated in models of myocardial injury and arrhythmias.17 In the rat model of myocardial infarction, mRNA levels of TGF-β118 are increased up to 50-fold in the infarcted area and up to 15 fold in the non-infarcted myocardium.19 The importance of TGF-β1 and other major cytokines is highlighted also by the fact that when released from myofibroblasts as paracrine factors they lead to cardiomyocyte hypertrophy, diastolic dysfunction, and apoptosis.20 In adition, TGF-β1 released by myofibroblasts can contribute to further fibroblast activation and proliferation in a positive feedback loop. However, knowledge is still incomplete regarding how TGF-β1 modifies the electrophysiological phenotype of myofibroblasts and whether it contributes to myofibroblast-cardiomyocyte cross talk and arrhythmogenesis in vitro or in vivo.
In an interesting and thought provoking article published in this issue of Circulation A&E, Salvarani et al21 have tested the hypothesis that TGF-β1 plays an important role in arrhythmogenesis by directly altering the electrophysiological phenotype of myofibroblasts through changes in ion channel gene expression, and by indirectly accentuating cardiomyocyte depolarization induced by myofibroblast-to-cardiomyocyte crosstalk. 21 Their model study is the neonatal myofibroblast exposed to TGF-β1 in vitro and co-cultured with neonatal cardiomyocytes. They first obtained patch-clamp recordings from cultured myofibroblasts while treating them with a clinically relevant concentration of TGF-β1 and its specific inhibitor SB431542, alone or in combination. As revealed by whole-cell current-voltage relation plots, TGF-β1 shifted the reversal potential in the depolarizing direction by about 7 mV and increased transmembrane currents. Conversely, the TGF-β1 inhibitor shifted the reversal potential by ~22 mV in the hyperpolarized direction. These changes in membrane potential could be explained by the decreased myofibroblast membrane resistance in TGF-β1 treated cells compared to control cells. The changes in the whole-cell current in TGF-β1 treated myofibroblasts could be explained also by changes in the expression of a variety of genes coding ion channels along with ion pumps and connexins.21 Surprisingly, in the absence of TGF-β1, myofibroblasts exerted no effects on the electrical properties of cardiomyocytes attached to them. In addition, in experiments where cardiomyocytes were cultured alone, TGF-β1 had no effect on the membrane potential, membrane resistance or membrane capacitance of the cardiomyocytes.21 On the other hand when cardiomyocytes were attached to myofibroblasts they showed a significant TGF-β1 dependent membrane potential depolarization, compared to cardiomyocytes co-cultured with myofibroblasts with no TGF-β1 treatment.21 These results clearly demonstrated that under the experimental conditions established by Salvarani et al,21 neonatal cardiomyocytes are depolarized by myofibroblasts only in the presence TGF-β1. Similarly, gap junctional conductance was increased in co-cultures only in the presence of TGF-β1, which increased the gap junctional conductance in cardiomyocyte-myofibroblast cell pairs, which led to cardiomyocytes depolarization.
To address the question of whether the electrophysiological effects seen in cardiomyocyte-myofibroblast pairs were due to either paracrine or direct molecular signaling between cells, Salvarani et al21 developed an ingenious ‘killing protocol’ where the myofibroblast of a heterologous cell pair was killed with a second patch pipette, while membrane voltage was being continuously recorded from the attached cardiomyocyte. Using this elegant “targeted disruption” protocol Salvarani et al21 demonstrated that upon killing the myofibroblast in control and TGF-β1 cell pairs, the now uncoupled cardiomyocytes hyperpolarized to rest and stopped beating spontaneously. These data suggested that electrotonic coupling between the cardiomyocyte and the myofibroblast in the heterologous cell pair mediated the cardiomyocyte depolarization. Further experiments using optical mapping in a cardiac fibrosis model showed that TGF-β1 treatment of the co-culture reduced both the maximal action potential upstroke velocity and the wave propagation velocity, and increased the rate of spontaneous discharge. As hypothesized, TGF-β1 signaling blockade completely abolished both arrhythmogenic conditions.21
Salvarani et al21 used transcriptome analysis to demonstrate TGF-β1-dependent changes in 29 ion channel/pump/connexin transcripts suggesting a multiplicity of cytokine actions related to the electrical phenotype of myofibroblasts. Of note, they also demonstrated a 275% upregulation in the transcript of consortin, a protein that is known to be involved in the efficient membrane targeting of connexinss,22 which might help explain the TGF-β1-induced increase in gap junctional conductance in the heterologous pairs. What remains to be determined is the mechanism whereby gap junction conductance is stronger in heterocellular pairs as compared to cardiomyocyte-cardiomyocyte pairs. Is the protein altered only in myofibroblasts but not cardiomyocytes? While it is reasonable to suggest that consortin may be one of the factors responsible for the increased gap junction conductance and thus electrical remodeling in heterocellular pairs treated with TGF-β1, it would be of interest to determine whether the change in transcript is followed by changes in consortin protein levels as well.
It is important to emphasize that, while the results of Salvarani et al21 are important in that they provide novel information on the electrophysiological consequences of TGF-β1 exposure, they were obtained in co-cultures of neonatal cells. Therefore, extrapolation to the in-vivo situation or to the adult heart should be made with extreme caution. Evidence in the literature suggests that the phenotypic changes produced by paracrine factors released by cardiac fibroblasts are different in the developing versus adult cardiomyocytes.23 Also a number of functional differences exist between neonatal and adult cardiomyocytes. For example, the resting membrane potential and the action potential morphology of the neonatal cardiomyocyte are significantly different from the adult cardiomyocyte, implying that large differences may exist also in terms of ion channel types, functional expression, localization and/or associated proteins between the adult and the neonatal cells.24
Prior studies using neonatal ventricular and atrial cells show changes in specific currents such as Ito, INa and IK1 upon TGF-β1 treatment.25 Further, fibroblast conditioned media was shown to produce dose-dependent reduction in conduction velocity, depolarization of the resting membrane potential, reduction of the action potential upstroke velocity and prolongation of action potential duration in neonatal rat ventricular myocytes,26 suggesting a direct paracrine effect of TGF-β1 on cardiomyocytes. The reason for such discrepancies with the data of Salvarani et al21 might be related to differences in experimental approach and/or culturing conditions, since some of the previous studies were conducted in serum free media,26 whereas as Salvarani et al21 used 5% FBS. It is possible that the residual cytokines and growth factors in serum may have lessened effect of added TGF-β1. Therefore, it will be important to understand what specific channels are being affected by TGF-β1 via myofibroblasts.
Despite accumulating evidence of potential heterocellular electrical coupling in vitro, electrical coupling between fully differentiated myocytes and fibroblasts has never been demonstrated in normal ventricular muscle. Camelliti et al27 suggested that fibroblast-myocyte coupling occurs in regions of isolated rabbit sinoatrial node preparations, but such data have yet to be confirmed by other investigators. In fact, whether in the normal, fully differentiated adult heart fibroblast-myocyte interactions also involve electrical connections through gap junctions continues to be a matter of debate. In fact, it makes sense to postulate that fully differentiated adult ventricular myocytes do not couple electrically with cardiac fibroblasts because heterocellular coupling would be detrimental to normal cardiac electro-mechanical function in the adult heart. We surmise that heterocellular electrical communication is a late process in the damaged myocardium, which likely occurs only after fibroblasts switch phenotypically to myofibroblasts, become contractile, express adhesive proteins and cytokines and attach to neighboring surviving cardiomyocytes forcing them to de-differentiate. This idea is based on experiments demonstrating that co-culturing terminally differentiated adult rabbit or rat cardiomyocytes with cardiac fibroblasts induces accelerated remodeling of the cardiomyocytes with morphological adaptation and de-differentiation.8
According to Driesen et al,8 structural adaptation begins the moment a healthy looking, fully differentiated cardiomyocyte comes in contact with a fibroblast. It starts at the distal end of the cardiomyocyte with disassembly of the intercalated disc and formation of cytoplasmic processes.8 It then extends to the whole membrane, changing the cardiomyocyte’s ultrastructure from a highly organized cylinder to a flat rounded cell, with extensive myofibrillar and mitochondrial rearrangement. Previously unpublished data from our laboratory, presented in Figure 1, reproduce the dramatic morphological changes described by Driesen et al.8 On attachment to a myofibroblast the originally rod-shaped cells flatten and spread all around (note different morphology of attached and unattached myocytes at 1 day in co-culture). On the third day, attached cardiomyocytes lose their striated appearance and begin to beat spontaneously. After 8 days, new myofibrils start being assembled and grow out into the expanding cell periphery in a crisscross pattern. In addition, myocytes develop large filopodia that extend toward, and attach to myofibroblasts, and they begin to express α-smooth muscle actin. By day 11 the surviving myocytes have doubled or tripled in size, beat vigorously and show significant α-smooth muscle staining interspaced with α-actinin-stained myofibers, completing the myofibroblast induced de-differentiation process of the cardiomyocyte. The significance of these data is underscored by the recent results of Quinn et al45 who used cell-specific voltage-sensitive fluorescent protein 2.3 (VSFP2.3) to monitor electrical activity in nonmyocytes of mouse hearts and showed cardiomyocyte action potential-like signals at the border of healed cryoinjuries. In addition, using electron tomography, they revealed multiple membrane protrusions originating from neighboring cardiomyocytes and non-myocytes, which formed nanotubes providing direct membrane heterocellular connections.28 Therefore, while heterocellular electrotonic connections are unlikely to occur in the normal human heart, after an infarct, myofibroblast infiltration of the ischemic region coincides with disruption of the cardiomyocyte sarcomeric structure followed by de-differentiation, or death.29 De-differentiated cardiomyocytes are more likely to make heterocellular electrotonic connections than fully differentiated cardiomyocytes. In addition, as shown by previous reports, a significant proportion of Cx43 expression is found in the myofibroblasts within the infarct.30 From the foregoing, it is reasonable to infer that paracrine, mechanical and even electrical myocyte-myofibroblast interactions leading to remodeling of injured myocardium may contribute to the mechanism of arrhythmias after myocardial injury.
Figure 1.
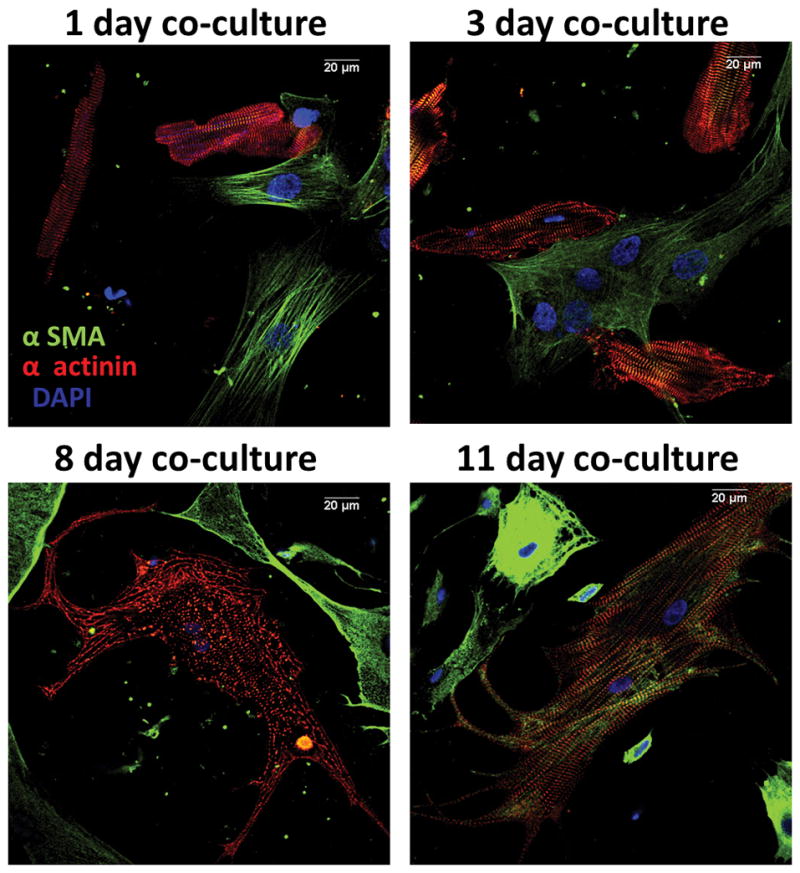
Evolution of cardiomyocyte trans-differentiation in co-culture with myofibroblasts. Fluorescence labeling for α-smooth muscle actin (α-sma, green), α-actinin (red) and DAPI (blue). At one day in co-culture, most cardiomyocytes express α-actinin and are rod-shaped and striated. At three days, many myocytes are no-longer rod shaped and acquire neonatal/fetal phenotype. On day 11, surviving myocytes beat spontaneously, are 4–5x larger than normal, show filopodia and stain strongly for α-sma in addition to α-actinin. Right, negative control. Calibration, 20 μm in all frames.
Salvarani et al21 have come close to their goal of understanding the mechanism(s) by which TGF-β1 alters the electrophysiology of myofibroblasts and cardiomyocytes.21 Their data suggest that in neonatal heterocellular pairs TGF-β1 indirectly alters cardiomyocyte transmembrane potentials by affecting myofibroblast electrophysiology. But there is more work to be done. Another very important way to answer the role of TGF-β1 would be to study these effects in cells from genetically engineered mice that lack or overexpress TGF-β receptors. Compared to wildtype, one should expect a dramatic difference in response to myocardial infarction in these hearts, if in fact the cytokine affects cardiomyocyte electrophysiology via TGF-β receptors on myofibroblasts. Experiments could also be performed using cardiomyocytes from wildtype mice and myofibroblasts from TGF-β receptor knockout or overexpressing animals. Moreover, the question still remains as to why TGF-β1 would affect myofibroblast electrophysiology and gap junctional conductance so effectively whereas at the same time failing to affect the membrane properties of cardiomyocytes and the intercellular conductance of cardiomyocyte pairs. While the interesting results reported by Salvarani et al21 will require confirmation by other laboratories, addressing the mechanisms underlying such cell specific effects might lead to novel strategies aimed at preventing the deleterious effects of the myofibroblast on cardiomyocyte membrane potential as a therapy for arrhythmias associated with myocardial injury.
Acknowledgments
Sources of Funding: Supported in part by National Heart, Lung and Blood Institute R01 grant HL122352 NIH/NHLBI, USA and by Fondos FEDER, Spain.
Footnotes
Disclosures: None
References
- 1.Pandit SV, Anumonwo J, Jalife J. Atrial Fibrillation Susceptibility in Obesity: An Excess Adiposity and Fibrosis Complicity? Circ Res. 2016;118:1468–1471. doi: 10.1161/CIRCRESAHA.116.308686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LaFramboise WA, Scalise D, Stoodley P, Graner SR, Guthrie RD, Magovern JA, Becich MJ. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am J Physiol Cell Physiol. 2007;292:C1799–808. doi: 10.1152/ajpcell.00166.2006. [DOI] [PubMed] [Google Scholar]
- 4.Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324:475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 5.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 7.Sepulveda JL, Gkretsi V, Wu C. Assembly and signaling of adhesion complexes. Curr Top Dev Biol. 2005;68:183–225. doi: 10.1016/S0070-2153(05)68007-6. [DOI] [PubMed] [Google Scholar]
- 8.Driesen RBVF, Dispersyn GD, Thoné F, Lenders MH, Ramaekers FC, Borgers M. Structural adaptation in adult rabbit ventricular myocytes: influence of dynamic physical interaction with fibroblasts. Cell Biochem Biophys. 2006;44:119–128. doi: 10.1385/CBB:44:1:119. [DOI] [PubMed] [Google Scholar]
- 9.Goshima K. Synchronized beating of myocardial cells mediated by FL cells in monolayer culture and its inhibition by trypsin-treated FL cells. Exp Cell Res. 1971;65:161–169. doi: 10.1016/s0014-4827(71)80062-9. [DOI] [PubMed] [Google Scholar]
- 10.Rook MB, vGA, de Jonge B, el Aoumari A, Gros D, Jongsma HJ. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am J Physiol Cell Physiol. 1992;263:C959–77. doi: 10.1152/ajpcell.1992.263.5.C959. [DOI] [PubMed] [Google Scholar]
- 11.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 12.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 13.Zlochiver S, Munoz V, Vikstrom KL, Taffet SM, Berenfeld O, Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chilton L, Ohya S, Freed D, George E, Drobic V, Shibukawa Y, Maccannell KA, Imaizumi Y, Clark RB, Dixon IM, Giles WR. K+ currents regulate the resting membrane potential, proliferation, and contractile responses in ventricular fibroblasts and myofibroblasts. Am J Physiol Heart Circ Physiol. 2005;288:H2931–9. doi: 10.1152/ajpheart.01220.2004. [DOI] [PubMed] [Google Scholar]
- 15.Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sundelacruz S, Levin M, Kaplan DL. Role of membrane potential in the regulation of cell proliferation and differentiation. Stem Cell Rev. 2009;5:231–246. doi: 10.1007/s12015-009-9080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaur K, Zarzoso M, Ponce-Balbuena D, Guerrero-Serna G, Hou L, Musa H, Jalife J. TGF-beta1, Released by Myofibroblasts, Differentially Regulates Transcription and Function of Sodium and Potassium Channels in Adult Rat Ventricular Myocytes. PLoS One. 2013;8:e55391. doi: 10.1371/journal.pone.0055391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duffy HS. Fibroblasts, Myofibroblasts, and Fibrosis: Fact, fiction, and the Future. J Cardiovasc Pharmacol. 2011;57:373–375. doi: 10.1097/FJC.0b013e3182155a38. [DOI] [PubMed] [Google Scholar]
- 19.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jalife J, Kaur K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc Med. 2015;25:475–484. doi: 10.1016/j.tcm.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salvarani N, Maguy A, De Simone SA, Miragoli M, Jousset F, Rohr S. Transforming Growth Factor-β1 (TGF-β1) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ Arrhythm Electrophysiol. 2017;10:e004567. doi: 10.1161/CIRCEP.116.004567. [DOI] [PubMed] [Google Scholar]
- 22.del Castillo FJ, Cohen-Salmon M, Charollais A, Caille D, Lampe PD, Chavrier P, Meda P, Petit C. Consortin, a trans-Golgi network cargo receptor for the plasma membrane targeting and recycling of connexins. Hum Mol Genet. 2010;19:262–275. doi: 10.1093/hmg/ddp490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, Srivastava D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell. 2009;16:233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank JS, Mottino G, Chen F, Peri V, Holland P, Tuana BS. Subcellular distribution of dystrophin in isolated adult and neonatal cardiac myocytes. Am J Physiol. 1994;267:C1707–16. doi: 10.1152/ajpcell.1994.267.6.C1707. [DOI] [PubMed] [Google Scholar]
- 25.Ramos-Mondragon R, Galindo CA, Garcia-Castaneda M, Sanchez-Vargas JL, Vega AV, Gomez-Viquez NL, Avila G. Chronic potentiation of cardiac L-type Ca(2+) channels by pirfenidone. Cardiovasc Res. 2012;96:244–254. doi: 10.1093/cvr/cvs248. [DOI] [PubMed] [Google Scholar]
- 26.Pedrotty DM, Klinger RY, Kirkton RD, Bursac N. Cardiac fibroblast paracrine factors alter impulse conduction and ion channel expression of neonatal rat cardiomyocytes. Cardiovasc Res. 2009;83:688–697. doi: 10.1093/cvr/cvp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res. 2004;94:828–835. doi: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]
- 28.Quinn TA, Camelliti P, Rog-Zielinska EA, Siedlecka U, Poggioli T, O’Toole ET, Knopfel T, Kohl P. Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc Natl Acad Sci U S A. 2016;113:14852–14857. doi: 10.1073/pnas.1611184114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–511. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Kanter EM, Yamada KA. Remodeling of cardiac fibroblasts following myocardial infarction results in increased gap junction intercellular communication. Cardiovasc Pathol. 2010;19:e233–40. doi: 10.1016/j.carpath.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]