

Abstract
In this study, a sensitive fluorescence sensor was developed for the detection of small, fluorescence-labeled particles dispersed in a solution. The prototype system comprises of a laser confocal optical system and a mechanical sample stage to detect photon bursting of fluorescence-labeled small particles in sample volumes less than 5 μL within 3 minutes. To examine the feasibility of the prototype system as a diagnostic tool, assemblages of rotavirus and fluorescence-labeled antibody were analyzed. The detection sensitivity for rotavirus was 1 × 104 pfu/mL. Rotavirus in stool samples from patients with acute gastroenteritis was also detected. The advantages and disadvantages of this immunosensor with respect to ELISA and RT-PCR, the current gold standards for virus detection, are discussed.
OCIS codes: (000.1430) Biology and medicine; (170.0170) Medical optics and biotechnology; (170.1610) Clinical applications; (170.3890) Medical optics instrumentation; (170.6280) Spectroscopy, fluorescence and luminescence
1. Introduction
Acute gastroenteritis is the principal cause of infant mortality worldwide with more than 1.8 million deaths per year in children of less than 5 years of age. Rotavirus is the most common pathogen found in acute gastroenteritis patients [1]. Rotavirus infection is an easily manageable disease in developed countries, but not in developing countries [2]. New rotavirus infections in adults are usually mild or even asymptomatic, usually because they have been previously exposed to the virus [3,4]. However, initial rotavirus infection in infants and young children can cause severe symptoms, sometimes resulting in death [5]. The cardinal symptoms of rotavirus infection are watery diarrhea, nausea, vomiting, fever, and abdominal pain. Additional complications may include spasms, hepatic dysfunction, acute renal failure, encephalopathy, and myocarditis.
Rotavirus is a double-stranded RNA virus belonging to the family Reoviridae. Viral particles are less than 100 nm in diameter and do not possess an envelope. The particle comprises a triple-layered icosahedral protein capsid composed of the capsid proteins VP4, VP6, and VP7, each of which exhibit independent neutralizing antigenicity. There are eight rotavirus species, A–H, based on the antigenic properties of VP6. Major group A and minor groups B and C cause gastroenteritis in humans.
Specific diagnosis of rotavirus A infection is based on the detection of viral antigen in the stool of patients using an immunochromatographic assay. This assay usually takes 20 min. Enzyme linked immunosorbent assay (ELISA) is used both for clinical diagnosis and research, but requires several hours of work in the laboratory. The reverse transcription-polymerase chain reaction (RT-PCR) has the greatest sensitivity in detecting rotavirus A and identifies all species and serotypes, but has only been employed in research laboratories [6]. In humans, rotavirus is transmitted via the fecal-oral route. Diarrheal stool from an infected patient contains 0.1–1 trillion viral particles per gram, and only 10–100 particles are sufficient to infect a susceptible individual [4,7]. These properties have made it very difficult to prevent rotavirus epidemics. To facilitate the point-of-care diagnosis of rotavirus infections, the development of a new method, which is both highly sensitive and rapid compared with conventional clinical test kits and current clinical examination devices, is considered highly desirable.
Recent studies demonstrate that several types of sensors can be used to detect viruses, including surface plasmon resonance and quartz-crystal microbalance [8]. These sensors are sensitive, but require regeneration for repeated use. In practice, the equilibration and maintenance of the sensor surface before and after use would be time-consuming and troublesome. Photon burst counting is a promising method for the rapid and sensitive detection of viruses that may overcome the inherent drawbacks of other methods.
In the early 1970s, the concept of fluorescence correlation spectroscopy (FCS) was introduced and applied to determine rates of diffusion and binding of ethidium bromide to double-stranded DNA by measuring time-dependent intensity fluctuations in the Brownian motion of ethidium bromide [9,10]. Subsequently, theoretical and quantitative studies of fluorescence fluctuation were conducted with the advent of confocal microscopy [11,12]. FCS has generated variants including those based on photon-counting histogram (PCH) [13], cross-correlation [14], scanning [15,16], imaging [17], and other approaches. However, the requirement for a substantial number of molecules to pass the focus restricts the applicability of correlation analysis when very rare targets need to be measured. The photon burst counting method, which uses confocal optics similar to those used in FCS, is more suitable for the detection of extremely large molecules and particles at low concentrations, such as DNA [18,19], bacteria [20], protein aggregates [21–24], quantum dots [25], gold nanoparticles [26], and viruses. The photon burst counting method is based on the analysis of time-dependent single-photon counts derived from the translational motion of fluorophores into and out of a small focal volume (10−15 L) defined by a focused laser beam and a confocal pinhole. As a molecule passes through this volume, it may emit a burst of fluorescence photons, which can then be collected and detected.
Here, we report the design and development of a compact immunosensor based on the photon burst counting method, which enables the detection of fluorescence signals derived from assemblages of rotavirus and fluorescence-labeled anti-VP6 antibody. The focal volume is defined by confocal illumination with a tightly focused laser beam that projects an image onto a small aperture in front of a fluorescence detector (Fig. 1). Importantly, the use of a small volume minimizes background signals originating from free fluorescent molecules, because the lower the volume, the lower the number of free fluorescent molecules. Viral particles bound to fluorescent antibodies emit a strong burst of fluorescence photons because their large assemblages will bind large numbers of antibody molecules, resulting in local concentrations much higher than those of unbound antibody. The signals of the detected photons are distinguishable from natural fluctuations in background noise. Of note, our instrument is equipped with a moving mechanical sample stage to permit scanning within a sample droplet, which is similar to the instrument used for scanning FCS [15,16]. By using a mobile stage, the focal point of the laser can be continuously moved during measurement, markedly improving detection sensitivity and reproducibility. A prototype instrument detected assemblages of rotavirus particles and fluorescence-labeled antibodies in stool suspensions within minutes without the need for a prior isolation step. Even including the incubation time for the antigen-antibody reaction, the operation can be completed within 15 minutes. Furthermore, this prototype is compact and easy to use. These properties meet the requirements for point-of-care testing and the instrument should contribute to the prevention and/or eradication of viral infections.
Fig. 1.
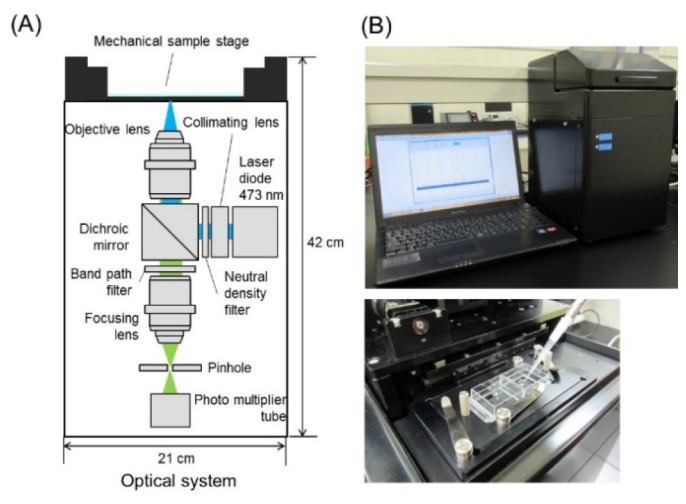
(A) A schematic overview of the fluorescence sensor system. (B) Front view of the prototype and enlarged view of the upper side (the mechanical sample stage).
2. Methods
2.1 Confocal optics for the immunosensor prototype
A confocal optics setup (a collaborative development with the R&D department, LIFETECH Co. Ltd, Saitama, Japan) is depicted in Fig. 1. A solid laser (NDA4116, Nichia Co., Ltd., Tokushima, Japan) was used as the excitation source. An excitation wavelength of 478 nm was used for all measurements, and the average power at the sample interface ranged from 15 to 25 mW. Laser light passing through a beam expander lens and a ND filter (48092, φ25 mm, Abs. 2.0 OD, Edmund Optics, NJ, USA) is reflected by a dichroic beam splitter (Semrock Di02-R488 Optical Filter, IDEX Health & Science LLC, WA) into an objective. Most experiments were performed with a water immersion objective (UPlanFLN40X/0.75, NA0.75, Olympus Co., Tokyo, Japan) and a 25 μm pinhole (S71-25, SURUGA SEIKI Co., Ltd., Shizuoka, Japan). In validation tests of the optical system, a water immersion objective (UAPON20XW340, NA0.7, Olympus Co.) and different pinhole sizes were used. The laser beam was focused approximately 0.2 mm above the bottom of a glass slide holding a 5 μL (using a custom-designed well glass slide) or 20 μL (using a normal glass slide) sample droplet. The emitted fluorescence in epi-direction was led to a band-path filter (Semrock FF01-525/45 Optical Filter) and focused onto a pinhole by a condenser lens. Photon counts were detected with a PMT (H10682-210, Hamamatsu Photonics K.K., Shizuoka, Japan) at the back of the pinhole.
2.2 Production of custom-designed well glass slides
Brosilicate glass (0.145 ± 0.015 mm thickness, cat. no. #100, Matsunami Glass IND., Ltd., Osaka, Japan) was washed by sonication with an alkaline detergent (pH 11) and rinsed with ultra-pure water and then ethanol. After drying, the glass was covered with a screen to transfer a slide surface design with eight wells of 3 mm in diameter. A hydrophobic film-forming ink paste was made from a solvent-soluble fluorine resin, tetrafluoride ethylene resin powder (ca. 5 μm in diameter), and a carbon pigment. The well pattern was printed by transfer coating of the film-forming ink onto the masked glass at a thickness of 40 μm. After removing the mask and hardening by heating at 280 °C for 15 min, the glass was cut into the shape of a slide. Glass slides were rinsed four times with ethanol and dried in a clean oven at 100 °C.
2.3 Culture of the human rotavirus KU strain
Stool specimens have been shown to contain rotaviruses by electron microscopic examination [27] and the serotypes of these rotaviruses have been shown to the correspond to those of the KU strain (VP7 serotype 1 and VP4 serotype 1A) by using a neutralizing antibody assay [28,29]. The rotaviruses with these serotypes were grown in rhesus monkey kidney (MA104) cells (ATTC #CRL-2378.1) that had been expanded in Eagle's minimal essential medium (MEM) containing 10% fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific Inc., MA). The same medium without fetal bovine serum was used for virus cultivation. Propagation of rotavirus KU strain in cell culture and quantification by the plaque assay were carried out using methods established by Urasawa and colleagues [30]. The viral titer of the rotavirus KU strain used in this experiment was 107 PFU/mL. Rotavirus inactivation with 25 kGy of γ-ray irradiation was carried out by a medical appliance company (Koga Isotope Co. Ltd., Shiga, Japan).
2.4 Preparation of anti-VP6 antibody (anti-HRV rabbit serum)
Antibody against rotavirus VP6 protein (rotavirus subgroup antigen) was prepared as previously reported [28]. Briefly, virus particles were purified and concentrated by polyethylene glycol precipitation, sucrose density gradient centrifugation, and cesium chloride (CsCl) density gradient banding [27]. Fractions containing single- and double-shelled virus particles were pooled, dialyzed against a buffered saline, and injected intravenously into weanling rabbits. Antisera were obtained 10–20 days after immunization. Antibody titers were determined by a 60% reduction of plaque formation using MA104 cells infected with approximately 150 PFU of rotavirus.
Anti-VP6 antibody was chemically labeled with a fluorescent dye (Alexa Fluor 488 Carboxylic Acid, Succinimidyl Ester, Thermo Fisher Scientific Inc.) for the measurements. Prior to reaction with the dye, IgG was purified from anti-HRV rabbit serum using the Mab Trap Kit (GE Healthcare UK Ltd., Buckinghamshire, UK). Purified IgG fraction (320 μg) was mixed with Alexa488 reagent (7 μg) in 20 μL of 1 M ammonium bicarbonate and gently shaken for 1 h at room temperature. The reagents were subsequently removed by gel filtration using PD Mini Trap G-25 (GE Healthcare UK Ltd.).
2.5 Collection and preparation of clinical samples
Between January 2012 and April 2013, fecal specimens were collected from patients who had been hospitalized for acute gastroenteritis at Kiambu County Hospital, one of the few main referral healthcare facilities in the central region of Kenya [31]. Patients enrolled in this study were children under 5 years of age who presented with acute diarrhea for not more than 7 days and had experienced an episode of three loose or watery stools in a 24 h period with or without episodes of vomiting. The samples were collected under the umbrella of a study approved by the Kenya Medical Research Institute (KEMRI)/National Ethical Review Committee (SSC No. 1324). Written informed consent was obtained from parents/guardians of the participants prior to sample collection.
A 10% fecal suspension (~1 ml) was prepared by adding about 1g of stool sample to 1ml of 0.01 M PBS (pH 7.2). Mixtures were vigorously vortexed for 40 sec followed by centrifugation at 10,000 rpm for 5 min. Supernatants were transferred into new tubes and stored at −30 °C until use in the photon counting, ELISA, and RT-PCR analyses.
2.6 Immunosensor prototype measurements
The rotavirus culture supernatant or clinical specimen was diluted using an immunoreaction enhancer (CanGetSignal solution one, Toyobo Co., Ltd., Osaka, Japan) at least ten times. Diluted samples were mixed with 0.4 μg of Alexa Fluor 488-labeled anti-VP6 antibody. After incubation for more than 10 min, 5 μL of the sample was placed onto a well glass slide set in the immunosensor prototype. The laser beam (λ = 473 nm; output power: 10 mW) was focused at 0.2 mm above the glass surface in the sample droplet. Fluorescence intensity data were collected for 3 min with spiral movement at 2 mm/sec of the mechanical sample stage. The software was coupled with the detector unit to monitor and visualize the sampling process. The sampling rate frequency was set to 1,000 samples/sec to ensure that sufficient data points were obtained for accurate analysis. The filter on the photodetector removed noise to record only signals related to the sample response.
2.7 ELISA for the detection of group A rotavirus (RVA)
Specimens were tested for group A rotavirus (RVA) antigen by ELISA as previously described [32] with slight modifications. Briefly, 100 µL of the Yo-156 monoclonal antibody, which recognizes the common RVA antigenic site in the inner capsid protein VP6, was coated onto plastic microtiter wells overnight at 4 °C. Unbound antibodies were washed with 10 mM PBS, and wells were blocked with 1% BSA in PBS with 0.05% Tween-20 (PBS-T). Samples (50 µL) were added to each well and incubated overnight at 4 °C. After removal of unbound components, 50 μL of anti-HRV rabbit serum, diluted (1:5,000) with PBS-T containing 2.5% skim milk, was added to each well and incubated for 1 h at 37 °C. After washing with PBS-T, 50 µL of peroxidase-conjugated donkey anti-rabbit IgG (H + L chains) (1:5,000) was added to each well and incubated for 1 h at 37 °C. After further washing with PBS-T, 100 µL of o-phenylenediamine was added to each well followed by incubation for 10–30 min at room temperature. Absorbance was subsequently measured at 490 nm using a microplate reader (Model 680, Bio-Rad Laboratories, Inc., CA). Specimens with an absorbance ≥ 0.3 were considered positive for group A human rotavirus. The KU strain (kindly donated by Fujita Health University, Department of Virology and Parasitology) was used as the positive control.
2.8 RT-PCR for the genotyping of rotavirus VP4 and VP7
The extracted RNA was reverse transcribed into complementary DNA (cDNA) using ReverTra Ace qPCR RT Kit (Toyobo Co., Ltd.). The cDNA was then amplified in a two-step multiplexed semi-nested RT-PCR to determine the G and P genotypes of RVA strains using the KOD-Plus-Ver.2 high fidelity DNA polymerase kit (Toyobo Co., Ltd.) as described previously with some modifications [33,34]. The amplified product was subjected to 1.2% agarose gel electrophoresis and visualized by staining with ethidium bromide.
3. Results and discussion
3.1 Quantification of fluorescent photon burst counts
To detect quantitatively the signal of labeled virus assemblages over the fluorescent background of unbound antibody molecules, emitted photons were counted and summed over time intervals of constant length (1ms). When intensely fluorescent targets are present, an additional component of high fluorescence photons is observed (Fig. 2), which is quantified by the number of channels above a threshold value to distinguish target signals from background photons. The threshold value (T) can be expressed as follows,
| (1) |
where B is the average value of the background photons, N is the coefficient, and σ represents the standard deviation of the background photons. When the value of N was varied from 3 to 7 in each experiment described below, the photon counts above a threshold gradually decreased with the increase in σ, which is shown by the reduction in sensitivity. But the error became larger at low σ, because certain noises in the data could not be completely eliminated. When N was set at 5, the errors of each point were small and the detection limit was the lowest for rotavirus detection (data not shown). Taking into account both sensitivity and reproducibility, we selected 5 as the value of σ, and the threshold value was set as B + 5 × σ. The number of channels in which photon numbers above the threshold were detected was defined as the burst counts for each data point in subsequent experiments.
Fig. 2.
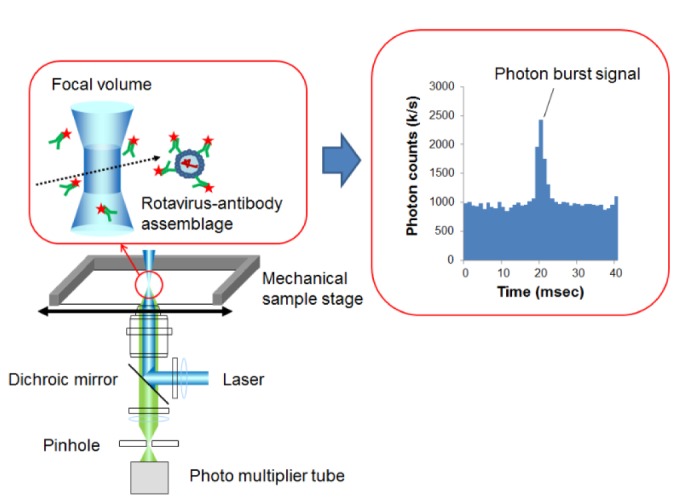
Principle of detection for rotavirus-antibody assemblages by the photon burst counting.
3.2 Installation of the mechanical sample stage
Slow diffusion is one of the factors limiting detection of large molecule complexes at low concentrations. This problem can be attenuated by increasing the frequency of particles passing through the observation volume either by making the sample solution flow or moving the sample droplet during data acquisition. Confocal detection of flowing samples is performed using a microfluidic channel that allows particle motion [35,36]. However, this method has disadvantages when it is used to handle clinical specimens; first, infectious samples may contaminate the flow channel and second, clinical samples with high viscosity, such as nasal swabs, throat swabs, and stool samples, will show high resistance to flow in a microfluidic channel. Instead of making the particles flow, an optical system equipped with a positioning table enabling scanning inside a sample droplet can be employed [16,17].
Our prototype device used a similar mechanism of scanning within a sample droplet by employing single-color fluorescence detection. The mechanical sample stage of the immunosensor prototype was controlled so that the focal volume moved in a spiral pattern from the center to the periphery of a sample droplet, thereby allowing it to be scanned. The mechanical sample stage was equipped with stepper motors operating at 5000 steps/mm. The spiral motion with a speed of over 1 mm/sec increased the total number of detected burst counts when analyzing fluorescent polystyrene (PS) beads (100 nm in diameter) (data not shown). Accordingly, the scanning speed was set to 2 mm/sec in the following experiments.
To save the sample volume, we produced a custom-designed well glass slide that was optimized for our instrument (Fig. 3). Eight wells of 3 mm in diameter were printed onto the glass slide (Fig. 3(A) and 3(B)). The glass surface, with the exception of the wells, was covered with a hydrophobic ink, showing a water contact angle of 130° (data not shown). The well can hold at its center a small amount of sample as a droplet by virtue of its internal hydrophilicity and external water repellent properties (Fig. 3(C)). Using our glass slide, we obtained results using a 5 μL sample volume that were equivalent those obtained using a 20 μL sample droplet on a normal glass slide (data not shown).
Fig. 3.

(A) A schema of the custom-designed well glass slide. (B) Upper view of the product. (C) Droplet shapes on normal glass (left) and the product (right).
3.3 Arrangement of confocal optics
A standard confocal optics setup (Fig. 1) equipped with a water immersion objective (UPAPLO 40 × W340 or UAPO 20 × W340, Olympus) was used. Optical fluorescence signals were collected by the photon-counting detector when fluorescence particles passed through the focal volume of the excitation laser beam. The size of the focal volume was optimized by varying both the objective lens magnification and the confocal pinhole diameter. An adequate confocal size defining the focal volume was determined using fluorescent PS beads (100 nm in diameter). Multiple combinations of these factors provided a confocal size of between 0.63 and 20 μm in diameter, and the largest burst counts were obtained using a confocal size of 10 μm (Fig. 4(A)). When fluorescent PS beads were synchronized with a small fluorescent molecule (fluorescein) as a noise-producing material, the signal-to-noise ratio (the ratio of detected photons of burst peaks to background photons) increased as the confocal size became narrower (Fig. 4(B)). This finding indicates that the optimal confocal size should be determined in each case depending on the size difference between the fluorescent particle of interest and the noise-producing fluorescent material. This was the case in the present study, where assemblages of virus and fluorescence-labeled antibody coexisted with the free fluorescent antibody. For rotavirus detection, a confocal size of 0.63 μm in diameter provided optimal results (data not shown).
Fig. 4.

(A) The burst counts of fluorescent polystyrene beads (106 particles/mL, 100 nm in diameter) with different confocal region sizes (n = 3). The confocal regions were formed by a combination of the pinhole size (diameter) and the objective lens magnification, indicated beneath each bar. (B) Estimation of the signal-to-noise ratio with different confocal region sizes. A solution containing fluorescent polystyrene beads (106 particles/mL) and fluorescein (10 nM) was assayed. Ratios of the photon burst intensity to the noise level were obtained.
3.4 Detection of assemblages of rotavirus and fluorescence-labeled antibody
To evaluate the sensitivity of rotavirus detection, the fluorescence-labeled anti-VP6 antibody was mixed with various concentrations of rotavirus-infected (KU strain) cell culture samples. A sample droplet was placed onto a well glass slide above the objective lens and moved with the mechanical sample stage at a speed of 2 mm/sec. The spiral motion of the mechanical sample stage generated a continuous inward movement of the observation volume.
Assemblages of rotavirus and fluorescence-labeled antibody displayed conspicuous fluorescence caused by multiple binding of the labeled antibody. We observed multiple peaks derived from virus-antibody assemblages passing through the observation volume (Fig. 5). Photon counts correlated well with the increase in rotavirus concentration, producing a smooth curve over three orders of magnitude (Fig. 6(A)). When the measurement was independently repeated three times for each sample with the same concentration, the standard deviation was within 20%, suggesting that the reproducibility was at least sufficient for semi-quantitative evaluation. No fluorescence photon bursts were detected when the motion of the sample stage was stopped, indicating that rotavirus particle diffusion did not occur rapidly enough for photon burst detection in the absence of scanning motion. Rotavirus inactivated by heating at 60 °C for 3 h or γ-ray irradiation with 25 kGy produced burst counts equivalent to those of untreated samples; however, samples treated with a detergent (0.02% Triton-X100) did not produce any photon bursts (Fig. 6(B)). This indicates that our method can detect intact viral particles, but not detergent-treated virus, most likely because the VP-6 antigen becomes disorganized in the presence of detergent. This means that the photon burst counts could not be detected without large virus-antibody assemblages, even though the antigen was recognized by the antibody.
Fig. 5.
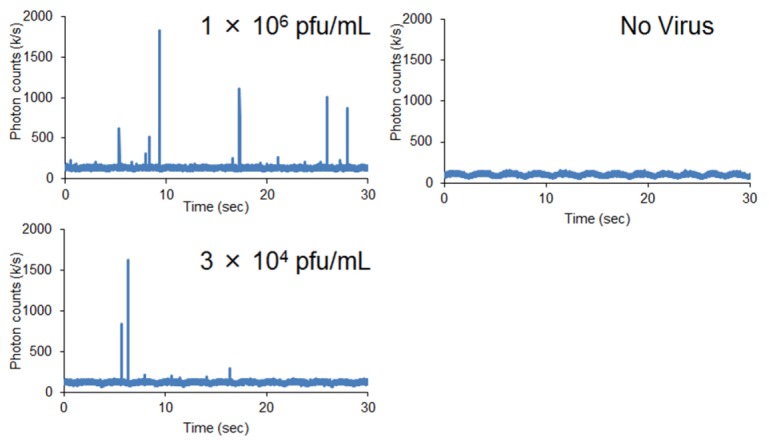
Voltage outputs produced by the emission of fluorescence photons in samples containing rotavirus (1 × 106, and 3 × 104 pfu/mL) (left) or no virus (right) and a fluorescence-labeled anti-VP6 antibody.
Fig. 6.

(A) The burst counts of rotavirus bound to a fluorescence-labeled anti-VP6 antibody with (red line) and without (blue line) motion of the sample stage (n = 3). (B) The burst counts of rotavirus inactivated by heating at 60 °C for 3 h (blue line), γ-ray irradiation with 25 kGy (red line), or treatment with a detergent (0.02% Triton-X100; green line) (n = 3).
The limit of quantitative detection of rotavirus was 1 × 104 pfu/mL, which resulted in burst counts significantly different from those of a negative control sample (n = 5, p<0.05). Evaluation of the sensitivity of the ELISA for the rotavirus KU strain resulted in a detection limit of 3.3 × 105 pfu/mL (data not shown). Although the same antibody was used in both the instrument and ELISA experiments, our system had a one order of magnitude higher detection sensitivity than that of ELISA.
3.5 Detection of rotavirus in stool specimens from patients with diarrhea
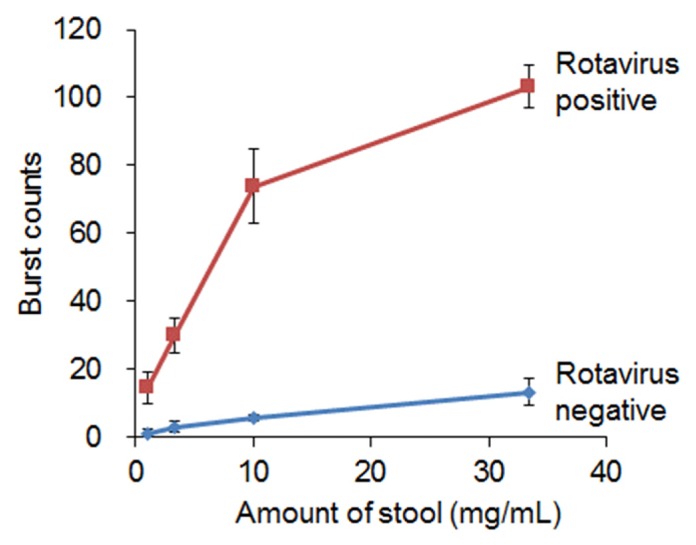
We next assessed whether the prototype instrument could detect burst counts in assemblages of fluorescent anti-VP6 antibody and rotavirus derived from stool specimens. Stool specimens included positive and negative rotavirus samples, both of which were confirmed by ELISA and RT-PCR. The limit of detection of RVA by semi-nested RT-PCR was 101 PFU/ml for the VP7 gene and 102 PFU/ml for the VP4 gene (data not shown). Serial dilution of rotavirus-positive samples produced burst counts that correlated with virus concentration, whereas rotavirus-negative samples produced very few burst counts (Fig. 7). Subsequently, we examined 183 stool specimens consisting of 120 rotavirus-positive samples and 63 negative samples (confirmed by ELISA and RT-PCR). In Fig. 8, samples are plotted according to the order of higher burst count, and rotavirus-positive and rotavirus-negative specimens (based on RT-PCR) are indicated in red and blue, respectively. The burst counts for all of the virus-negative specimens, with the exception of one sample, were below 15, while most of the virus-positive specimens (37 of 120) showed the burst counts above 15, suggesting that the detection limit of our method for rotavirus in stool specimens is approximately 15 burst counts, which is equivalent to 1 × 105 pfu/mL (Fig. 6(A)). The sensitivity of virus detection in stool samples was 10-fold lower than that in culture samples. This discrepancy may be due to the thermal stability of rotavirus particles, as our system can only detect intact viral particles (Fig. 6(B)). This feature has both advantages and disadvantages. Our system can determine the quantity of active viruses with intact envelopes, but is unable to detect all existing antigens in a sample. Clinically relevant ranges of virus detection are 103–106 viral particles per mL for HIV-1 [37], 101−106 pfu/mL for Ebola virus [38], and 101−104 pfu/mL for dengue virus [39], which are, at least partially, within the range of detection of our system.
Fig. 7.
Burst counts of rotavirus bound to a fluorescence-labeled anti-VP6 antibody in a positive stool specimen (red line) and a rotavirus-negative stool specimen (blue line) (n = 3).
Fig. 8.
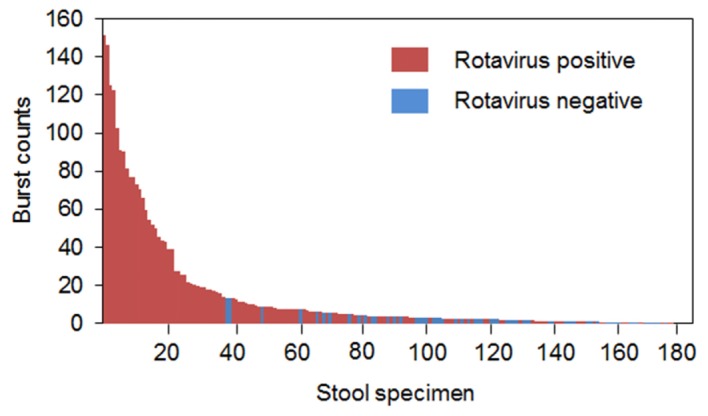
A waterfall plot of the burst counts of each of 183 stool specimens (diluted 5-fold). Red and blue bars indicate rotavirus-positive and rotavirus-negative samples, respectively.
4. Conclusions
In the present study, we designed and developed an optical sensor that detects photon bursting derived from fluorescent particles. The instrument comprises a laser confocal system and a mechanical sample stage. The high sensitivity of the photomultiplier tube allowed for the detection of particles in the submicron range. Assemblages of rotavirus mixed with fluorescence-labeled antibody were analyzed. Our method requires less than 5 μL of sample, uses a custom-designed well glass slide, and is rapid, requiring only an assay time of 3 minutes. We successfully constructed a compact and relatively low-cost prototype. Using this instrument, viral signals from clinical specimens were also successfully detected.
The new immunosensor has several advantages. First, less than 100 virus particles can be detected in a 5 μL droplet on a custom-designed well glass slide. Second, this sensor does not come into contact with the sample and is therefore applicable to clinical specimens containing infectious pathogens. Third, the assay is rapid because there is no need for prior separation of fluorescent antibody-bound virus from the free fluorescent antibody, which makes the method suitable for automation. These features make the method superior to ELISA in terms of sensitivity, rapidness, ease-of-use, and cost. The next step will be to design a compact, automated system for the diagnosis of other diseases. This would be particularly useful for point-of-care testing and for epidemic prevention.
Disclosures
O.T.: LIFETECH Co. Ltd. (F,E,P), H.S.: Matsunami Glass IND. Ltd. (F,E,P)
Acknowledgments
We thank the Director of Kenya Medical Research Institute (KEMRI) for providing technical support for our project activities.
Funding
This work was supported by the Adaptable and Seamless Technology Transfer Program through Target-driven R&D, Japan Science and Technology Agency. The collaborative work with Kenya Station, Nagasaki University was supported by the Joint Usage / Research Center on Tropical Disease, Institute of Tropical Medicine, Nagasaki University (2014-Ippan-8, 2015-Ippan-13).
References and links
- 1.Bernstein D. I., “Rotavirus overview,” Pediatr. Infect. Dis. J. 28(3 Suppl), S50–S53 (2009). 10.1097/INF.0b013e3181967bee [DOI] [PubMed] [Google Scholar]
- 2.Simpson E., Wittet S., Bonilla J., Gamazina K., Cooley L., Winkler J. L., “Use of formative research in developing a knowledge translation approach to rotavirus vaccine introduction in developing countries,” BMC Public Health 7(1), 281 (2007). 10.1186/1471-2458-7-281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glass R. I., Parashar U. D., Bresee J. S., Turcios R., Fischer T. K., Widdowson M. A., Jiang B., Gentsch J. R., “Rotavirus vaccines: current prospects and future challenges,” Lancet 368(9532), 323–332 (2006). 10.1016/S0140-6736(06)68815-6 [DOI] [PubMed] [Google Scholar]
- 4.Bishop R. F., “Natural history of human rotavirus infection,” Arch. Virol. Suppl. 12, 119–128 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Tate J. E., Burton A. H., Boschi-Pinto C., Steele A. D., Duque J., Parashar U. D., WHO-coordinated Global Rotavirus Surveillance Network , “2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis,” Lancet Infect. Dis. 12(2), 136–141 (2012). 10.1016/S1473-3099(11)70253-5 [DOI] [PubMed] [Google Scholar]
- 6.Fischer T. K., Gentsch J. R., “Rotavirus typing methods and algorithms,” Rev. Med. Virol. 14(2), 71–82 (2004). 10.1002/rmv.411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimwood K., Lambert S. B., “Rotavirus vaccines: opportunities and challenges,” Hum. Vaccin. 5(2), 57–69 (2009). 10.4161/hv.5.2.6924 [DOI] [PubMed] [Google Scholar]
- 8.Amano Y., Cheng Q., “Detection of influenza virus: traditional approaches and development of biosensors,” Anal. Bioanal. Chem. 381(1), 156–164 (2005). 10.1007/s00216-004-2927-0 [DOI] [PubMed] [Google Scholar]
- 9.Magde D., Elson E. L., Webb W. W., “Fluorescence correlation spectroscopy. II. An experimental realization,” Biopolymers 13(1), 29–61 (1974). 10.1002/bip.1974.360130103 [DOI] [PubMed] [Google Scholar]
- 10.Elson E. L., Magde D., “Fluorescence correlation spectroscopy. I. Conceptual basis and theory,” Biopolymers 13(1), 1–27 (1974). 10.1002/bip.1974.360130102 [DOI] [PubMed] [Google Scholar]
- 11.Qian H., Elson E. L., “Analysis of confocal laser-microscope optics for 3-D fluorescence correlation spectroscopy,” Appl. Opt. 30(10), 1185–1195 (1991). 10.1364/AO.30.001185 [DOI] [PubMed] [Google Scholar]
- 12.Rigler R., Mets Ü., Widengren J., Kask P., “Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion,” Eur. Biophys. J. 22(3), 169–175 (1993). 10.1007/BF00185777 [DOI] [Google Scholar]
- 13.Chen Y., Müller J. D., So P. T., Gratton E., “The photon counting histogram in fluorescence fluctuation spectroscopy,” Biophys. J. 77(1), 553–567 (1999). 10.1016/S0006-3495(99)76912-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koltermann A., Kettling U., Bieschke J., Winkler T., Eigen M., “Rapid assay processing by integration of dual-color fluorescence cross-correlation spectroscopy: high throughput screening for enzyme activity,” Proc. Natl. Acad. Sci. U.S.A. 95(4), 1421–1426 (1998). 10.1073/pnas.95.4.1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petersen N. O., “Scanning fluorescence correlation spectroscopy. I. Theory and simulation of aggregation measurements,” Biophys. J. 49(4), 809–815 (1986). 10.1016/S0006-3495(86)83709-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen N. O., Johnson D. C., Schlesinger M. J., “Scanning fluorescence correlation spectroscopy. II. Application to virus glycoprotein aggregation,” Biophys. J. 49(4), 817–820 (1986). 10.1016/S0006-3495(86)83710-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petersen N. O., Höddelius P. L., Wiseman P. W., Seger O., Magnusson K. E., “Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application,” Biophys. J. 65(3), 1135–1146 (1993). 10.1016/S0006-3495(93)81173-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Ying L., Green J. J., Balasubramanian S., Klenerman D., “Ultrasensitive Coincidence Fluorescence Detection of Single DNA Molecules,” Anal. Chem. 75(7), 1664–1670 (2003). 10.1021/ac026367z [DOI] [PubMed] [Google Scholar]
- 19.Zhang C.-Y., Johnson L. W., “Homogenous rapid detection of nucleic acids using two-color quantum dots,” Analyst (Lond.) 131(4), 484–488 (2006). 10.1039/b514309h [DOI] [PubMed] [Google Scholar]
- 20.Edel J. B., Lahoud P., Cass A. E. G., deMello A. J., “Discrimination Between Single Escherichia Coli Cells Using Time-Resolved Confocal Spectroscopy,” J. Phys. Chem. B 111(5), 1129–1134 (2007). 10.1021/jp066267n [DOI] [PubMed] [Google Scholar]
- 21.Nalefski E. A., D’Antoni C. M., Ferrell E. P., Lloyd J. A., Qiu H., Harris J. L., Whitney D. H., “Single-Molecule Detection for Femtomolar Quantification of Proteins in Heterogeneous Immunoassays,” Clin. Chem. 52(11), 2172–2175 (2006). 10.1373/clinchem.2006.072850 [DOI] [PubMed] [Google Scholar]
- 22.Bieschke J., Giese A., Schulz-Schaeffer W., Zerr I., Poser S., Eigen M., Kretzschmar H., “Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets,” Proc. Natl. Acad. Sci. U.S.A. 97(10), 5468–5473 (2000). 10.1073/pnas.97.10.5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giese A., Bader B., Bieschke J., Schaffar G., Odoy S., Kahle P. J., Haass C., Kretzschmar H., “Single particle detection and characterization of synuclein co-aggregation,” Biochem. Biophys. Res. Commun. 333(4), 1202–1210 (2005). 10.1016/j.bbrc.2005.06.025 [DOI] [PubMed] [Google Scholar]
- 24.Kostka M., Högen T., Danzer K. M., Levin J., Habeck M., Wirth A., Wagner R., Glabe C. G., Finger S., Heinzelmann U., Garidel P., Duan W., Ross C. A., Kretzschmar H., Giese A., “Single Particle Characterization of Iron-Induced Pore-Forming α-Synuclein Oligomers,” J. Biol. Chem. 283(16), 10992–11003 (2008). 10.1074/jbc.M709634200 [DOI] [PubMed] [Google Scholar]
- 25.Zhang C. Y., Johnson L. W., “Simple and accurate quantification of quantum dots via single-particle counting,” J. Am. Chem. Soc. 130(12), 3750–3751 (2008). 10.1021/ja711493q [DOI] [PubMed] [Google Scholar]
- 26.Lan T., Wang J., Dong C., Huang X., Ren J., “Homogeneous immunoassays by using photon burst counting technique of single gold nanoparticles,” Talanta 132, 698–704 (2015). 10.1016/j.talanta.2014.10.004 [DOI] [PubMed] [Google Scholar]
- 27.Taniguchi K., Urasawa S., Urasawa T., “Electrophoretic analysis of RNA segments of human rotaviruses cultivated in cell culture,” J. Gen. Virol. 60(1), 171–175 (1982). 10.1099/0022-1317-60-1-171 [DOI] [PubMed] [Google Scholar]
- 28.Urasawa S., Urasawa T., Taniguchi K., “Three human rotavirus serotypes demonstrated by plaque neutralization of isolated strains,” Infect. Immun. 38(2), 781–784 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urasawa S., Urasawa T., Taniguchi K., Chiba S., “Serotype determination of human rotavirus isolates and antibody prevalence in pediatric population in Hokkaido, Japan,” Arch. Virol. 81(1-2), 1–12 (1984). 10.1007/BF01309292 [DOI] [PubMed] [Google Scholar]
- 30.Urasawa T., Urasawa S., Taniguchi K., “Sequential passages of human rotavirus in MA-104 cells,” Microbiol. Immunol. 25(10), 1025–1035 (1981). 10.1111/j.1348-0421.1981.tb00109.x [DOI] [PubMed] [Google Scholar]
- 31.Wandera E. A., Mohammad S., Komoto S., Maeno Y., Nyangao J., Ide T., Kathiiko C., Odoyo E., Tsuji T., Taniguchi K., Ichinose Y., “Molecular epidemiology of rotavirus gastroenteritis in Central Kenya before vaccine introduction, 2009-2014,” J. Med. Virol. 89(5), 809–817 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Taniguchi K., Urasawa T., Morita Y., Greenberg H. B., Urasawa S., “Direct serotyping of human rotavirus in stools by an enzyme-linked immunosorbent assay using serotype 1-, 2-, 3-, and 4-specific monoclonal antibodies to VP7,” J. Infect. Dis. 155(6), 1159–1166 (1987). 10.1093/infdis/155.6.1159 [DOI] [PubMed] [Google Scholar]
- 33.Gouvea V., Glass R. I., Woods P., Taniguchi K., Clark H. F., Forrester B., Fang Z. Y., “Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens,” J. Clin. Microbiol. 28(2), 276–282 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taniguchi K., Urasawa T., Kobayashi N., Gorziglia M., Urasawa S., “Nucleotide sequence of VP4 and VP7 genes of human rotaviruses with subgroup I specificity and long RNA pattern: implication for new G serotype specificity,” J. Virol. 64(11), 5640–5644 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson J., Chen Y., Mueller J. D., “Characterization of brightness and stoichiometry of bright particles by flow-fluorescence fluctuation spectroscopy,” Biophys. J. 99(9), 3084–3092 (2010). 10.1016/j.bpj.2010.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magde D., Webb W. W., Elson E. L., “Fluorescence correlation spectroscopy. III. Uniform translation and laminar flow,” Biopolymers 17(2), 361–376 (1978). 10.1002/bip.1978.360170208 [DOI] [Google Scholar]
- 37.Mellors J. W., Rinaldo C. R., Jr, Gupta P., White R. M., Todd J. A., Kingsley L. A., “Prognosis in HIV-1 infection predicted by the quantity of virus in plasma,” Science 272(5265), 1167–1170 (1996). 10.1126/science.272.5265.1167 [DOI] [PubMed] [Google Scholar]
- 38.Towner J. S., Rollin P. E., Bausch D. G., Sanchez A., Crary S. M., Vincent M., Lee W. F., Spiropoulou C. F., Ksiazek T. G., Lukwiya M., Kaducu F., Downing R., Nichol S. T., “Rapid diagnosis of Ebola hemorrhagic fever by reverse transcription-PCR in an outbreak setting and assessment of patient viral load as a predictor of outcome,” J. Virol. 78(8), 4330–4341 (2004). 10.1128/JVI.78.8.4330-4341.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de la Cruz-Hernández S. I., Flores-Aguilar H., González-Mateos S., López-Martinez I., Alpuche-Aranda C., Ludert J. E., del Angel R. M., “Determination of viremia and concentration of circulating nonstructural protein 1 in patients infected with dengue virus in Mexico,” Am. J. Trop. Med. Hyg. 88(3), 446–454 (2013). 10.4269/ajtmh.12-0023 [DOI] [PMC free article] [PubMed] [Google Scholar]