

Abstract
The catalytic activation of protein kinases requires precise positioning of key conserved catalytic and regulatory motifs in the kinase core. The Regulatory Spine (RS) is one such structural motif that is dynamically assembled upon kinase activation. The RS is also a mutational hotspot in cancers; however, the mechanisms by which cancer mutations impact RS assembly and kinase activity are not fully understood. In this study, through mutational analysis of patient derived mutations in the RS of EGFR kinase, we identify an activating mutation, M766T, at the RS3 position. RS3 is located in the regulatory αC-helix, and a series of mutations at the RS3 position suggest a strong correlation between the amino acid type present at the RS3 position and ligand (EGF) independent EGFR activation. Small polar amino acids increase ligand independent activity, while large aromatic amino acids decrease kinase activity. M766T relies on the canonical asymmetric dimer for full activation. Molecular modeling and molecular dynamics simulations of WT and mutant EGFR suggest a model in which M766T activates the kinase domain by disrupting conserved autoinhibitory interactions between M766 and hydrophobic residues in the activation segment. In addition, a water mediated hydrogen bond network between T766, the conserved K745-E762 salt bridge, and the backbone amide of the DFG motif is identified as a key determinant of M766T-mediated activation. M766T is resistant to FDA approved EGFR inhibitors such as gefitinib and erlotinib, and computational estimation of ligand binding free energy identifies key residues associated with drug sensitivity. In sum, our studies suggest an unusual mode of RS assembly and oncogenic EGFR activation, and provide new clues for the design of allosteric protein kinase inhibitors.
Graphical Abstract
Eukaryotic protein kinases (EPKs) are a large family of signaling proteins that propagate cellular signals through the controlled phosphorylation of serine, threonine, and tyrosine residues on protein substrates.1,2 EPKs share a conserved structural fold consisting of an N-terminal ATP binding lobe (N-lobe) and a C-terminal substrate binding lobe (C-lobe).2 The N-lobe of the kinase domain contains five β-strands and the regulatory αC-helix,1–3 while the C-lobe of the kinase domain is mainly helical and serves as a scaffold for protein docking and substrate recognition.1–4 ATP binding and phosphoryl-transfer occur at the catalytic cleft located between the N and C-lobes.2,3 Drug discovery efforts focused on the catalytic cleft of protein kinases have identified several ATP-competitive small molecule inhibitors, some of which are currently used in a clinical setting to target abnormally regulated kinases in human cancers.5,6 While some cancer patients respond better to kinase targeted small molecule therapy, others display resistance due, in part, to accumulation of mutations in the protein kinase domain.7–10 Indeed, large scale cancer genome sequencing studies have revealed thousands of nonsynonymous (amino acid altering) mutations in the protein kinase domain.11–14 However, an incomplete understanding of how (or if) these mutations impact kinase activity, regulation, and drug sensitivity have hindered progress in translating genomic discoveries into therapeutic strategies.
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase (RTK) that is frequently mutated in many human cancers.1 Nearly 800 unique EGFR mutations have been catalogued in various databases.15–17 Although the structural and functional impacts of many of these mutations are poorly understood, a detailed mechanistic understanding of EGFR activation in normal cells is beginning to provide a conceptual framework for investigating mutational EGFR regulation in diseases.18 Structural and functional studies have shown that EGFR is activated by dimerization in which the C-lobe of one kinase (activator) activates the other (receiver) by docking to its N-lobe.18 Upon dimerization, the receiver kinase is activated through stabilization of key flexible elements in the catalytic core. In particular, the flexible αC-helix, which is part of the dimer interface, undergoes a conformational change such that a conserved glutamate (E768) from the αC-helix forms a salt bridge with the ATP coordinating catalytic lysine (K745).18 Likewise, a long flexible loop connecting the ATP and substrate binding lobes, termed the activation loop (A-loop), undergoes conformational changes to prime the kinase domain for substrate binding and phosphorylation.18 These conformational changes are critical for kinase activation, and some of the frequently occurring oncogenic mutations in EGFR, such as L858R (numbering scheme corresponds to human EGFR with 24aa signaling peptide), contribute to abnormal activation and drug sensitivity by altering these conformational changes.10,18,19
Advances in conformational sampling algorithms and computer hardware have enabled long-time-scale simulations on kinases, providing insights into EGFR mutational activation and drug sensitivity.20–28 For example, microsecond-time-scale simulations revealed that the regulatory αC-helix in EGFR is intrinsically disordered and the L858R oncogenic mutation activates the kinase domain by quenching intrinsic disorder.20 Likewise, ensemble molecular dynamics simulations suggest that oncogenic mutations contribute to drug sensitivity and EGFR activation by altering the relative stability and conformational transition of the kinase domain.25 Free energy based methods have also provided insights into kinase mutational activation and drug sensitivity. Computational estimation of free energy difference in various receptor tyrosine kinases suggests that the inactive states are destabilized more than the active state by oncogenic mutations.29 Analysis of the free energy landscape of the drug resistant gatekeeper mutation (T790M) identified the χ1 angle dynamics of the gatekeeper residue as the key determinant of drug binding preference.24 More recently, a repository of computationally derived EGFR mutant structures and their associated affinity for gefitinib and erlotinib have been reported.28
The wealth of crystal structure data available on protein kinases has also enabled structural bioinformatics approaches in the study of kinase activation and regulation. In particular, comparisons of the active and inactive conformations of various kinases using spatial pattern matching techniques identified the Regulatory Spine (RS), a set of 4 nonconsecutive residues spanning the ATP and substrate binding regions, as a key spatial motif for kinase regulation.30–32 The RS is formed by a contiguous network of hydrophobic interactions (RS1, RS2, RS3, and RS4) that are structurally assembled in the active state, but disassembled in the inactive state (Figure 1),30,32,33 though some exceptions to this rule have been noted.34,35 Large-scale statistical comparisons of protein kinase sequences and crystal structures have also identified additional structural and conformational features associated with RS assembly and kinase activity. For example, a conformational “strain” switch in the catalytic site was shown to be correlated with RS assembly and kinase activity.34 Likewise, extension of the RS through family and group- specific residues have been suggested to contribute to unique modes of allosteric regulation in some kinases.36,37 A molecular dynamics (MD) based structural network modeling approach identified the RS as a central hub for structural stability and allosteric communication.38
Figure 1.
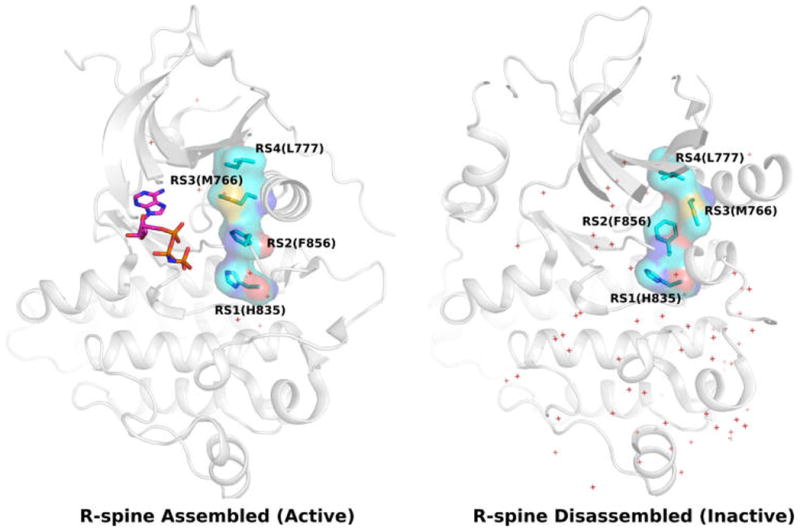
R-spine assembly/disassembly in active (PDB: 2GS6) and inactive (PDB: 3W32) state of EGFR.
The RS residues are also frequently mutated in human cancers. Mining of cancer mutations in the context of kinase sequence and structural motifs conceptualized in the Protein Kinase Ontology (ProKinO) revealed the RS as a mutational hotspot in cancers.15,16 Experimental characterization of some of these mutations in the oncogenic kinase B-Raf revealed that oncogenic variants that introduce additional aromatic interactions in the RS result in constitutive B-Raf activity.39,40 Here, we computationally and experimentally screen patient-derived RS mutation in EGFR and identify M766T, a mutation at the RS3 position, as a novel activating mutation. We find that the nature and size of amino acids present at the RS3 position strongly correlate with ligand independent EGFR activation. Based on molecular modeling and molecular dynamics simulation studies, we propose a model in which M766T activates the kinase domain by both destabilizing the inactive hydrophobic packing between αC-helix and A-loop, and stabilizing the active state through a novel water mediated interaction in the dynamic core. We show that EGFR harboring the M766T mutation is resistant to gefitinib and erlotinib treatment, but not to lapatinib treatment. Since M766 is part of the binding site for some allosteric protein kinase inhibitors,41 our findings are relevant for the design of structure guided protein kinase inhibitors as well.
EXPERIMENTAL PROCEDURES
EGF Stimulation, Cell Lysis, and Immunoblotting
The pEGFP-N1-EGFR plasmid from our previous studies15,17 was used to generate point mutations. Before transfection, Chinese Hamster Ovary (CHO) cells were grown in high-glucose Dubecco’s Modified Eagle Medium (DMEM) (Cellgro, Manassas, VA, USA) with 10% fetal bovine serum (Bioexpress, UT, USA) without antibiotics at 30% confluency. Transient transfection was then performed using lipofectamine-2000 (Invitrogen, Carlsbad, CA) according to manufacturer’s protocol with WT and mutant EGFR. To detect autophosphorylation, transfected cells were serum-starved in Ham’s F-12 media for 18 h followed by ligand stimulation with EGF (100 ng/mL) (Sigma, St. Louis, MO) for 5 min. Cells were washed with PBS, and lysed with lysis buffer (50 mM Tris-HCL, pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 1 mM sodium orthovanadate, 1% Triton X-100, 1 mM PMSF, and 1× Protease Inhibitor Cocktail Set V, EDTA-free). Total protein was resolved on 10% SDS-PAGE and transferred onto polyvinylidenedifluoride (PVDF) membrane. Western blotting was done using anti-GFP, anti-pY1197-EGFR, anti-FLAG, anti-STAT3, and anti-pY705-STAT3 antibodies (Cell signaling, Danvers, MA). Proteins were detected using chemiluminescent substrate (ECL substrate, Pierce, Rockford, IL).
Drug Sensitivity Assays
For drug sensitivity assays, CHO cells were cultured, transfected, and starved as described above. After 18 h of starvation, cells were treated with 0, 0.01, 0.1, 1.0, and 10 μM lapatinib, gefitinib, or erlotinib for 2 h in Ham’s 12 media without fetal bovine serum followed by EGF (100 ng/mL) stimulation for 5 min. Immunoblotting was performed as described above.
Structural Modeling
Inactive and active states of EGFR were modeled based on the crystal structures of EGFR kinase domain in the inactive (3W32)42 and active conformations (2GS6),18 respectively. Gefitinib, lapatinib, and erlotinib bound structure of EGFR were modeled based on PDB entries 4WKQ (gefitinib), 1XKK (lapatinib), 1M17 (erlotinib active), and 4HJO (erlotinib inactive), respectively.43–45 PDB entry 5D41 was used to model EGFR bound to the allosteric inhibitor EAI045.41 Disordered regions were modeled using the MODELLER program (version 9.12).46 Mutations introduced during crystallization were reverted to WT EGFR sequence during the modeling step. RS mutations were introduced in the modeled WT structure using the loop refine module and all modeled structures were manually inspected for steric clashes.
Molecular Dynamics Simulation
Molecular dynamics (MD) simulations were done using GROMACS 5.0.447 with AMBER ff99SB-ildn force field48 and TIP3P water model. Force field parameter for various ligands were generated using acpype49 based on General Amber Force Field (GAFF).50 Long-range electrostatics were computed with PME method and a cutoff of 0.9 nm. Steepest descent and conjugate-gradient energy minimization was performed on the solvated protein for 20 000 steps until Fmax was less than 100 kcal/mol. NVT simulations were performed to heat the system from 0 to 310 K by coupling it to berendsen thermostat for 200 ps with a restraint on the non-hydrogen atoms of the protein and the ligand, if present. Then, constant pressure dynamics (P = 1 atm, T = 310 K) were carried out under Parrinello–Rahman barostat to maintain pressure and density with the same restraint. The unrestrained MD productions were run for 300 ns for EGFR monomer and 150 ns for EGFR dimer with a time step of 3 fs. Trajectories were determined to be stable based on root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) measurements (Figure S1–S6). Analysis of MD trajectories was carried out using programs in the GROMACS suite.47 All structure visualization was done using PyMOL 1.7.6.51
MM/PBSA Calculations
Molecular mechanics Poisson Boltzmann surface area (MM/PBSA) method has been widely used to estimate ligand-binding free energy.52–54 In this study, MM/PBSA calculation was performed using the g_mmpbsa program.55–56 Specifically, MD simulations with various ligands were performed for 300 ns following the protocol described above. To ensure equilibrium, the first 50 ns trajectory was omitted and a total of 210 snapshots were extracted from the remaining 250 ns MD data. Molecular mechanics potential energy (Gmm) for protein and ligand was calculated based on the force field parameters. Polar solvation energy (Gpolar) was calculated using the linear Poisson Boltzmann equation solver in APBS.57 Nonpolar solvation energy (Gnonpolar) was generated using SASA-only model (Gnonpolar = 0.0226778A + 3.84928). Entropic contribution was not considered, as previous studies showed that it does not improve prediction accuracy.58,59 The energy decomposition was analyzed by summing the energy component (Emm, Gpolar, Gnonpolar) of each residue under consideration.
Kullback-Leibler (KL) Divergence Analysis
Kullback–Leibler divergence expansion has recently been proposed as a powerful approach to compare conformational ensembles of biomolecules.60,61 The MutInf package was used to perform KL divergence analysis on MD trajectories.61,62 In this study, we primarily focus on the local KL divergence to identify residues perturbed by a given mutation. Specifically, we split the 2 independent 300 ns MD trajectories (150 ns for EGFR dimer) into 4 equal-sized blocks (ignoring the first 50 ns to ensure equilibrium) and quantify the residue torsion angles for each snapshot. The distribution of each torsion angle is approximated using bins with a size of 15°. The KL divergence was used to identify residues that show divergence in their torsion angle distributions. The identified residues were mapped to structures (Figure S8–S10) and manually inspected for divergence in dynamics.
RESULTS
Identification and Mutational Analysis of Patient Derived EGFR RS Mutations
To investigate the impact of mutations mapping to the RS of EGFR, we queried the protein kinase ontology, ProKinO, which integrates and conceptualizes the relationships connecting protein kinase sequence, structure, function, evolution, and disease in a human and machine readable format.15,16 Queries requesting mutations mapping to the RS in EGFR revealed a total of 18 distinct samples containing 7 unique mutations (Table S1). To understand how these mutations alter kinase activity, we generated all 7 RS mutations in EGFR and monitored the extent of Y1197 autophosphorylation in the absence/presence of EGF as a measure of kinase activation (see experimental procedures). Ligand dependent and independent activity of H835L (RS1 mutation) is less than WT (Figure 2a, lane 7–8; Figure S10, lane 3–4). H835L tends to co-occur with the L833V mutation (9 out of 11 patient samples). Western blot analysis shows that L833V itself is highly active, but the double mutant H835L/L833V is less active than H835L (Figure S10). The DFG-Phe (RS2) is mutated to a leucine (F856L) or serine (F856S) in cancer samples. F856L shows reduced Y1197 autophosphorylation (Figure 2a, lane 4) whereas F856S completely abrogates Y1197 autophosphorylation (Figure 2a, lane 6). M766T at the RS3 position is the only mutation more active than WT EGFR in the absence of EGF (Figure 2a, lane 11), whereas M766V is less active (Figure 2a, lane 13). Mutations at the RS4 position, L777Q and L777P, reduced Y1197 phosphorylation in the presence and absence of EGF (Figure 2a, lane 16, 18). Thus, our initial screening identified M766T as an activating mutation in the RS of EGFR. Downstream signaling of EGFR is also altered by the M766T mutation, as the phosphorylation status of STAT3 is increased in M766T relative to WT (Figure S11).
Figure 2.
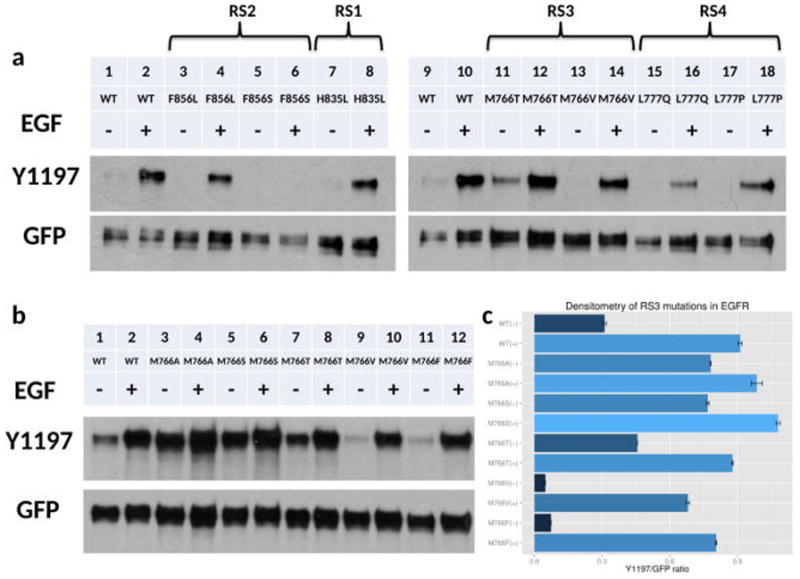
Western blot analyses and screening of RS3 mutations in EGFR. (a) Lanes from left to right: WT EGFR (−), WT EGFR (+), F856L (−), F856L (+), F856S (−), F856S (+), H835L (−), H835L (+), WT EGFR (−), WT EGFR (+), M766T (−), M766T (+), M766V (−), M766V (+), L777Q (−), L777Q (+), L777P (−), L777P (+). − and + indicate the absence and presence of EGF stimulation. (b) Series of mutations at RS3 residue. Lanes from left to right: WT EGFR (−), WT EGFR (+), M766A (−), M766A (+), M766S (−), M766S (+), M766T (−), M766T (+), M766V (−), M766V (+), M766F (−), M766F (+). − and + indicate the absence and presence of EGF stimulation. (c) Densitometry of three independent experiments of RS3 residue mutations.
Activity of EGFR is Correlated with Side-Chain Size at the RS3 Position
To understand the activation mechanism of M766T in EGFR, we made a series of mutations (M766A/T/S/V/F) at the RS3 position (Figure 2b). The phosphorylation levels of WT and mutant EGFR are comparable in the presence of EGF. However, ligand independent activity of EGFR differs for the various RS3 mutants. In general, when the RS3 methionine is replaced by a smaller side-chain residue (M766T/S/A), the mutants display higher ligand independent activity compared to WT (Figure 2b, lane 3–8), except for M766V, which displays lower activity in comparison to WT (Figure 2b, lane 9–10). M766T is 46% more active than WT EGFR in the absence of EGF, as indicated by densitometry quantification (Figure 2c). However, when RS3 methionine is replaced by a phenylalanine, activity of M766F is significantly reduced in comparison to WT (Figure 2b, lane 11–12), suggesting that the nature and size of amino acid side chain at the RS3 position is critical for normal EGFR functions.
M766T Mediated EGFR Activation Is Dimerization Dependent
It is well established that WT EGFR requires the formation of the asymmetric dimer to be active.18 To test if M766T mediated activation of EGFR also requires the asymmetric dimer, we performed complementation assays, as described in a previous study.18 Specifically, we introduced kinase C-lobe dimerization deficient mutation (V948R) and N-lobe dimerization deficient mutation (L760R)18 in the M766T background and probed for Y1197 phosphorylation. Y1197 phosphorylation cannot be detected once the dimerization deficient mutation is introduced (M766T/V948R and M766T/L760R) (Figure 3, lane 5–8). However, Y1197 phosphorylation can be rescued by cotransfection of M766T/V948R (receiver kinase) and M766T/L760R (activator kinase) in the presence of EGF (Figure 3, lane 9–10), since the asymmetric dimer can still form when both constructs are present. These data demonstrate that M766T mediated EGFR activation requires the asymmetric dimer.
Figure 3.
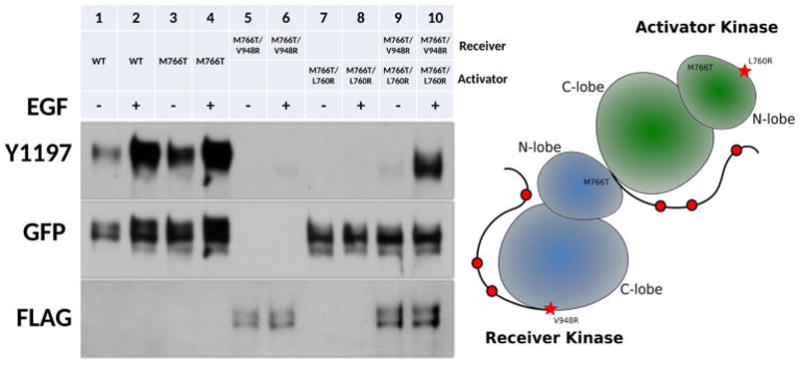
Dimerization dependency of M766T mutation. Right panel: Lanes from left to right: WT EGFR (−), WT EGFR (+), M766T (−), M766T (+), M766T/V948R (−), M766T/V948R (+), M766T/L760R (−), M766T/L760R (+), M766T/V948R+M766T/L760R (−), M766T/V948R +M766T/L760R (+). − and + indicate the absence and presence of EGF stimulation. Left panel: a cartoon representation of EGFR asymmetric dimer with N-lobe dimerization deficient mutation (L760R) and C-lobe dimerization deficient mutation (V948R).
Molecular Dynamics Simulations of WT and Mutant EGFR
To investigate the activation mechanism of M766T mediated activation, we performed MD simulations of WT and mutant (M766T/S/A/V/F) EGFR in the active, inactive, and asymmetric dimer forms with two independent replicates, accumulating a total of 7.5 μs MD data (see experimental procedures). Although we performed Kullback Leibler (KL) divergence comparison for all RS3 mutants (Figure S7–S9, Table S2), we primarily focus our discussions on M766T since it is a novel activating mutation.
M766T Relieves Autoinhibitory Packing Interactions Between αC-helix and the Activation Segment
Previous studies have shown that the autoinhibitory hydrophobic packing interactions between the regulatory αC-helix and the activation segment are critical for EGFR mutational activation.18,64 M766 is part of the hydrophobic network connecting the αC-helix and activation segment in the inactive state and makes van der Waals interactions with residues F856, L858, and L861 in the activation segment (Figure 4a). In our inactive MD simulation, the interaction between the two turn helix in the A-loop and the RS3 residue is weaker in M766T/S/A/V compared to WT EGFR (Figure 4b). In addition, the sulfur group of M766 is optimally positioned to form an S-π interaction with the DFG-Phe (F856) (Figure 4c).65 Quantification of the contact area between the A-loop and the RS3 residue also suggests that M766T/S/A/V decreases hydrophobic packing between these two regions (Figure 4d) and can potentially activate EGFR by destabilizing the inactive conformation. M766F, on the other hand, makes a strong van der Waals interaction with the A-loop, and increases the contact area between A-loop and αC-helix (Figure 4b, 4d). Notably, M766F shows less activity in comparison to WT, suggesting mutational stabilization of the inactive conformation. M766T also displays the highest RMSF value at the C-terminal region of the αC-helix among all the mutants investigated (Figure S12). This is likely due to an intrahelical hydrogen bond between the hydroxyl group of T766 and the carbonyl oxygen of E762 or A763 (92.6% occupancy), which introduces a bend in the αC-helix conformation (Figure 4e). Although S766, in theory, can also initiate such αC-helix bend, our MD data suggests that S766 is very flexible and does not form stable intrahelical hydrogen bonds (27.7% occupancy).
Figure 4.
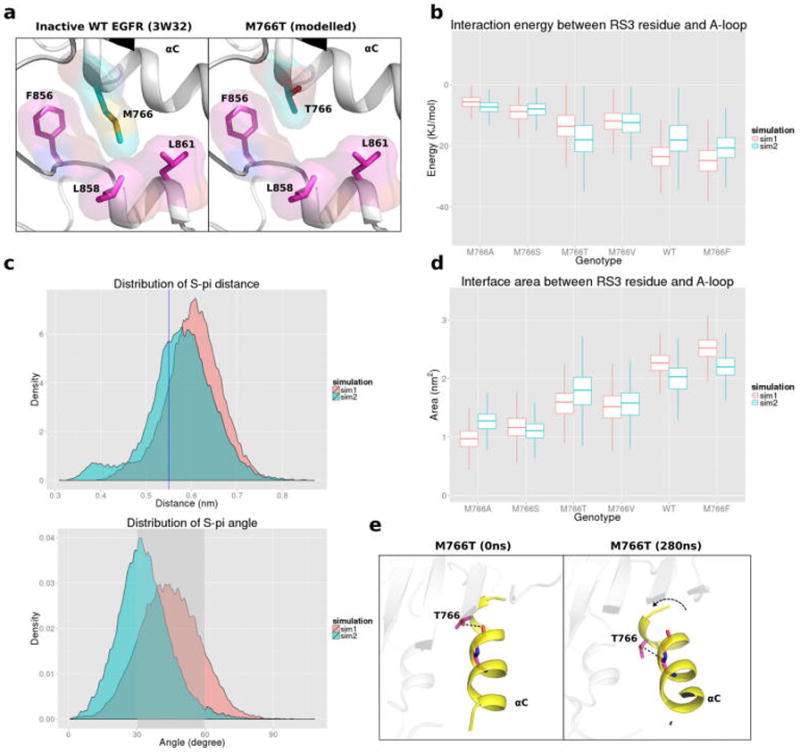
(a) Packing interaction between M766 in the αC-helix and hydrophobic residues in the activation loop. (b) Boxplot representing potential (VDW and couloumbic) interaction energy distributionbetween RS3 residue and A-loop helix. Distribution quantile (5%, 25%, 50%, 75%, and 95%) is shown in each vertical box plot. (c) Distance distribution of of M766 sulfur atom and the center of F856 benzyl group (upper panel) and angle distribution of M766 sulfur atom and the normal vector of F856 benzyl group (bottom panel). Blue line or shaded area indicate optimal interaction region. (d) Boxplot representing the surface area between RS3 residue and A-loop in different RS3 mutations. (e) MD snapshots showing the intrahelix hydrogen bonding interactions between the hydroxyl group of T766 and carbonyl oxygen of E762 or A763, resulting in a bent αC-helix conformation.
Conserved Water Mediated Interactions May Contribute to M766T Mediated Activation of EGFR
To further investigate the structural basis of M766T mediated activation, we performed two independent 300 ns MD simulations of WT and mutant (M766T/A/S/V/F) EGFR in the active monomeric conformation. Active monomeric EGFR is relatively unstable as the average RMSD value of the MD simulation is around 4 Å, whereas the RMSD of inactive monomeric EGFR is less than 3 Å (Figure S2). In addition, a previous MD study noted that the αC-helix is intrinsically disordered in the active monomeric state, and oncogenic mutations promote receptor dimerization by quenching intrinsic disorder.20 Consistent with this, our active monomeric simulations identified different degrees of αC-helix disorder for the various RS3 mutants (Figure 5a, Figure S13). Notably, the αC-helix is relatively stable in both simulation replicates for the M766T mutant (Figure 5a, Figure S13). We also quantified the K745-E762 salt bridge distance of M766T and other RS3 mutants (Figure 5b). Surprisingly, the largest variance in K745-E762 salt bridge distance is observed for M766V (Figure 5b). Although threonine and valine are similar in terms of their side-chain size, their physicochemical properties are different. Unlike valine, threonine is capable of forming hydrogen bonds. Indeed, in the M766T simulation, the hydroxyl group of T766 hydrogen bonds to the E762 side-chain and the DFG-Phe backbone through a water molecule, which is absent in other RS3 mutant simulations (Figure 5c). Such an interaction might help maintain the catalytically important K745-E762 salt bridge and rigidify αC-helix in M766T, but not in M766V. Furthermore, comparison of the torsion angle dynamics between M766T and WT EGFR identified other residues (L833, R836, and H893) that are allosterically perturbed by M766T (Table S2).
Figure 5.
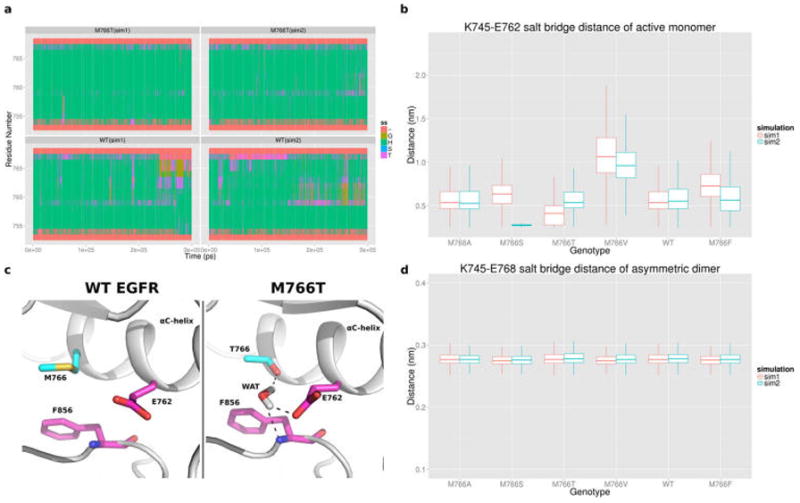
(a) Secondary structure assignment of the αC-helix region in WT and M766T EGFR simulation. Coil: ~3-Helix: G, A-helix: H, 5-Helix: I, Bend: S, Turn: T. (b) Boxplot showing the K745-E768 salt bridge distance in the active monomer simulation. (c) Representative MD snapshots of WT EGFR and M766T mutation in the active state. (d) Boxplot showing the K745-E766 salt bridge distance in the active asymmetric dimer simulation.
Our experimental studies show that M766T mediated EGFR activation requires the asymmetric dimer (Figure 3). To determine how RS3 mutations are coupled to dimer assembly, we performed 150 ns MD simulations for each mutant in the active asymmetric dimer and compared the RMSF values of αC-helix residues in the receiver and activator. As observed in Figure 6, the RMSF values for the αC-helix residues in the receiver and activator kinase are comparable for the activating mutations (M766T/A/S). In contrast, for the inactivating mutants (M766F and M766V), αC-helix residue fluctuations in the activator are significantly higher than the receiver. A similar trend is observed when the active monomer is compared against the receiver in the asymmetric dimer (Figure S14). Analysis of the K745-E762 salt bridge distance indicates that the salt bridge is well maintained in the activating M766T mutant (Figure 5d) and the conserved water molecule that mediates hydrogen bonding network with T766 and E762 is present in 49.2% of the MD trajectories, suggesting that the identified water-mediated network is functional and relevant for M766T mediated activation. Together, these data support the hypothesis that M766T reduces intrinsic flexibility of the αC-helix to promote dimerization and kinase activation.
Figure 6.
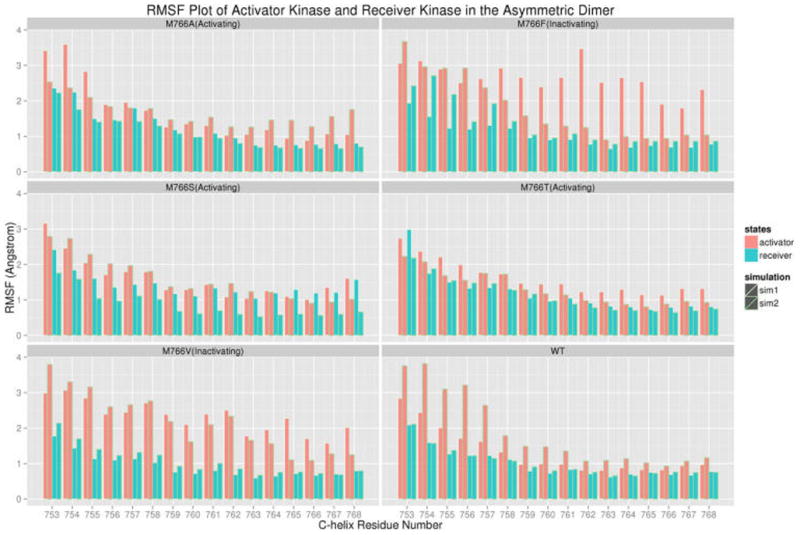
Root mean square fluctuation (RMSF) plot of αC-helix residues of activator kinase (red) and receiver kinase (cyan). For each RS3 mutants, two independent replicates are shown.
M766T is Resistant to Gefitinib/Erlotinib Treatment, But Not Lapatinib Treatment
Because M766 is part of the inhibitor binding site in the lapatinib/gefitinib/erlotinib bound structure of EGFR,43,44 we next wanted to explore the efficacy of clinically approved EGFR inhibitors on WT and mutant EGFR activity. To this end, we performed drug binding assays using inhibitors that target the inactive (lapatinib),44 active (gefitinib),19 and active/inactive conformations (erlotinib)43,45 of the kinase domain (Figure 7b). CHO cells were treated with increasing doses of EGFR inhibitors, and Y1197 phosohorylation was probed as readout for kinase inhibition. As observed in Figure 7a, M766T is slightly more sensitive to lapatinib in comparison to WT EGFR (Figure 7a, upper panel). The half inhibition concentration (IC50) of gefitinib (Figure 7a, middle panel) and erlotinib (Figure 7a, bottom panel) for WT EGFR is in the 0.01–0.1 μM range, whereas the IC50 for M766T is in the range of 1.0–10.0 μM, suggesting that M766T is more resistant to gefitinib and erlotinib compared to WT. These findings are consistent with previous IC50 estimates for various EGFR drug resistance mutations.63
Figure 7.
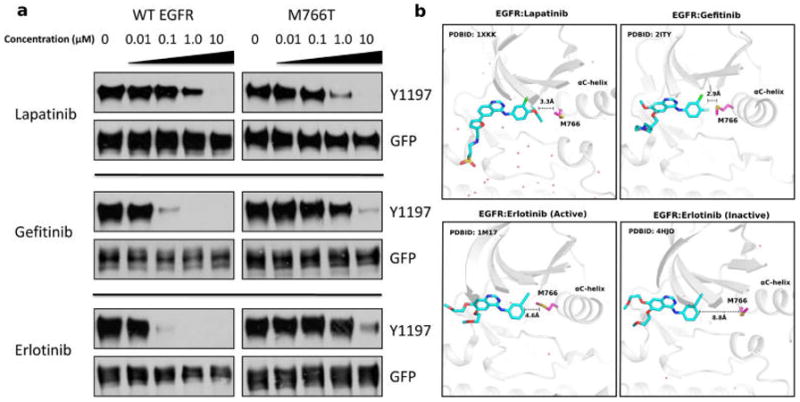
(a) Drug sensitivity of WT and mutant (M766T) EGFR. Drug concentration from left to right: 0 μM, 0.01 μM, 0.1 μM, 1.0 μM, and 10.0 μM. Upper panel (lapatinib),44 middle panel (gefitinib),19 lower panel (erlotinib).43–45 (b) Structural binding mode of EGFR M766 with lapatinib/gefitinib/erloti nib.
To understand the mechanisms by which M766T contributes to drug resistance, we performed modeling and 300 ns MD simulations of WT and mutant EGFR in the presence of EGFR inhibitors (lapatinib/gefitinib and erlotinib). Binding free energy estimates indicate a more favorable binding of lapatinib to M766T versus WT EGFR (Table 1). However, M766T binds less stably to gefitinib/erlotinib compared to WT EGFR, as indicated by positive ΔΔG values (Table 1). To pinpoint residues that confer the binding free energy difference, we performed energy decomposition analysis on both WT EGFR and M766T mutant and mapped the energy difference of each residue in available crystal structures (Figure S15). The analysis picked up residue clusters in the ligand binding pocket (Figure S15). Residues are colored based on energy contribution difference (ΔGM766T − ΔGWT < 0: red, ΔGM766T − ΔGWT > 0: blue) and a stick representation is shown if the energy difference is above a threshold. Residues that contribute most to the binding free energy difference in the lapatinib bound MD simulation includes M766 (RS3), K745, V762, T790, D800, D855, T854, and L1001 (Figure S16). Although M766, T854, T790, V726, and L1001 positively affect the binding free energy, this is compensated by several other charged residues (K745, D855, and D800) in the ligand binding pocket. The overall binding free energy for M766T:lapatinib is more negative than WT:lapatinib (Table 1). The gefitinib bound simulation identified the K745-E762 salt bridge residues as contributing most to the binding free energy (Figure S15). This difference might relate to the water mediated interaction between M766T and E762, as the hydrophobic local environment created by M766 is replaced with a water molecule, destabilizing the protein ligand interaction. Although MD simulation of erlotinib in both active and inactive states suggests a destabilizing effect of M766T (Table 1), the residues contributing to binding free energy differences are different (Figure S15). In the active state, the salt bridge residue E762 carries the most destabilizing effect, similar to gefitinib simulation (Figure S15). In the inactive state, the binding mode of lapatinib is distal from the RS3 residue (Figure 7b). Our analysis suggest that residues in the ligand binding pocket (K728, L792, T790, K745, D855, N842, R841, D800, R803) are allosterically perturbed by the M766T mutation (Figure S15). D855 and D800, in particular, exert the most positive impact on the binding free energy (Figure S15). Thus, RS3 is not the only residue that affects drug binding. M766T communicates with other residues in the ligand binding pocket to allosterically perturb the specific mode of binding, leading to the observed resistance/sensitivity.
Table 1.
Binding Free Energy Estimates of WT and Mutant (M766T) EGFR for Various Inhibitors Using MM/PBSA Method
| WT | M766T | ΔΔGM766T–WT | |
|---|---|---|---|
| Lapatinib | −171.63 ± 1.37 kJ/mol | −176.23 ± 1.20 kJ/mol | −4.60 kJ/mol |
| Gefitinib | −138.05 ± 1.42 kJ/mol | −120.38 ± 1.47 kJ/mol | 17.67 kJ/mol |
| Erlotinib (Active) | −121.01 ± 1.17 kJ/mol | −116.77 ± 1.55 kJ/mol | 4.24 kJ/mol |
| Erlotinib (Inactive) | −126.13 ± 1.17 kJ/mol | −118.32 ± 1.08 kJ/mol | 7.81 kJ/mol |
DISCUSSION
RS assembly is considered a key event in kinase activation.30,32,39,40 Although most RS mutations inactivate EGFR, our screening identified an activating mutation, M766T, at the RS3 position. Our studies demonstrate that M766T mediated allosteric activation involves multiple aspects (Figure 8). First, M766T activates EGFR by destabilizing the inactive conformation, the conformation that EGFR predominantly adopts in the absence of EGF. Second, M766T can stabilize the active conformation through water mediated interactions with the conserved K745-E762 salt bridge and backbone of the DFG motif, the precise positioning of which is critical for kinase activation.2,3 Third, as suggested by other MD studies, M766T can promote dimerization by rigidifying αC-helix conformation. It is worth mentioning that L833V also significantly increases the autophosphorylation of EGFR (Figure S10). L833 resides beneath the RS3 residue and packs against the A-loop, which is also part of the autoinhibitory network. The fact that L833V is activating further enforces the importance of RS3 associated interactions in the inactive state.
Figure 8.
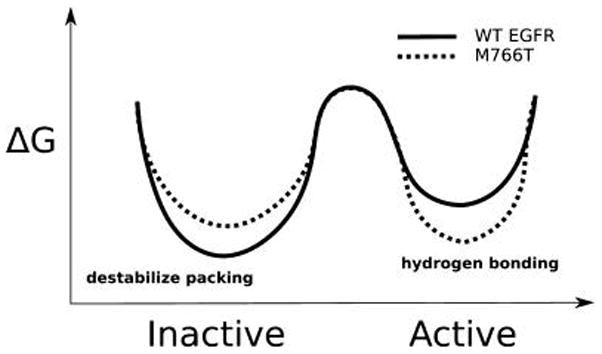
M766T activates EGFR by lowering the energy barrier for transition between active and inactive conformations.
Although the RS is conserved across diverse kinases, the impact of RS mutations is different in each kinase. For example, M766F inactivates EGFR by stabilizing the hydrophobic packing in the inactive conformation. This is in contrast to B-Raf, where introducing a phenylalanine residue at the RS3 position activates the kinase.39,40 Comparing the local structural environment of RS3 in the active and inactive states of EGFR and B-Raf provides some clues. First, the DFG-Phe adopts a canonical “out” conformation in the inactive state of B-Raf while in EGFR the DFG-Phe still adopts an “in” conformation (Figure S16). Second, the nature of residues conserved at the RS4 position is different in EGFR and B-Raf. In B-Raf, the RS4 position is an aromatic (phenylalanine) residue, which can potentially stabilize the active conformation through π–π aromatic stacking interactions with the phenylalanine at the RS3 position.66 Such aromatic stabilization is not possible in EGFR since a leucine is present at the RS4 position (Figure S16). Replacing RS4 leucine in EGFR by phenylalanine (L777F) activates EGFR (Figure S17), supporting the critical role of aromatic interactions in RS assembly and kinase activation.
Our study also suggests that the hydrophobic nature of RS residues is not a strict requirement for RS assembly and kinase activity. The active state of a kinase can be achieved in multiple ways. Most protein kinases conserve a hydrophobic residue at the RS3 position that mediates van der Waals interaction with the DFG-Phe (RS2 position). However, there are many exceptions to this rule. For example, Aurora kinase A and CHK1 conserve an RS3 glutamine, which coordinates with the backbone of the DFG motif via a water molecule.67,68 In addition, substitution of RS3 (L95) to hydrophilic residues (asparagine) in PKA only has a small effect on catalytic activity.32 Together, these data suggest that hydrophilic residues at the RS3 position can also facilitate RS assembly with the help of water mediated interactions.
The pocket between the αC-helix and activation segment is emerging as an important site for the design of allosteric protein kinase inhibitors.41 The RS3 methionine is part of the drug binding pocket in several inhibitor bound crystal structures of EGFR.10,19,43,44 In fact, in one of the recently solved crystal structures of EGFR bound to the allosteric inhibitor, EAI045, M766 is part of the allosteric binding pocket (Figure S18). We also modeled the binding mode of the allosteric inhibitor in M766T, which drastically reduced the contact between αC-helix and EAI045 (Figure S18). Based on our findings, we hypothesize that patients harboring M766T mutation would be resistant to EAI045. This hypothesis, however, needs to be tested in future studies.
The RS3 position is a mutational hotspot in the kinase domain. A total of 70 different mutations spanning 55 different kinases have been identified at the RS3 position (Table S3). EphA2, EphA7, FRK, MET, and ZAP70 harbor the exact same methionine to threonine mutation as seen in EGFR. MET and ZAP70 also adopt an inactive conformation similar to EGFR.69,70 Thus, although it is tempting to speculate that methionine to threonine substitution in these kinases will be activating, a detailed analysis of the local structural environment and family specific variations in the hydrophobic core will be essential to accurately predict the structural and functional impact of RS3 mutations. We also note the preponderance of hydrophilic mutations at the RS3 position in cancer samples (Table S3). Analogous to the M766T mutation studied here, hydrophilic residues at the RS3 position may involve a water molecule to stabilize the regulatory spine. Additional support for this notion comes from haspin kinase, where the RS3 residue is naturally conserved as a serine, which makes water mediated hydrogen bonds with the K511-E535 salt bridge and backbone amide of DYG-Y688 in a manner similar to the model proposed for T766 in EGFR (Figure S19).71 Thus, a deeper understanding of how naturally occurring variations contribute to protein functions and dynamics will be critical in accurately predicting the structural and functional impact of oncogenic mutations.
Mutation impact prediction is an important bioinformatics problem and several specialized web servers are available online for predicting mutation impact on protein structure and function. Our experimental studies on EGFR RS3 mutations provide a good data set to test the performance of existing algorithms. We employed a total of 5 different mutation impact prediction algorithms (PANTHER-PSEP,72 PolyPhen2,73 I-Mutant2.0,74 PROVEAN,75,76 MutPred77) to predict the impact of EGFR RS3 mutations. The result is summarized in Table S4. PANTHER-PSEP, PolyPhen2, and PROVEAN predict all 5 mutants (M766A/S/T/V/F) as “probably damaging” or “deleterious”. I-Mutant2.0 predicts all 5 mutants to “decrease stability”. MutPred predicted all five RS3 mutations to be deleterious with a probability more than 0.8. In addition, several predefined features in the MutPred algorithm annotate RS3 mutations as “Loss of stability”, “Gain of disorder”, “Loss of catalytic residue at M766, and “Loss of phosphorylation at Y764”. However, none of the prediction methods annotate M766T as potentially activating or gain of function. Thus, improved methods based on a deeper understanding of kinase structure, function and evolution will be essential to accurately predict the structural and functional impact of somatic variants identified in cancer genome sequencing studies.
Supplementary Material
Acknowledgments
Funding
Funding for N.K. is provided by the National Institutes of Health (GM114409–01). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Z.R. is the recipient of Grimes Family Distinguished Graduate Fellowship in Natural Sciences.
This study was supported in part by resources and technical expertise from the Georgia Advanced Computing Resource Center, a partnership between the University of Georgia’s Office of the Vice President for Research and Office of the Vice President for Information Technology. Members of the N.K. lab are acknowledged for helpful discussions and constructive comments on the manuscript. We appreciate Dr Donald Hamelbergs for his suggestions on carrying out MM/PBSA analysis.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.6b00572.
RMSD plots, RMSF plots, Western blots, αC-helix secondary structure assignment, structural mapping and comparisons (PDF)
RS residue mutations and sample ids from cosmic database (XLSX)
Cancer mutations at the RS3 position in all human kinases. Data Source: COSMIC database (XLSX)
Mutation impact prediction for EGFR RS3 mutants (XLSX)
Kullback Leibler (KL) divergence score of RS3 mutants in various states (inactive monomer, active monomer and asymmetric dimer) (XLSX)
References
- 1.Roskoski R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Endicott JA, Noble ME, Johnson LN. The structural basis for control of eukaryotic protein kinases. Annu Rev Biochem. 2012;81:587–613. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- 3.Hanks SK, Hunter T. The eukaryotic protein kinase superfamily: (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 4.Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 5.Sattler M, et al. Mutated tyrosine kinases as therapeutic targets in myeloid leukemias. Adv Exp Med Biol. 2003;532:121–140. doi: 10.1007/978-1-4615-0081-0_11. [DOI] [PubMed] [Google Scholar]
- 6.Banerji L, Sattler M. Targeting mutated tyrosine kinases in the therapy of myeloid leukaemias. Expert Opin Ther Targets. 2004;8:221–239. doi: 10.1517/14728222.8.3.221. [DOI] [PubMed] [Google Scholar]
- 7.Marchetti A, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29:3574–3579. doi: 10.1200/JCO.2011.35.9638. [DOI] [PubMed] [Google Scholar]
- 8.Minami Y, et al. The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene. 2007;26:5023–5027. doi: 10.1038/sj.onc.1210292. [DOI] [PubMed] [Google Scholar]
- 9.Heinrich MC, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 10.Yun CH, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyner JW, et al. Functional characterization of an activating TEK mutation in acute myeloid leukemia: a cellular context-dependent activating mutation. Leukemia. 2009;23:1345–1348. doi: 10.1038/leu.2009.66. [DOI] [PubMed] [Google Scholar]
- 12.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephens P, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005;37:590–592. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 14.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McSkimming DI, et al. ProKinO: A Unified Resource for Mining the Cancer Kinome. Hum Mutat. 2015;36:175–186. doi: 10.1002/humu.22726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gosal G. ProKinO: an ontology for integrative analysis of protein kinases in cancer. PLoS One. 2011;6:e28782. doi: 10.1371/journal.pone.0028782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruan Z, Kannan N. Mechanistic insights into R776H mediated activation of epidermal growth factor receptor kinase. Biochemistry. 2015;54:4216–4225. doi: 10.1021/acs.biochem.5b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 19.Yun CH, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shan Y, et al. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;149:860–870. doi: 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 21.Shan Y, et al. Transitions to catalytically inactive conformations in EGFR kinase. Proc Natl Acad Sci U S A. 2013;110:7270–7275. doi: 10.1073/pnas.1220843110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diwakar S. Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat Commun. 2014;5:1. doi: 10.1038/ncomms4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shih AJ, et al. Molecular dynamics analysis of conserved hydrophobic and hydrophilic bond-interaction networks in ErbB family kinases. Biochem J. 2011;436:241–251. doi: 10.1042/BJ20101791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park J, et al. Molecular Dynamics Analysis of Binding of Kinase Inhibitors to WT EGFR and the T790M Mutant. J Chem Theory Comput. 2016;12:2066–2078. doi: 10.1021/acs.jctc.5b01221. [DOI] [PubMed] [Google Scholar]
- 25.Wan S, Coveney PV. Molecular dynamics simulation reveals structural and thermodynamic features of kinase activation by cancer mutations within the epidermal growth factor receptor. J Comput Chem. 2011;32:2843–2852. doi: 10.1002/jcc.21866. [DOI] [PubMed] [Google Scholar]
- 26.Songtawee N, Bevan DR, Choowongkomon K. Molecular dynamics of the asymmetric dimers of EGFR: Simulations on the active and inactive conformations of the kinase domain. J Mol Graphics Model. 2015;58:16–29. doi: 10.1016/j.jmgm.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Songtawee N, Gleeson MP, Choowongkomon K. Computational study of EGFR inhibition: molecular dynamics studies on the active and inactive protein conformations. J Mol Model. 2013;19:497–509. doi: 10.1007/s00894-012-1559-0. [DOI] [PubMed] [Google Scholar]
- 28.Ma L. EGFR Mutant Structural Database: computationally predicted 3D structures and the corresponding binding free energies with gefitinib and erlotinib. BMC Bioinf. 2015;16:1. doi: 10.1186/s12859-015-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hashimoto K, Rogozin IB, Panchenko AR. Oncogenic potential is related to activating effect of cancer single and double somatic mutations in receptor tyrosine kinases. Hum Mutat. 2012;33:1566–1575. doi: 10.1002/humu.22145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kornev AP, et al. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci U S A. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meharena HS. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 2013;11:e1001680. doi: 10.1371/journal.pbio.1001680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci U S A. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oruganty K, et al. Identification of a hidden strain switch provides clues to an ancient structural mechanism in protein kinases. Proc Natl Acad Sci U S A. 2013;110:924–929. doi: 10.1073/pnas.1207104110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mobitz H, et al. The ABC of protein kinase conformations. Biochim Biophys Acta, Proteins Proteomics. 2015;1854:1555–1566. doi: 10.1016/j.bbapap.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Oruganty K, Kannan N. Evolutionary variation and adaptation in a conserved protein kinase allosteric network: implications for inhibitor design. Biochim Biophys Acta, Proteins Proteomics. 2013;1834:1322–1329. doi: 10.1016/j.bbapap.2013.02.040. [DOI] [PubMed] [Google Scholar]
- 37.Mohanty S. Hydrophobic Core Variations Provide a Structural Framework for Tyrosine Kinase Evolution and Functional Specialization. PLoS Genet. 2016;12:e1006265. doi: 10.1371/journal.pgen.1005885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.James KA, Verkhivker GM. Structure-based network analysis of activation mechanisms in the ErbB family of receptor tyrosine kinases: the regulatory spine residues are global mediators of structural stability and allosteric interactions. PLoS One. 2014;9:e113488. doi: 10.1371/journal.pone.0113488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu J, et al. Kinase regulation by hydrophobic spine assembly in cancer. Mol Cell Biol. 2015;35:264–276. doi: 10.1128/MCB.00943-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu J, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036–1046. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jia Y, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawakita Y, et al. Design and synthesis of novel pyrimido[4,5-b]azepine derivatives as HER2/EGFR dual inhibitors. Bioorg Med Chem. 2013;21:2250–2261. doi: 10.1016/j.bmc.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 43.Park JH, et al. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem J. 2012;448:417–423. doi: 10.1042/BJ20121513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wood ER, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 45.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 46.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pronk S, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct, Funct, Genet. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sousa da Silva AW, Vranken WF. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res Notes. 2012;5:367–367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J, et al. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 51.Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010. [Google Scholar]
- 52.Velazquez HA Hamelberg Conformation-Directed, D. Catalysis and Coupled Enzyme-Substrate Dynamics in Pin1 Phosphorylation-Dependent Cis-Trans Isomerase. J Phys Chem B. 2013;117:11509–11517. doi: 10.1021/jp405271s. [DOI] [PubMed] [Google Scholar]
- 53.Barman A, Hamelberg D. Coupled Dynamics and Entropic Contribution to the Allosteric Mechanism of Pin1. J Phys Chem B. 2016;120:8405–8415. doi: 10.1021/acs.jpcb.6b02123. [DOI] [PubMed] [Google Scholar]
- 54.Li M, et al. Predicting the Impact of Missense Mutations on Protein-Protein Binding Affinity. J Chem Theory Comput. 2014;10:1770–1780. doi: 10.1021/ct401022c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumari R, et al. g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 56.Genheden S, et al. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discovery. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baker NA, et al. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spackova N, et al. Molecular dynamics simulations and thermodynamics analysis of DNA-drug complexes. Minor groove binding between 4′,6-diamidino-2-phenylindole and DNA duplexes in solution. J Am Chem Soc. 2003;125:1759–1769. doi: 10.1021/ja025660d. [DOI] [PubMed] [Google Scholar]
- 59.Yang T, et al. Virtual screening using molecular simulations. Proteins: Struct, Funct, Genet. 2011;79:1940–1951. doi: 10.1002/prot.23018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stefely JA, et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol Cell. 2016;63:608–620. doi: 10.1016/j.molcel.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McClendon CL, et al. Comparing Conformational Ensembles Using the Kullback-Leibler Divergence Expansion. J Chem Theory Comput. 2012;8:2115–2126. doi: 10.1021/ct300008d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McClendon CL, et al. Quantifying Correlations Between Allosteric Sites in Thermodynamic Ensembles. J Chem Theory Comput. 2009;5:2486–2502. doi: 10.1021/ct9001812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Avizienyte E, Ward RA, Garner AP. Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug. Biochem J. 2008;415:197–206. doi: 10.1042/BJ20080728. [DOI] [PubMed] [Google Scholar]
- 64.Choi SH, Mendrola JM, Lemmon MA. EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene. 2007;26:1567–1576. doi: 10.1038/sj.onc.1209957. [DOI] [PubMed] [Google Scholar]
- 65.Valley CC, et al. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J Biol Chem. 2012;287:34979–34991. doi: 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kannan N, Vishveshwara S. Aromatic clusters: a determinant of thermal stability of thermophilic proteins. Protein Eng, Des Sel. 2000;13:753–761. doi: 10.1093/protein/13.11.753. [DOI] [PubMed] [Google Scholar]
- 67.Nowakowski J, et al. Structures of the cancer-related Aurora-A, FAK, and EphA2 protein kinases from nanovolume crystallography. Structure. 2002;10:1659–1667. doi: 10.1016/s0969-2126(02)00907-3. [DOI] [PubMed] [Google Scholar]
- 68.Oza V, et al. Synthesis and evaluation of triazolones as checkpoint kinase 1 inhibitors. Bioorg Med Chem Lett. 2012;22:2330–2337. doi: 10.1016/j.bmcl.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 69.Buchanan SG, et al. SGX523 is an exquisitely selective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol Cancer Ther. 2009;8:3181–3190. doi: 10.1158/1535-7163.MCT-09-0477. [DOI] [PubMed] [Google Scholar]
- 70.Deindl S, et al. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell. 2007;129:735–746. doi: 10.1016/j.cell.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 71.Eswaran J, et al. Structure and functional characterization of the atypical human kinase haspin. Proc Natl Acad Sci U S A. 2009;106:20198–20203. doi: 10.1073/pnas.0901989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang H, Thomas PD. PANTHER-PSEP: predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics. 2016;32:2230–2232. doi: 10.1093/bioinformatics/btw222. [DOI] [PubMed] [Google Scholar]
- 73.Adzhubei I, Jordan DM, Sunyaev SR. Current Protocols in Human Genetics. Wiley Online Library; Predicting functional effect of human missense mutations using PolyPhen-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choi Y. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li B, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.