

Abstract
Pre-clinical and epidemiologic studies provide rationale for evaluating lipophilic statins for breast cancer prevention. We conducted a single-arm, biomarker modulation trial of lovastatin among women with increased risk of breast cancer. Eligibility criteria included a deleterious germline mutation in BRCA1, BRCA2, CDH1, or TP53; lifetime breast cancer risk of ≥20 % as estimated by the Claus model; or personal history of estrogen receptor and progesterone receptor-negative breast cancer. Participants received 40 mg of lovastatin orally twice daily for 6 months. We evaluated the following biomarkers before and after lovastatin use: breast duct cytology (primary endpoint), serum lipids, C-reactive protein, insulin-like growth factor-1, IGF binding protein-3, lipid peroxidation, oxidative DNA damage, 3-hydroxy-3-methylglutaryl CoA reductase genotype, and mammographic density. Thirty women were enrolled, and 26 (86.7 %) completed the study. For the primary endpoint of changes in breast duct cytology sampled by random periareolar fine needle aspiration, most participants [57.7 %, 95 % confidence interval (CI) 38.9–74.5 %] showed no change after lovastatin; 19.2 % (CI 8.1–38.3 %) had a favorable change in cytology, 7.7 % (95 % CI 1.0–25.3 %) had an unfavorable change, and 15.4 % (95 % CI 5.5–34.2 %) had equivocal results due to acellular specimens, usually after lovastatin. No significant changes were observed in secondary biomarker endpoints. The study was generally well-tolerated: 4 (13.3 %) participants did not complete the study, and one (3.8 %) required a dose reduction. This trial was technically feasible, but demonstrated no significant biomarker modulation; contributing factors may include insufficient sample size, drug dose and/or duration. The results are inconclusive and do not exclude a favorable effect on breast cancer risk.
Keywords: Breast cancer, Cancer prevention, Cancer risk reduction, BRCA1/2, Lovastatin, Clinical trial, Biomarkers, Random periareolar fine needle aspiration
Introduction
Breast cancer is the most common cancer among women in the United States, and is the second leading cause of cancer deaths in women [1]. The burden of disease is also high worldwide, with an estimated 1.4 million new cases and 458,000 deaths in 2008 [2]. Currently approved drugs for prevention of breast cancer, such as tamoxifen and raloxifene, significantly reduce a woman’s risk of developing hormone-receptor positive [estrogen receptor (ER) and/or progesterone receptor (PR)-positive] breast cancer, but not hormone-receptor negative (ER/PR-negative) breast cancer [3–5]. There is an urgent need for risk-reduction strategies directed against ER/PR-negative breast cancer, given its high incidence among young women who carry inherited cancer susceptibility gene mutations, especially BRCA1 and BRCA2 (BRCA1/2), and its poor prognosis. In lieu of large, phase III cancer prevention trials that require thousands of participants and multiple years of follow-up, short-term biomarker-based trials evaluating surrogate markers, such as cytology, have been proposed [6]. If a drug appears to be promising based on these proof-of-biological concept trials, then the substantial resource investment required for phase III trials may be worthwhile.
Chemoprevention generally requires treatment of healthy patients over several years, such that long-term tolerability must be considered when choosing a candidate drug [3, 5]. 3-Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, known as “statins,” are appealing candidates, given their proven safety profile and tolerability [7]. Pre-clinical studies have shown that lipophilic statins have a more potent effect on growth inhibition of ER-negative cell lines as opposed to ER-positive cell lines [8]. Furthermore, some epidemiologic studies reported an association of breast cancer risk reduction among lipophilic statin users [9]. Based on these findings, we conducted a prevention trial using lovastatin for modification of breast-cancer associated biomarkers in high-risk women. Our main hypothesis was that oral lovastatin, given daily at a total dose of 80 mg for 6 months, would result in a 25 % decrease in the rate of abnormal breast duct cytology (either proliferative without atypia or proliferative with atypia as measured by random periareolar fine needle aspiration (RPFNA) of breast duct cells) in women at high inherited breast cancer risk.
We studied several breast-cancer associated biomarkers related to inflammation, oxidative stress, and breast density in this clinical trial. In various epidemiologic studies, serum C-reactive protein (CRP) has been reported to be weakly associated with breast cancer risk [10], but the results have not been consistent [11]. In addition, an association between circulating insulin-like growth factor-1 (IGF-1) and breast cancer risk has been reported by several studies [12]. This relationship could possibly be modified by serum IGF binding protein-3 (IGFBP-3). Therefore, we evaluated all three inflammatory markers including CRP, IGF-1, and IGFBP-3. [13]. As surrogate markers of oxidative stress, we aimed to assay a marker of lipid peroxidation (isoprostane) and of oxidative DNA damage [8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG)], both of which can be measured in the urine [14, 15]. Mammographic density has been associated with an increase in breast cancer risk [16]. There is a reported association between mammographic density and abnormal breast duct cytology [17, 18]. For our trial, mammographic density was evaluated via methods previously described by Boyd et al. [19].
Materials and methods
Study design
We conducted a single-arm, non-randomized trial of lovastatin among women with an increased risk of developing breast cancer. Figure 1 presents the study schema. Patients underwent pre-treatment evaluation with RPFNA (described below), serum studies including complete blood count, metabolic comprehensive panel and cholesterol panel, urine and serum collection for biomarker evaluation, and a mammogram (for exclusion of suspicious findings and evaluation of density). All participants received a prescription for lovastatin, at a dose of 40 mg, which they were advised to take twice daily (a total dose of 80 mg daily) for 6 months. They subsequently underwent a post-treatment evaluation, which repeated all the tests performed pre-treatment. The primary study endpoint was breast ductal cytology, as collected by RPFNA. Secondary endpoints included serum cholesterol [total and low density lipoprotein (LDL), serum CRP, serum IGF-1, serum IGFBP-3, urine lipid peroxidation (isoprostane), urine oxidative DNA damage (8-oxo-dG), mammographic density, and HMG-CoA reductase (HMGCR) genotype]. To monitor for lovastatin toxicity, serum transaminases (aspartate aminotransferase, AST and alanine aminotransferase, ALT) and creatine phosphokinase (CPK) were evaluated pre-treatment, post-treatment, and after 3 months of lovastatin use. Lovastatin dose modification or discontinuation was required for persistently elevated serum transaminases and evidence of muscle dysfunction with or without CPK elevation. Adherence was monitored by pill counting at the end of the study. The clinical trial was registered at http://www.clinicaltrials.gov/ (ClinicalTrials.gov Identifier: NCT00285857).
Fig. 1.
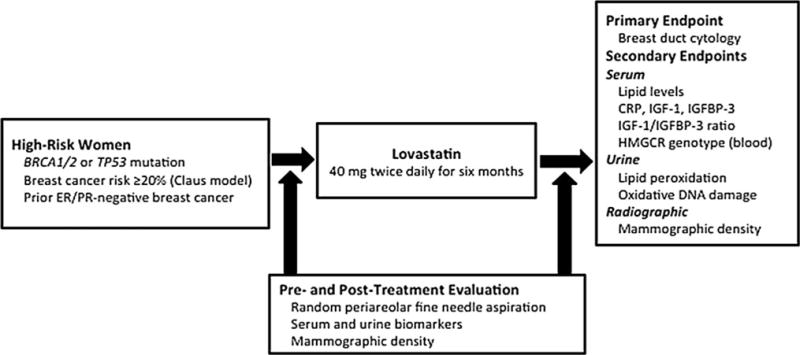
Study schema. BRCA1 and BRCA2 (BRCA1/2), estrogen receptor and progesterone receptor (ER/PR), C-reactive protein (CRP), insulin-like growth factor-1 (IGF-1), IGF binding protein-3 (IGFBP-3), 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR)
Eligibility criteria
Women were eligible to participate in the trial if they had increased risk of developing breast cancer, defined as having any of the following: (1) a deleterious germline mutation in a cancer susceptibility gene, including BRCA1/2, TP53, or CDH1; (2) a lifetime breast cancer risk of 20 % or greater, estimated by the Claus model [20]; (3) a risk-associated breast lesion including atypical hyperplasia or lobular carcinoma in situ; (4) a prior history of ER/PR-negative breast cancer, given evidence that such patients have increased risk of contralateral ER/PR-negative breast tumors [21]. Additional criteria included good performance status (Eastern Cooperative Oncology Group 0–1 [22]), normal bone marrow, renal, hepatic and muscle function, normal CPK, and no evidence of breast abnormality on mammography, magnetic resonance imaging (MRI, if clinically indicated [23]) and clinical breast examination. Patients with cancer history were eligible if they had survived at least 5 years without recurrence. Exclusion criteria were as follows: (1) evidence of breast mass or other finding suspicious for malignancy on pre-study physical exam, MRI or mammogram; (2) use of tamoxifen or raloxifene within the last 5 years; or (3) evidence of malignant cytology on RPFNA. The study protocol and informed consent document were approved by the Stanford University Institutional Review Board.
Random periareolar fine needle aspiration and cytologic evaluation
The RPFNA technique was used as previously described with certain modifications [6, 24]. All RPFNA procedures were conducted jointly by a clinical cytopathologist (EJS) and medical oncologist (AWK and SV). After local anesthesia, we performed fine needle aspiration to sample periareolar cells in the bilateral upper inner and upper outer breast quadrants (four sites in total), using either 23- or 25-gauge needles. We used a smaller gauge needle than in prior studies in order to minimize patient discomfort. Depending on the patient’s tolerance of the procedure, several passes were made in each quadrant to obtain an adequate sample. We evaluated the samples from upper and inner quadrants separately. For all samples, part of the aspirate smears was air-dried, while the remainder was fixed in methanol with 3 % acetic acid for Diff-Quik and Papanicolaou staining, respectively. A cytopathologist (EJS) examined the slides immediately after each procedure to evaluate for cellularity. We assessed cytology qualitatively and quantitatively, using the Masood semi-quantitative scale to assign a number to each specimen, with higher numbers indicating increasing degrees of abnormality, as follows: non-proliferative breast disease, 6–10; proliferative breast disease without atypia, 11–14; proliferative breast disease with atypia, 15–18; and carcinoma in situ and invasive cancer, 19–24 [25]. If no cells could be obtained after multiple RPFNA attempts, the classification was acellular. Slides were processed by the Stanford Cytopathology Laboratory. Independent cytologic review and scoring was performed for all samples by a second pathologist (KJ).
3-Hydroxy-3-methylglutaryl CoA reductase (HMGCR) receptor genotyping
Whole blood was collected at specified time points (Fig. 1) and frozen at −80 °C until batched analysis of all study samples. Genomic DNA was isolated from whole blood samples using the Gentra Puregene Blood kit (Qiagen Inc., Valencia, CA). Given prior reports that HMGCR genotype may affect serum lipid response to statin therapy, we tested participants for carriage of the A/A, A/T, or T/T genotype, with the T/T genotype associated with resistance to statins [26]. A ready-to-use single nucleotide polymorphism (SNP) genotyping assay for detecting the rs12654264 polymorphism in HMGCR was purchased from Applied Biosystems. The assay was performed using TaqMan® Universal PCR Master mix on an Applied Biosystems 7900HT Real-Time PCR System.
Serum biomarkers
Serum was collected at specified time points (Fig. 1) and frozen at −80 °C until batched analysis of all study samples. Lipid levels were measured as a drug-specific marker, since they are modulated by statins and were used as a functional pharmacodynamic marker of lovastatin delivery. Serum lipid levels, including total cholesterol, LDL, high density lipoprotein (HDL), and very low density lipoprotein (VLDL), were measured by Stanford University Hospital clinical laboratory.
In addition, CRP, IGF-1, and IGFBP-3 were evaluated as breast cancer risk-associated biomarkers, which may be modulated by lovastatin. Commercially available human enzyme-linked immunosorbent assay (ELISA) kits were used (Raybiotech Inc.) according to the manufacturer’s instructions for CRP, IGF-1, and IGFBP-3.
Urine biomarkers
Urine was collected at specified time points (Fig. 1) and frozen at −80 °C until batched analysis of all study samples. Urine lipid peroxidation was measured using the urinary isoprostane ELISA kit (Oxford Biomedical Research), which was normalized using the colorimetric microplate assay for creatinine (Oxford Biomedical Research) according to the manufacturer’s instructions. Oxidative DNA damage levels were determined using the HT 8-oxo-dG ELISA Kit (Trevigen) according to the manufacturer’s instructions.
Mammographic density
Bilateral mammography was performed in all participants at study entry and at study conclusion. Mammograms before and after lovastatin therapy were compared in order to assess for a decline in mean breast density before and after therapy. All pre- and post-lovastatin mammograms were evaluated at the same time by a radiologist specialized in breast imaging (JL). We used the American College of Radiology Breast Imaging Reporting and Data System (BI-RADS) composition system for mammographic density assessment: 1 (predominantly fatty), 2 (scattered fibroglandular densities), 3 (heterogeneously dense), and 4 (extremely dense) [27]. We used the computer-assisted Cumulus method [28, 29] for assessing mammographic breast density on a quantitative continuous scale on a subset of our patients.
Statistical methods
A pre-study sample size calculation was performed. We justified the sample size in terms of power against null hypotheses defined by published reports of historical controls. A prior randomized trial using RPFNA reported a change in cytologic category of approximately 25 % in the control arm [18]; accordingly, we concluded that a decrease of greater than 25 % would provide evidence of a treatment effect. The statistical design for the trial included two study populations: (1) women without cytologic abnormality at baseline and (2) women with cytologic abnormality at baseline. The intent was to enroll 60 women in two cohorts: women without cytologic abnormality at baseline would form the prevention cohort, and women with cytologic abnormality, defined as proliferative cytology with or without atypia, would form the treatment cohort. Twenty-four women in the atypia “treatment” cohort and 36 women in the atypia “prevention” cohort, for a total of 60 women in the study, would provide 90 % power to detect a 20–30 % absolute improvement in cytologic category (29 % for improvement from atypical to non-proliferative or proliferative without atypical cytology, and 22 % for prevention of progression from non-proliferative or proliferative without atypical to atypical cytology) after treatment with lovastatin. Given our lower than expected accrual to the trial, the endpoints were evaluated for the entire study population, instead of splitting between prevention and treatment cohorts as planned. Participant characteristics were tabulated, with descriptive statistics including means and standard deviations (SD) (for continuous data) or proportions and 95 % confidence intervals (CI) (for categorical data). Means were compared using the t test, two-sided.
Results
Participant characteristics
The trial opened to enrollment at Stanford University Hospital and Clinics in November 2005, and closed in May 2010. A total of 30 participants were enrolled; the trial was closed due to slow accrual in later years of the study, at which point we concluded that reaching the initial goal of 60 appeared infeasible. Participant characteristics are presented in Table 1. The median age was 45 years, with a range from 25 to 62 years; 54 % carried a BRCA1/2 mutation. One-third of patients had prior breast cancer, mostly ER/PR-negative. Forty percent were pre-menopausal; 10 % were taking oral contraceptive pills, 3 % were taking hormone replacement therapy, and 7 % had previously taken tamoxifen.
Table 1.
Participant characteristics
| Characteristics (N = 30) | Number | Range or % |
|---|---|---|
| Median age (years) | 45 | 25–62 |
| Ethnicity | ||
| Non-Hispanic White | 27 | 90.0 |
| NH White, Ashkenazi Jewish | 9 | 30.0 |
| NH Asian/Pacific Islander | 1 | 3.3 |
| NH Black | 1 | 3.3 |
| Hispanic | 1 | 3.3 |
| Personal history of breast cancer | 10 | 33.3 |
| Prior ER and PR-negative breast cancer | 8 | 26.7 |
| BRCA1 mutation carriera | 11 | 36.7 |
| BRCA2 mutation carriera | 5 | 16.7 |
| TP53 mutation carrier | 1 | 3.3 |
| No mutation, lifetime risk ≥20 % estimated by the Claus model | 8 | 26.7 |
| Pre-menopausal | 12 | 40.0 |
| Post-menopausal, taking hormone replacement therapy (HRT) | 1 | 3.3 |
| Post-menopausal, no HRT | 17 | 56.7 |
| Current use of oral contraceptive pills | 3 | 10.0 |
| Prior tamoxifen treatment for breast cancer | 2 | 6.7 |
One participant had both BRCA1 and BRCA2 mutations
Primary endpoint: breast duct cytology
Four participants (13.3 %) underwent an initial RPFNA and were provided with lovastatin, but never returned for evaluation. Twenty-six participants (86.7 %) had paired pre- and post-treatment cytology specimens available for analysis (Table 2). There was a median delay of 41 days (range 5–169 days) between stopping lovastatin and returning for post-treatment RPFNA. An independent cytologic review by a second pathologist (KJ) yielded no significant difference from the primary pathologist’s (EJS) evaluation. At study entry, 13 (50 %, 95 % CI 32.1–67.9 %) participants had specimens that were non-proliferative, 12 (46.2 %, 95 % CI 28.8–64.6 %) had specimens that were proliferative without atypia, and one (3.8 %, 95 % CI 0–20.5 %) had an acellular specimen. At study conclusion, 14 (53.8 %, 95 % CI 34.5–71.3 %) participants had specimens that were non-proliferative, 9 (34.6 %, 95 % CI 19.3–53.9 %) had specimens that were proliferative without atypia, and 3 (11.5 %, 95 % CI 3.2–29.8 %) had acellular specimens. Using the semi-quantitative Masood cytology scale [25], the mean score at study entry was 10.5 (SD 1.56), and the mean score at study conclusion was 10.4 (SD 1.47). The difference in post-treatment, compared to pre-treatment, cytology category was coded as unchanged, favorable (consisting of a change from proliferative without atypia to non-proliferative), unfavorable (consisting of a change from non-proliferative to proliferative without atypia), or equivocal (consisting of an acellular specimen at any time point, which could not be evaluated). Most participants (57.7 %, 95 % CI 38.9–74.5 %) had unchanged cytology; 19.2 % (95 % CI 8.1–38.3 %) had a favorable change in cytology category, 7.7 % (95 % CI 1.0–25.3 %) had an unfavorable change, and 15.4 % (95 % CI 5.5–34.2 %) were equivocal due to an acellular specimen, mostly at the post-treatment time point. No participant had atypical or malignant cytology. Examples of non-proliferative and proliferative without atypia cytology from study participants are shown in Fig. 2.
Table 2.
Primary study endpoint: breast duct cytology pre- and post-treatment with lovastatin
| ID | Entry criterion | Pre-treatment cytology (Masood score)a | Post-treatment cytology (Masood score)a | Change in cytology |
|---|---|---|---|---|
| 1 | BRCA1 mutation carrier | Non-proliferative (10) | Non-proliferative (9) | Unchanged |
| 2 | BRCA1 mutation carrier | Non-proliferative (10) | Non-proliferative (9) | Unchanged |
| 3 | BRCA1 mutation carrier | Non-proliferative (10) | Non-proliferative (9) | Unchanged |
| 4 | BRCA2 mutation carrier | Non-proliferative (10) | Non-proliferative (10) | Unchanged |
| 5 | BRCA2 mutation carrier | Proliferative without atypia (11) | Proliferative without atypia (12) | Unchanged |
| 6 | BRCA2 mutation carrier | Proliferative without atypia (11) | Proliferative without atypia (12) | Unchanged |
| 7 | BRCA1 mutation carrier | Proliferative without atypia (11) | Non-proliferative (9) | Favorable |
| 8 | BRCA1 mutation carrier | Proliferative without atypia (12) | Non-proliferative (10) | Favorable |
| 9 | BRCA2 mutation carrier | Proliferative without atypia (13) | Non-proliferative (9) | Favorable |
| 10 | BRCA1 mutation carrier | Non-proliferative (9) | Proliferative without atypia (11) | Unfavorable |
| 11 | BRCA1 and BRCA2 mutation carrier | Non-proliferative (8) | Acellular | Equivocal |
| 12 | Lifetime risk ≥20 %, Claus model | Non-proliferative (9) | Non-proliferative (10) | Unchanged |
| 13 | Lifetime risk ≥20 %, Claus model | Proliferative without atypia (13) | Proliferative without atypia (14) | Unchanged |
| 14 | Lifetime risk ≥20 %, Claus model | Proliferative without atypia (12) | Proliferative without atypia (12) | Unchanged |
| 15 | Lifetime risk ≥20 %, Claus model | Non-proliferative (9) | Non-proliferative (10) | Unchanged |
| 16 | Lifetime risk ≥20 %, Claus model | Proliferative without atypia (12) | Proliferative without atypia (11) | Unchanged |
| 17 | Lifetime risk ≥20 %, Claus model | Proliferative without atypia (13) | Non-proliferative (10) | Favorable |
| 18 | Lifetime risk ≥20 %, Claus model | Non-proliferative (10) | Proliferative without atypia (12) | Unfavorable |
| 19 | Lifetime risk ≥20 %, Claus model | Non-proliferative (8) | Acellular | Equivocal |
| 20 | Personal history of breast cancer | Non-proliferative (10) | Non-proliferative (9) | Unchanged |
| 21 | TP53 mutation carrier | Proliferative without atypia (11) | Proliferative without atypia (12) | Unchanged |
| 22 | ER/PR-negative breast cancer | Proliferative without atypia (12) | Proliferative without atypia (11) | Unchanged |
| 23 | ER/PR-negative breast cancer | Non-proliferative (8) | Non-proliferative (8) | Unchanged |
| 24 | ER/PR-negative breast cancer | Proliferative without atypia (11) | Non-proliferative (10) | Favorable |
| 25 | ER/PR-negative breast cancer | Non-proliferative (9) | Acellular | Equivocal |
| 26 | ER/PR-negative breast cancer | Acellular | Non-proliferative (9) | Equivocal |
| Summary of cytology change, comparing post-treatment to pre-treatment cytology category | ||||
|---|---|---|---|---|
|
| ||||
| Change | Number of participants (percentage, 95 % CI) | |||
| Unchanged | 15/26 (57.7 %, 38.9–74.5 %) | |||
| Favorable | 5/26 (19.2 %, 8.1–38.3 %) | |||
| Unfavorable | 2/26 (7.7 %, 1.0–25.3 %) | |||
| Equivocal | 4/26 (15.4 %, 5.5–34.2 %) | |||
Masood scale: non-proliferative breast disease, 6–10; proliferative breast disease without atypia, 11–14; proliferative breast disease with atypia, 15–18; and carcinoma in situ and invasive cancer, 19–24 [25]
Fig. 2.
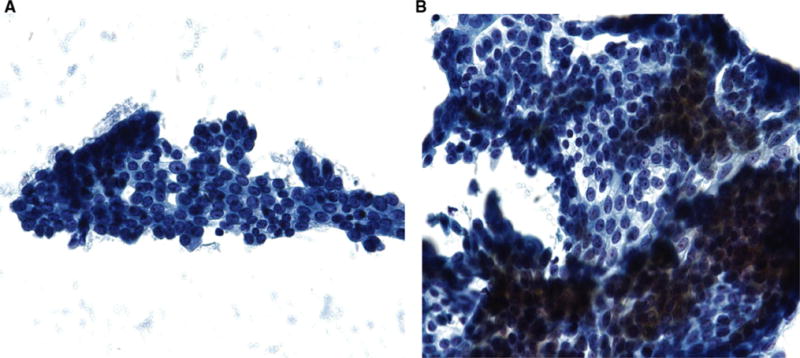
a Example of non-proliferative breast disease (Masood score 6–10) from Participant #20, and b Example of proliferative breast disease without atypia (Masood score 11–14) from Participant #18
Study adherence and lovastatin toxicity
Study adherence and lovastatin toxicity are reported in Table 3. As described above, 26/30 (86.7 %, 95 % CI 69.7–95.3 %) participants completed the study, with 4/30 (13.3 %, 95 % CI 4.7–30.3 %) who did not return after pre-treatment RPFNA and initiating lovastatin. Post-study pill counts determined that 25/26 (96.1 %, 95 % CI 79.6–100 %) participants who completed the study had taken ≥5 months of lovastatin therapy. One patient required a 50 % dose reduction because of myalgias despite normal CPK (this was a protocol-specified dose reduction, given concern for statin-associated rhabdomyolysis), and two reported dyspepsia and joint pain, which resolved spontaneously without any change in lovastatin dose. No patient required lovastatin discontinuation due to toxicity.
Table 3.
Study adherence and toxicity
| Participant numbers, N = 30 (%) | |
|---|---|
| Adherence | |
| Completed the study according to the protocol | 26 (86.7 %) |
| Did not return for post-treatment study evaluationa | 4 (13.3 %)a |
| Drug compliance (≥5 months, by pill count)b | 25 (96.1 %)b |
| Toxicity | |
| Myalgias | 1 (3.3 %) |
| CPK elevation | 0 |
| Liver transaminase elevation | 0 |
| Otherc | 2c (6.7 %) |
| Lovastatin dose-reduced per study protocold | 1d (3.3 %) |
| Lovastatin discontinued per study protocol | 0 |
Two participants were lost to follow-up despite multiple contact attempts. For the two others, one participant was dropped from the study due to inadequate initial breast cytology, and the other did not return for post-treatment evaluation, after having discontinued lovastatin due to unrelated health problems
Drug compliance for ≥5 months reported for participants who completed the study (N = 26)
Dyspepsia and joint pain in two different participants; resolved spontaneously
Dose reduced by 50 % due to myalgias
Secondary endpoints
Table 4 presents secondary endpoints, including serum and urine biomarkers, mammographic density, and HMGCR genotype. We observed little change in total cholesterol (pre-treatment mean 202, SD 45.7; post-treatment mean 194, SD 39.6) or LDL (pre-treatment mean 121, SD 34.3; post-treatment mean 115, SD 32.1). Similarly, we observed no significant post-treatment versus pre-treatment change in mean levels of CRP, IGF-1, IGFBP-3, or IGF-1/IGFBP-3 ratio. We found no difference in mean levels of urine lipid peroxidation or oxidative DNA damage post-treatment versus pre-treatment. Mean mammographic density was 2.77 (SD 1.11) pre-treatment, and 2.67 (SD 1.08) post on the BI-RADS scale. There was also no difference in the quantitative mammographic density using the computer-assisted Cumulus method post-treatment versus pre-treatment. The HMGCR genotype distribution (50.0 % A/A, 38.5 % A/T, 11.5 % T/T) was consistent with previously published reports [26], and we observed no correlation between HMGCR genotype and changes in cholesterol or LDL levels.
Table 4.
Secondary endpoints: serum, urine, and radiographic correlative studies
| Secondary endpoints | Pre-treatment Mean (SD) | Post-treatment Mean (SD) | p Value (t test) |
|---|---|---|---|
| Serum lipid levels | |||
| Total cholesterol (mg/dl) | 202 (45.7) | 194 (39.6) | 0.51 |
| LDL (mg/dl) | 121 (34.3) | 115 (32.1) | 0.53 |
| Serum markers | |||
| CRP (pg/mL | 1.22 (1.77) | 0.973 (1.22) | 0.61 |
| IGF-1 (ng/mL) | 17.4 (4.38) | 15.3 (4.96) | 0.16 |
| IGFBP-3 (ng/mL) | 19.9 (5.45) | 20.6 (5.62) | 0.69 |
| IGF-1/IGFBP-3 ratio | 0.915 (0.313) | 0.778 (0.274) | 0.15 |
| Urine markers | |||
| Lipid peroxidation (pg/mg) | 8,422 (5451) | 8,241 (6537) | 0.91 |
| Oxidative DNA damage (8-oxo-dG) (ng/mL) | 104 (84.3) | 138 (97.3) | 0.18 |
| Mammographic density, BI-RADSa | 2.77 (1.11) | 2.67 (1.08) | 0.79 |
| Genotype (%, 95 % CI) | |
|---|---|
| HMGCR genotypeb | A/A (50.0 %, 32.1–67.9 %)b |
| A/T (38.5 %, 22.4–57.5 %) | |
| T/T (11.5 %, 3.2–29.8 %) |
Lipid levels were available for 25 participants, serum markers for 20 participants, and mammographic density for 18 participants. Lipid peroxidation, oxidative DNA damage, and HMGCR genotypes were available for all 26 participants
BI-RADS composition system for mammographic density assessment: 1 (predominantly fatty), 2 (scattered fibroglandular densities), 3 (heterogeneously dense), and 4 (extremely dense) [27]
No correlation was observed with HMGCR genotype and changes in lipid levels
Discussion
We conducted a 6-month, non-randomized, biomarker modulation trial of lovastatin as a breast cancer prevention agent among women with an increased risk of developing breast cancer. The study drug and primary endpoint procedure, RPFNA for assessment of breast duct cytology, were well tolerated, with 87 % of participants completing the trial; however, enrollment proved slow, and the trial ultimately closed without reaching its accrual goal. We observed no significant change in the primary study endpoint, breast duct cytology, or in the secondary endpoints of serum and urine biomarkers involved in inflammatory and oxidative DNA damage pathways [15], or the imaging biomarker of mammographic density.
Epidemiologic and pre-clinical laboratory studies have suggested a role for statins in breast cancer risk reduction. Early observational studies reported large relative risk reductions for breast cancer among statin users [30]; however, subsequent meta-analyses have not generally confirmed such an effect [31–34]. Notably, a study in the Kaiser Permanente Northern California cohort reported a statistically significant 37 % reduction in the odds of ER/PR-negative breast cancer with lipophilic statin use [35]; two other observational studies, including the Three States Cancer Registries [36] and the Women’s Health Initiative cohort [9], reported breast cancer risk reduction with lipophilic statin use, although others did not [37]. In the laboratory, lipophilic statins selectively inhibit the growth of ER-negative cell lines as opposed to ER-positive cell lines [8]. These intriguing pre-clinical findings, coupled with a drug safety profile acceptable to healthy patients, led us and others to conduct early-stage clinical trials of statins for breast cancer risk reduction [38, 39].
Given the substantial participant numbers and lengthy follow-up period required of definitive phase III cancer prevention trials [3, 5, 40], early-stage trials with surrogate endpoints of breast duct cytology on RPFNA or ductal lavage [6, 24] or mammographic density might offer an early efficacy signal, prior to more substantial resource investment. A caveat to this approach is the potential for uncertainty in interpretation of trial results. Our findings of no effect on breast duct cytology may result from inadequate statistical power, from inadequate drug dose or treatment duration, secondary to heterogeneity in patient characteristics that affect breast cellularity including age, menopausal status, prior oral contraceptive pill or hormone replacement therapy use, prior treatment for cancer and prior oophorectomy; or, more broadly, from selection of surrogate endpoints whose modulation (or lack thereof) does not predict an agent’s impact on breast cancer development [41]. As heterogeneous patient characteristics may confound results, we would recommend excluding subjects with concomitant use of hormones or prior oophorectomy for future prevention studies. Of note, we anticipated a higher prevalence of pre-treatment atypical cytology, as has been reported among women at increased breast cancer risk [42, 43]. The absence of atypical cytology in our study may relate to the high prevalence of early menopause due to prophylactic oophorectomy or prior cancer treatment, and might have reduced our ability to observe a drug effect. To date, other biomarker-based statin trials have reported a decrease in serum estrone sulfate concentrations with statin use but no change in other biomarkers, including mammographic density [39]. An additional biomarker-based prevention trial, evaluating the use of atorvastatin is ongoing (ClinicalTrials.gov Identifier: NCT00637481).
This study illustrates several challenges in the clinical development of cancer prevention agents. Although we initially proposed accruing 60 participants, enrollment proved less brisk than we had expected despite intensive recruitment attempts, and after 5 years the trial was closed with 30 participants enrolled. This experience is consistent with under-utilization of FDA-approved breast cancer prevention agents: a recent study using the National Health Interview Survey found that only 0.08 % of women accepted prophylactic tamoxifen [44], whereas 16 % were eligible according to FDA-approval guidelines [45]. One barrier to patient and physician acceptance of prevention drugs may be the term “chemoprevention”; some have recommended that “preventive therapy” should be used instead, to minimize any association with chemotherapy [46]. The absence of a well-established surrogate endpoint may decrease enthusiasm for breast cancer prevention drugs among physicians and patients, since a patient’s benefit cannot readily be monitored [47]. In our study, it is possible that we chose a primary surrogate endpoint, breast cytomorphology, which is not modulated by lovastatin. For future prevention trials, we recommend that investigators consider preliminary pilot studies of biomarker modulation by an investigational agent. Moreover, we found that the study RPFNA procedure to collect breast duct cytology was not acceptable to many eligible women. A surprising result was the lack of significant change in cholesterol levels, which we measured as an indirect marker of lovastatin adherence and effect. Although post-study pill counts were consistent with high adherence, we experienced difficulty in insuring that participants return for timely follow-up RPFNA, such that the delay between stopping lovastatin and post-treatment evaluation was a median of 41 days (range 5–169 days). We aimed for a uniform duration of lovastatin use among participants, and therefore we did not specify that lovastatin should be continued until the post-treatment visit. This finding again reflects the challenges of obtaining protocol adherence and timely follow-up in a cancer-free study population; future cancer prevention trials would benefit from requiring post-treatment evaluation before cessation of the study drug.
In summary, a 6-month biomarker-based trial of lovastatin was technically feasible and generally well-tolerated in women at increased risk of developing breast cancer, but accrual proved difficult, and we observed no effect on the primary or secondary endpoints. These results do not exclude a potential benefit of lovastatin for breast cancer prevention. The challenges and limitations encountered may inform the design of future biomarker-based cancer prevention trials.
Acknowledgments
Support for this study was provided by a 2005 American Society of Clinical Oncology Young Investigator Award to A.W.K., a 2005 Cancer Research and Prevention Foundation Post-Doctoral Fellowship Award to A.W.K. and J.M.F., an award from the Breast Cancer Research Foundation to J.M.F, and by the Jan Weimer Memorial Research Fund at Stanford University. The authors thank all study participants.
Footnotes
Conflict of interest The authors report no conflicts of interest.
A preliminary report was presented in partial form at the American Society of Clinical Oncology Annual Meeting, Chicago IL, June 2007.
Contributor Information
Shaveta Vinayak, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA.
Erich J. Schwartz, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA
Kristin Jensen, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA.
Jafi Lipson, Department of Radiology, Stanford University School of Medicine, Stanford, CA, USA.
Elizabeth Alli, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA.
Lisa McPherson, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA.
Adrian M. Fernandez, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA
Vandana B. Sharma, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA
Ashley Staton, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA.
Meredith A. Mills, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA
Elizabeth A. Schackmann, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA
Melinda L. Telli, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA
Ani Kardashian, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA.
James M. Ford, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USADepartment of Genetics, Stanford University School of Medicine, Stanford, CA, USA
Allison W. Kurian, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA Department of Health Research and Policy, Stanford University School of Medicine, HRP Redwood Building, Room T254A, 150 Governor’s Lane, Stanford, CA 94305-5405, USA.
References
- 1.American Cancer Society. Cancer Facts and Figures. 2013 http://www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2013. Accessed Jan 2013.
- 2.Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- 3.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90(18):1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Brown PH. Translational approaches for the prevention of estrogen receptor-negative breast cancer. Eur J Cancer Prev. 2007;16(3):203–215. doi: 10.1097/CEJ.0b013e328011ed9800008469-200706000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Jr, Wade JL, 3rd, Robidoux A, Margolese RG, James J, Lippman SM, Runowicz CD, Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan VC, Wolmark N. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295(23):2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 6.Fabian CJ, Kimler BF, Mayo MS, Khan SA. Breast-tissue sampling for risk assessment and prevention. Endocr Relat Cancer. 2005;12(2):185–213. doi: 10.1677/erc.1.01000. [DOI] [PubMed] [Google Scholar]
- 7.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 8.Campbell MJ, Esserman LJ, Zhou Y, Shoemaker M, Lobo M, Borman E, Baehner F, Kumar AS, Adduci K, Marx C, Petricoin EF, Liotta LA, Winters M, Benz S, Benz CC. Breast cancer growth prevention by statins. Cancer Res. 2006;66(17):8707–8714. doi: 10.1158/0008-5472.CAN-05-4061. [DOI] [PubMed] [Google Scholar]
- 9.Cauley JA, McTiernan A, Rodabough RJ, LaCroix A, Bauer DC, Margolis KL, Paskett ED, Vitolins MZ, Furberg CD, Chlebowski RT. Statin use and breast cancer: prospective results from the Women’s Health Initiative. J Natl Cancer Inst. 2006;98(10):700–707. doi: 10.1093/jnci/djj188. [DOI] [PubMed] [Google Scholar]
- 10.Heikkila K, Harris R, Lowe G, Rumley A, Yarnell J, Gallacher J, Ben-Shlomo Y, Ebrahim S, Lawlor DA. Associations of circulating C-reactive protein and interleukin-6 with cancer risk: findings from two prospective cohorts and a meta-analysis. Cancer Causes Control. 2009;20(1):15–26. doi: 10.1007/s10552-008-9212-z. [DOI] [PubMed] [Google Scholar]
- 11.Zhang SM, Lin J, Cook NR, Lee IM, Manson JE, Buring JE, Ridker PM. C-reactive protein and risk of breast cancer. J Natl Cancer Inst. 2007;99(11):890–894. doi: 10.1093/jnci/djk202. [DOI] [PubMed] [Google Scholar]
- 12.Key TJ, Appleby PN, Reeves GK, Roddam AW. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: pooled individual data analysis of 17 prospective studies. Lancet Oncol. 2010;11(6):530–542. doi: 10.1016/S1470-2045(10)70095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49(11):1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooke MS, Olinski R, Evans MD. Does measurement of oxidative damage to DNA have clinical significance? Clin Chim Acta. 2006;365(1–2):30–49. doi: 10.1016/j.cca.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Olinski R, Rozalski R, Gackowski D, Foksinski M, Siomek A, Cooke MS. Urinary measurement of 8-OxodG, 8-OxoGua, and 5HMUra: a noninvasive assessment of oxidative damage to DNA. Antioxid Redox Signal. 2006;8(5–6):1011–1019. doi: 10.1089/ars.2006.8.1011. [DOI] [PubMed] [Google Scholar]
- 16.Ziv E, Shepherd J, Smith-Bindman R, Kerlikowske K. Mammographic breast density and family history of breast cancer. J Natl Cancer Inst. 2003;95(7):556–558. doi: 10.1093/jnci/95.7.556. [DOI] [PubMed] [Google Scholar]
- 17.Lee MM, Petrakis NL, Wrensch MR, King EB, Miike R, Sickles E. Association of abnormal nipple aspirate cytology and mammographic pattern and density. Cancer Epidemiol Biomarkers Prev. 1994;3(1):33–36. [PubMed] [Google Scholar]
- 18.Fabian CJ, Kimler BF, Brady DA, Mayo MS, Chang CH, Ferraro JA, Zalles CM, Stanton AL, Masood S, Grizzle WE, Boyd NF, Arneson DW, Johnson KA. A phase II breast cancer chemoprevention trial of oral alpha-difluoromethylornithine: breast tissue, imaging, and serum and urine biomarkers. Clin Cancer Res. 2002;8(10):3105–3117. [PubMed] [Google Scholar]
- 19.Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E, Jong RA, Hislop G, Chiarelli A, Minkin S, Yaffe MJ. Mammographic density and the risk and detection of breast cancer. N Engl J Med. 2007;356(3):227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- 20.Claus EB, Risch N, Thompson WD. Autosomal dominant inheritance of early-onset breast cancer. Implications for risk prediction. Cancer. 1994;73(3):643–651. doi: 10.1002/1097-0142(19940201)73:3<643::aid-cncr2820730323>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Kurian AW, McClure LA, John EM, Horn-Ross PL, Ford JM, Clarke CA. Second primary breast cancer occurrence according to hormone receptor status. J Natl Cancer Inst. 2009;101(15):1058–1065. doi: 10.1093/jnci/djp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, Carbone PP. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5(6):649–655. [PubMed] [Google Scholar]
- 23.Saslow D, Boetes C, Burke W, Harms S, Leach MO, Lehman CD, Morris E, Pisano E, Schnall M, Sener S, Smith RA, Warner E, Yaffe M, Andrews KS, Russell CA. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin. 2007;57(2):75–89. doi: 10.3322/canjclin.57.2.75. [DOI] [PubMed] [Google Scholar]
- 24.Fabian CJ, Kimler BF, Zalles CM, Klemp JR, Kamel S, Zeiger S, Mayo MS. Short-term breast cancer prediction by random periareolar fine-needle aspiration cytology and the Gail risk model. J Natl Cancer Inst. 2000;92(15):1217–1227. doi: 10.1093/jnci/92.15.1217. [DOI] [PubMed] [Google Scholar]
- 25.Masood S. Cytomorphology of fibrocystic change, high-risk proliferative breast disease, and premalignant breast lesions. Clin Lab Med. 2005;25(4):713–731. vi. doi: 10.1016/j.cll.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Lipkin SM, Chao EC, Moreno V, Rozek LS, Rennert H, Pinchev M, Dizon D, Rennert G, Kopelovich L, Gruber SB. Genetic variation in 3-hydroxy-3-methylglutaryl CoA reductase modifies the chemopreventive activity of statins for colorectal cancer. Cancer Prev Res (Phila) 2010;3(5):597–603. doi: 10.1158/1940-6207.CAPR-10-0007. [DOI] [PubMed] [Google Scholar]
- 27.American College of Radiology. ACR BI-RADS breast imaging and reporting data system: breast imaging atlas. 4th. American College of Radiology; Reston: 2003. [Google Scholar]
- 28.Byng JW, Boyd NF, Fishell E, Jong RA, Yaffe MJ. The quantitative analysis of mammographic densities. Phys Med Biol. 1994;39(10):1629–1638. doi: 10.1088/0031-9155/39/10/008. [DOI] [PubMed] [Google Scholar]
- 29.Heine JJ, Carston MJ, Scott CG, Brandt KR, Wu FF, Pankratz VS, Sellers TA, Vachon CM. An automated approach for estimation of breast density. Cancer Epidemiol Biomarkers Prev. 2008;17(11):3090–3097. doi: 10.1158/1055-9965.EPI-08-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cauley JA, Zmuda JM, Lui LY, Hillier TA, Ness RB, Stone KL, Cummings SR, Bauer DC. Lipid-lowering drug use and breast cancer in older women: a prospective study. J Womens Health (Larchmt) 2003;12(8):749–756. doi: 10.1089/154099903322447710. [DOI] [PubMed] [Google Scholar]
- 31.Bonovas S, Filioussi K, Tsavaris N, Sitaras NM. Statins and cancer risk: a literature-based meta-analysis and meta-regression analysis of 35 randomized controlled trials. J Clin Oncol. 2006;24(30):4808–4817. doi: 10.1200/JCO.2006.06.3560. [DOI] [PubMed] [Google Scholar]
- 32.Browning DR, Martin RM. Statins and risk of cancer: a systematic review and metaanalysis. Int J Cancer. 2007;120(4):833–843. doi: 10.1002/ijc.22366. [DOI] [PubMed] [Google Scholar]
- 33.Dale KM, Coleman CI, Henyan NN, Kluger J, White CM. Statins and cancer risk: a meta-analysis. JAMA. 2006;295(1):74–80. doi: 10.1001/jama.295.1.74. [DOI] [PubMed] [Google Scholar]
- 34.Kuoppala J, Lamminpaa A, Pukkala E. Statins and cancer: a systematic review and meta-analysis. Eur J Cancer. 2008;44(15):2122–2132. doi: 10.1016/j.ejca.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 35.Kumar AS, Benz CC, Shim V, Minami CA, Moore DH, Esserman LJ. Estrogen receptor-negative breast cancer is less likely to arise among lipophilic statin users. Cancer Epidemiol Biomarkers Prev. 2008;17(5):1028–1033. doi: 10.1158/1055-9965.EPI-07-0726. [DOI] [PubMed] [Google Scholar]
- 36.Pocobelli G, Newcomb PA, Trentham-Dietz A, Titus-Ernstoff L, Hampton JM, Egan KM. Statin use and risk of breast cancer. Cancer. 2008;112(1):27–33. doi: 10.1002/cncr.23129. [DOI] [PubMed] [Google Scholar]
- 37.Woditschka S, Habel LA, Udaltsova N, Friedman GD, Sieh W. Lipophilic statin use and risk of breast cancer subtypes. Cancer Epidemiol Biomarkers Prev. 2010;19(10):2479–2487. doi: 10.1158/1055-9965.EPI-10-0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garwood ER, Kumar AS, Baehner FL, Moore DH, Au A, Hylton N, Flowers CI, Garber J, Lesnikoski BA, Hwang ES, Olopade O, Port ER, Campbell M, Esserman LJ. Fluvastatin reduces proliferation and increases apoptosis in women with high grade breast cancer. Breast Cancer Res Treat. 2010;119(1):137–144. doi: 10.1007/s10549-009-0507-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higgins MJ, Prowell TM, Blackford AL, Byrne C, Khouri NF, Slater SA, Jeter SC, Armstrong DK, Davidson NE, Emens LA, Fetting JH, Powers PP, Wolff AC, Green H, Thibert JN, Rae JM, Folkerd E, Dowsett M, Blumenthal RS, Garber JE, Stearns V. A short-term biomarker modulation study of simvastatin in women at increased risk of a new breast cancer. Breast Cancer Res Treat. 2012;131(3):915–924. doi: 10.1007/s10549-011-1858-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goss PE, Ingle JN, Ales-Martinez JE, Cheung AM, Chlebowski RT, Wactawski-Wende J, McTiernan A, Robbins J, Johnson KC, Martin LW, Winquist E, Sarto GE, Garber JE, Fabian CJ, Pujol P, Maunsell E, Farmer P, Gelmon KA, Tu D, Richardson H. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med. 2011;364(25):2381–2391. doi: 10.1056/NEJMoa1103507. [DOI] [PubMed] [Google Scholar]
- 41.Brenner DE, Hawk E. Trials and tribulations of interrogating biomarkers to define efficacy of cancer risk reductive interventions. Cancer Prev Res (Phila) 2013;6(2):71–73. doi: 10.1158/1940-6207.CAPR-12-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fabian CJ, Zalles C, Kamel S, McKittrick R, Moore WP, Zeiger S, Simon C, Kimler B, Cramer A, Garcia F, et al. Biomarker and cytologic abnormalities in women at high and low risk for breast cancer. J Cell Biochem Suppl. 1993;17G:153–160. doi: 10.1002/jcb.240531129. [DOI] [PubMed] [Google Scholar]
- 43.Fabian CJ, Zalles C, Kamel S, Zeiger S, Simon C, Kimler BF. Breast cytology and biomarkers obtained by random fine needle aspiration: use in risk assessment and early chemoprevention trials. J Cell Biochem Suppl. 1997;28–29:101–110. [PubMed] [Google Scholar]
- 44.Waters EA, Cronin KA, Graubard BI, Han PK, Freedman AN. Prevalence of tamoxifen use for breast cancer chemoprevention among U.S. women. Cancer Epidemiol Biomarkers Prev. 2010;19(2):443–446. doi: 10.1158/1055-9965.EPI-09-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freedman AN, Graubard BI, Rao SR, McCaskill-Stevens W, Ballard-Barbash R, Gail MH. Estimates of the number of US women who could benefit from tamoxifen for breast cancer chemoprevention. J Natl Cancer Inst. 2003;95(7):526–532. doi: 10.1093/jnci/95.7.526. [DOI] [PubMed] [Google Scholar]
- 46.Cuzick J, DeCensi A, Arun B, Brown PH, Castiglione M, Dunn B, Forbes JF, Glaus A, Howell A, von Minckwitz G, Vogel V, Zwierzina H. Preventive therapy for breast cancer: a consensus statement. Lancet Oncol. 2011;12(5):496–503. doi: 10.1016/S1470-2045(11)70030-4. [DOI] [PubMed] [Google Scholar]
- 47.Meyskens FL, Jr, Curt GA, Brenner DE, Gordon G, Herberman RB, Finn O, Kelloff GJ, Khleif SN, Sigman CC, Szabo E. Regulatory approval of cancer risk-reducing (chemopreventive) drugs: moving what we have learned into the clinic. Cancer Prev Res (Phila) 2011;4(3):311–323. doi: 10.1158/1940-6207.CAPR-09-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]