

Abstract
We have used solid-state NMR to characterize the exact nature of the dual mode of action of oritavancin in preventing cell-wall assembly in Staphylococcus aureu. Measurements performed on whole cells labeled selectively in vivo have established that des-N-methylleucyl-N-4-(4-fluorophenyl)benzyl-chloroeremomycin, an Edman degradation product of [19F]oritavancin, which has a damaged D-Ala-D-Ala binding aglycon, is a potent inhibitor of the transpeptidase activity of cell-wall biosynthesis. The desleucyl-drug binds to partially cross-linked peptidoglycan by a cleft formed between the drug aglycon and its biphenyl hydrophobic sidechain. This type of binding site is present in other oritavancin-like glycopeptides which suggests that for these drugs a similar transpeptidase inhibition occurs.
Keywords: Glycopeptide antibiotic, peptidoglycan, oritavancin, transglycosylase, transpeptidation
Graphical Abstract
Introduction
Mono-alkylation of glycopeptides by hydrophobic moieties significantly improve drug activity against glycopeptide-resistant pathogens (1,2). Oritavancin (Orbactiv®,The Medicines Company, Montreal Canada) is a semi-synthetic chloroeremomycin (3) developed by Eli-Lilly and Company (Indianapolis, IN). Oritavancin exhibits rapid concentration-dependent bactericidal activity (4) against a broad spectrum of Gram-positive pathogens including vancomycin-resistant Staphylococcus aureus and vancomycin-resistant enterococci (5). Oritavancin however does not exhibit improved drug binding affinity to the depsipeptide (D-Ala-D-Lac) terminated peptidoglycan precursors found in high-level vancomycin-resistant pathogens. Thus, the hydrophobic sidechain of oritavancin was originally proposed to mediate the formation of drug dimers and membrane anchors at the bacterial cytoplasmic membrane (6,7). The drug dimer presumably would compensate for the weak monomeric-drug binding to the D-Ala-D-Lac of lipid II through positive cooperativity, while the membrane anchoring would enhance drug activity by co-localizing oritavancin with lipid II at the membrane.
The possible formation of [19F]oritavancin (Figure 1, middle) dimers and membrane anchors in intact whole cells of Staphylococcus aureus was investigated using solid-state NMR (8). The fluorine in [19F]oritavancin has no substantive pharmacological effect determined by minimal inhibitory concentration (MIC) measurements, but serves as an-NMR active probe. In situ only monomers of [19F]oritavancin were found bound to the peptidoglycan in the cell wall (9) and the hydrophobic side chain of [19F]oritavancin did not partition to the membrane (10).
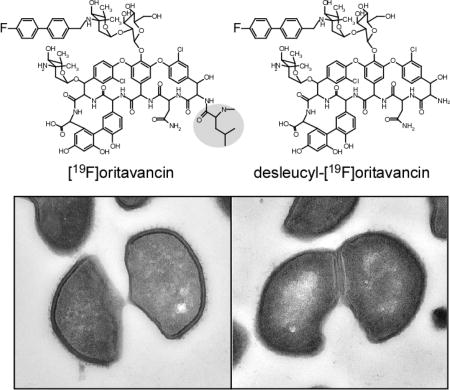
Figure 1.

Chemical structures of chloroeremomycin (left), [19F]oritavancin (center), and des-N-methylleucyl-4-(4-fluorophenyl)benzyl-chloroeremomycin (desleucyl-[19F]oritavancin) (right). The terminal leucyl residue of the aglycon core of [19F]oritavancin is highlighted.
An alternative proposed mode of action for oritavancin (11–13) argued that the oritavancin hydrophobic sidechain (the biphenyl-disaccharide moiety) directly targets a penicillin-binding protein to inhibit transglycosylase activity of peptidoglycan biosynthesis. This proposed mode of action was based on the observation that the Edman degradation product of vancomycin, a hexapeptide with N-terminal N-methyl-leucine cleaved, is devoid of all antimicrobial activity due to the damaged vancomycin aglycon D-Ala-D-Ala binding cleft (13), whereas the Edman degradation products of oritavancin and its analogues retain significant antimicrobial activity exhibiting in vitro transglycosylase inhibition of peptidoglycan biosynthesis (12,13). Thus the hydrophobic sidechain of desleucyl-oritavancin was thought to target an enzyme directly and inhibit glycan chain extension.
Solid-state NMR investigation of the Edman degradation products of both [19F]oritavancin and 4-(4-fluorophenyl)benzyl-vancomycin complexed to whole cells of S. aureus indicated, however, that penicillin binding protein is probably not an important target for these drugs in vivo. Both glycopeptides are bound to the peptidoglycan of whole cells as monomers, despite their damaged D-Ala-D-Ala binding cleft (8,14) Analysis by rotational-echo double resonance (REDOR) 13C{19F} NMR (15) confirmed that the peptidoglycan-bound conformations of desleucyl-[19F]oritavancin and desleucyl-4-(4-fluorophenyl)benzyl-vancomycin were similar to those of the corresponding undamaged heptapeptide glycopeptides (8,14). Models of these complexes suggested that the hydrophobic sidechains of the desleucyl-drugs form a cleft (a secondary binding site) with the aglycon that targets the cross-linked interbridge structure of peptidoglycan (9). (The primary and secondary binding targets of S. aureus peptidoglycan are highlighted in Figure S1.)
Oritavancin exhibits a unique dual mode of action by inhibiting both transglycosylase and transpeptidase activity of peptidoglycan biosynthesis in S. aureus (8). Like vancomycin, oritavancin inhibits transglycosylase activity by binding to the D-Ala-D-Ala terminus of lipid II. Previously we proposed that the transpeptidase inhibition by oritavancin could be attributed to its secondary binding site. We suspected that the secondary binding site of oritavancin enabled the drug to target partially cross-linked peptidoglycan stem and bridge structures found in the nascent and the template peptidoglycan and so interfere with both transglycosylase and transpeptidase activity (8). (The nascent peptidoglycan refers to a newly formed glycan chain that remains tethered to the membrane, while the template peptidoglycan is the nearest glycan chain not attached to the membrane surface.)
In this report we present experimental evidence that the transpeptidase inhibition by oritavancin is indeed due to its secondary binding site. We base this conclusion on measurements of glycine metabolism, Park’s nucleotide accumulation, peptidoglycan cross-linking, and cell-wall morphology of whole cells of S. aureus treated with either [19F]oritavancin or desleucyl-[19F]oritavancin during exponential growth. We believe that the resulting validation of the role of the secondary binding site of oritavancin, and the recently published architectural framework of the S. aureus peptidoglycan lattice (16), may encourage efforts to generate improved three-dimensional structures of complexes at the secondary binding site to gain insights for future drug development.
Materials and Methods
Growth and Labeling of S. aureus
Cells of S. aureus (ATCC 6538P) were grown in a Staphylococcus aureus Standard Medium (SASM) as described before (17). The cells were labeled by replacing an unlabeled amino acid of the SASM by the corresponding labeled version. Typically, 1 L flasks with 300 ml SASM were inoculated 1:100 with an overnight cell culture. For samples shown in Figures 2 and 3, S. aureus was grown in SASM containing the labeled amino acids [1-13C]glycine and L-[ε-15N]lysine. To each flask glycopeptide antibiotics were added to the growing cells at OD660 0.5 to yield concentrations of 3.4, 6.7, and 10.1 µg/ml for [19F]oritavancin, and 3.5, 6.8, and 10.4 µg/ml for desleucyl-[19F]oritavancin. For samples shown in Figure 4, S. aureus was grown in SASM in the presence of the racemase inhibitor alaphosphin (8) and containing the labeled amino acids [15N]glycine and D-[1-13C]alanine. To each flask glycopeptide antibiotics were added to the growing cells at OD660 0.5 to yield concentrations of 3.5, 7.0, and 10.4 µg/ml for [19F]oritavancin, and 3.3, 6.7, and 10.0 µg/ml for desleucyl-[19F]oritavancin. Cells with and without added antibiotic were harvested 60 min after the drug addition by centrifugation at 10,000g for 10 min at 4 °C, washed twice with 40 mM triethanolamine (pH 7.0), re-suspended in 12 ml of the same buffer, frozen, and lyophilized.
Figure 2.

13C full-echo NMR spectra of S. aureus grown in SASM containing [1-13C]glycine and L-[ε-15N]lysine treated with [19F]oritavancin (left) and desleucyl-[19F]oritavancin (right). The spectra are normalized to equal integrated peak intensity between 10 and 30 ppm to approximate equal aliphatic-carbon content. The [1-13C]glycine incorporation into proteins and peptidoglycan appears at 172 ppm, and the metabolism of glycine via purine biosynthesis, at 153 ppm. The addition of [19F]oritavancin and desleucyl-[19F]oritavancin to S. aureus during growth inhibited purine biosynthesis.
Figure 3.

(Top) 15N full-echo NMR spectra of S. aureus grown in media containing [1-13C]glycine and L-[ε-15N]lysine in the presence of varying concentrations of [19F]oritavancin (low, 3.4 µg/ml; intermediate, 6.7 µg/ml; high, 10.1 µg/ml). The spectra were scaled using the normalization factors of Figure 2 (left). Spectra of S. aureus grown in the presence of [19F]oritavancin show a reduced ε-15N lysyl-amide peak at 95 ppm (cell wall) and an increased ε-15N lysyl-amine peak at 10 ppm (cytoplasmic peptidoglycan precursors), consistent with transglycosylase inhibition. (Bottom) 15N full-echo NMR spectra of S. aureus grown in media containing [1-13C]glycine and L-[ε-15N]lysine in the presence of varying concentrations of desleucyl-[19F]oritavancin (low, 3.5 µg/ml; intermediate, 6.9 µg/ml; high, 10.4 µg/ml). The spectra were scaled using the normalization factors of Figure 2 (right). At low drug concentrations, the level of transglycosylase inhibition by desleucyl-[19F]oritavancin is reduced relative to that by [19F]oritavancin (lower amine-to-amide ratio). Slowed growths (Figure S2, top right) suggest the possibility of some accumulation of cell-wall debris from dead cells. Chemical shifts are referenced to solid ammonium sulfate.
Figure 4.

A comparison of inhibition of cross-linking from the 15N{13C} REDOR difference spectra of whole cells of S. aureus grown in media containing [15N]glycine and D-[1-13C]alanine, plus therapeutic doses of [19F]oritavancin (left) or desleucyl-[19F]oritavancin (right) after 1.6 ms of dipolar evolution. Full-echo spectra normalized to the control are shown at the bottom and REDOR differences are at the top of the figure. The REDOR difference measures the relative number of cross-links per peptidoglycan pentaglycyl bridging segment. At low concentrations, both drugs have a transpeptidase inhibitory effect. Chemical shifts are referenced to solid ammonium sulfate.
Synthesis of [19F]oritavancin and des-N-methylleucyl-[19F]oritavancin
4’-Fluorobiphenyl-4-carbaldehyde
To a solution of 1-bromo-4-fluorobenzene (1.25 ml, 11.4 mmol) in 1,2-dimethoxyethane (80 ml) was added tetrakis(triphenylphosphine)palladium (659 mg, 0.570 mmol). The mixture was stirred for 5 minutes and a solution of 4-formylphenylboronic acid (2.56 g, 17.1 mmol) in absolute ethanol (25 ml) was added followed by an aqueous solution of Cs2CO3 (11.4 ml, 2M, 22.8 mmol). The mixture was refluxed for 18h, filtered and concentrated. The crude mixture was diluted with CH2Cl2, washed with H2O (1×) and saturated aqueous NaCl solution (1×), dried over MgSO4, filtered and concentrated. Purification by flash chromatography on silica gel using a gradient of 0–20% EtOAc in hexanes provided 4’-fluorobiphenyl-4-carbaldehyde as a white solid (1.14 g, 50 % yield). 1H NMR (CDCl3, 400 MHz) δ 10.06 (s, 1H), 7.94–7.97 (m, 2H), 7.71 (d, J=8.2 Hz, 2H), 7.59–7.63 (m, 2H), 7.15–7.20 (m, 2H).
N-4-(4-fluorophenyl)benzyl chloroeremomycin ([19F]oritavancin)
To a suspension of chloroeremomycin acetate (443 mg, 0.250 mmol) in a mixture of DMF and methanol (2 ml and 12 ml) was added 4’-fluorobiphenyl-4-carbaldehyde (65 mg, 0.32 mmol). The reaction mixture was stirred for 2.5 h at 75 °C after which sodium cyanoborohydride (79 mg, 1.25 mmol) was added. Stirring was continued at 75 °C for an additional 2.5 h. The mixture was then cooled to room temperature and the crude product was precipitated upon addition of diethyl ether. The solids were filtered, washed with ether and dried under vacuum. The crude material was purified by reverse-phase C18 chromatography using a Biotage Horizon™ flash system. Elution was carried out with a gradient of 15–80% MeOH in aqueous Et3N/H3PO4 buffer (0.2% Et3N/H2O + H3PO4, pH = 3). The crude material was suspended in a mixture of MeOH and aqueous buffer and treated with 3.5 eq of 1N HCl to obtain a clear solution before purification. Pure fractions were combined, concentrated and lyophilized, then desalted by reverse-phase C18 chromatography, using a gradient of 15–80% MeCN in aqueous 0.1% HCO2H buffer. Pure fractions were combined, concentrated and lyophilized to provide [19F]oritavancin tris(hydrochloride) (123 mg, 26% yield). LCMS 98.2% (254 nm), 97.9% (220 nm), 98.0% (290 nm). MS: 1777.3 (M+H)+. Alternatively, desalting was completed on a reverse-phase C18 column, using a gradient of 15–80% 0.5% NH4OH/MeCN in 0.5% NH4OH/H2O. Pure fractions from this basic column were combined, concentrated and lyophilized to provide [19F]oritavancin as an ammonium salt in a similar yield. LCMS 98.9% (254 nm), 98.7% (220 nm), 98.9% (290 nm). MS: 1777.4 (M+H)+.
Des-N-methylleucyl N-4-(4-fluorophenyl)benzyl chloroeremomycin (desleucyl-[19F]oritavancin)
To [19F]oritavancin tris(hydrochloride) (160 mg, 0.085 mmol) in a 1:1 mixture of pyridine and H2O (8ml) was added phenylisothiocyanate (15 µL, 0.127 mmol). The reaction mixture was stirred for 1.5 h at room temperature and concentrated to dryness. The residue was suspended in CH2Cl2 at 0 °C and TFA (260 µL, 3.4 mmol) was added. The reaction mixture was stirred for 1 h 45 min at 0 °C then 4 h at room temperature, after which it was concentrated to dryness and purified by reverse-phase on a C18 column using the Biotage Horizon™ flash system. Product elution was accomplished with a gradient of 10–40% MeCN in 0.05% TFA/H2O. Pure fractions were combined, concentrated and lyophilized, then repurified on a second reverse-phase C18 column to remove TFA, using a gradient of 15–80% 0.5% NH4OH/MeCN in 0.5% NH4OH/H2O. Fractions from this second column were combined, concentrated and lyophilized to provide des N-methylleucyl [19F]oritavancin (54 mg, 38% yield). LCMS 98.4% (254 nm), 98.1% (220 nm), 97.7% (290 nm). MS: 1650.2 (M+H)+.
Dipolar Recoupling
REDOR was used to restore the dipolar coupling between heteronuclear pairs of spins that is removed by magic-angle spinning (18) REDOR experiments are done in two parts, once with rotor-synchronized dephasing pulses (S) and once without (S0). The dephasing pulses change the sign of the heteronuclear dipolar coupling and this interferes with the spatial averaging resulting from the motion of the rotor. The difference in signal intensity (ΔS = S0 − S) for the observed spin in the two parts of the REDOR experiment is directly related to the distance to the dephasing spin (18).
Solid-state NMR Spectrometer
The 15N and 13C REDOR full-echo (S0) spectra, and 15N{13C} REDOR difference (ΔS) spectra were obtained using a 4-frequency transmission-line probe with a 17-mm long, 8.6-mm inside-diameter analytical coil and a Chemagnetics/Varian magic-angle spinning ceramic stator. Lyophilized whole-cell samples were spun in Chemagnetics/Varian 7.5-mm outside-diameter zirconia rotors at 5 kHz with the spinning speed under active control to within ±2 Hz. A 4.7-T static magnetic field was provided by an 89-mm bore Oxford superconducting solenoid. A Tecmag (Houston, TX) pulse programmer controlled the spectrometer. Radio frequency pulses for 13C and 15N were produced by 1-kW ENI (Andover, MA) LPI-10 power amplifiers. Radio frequency pulses for 1H (200 MHz) were produced by a 1-kW Kalmus Engineering Int. Ltd (Valencia, CA) power amplifier, and the 19F pulses by a 1-kW Dressler Hochfrequenztechnik Gmbh (Stolberg-Vicht, Germany) power amplifier. All four amplifiers were under active control (19) Standard XY-8 phase cycling (20) was used for all pulses. The π-pulse lengths were 10 µsec for 13C and 15N. Proton-carbon and proton-nitrogen matched cross-polarization magic-angle-spinning (CPMAS) transfers were made in 2 msec at 50 kHz. Proton dipolar decoupling was 100 kHz during dipolar evolution and data acquisition. Spectra were typically the result of 20,000 scans with magic-angle spinning at 5 kHz.
Transmission Electron Microscopy
Overnight culture of S. aureus grown in brain-heart infusion (BHI) media was harvested at an OD660 nm of 1.1 by centrifugation at 2,750 g for 20 min at 4 °C (Eppendorf Centrifuge 5810R). The pellet was resuspended in fresh BHI of twice the volume and glycopeptide antibiotics were added to 1 ml of S. aureus suspension to the final concentrations of 740 µg/ml for [19F]oritavancin, 640 µg/ml for desleucyl-[19F]oritavancin, and 640 µg/ml for vancomycin. After one hour incubation at 37 °C, the drug-treated S. aureus cells were pelleted, and fixed in 0.5 ml of 2% paraformaldehyde / 2.5% glutaraldehyde (Polysciences Inc.) in 100 mM phosphate buffer, pH 7.2 for 3 h at room temperature. Samples were washed in phosphate buffer and post-fixed in 1% osmium tetroxide (Polysciences Inc.) for 1 h. Samples were then rinsed extensively in distilled water prior to enbloc staining with 1% aqueous uranyl acetate (Ted Pella Inc.) for 1 h. Following several rinses in distilled water, samples were dehydrated in a graded series of ethanol and embedded in Eponate 12 resin (Ted Pella Inc.). Sections of 95 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc.), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA Inc.).
Results
Inhibition of S. aureus growth by [19F]oritavancin and desleucyl-[19F]oritavancin
The addition of [19F]oritavancin and desleucyl-[19F]oritavancin during the mid-exponential-growth phase of S. aureus shows a concentration-dependent inhibition of growth, as measured by optical density at 660 nm (OD660nm) (Figure S2). Desleucyl-[19F]oritavancin, despite the damaged D-Ala-D-Ala binding site, has antimicrobial activity comparable to that of [19F]oritavancin.
Glycine metabolism and purine biosynthesis
The 13C-NMR spectra of lyophilized whole cells of S. aureus grown in SASM containing [1-13C]glycine and L-[15N]lysine, exposed to varying sub-lethal concentrations of [19F]oritavancin (Figure 2, left) and desleucyl-[19F]oritavancin (Figure 2, right) are dominated by the labeled peptide glycine peak. These spectra have been normalized for equal integrated intensity of the natural-abundance protein peaks in the 10–30 ppm region. The peak due to [1-13C]glycine in the cell-wall peptidoglycan bridge appears at 172 ppm, and that due to metabolized [1-13C]glycine incorporated into RNA through purine biosynthesis, at 153 ppm. Both peaks are reduced in intensity with increasing [19F]oritavancin concentration in the growth media. The reduction of the 149-ppm peak indicates a [19F]oritavancin-induced stress that re-routes all available glycine to attempt peptidoglycan biosynthesis, thereby indirectly inhibiting purine biosynthesis. The reduction of the 172-ppm peak (pentaglycyl bridge) indicates a decreased peptidoglycan production and cell-wall thinning. These effects are identical to those of vancomycin (21), a known transglycosylase inhibitor. Desleucyl-[19F]oritavancin also inhibited purine biosynthesis (reduced 153-ppm peak) but showed no substantial change in the 172-ppm peak intensity except at low drug concentration (Figure 2, right, red highlight). At higher concentrations, desleucyl-[19F]oritavancin resulted in no apparent decrease in the 172-ppm peak intensity (Figure 2, right), an anomaly which we attribute to the normalization of the spectra (see immediately below).
Park’s nucleotide accumulation and transglycosylase activity
15N-NMR spectra of S. aureus grown in SASM containing L-[15N]lysine and [1-13C]glycine in the presence of either [19F]oritavancin or desleucyl-[19F]oritavancin are shown in Figure 3. The spectra have been normalized to equal number of scans and scaled by the same factors used for the 13C-NMR spectra of Figure 2. The ε-15N lysyl-amide peak (95 ppm) is unique to cell walls and arises only from bridge-links of the peptidoglycan (Figure S1). This peak intensity is therefore directly proportional to the concentration of cell walls in whole cells. The ε-15N lysyl-amine peak intensity (10 ppm) is a combined contribution from the L-[15N]lysine in proteins, cytoplasmic peptidoglycan precursors, and peptidoglycan stems without attached bridges (21). An increasing concentration of [19F]oritavancin in the growth media resulted in a decreasing lysyl-amide-peak intensity (Figure 3, top), consistent with the cell-wall thinning detected by the reduction of the 172-ppm glycyl peak intensity in 13C-spectra (Figure 2, left). The cell-wall thinning was accompanied by an increase in concentration of cytoplasmic peptidoglycan precursors, in particular Park’s nucleotide, which resulted in an increase in ε-15N lysyl-amine intensity for low and intermediate [19F]oritavancin concentrations but not for the highest concentration.
For S. aureus treated with desleucyl-[19F]oritavancin at 3.5 µg/ml (“low” concentration), the 15N-NMR spectra also show decreased lysyl-amide peak intensity at 95 ppm (Figure 3, bottom), consistent with some cell-wall thinning, but not much of an amine-peak increase, which suggests no accumulation of Park’s nucleotide. Inhibition of transglycosylase activity therefore does not appear to be the primary target for desleucyl-[19F]oritavancin. At higher desleucyl-[19F]oritavancin concentrations the amide-peak intensities increase. We attribute this increase to an accumulation of some cell-wall fragments but mostly cytoplasmic proteins from dead cells. Normalization by the factors derived from the 10–30 ppm natural-abundance 13C region in Figure 2 therefore exaggerates the 15N-labeled lysine peak intensities, consistent with the low signal-to-noise ratios for these spectra (Figure 3, bottom right).
Peptidoglycan cross-linking and transpeptidase activity
Figure 4 shows 15N{13C} REDOR spectra following 1.6 ms of dipolar evolution of the lyophilized whole cells of S. aureus grown in SASM containing D-[1-13C]alanine and [15N]glycine, and exposed to sub-inhibitory concentrations of [19F]oritavancin (left) and desleucyl-[19F]oritavancin (right). The full-echo spectra were normalized for equal 95-ppm glycyl-amide peak intensity and are identical for all samples (Figure 4, bottom). The REDOR-difference spectra of drug-treated S. aureus are color coded and are overlaid (Figure 4, top). During 1.6 ms of dipolar evolution, only the 13C-15N peptide bond between D-[1-13C]alanine and [15N]glycine (the cross-link, Figure S1) contributes to the REDOR dephasing (18). That is, a reduced 95-ppm difference-peak intensity is directly proportional to the reduced cross-linking in the cell-wall peptidoglycan.
Both [19F]oritavancin and desleucyl-[19F]oritavancin-treated S. aureus show a reduction in the 95-ppm REDOR difference peak intensity at low drug concentration (Figure 4, red), consistent with inhibition of transpeptidase activity. The level of inhibition for desleucyl-[19F]oritavancin was about double that of [19F]oritavancin, although the latter is also associated with inhibition of transpeptidase. Thus, the similarity of the maximum growth measured by OD660nm for the two drugs at low concentration (Figure S2, 9 hours) seems reasonable. At higher drug concentrations, the REDOR differences for desleucyl-[19F]oritavancin treated cells decrease (Figure 4, top right). We attribute this anomaly to the normalization based on total 15N-labeled glycine incorporation, which includes incorporation of debris generated by the drug into cytoplasmic protein and so a reduced presence of cell-wall D-[1-13C]alanine.
Cell-wall morphology
We synchronized the growth phase of S. aureus by harvesting during stationary growth, followed by resuspension in fresh media, just prior to glycopeptide-drug addition. Transmission electron micrographs of S. aureus whole cells exposed to high concentrations of glycopeptide antibiotics for one doubling time show that a majority of the bacteria have just undergone cell division, or are at the point of cell division. In vancomycin-treated S. aureus, newly formed cell walls from the daughter cells, what were previously the cell-wall division planes, show reduced cell-wall thickness with diffused staining (Figure 5, left column, bottom). Daughter cells from vancomycin-treated S. aureus however resume the shape of the parent form without significant deviation in morphology. For [19F]oritavancin and desleucyl-[19F]oritavancin-treated S. aureus, cells were unable to regenerate the parent form after division. Instead, these cells had a characteristic half-moon-shape morphology (Figure 5, bottom, middle and right columns) and cell-wall thinning was observed at the septum.
Figure 5.

Transmission electron micrographs of S. aureus treated with vancomycin (left column), [19F]oritavancin (middle column), and desleucyl-[19F]oritavancin (right column). The top row shows a representative, large field of view. Vancomycin-treated S. aureus shows no significant change in cell shape, but general thinning of cell wall with diffused cell-wall staining for newly formed cell walls at the plane of cell division. Oritavancin- and desleucyl-[19F]oritavancin-treated S. aureus show thinning of cell wall at the septum of dividing cells and an aberrant cell shape.
Discussion
Inhibition by [19F]oritavancin
[19F]oritavancin addition during the exponential-growth phase of S. aureus resulted in purine biosynthesis inhibition, cell-wall thinning, and an accumulation of cytoplasmic peptidoglycan precursors (Figures 2–3). These effects are identical to those associated with transglycosylase inhibition by vancomycin (8,21,22). Like vancomycin, [19F]oritavancin targets lipid II to prevent the regeneration of the lipid transporter C55 (pyrophosphoryl-undecaprenol), which can be only released by transglycosylase. The sequestration of scarce C55 by [19F]oritavancin effectively inhibits all C55-mediated transport of cytoplasmic peptidoglycan precursor to the membrane (22). This results in an accumulation of Park’s nucleotide in the cytoplasm and concomitant cell-wall thinning. Pressure to synthesize cell walls routes glycine to Park’s nucleotide and away from purine synthesis. However, unlike vancomycin, [19F]oritavancin has a dual mode of action inhibiting both translgycosylase and transpeptidase activity (8). The latter is revealed by a drug-concentration-dependent diminution of peptidoglycan cross-linking (Figure 4, left) not present in vancomycin-treated cells (21).
Inhibition by desleucyl-[19F]oritavancin
The damaged D-Ala-D-Ala binding site limits lipid II targeting by desleucyl-[19F]oritavancin and a pronounced accumulation of Park’s nucleotide (high amine-to-amide ratio, Figure 3) is not observed. This indicates that transglycosylase inhibition is not the primary mode of action for desleucyl-[19F]oritavancin. Instead, the drug’s potent antibacterial activity results from its ability to inhibit transpeptidase activity (Figure 4, top right, red).
Given these data and the known proximity of the fluorine of desleucyl-[19F]oritavancin to the peptidoglycan pentaglycyl bridge (8), desleucyl-[19F]oritavancin must be bound to the cell wall through its undamaged secondary binding site. We conclude therefore that the mode of action associated with the secondary binding site of oritavancin-like drugs is transpeptidase inhibition. This site enables desleucyl-[19F]oritavancin (and oritavancin) to target partially cross-linked pentaglycyl segments of peptidoglycan (8,10,14). Such sites can occur throughout the cell wall, but high concentrations are found at the site of peptidoglycan biosynthesis near the cytoplasmic membrane in nascent and template peptidoglycan. An under-cross-linked peptidoglycan chain is then propagated and becomes a faulty template for subsequent growing layers of nascent peptidoglycan. The fact that desleucyl-oritavancin-like drugs are nearly exclusively bound to the cell wall supports this mechanism over the action of a minor drug fraction which partitions to the membrane and targets an enzyme. A cell-wall-bound killing agent is dependent on peptidoglycan architecture, which is consistent with differing modes of action of oritavancin in S. aureus and E. faecium (23). All of the membrane targets for these two Gram-positive bacteria are similar but their long-bridge and short-bridge peptidoglycan architectures differ significantly (16,24).
Acknowledgments
The authors thank Adel Rafai Far (The Medicines Company, Montreal, Quebec, Canada) for the generous gift of [19F]oritavancin and desleucyl-[19F]oritavancin, and W. Beatty of the Microbiology Imaging Facility at Washington University School of Medicine for the transmission electron micrographs.
Funding
This paper is based on work supported by the National Institutes of Health grant numbers GM116130.
Abbreviations
- [19F]oritavancin
N-4-(4-fluorophenyl)benzyl-chloroeremomycin
- desleucyl-oritavancin
des-N-methylleucyl-N-4-(4-chlorophenyl)benzyl-chloroeremomycin
- Lipid II
N-acetylglucosamine-N-acetyl-muramyl-pentapeptide-pyrophosphoryl-undecaprenol
- MIC
minimum inhibitory concentration
- NMR
nuclear magnetic resonance
- oritavancin
N-4-(4-chlorophenyl)benzyl-chloroeremomycin
- REDOR
rotational-echo double resonance
- SASM
Staphylococcus aureus standard medium
Footnotes
Supporting Information
(i) Chemical structure of the repeating unit of the peptidoglycan of S. aureus with the location of its primary and secondary binding sites; and (ii) growth curves of S. aureus showing absorbance at 660 nm as a function of time for three different concentrations of [19F]oritavancin and desleucyl-[19F]oritavancin. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Printsevskaya SS, Pavlov AY, Olsufyeva EN, Mirchink EP, Isakova EB, Reznikova MI, Goldman RC, Branstrom AA, Baizman ER, Longley CB, Sztaricskai F, Batta G, Preobrazhenskaya MN. Synthesis and mode of action of hydrophobic derivatives of the glycopeptide antibiotic eremomycin and des-(N-methyl-D-leucyl)eremomycin against glycopeptide-sensitive and -resistant bacteria. J. Med. Chem. 2002;45:1340–1347. doi: 10.1021/jm010460i. [DOI] [PubMed] [Google Scholar]
- 2.Cooper RD, Snyder NJ, Zweifel MJ, Staszak MA, Wilkie SC, Nicas TI, Mullen DL, Butler TF, Rodriguez MJ, Huff BE, Thompson RC. Reductive alkylation of glycopeptide antibiotics: synthesis and antibacterial activity. J. Antibiot. 1996;49:575–581. doi: 10.7164/antibiotics.49.575. [DOI] [PubMed] [Google Scholar]
- 3.Barrett JF. Oritavancin. Eli Lilly & Co. Curr. Opin. Investig. Drugs. 2001;2:1039–1044. [PubMed] [Google Scholar]
- 4.Coyle EA, Rybak MJ. Activity of oritavancin (LY333328), an investigational glycopeptide, compared to that of vancomycin against multidrug-resistant Streptococcus pneumoniae in an in vitro pharmacodynamic model. Antimicrob. Agents Chemother. 2001;45:706–709. doi: 10.1128/AAC.45.3.706-709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westwell MS, Bardsley B, Dancer RJ, Try AC, Williams DH. Cooperativity in ligand binding expressed at a model cell membrane by the vancomycin group antibioitcs. Chem. Commun. 1996:580–590. [Google Scholar]
- 6.Sharman GJ, Try AC, Dancer RJ, Cho YR, Staroske T, Bardsley B, Maguire AJ, Cooper MA, O'Brien DP, Williams DH. The roles of dimerization and membrane anchoring in activity of glycopeptide antibiotics against vancomycin-resistant bacteria. J. Am. Chem. Soc. 1997;119:12041–12047. [Google Scholar]
- 7.Beauregard DA, Williams DH, Gwynn MN, Knowles DJ. Dimerization and membrane anchors in extracellular targeting of vancomycin group antibiotics. Antimicrob. Agents Chemother. 1995;39:781–785. doi: 10.1128/AAC.39.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim SJ, Cegelski L, Stueber D, Singh M, Dietrich E, Tanaka KS, Parr TR, Far AR, Schaefer J. Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus. J. Mol. Biol. 2008;377:281–293. doi: 10.1016/j.jmb.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SJ, Cegelski L, Preobrazhenskaya M, Schaefer J. Structures of Staphylococcus aureus cell-wall complexes with vancomycin, eremomycin, and chloroeremomycin derivatives by 13C{19F} and 15N{19F} rotational-echo double resonance. Biochemistry. 2006;45:5235–5250. doi: 10.1021/bi052660s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SJ, Singh M, Schaefer J. Oritavancin binds to isolated protoplast membranes but not intact protoplasts of Staphylococcus aureus. J. Mol. Biol. 2009;391:414–425. doi: 10.1016/j.jmb.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinha Roy R, Yang P, Kodali S, Xiong Y, Kim RM, Griffin PR, Onishi HR, Kohler J, Silver LL, Chapman K. Direct interaction of a vancomycin derivative with bacterial enzymes involved in cell wall biosynthesis. Chem. Biol. 2001;8:1095–1106. doi: 10.1016/s1074-5521(01)00075-8. [DOI] [PubMed] [Google Scholar]
- 12.Ge M, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding D-Ala-D-Ala. Science. 1999;284:507–511. doi: 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- 13.Goldman RC, Baizman ER, Longley CB, Branstrom AA. Chlorobiphenyl-desleucyl-vancomycin inhibits the transglycosylation process required for peptidoglycan synthesis in bacteria in the absence of dipeptide binding. FEMS Microbiol. Lett. 2000;183:209–214. doi: 10.1111/j.1574-6968.2000.tb08959.x. [DOI] [PubMed] [Google Scholar]
- 14.Kim SJ, Matsuoka S, Patti GJ, Schaefer J. Vancomycin derivative with damaged D-Ala-D-Ala binding cleft binds to cross-linked peptidoglycan in the cell wall of Staphylococcus aureus. Biochemistry. 2008;47:3822–3831. doi: 10.1021/bi702232a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Kim SJ, Singh M, Preobrazhenskaya M, Schaefer J. Staphylococcus aureus peptidoglycan stem packing by rotational-echo double resonance NMR spectroscopy. Biochemistry. 2013;52:3651–3659. doi: 10.1021/bi4005039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong G, Pan Y, Dong H, Pryor R, Wilson GE, Schaefer J. Structure and dynamics of pentaglycyl bridges in the cell walls of Staphylococcus aureus by 13C-15N REDOR NMR. Biochemistry. 1997;36:9859–9866. doi: 10.1021/bi970495d. [DOI] [PubMed] [Google Scholar]
- 18.Gullion T, Schaefer J. Detection of weak heteronuclear dipolar coupling by rotational-echo double-resonance nuclear magnetic resonance. Adv. Magn. Reson. 1989;13:57–83. [Google Scholar]
- 19.Stueber D, Mehta AK, Chen Z, Wooley KL, Schaefer J. Local order in polycarbonate glasses by 13C{19F} Rotational-Echo Double-Resonance NMR. J. Polym. Sci., Part B: Polym. Phys. 2006;44:2760–2775. [Google Scholar]
- 20.Gullion T, Baker DB, Conradi MS. New, compensated Carr-Purcell sequences. J. Magn. Reson. 1990;89:479–484. [Google Scholar]
- 21.Cegelski L, Kim SJ, Hing AW, Studelska DR, O'Connor RD, Mehta AK, Schaefer J. Rotational-echo double resonance characterization of the effects of vancomycin on cell wall synthesis in Staphylococcus aureus. Biochemistry. 2002;41:13053–13058. doi: 10.1021/bi0202326. [DOI] [PubMed] [Google Scholar]
- 22.Brotz H, Bierbaum G, Reynolds PE, Sahl HG. The lantibiotic mersacidin inhibits peptidoglycan biosynthesis at the level of transglycosylation. Eur. J. Biochem. 1997;246:193–199. doi: 10.1111/j.1432-1033.1997.t01-1-00193.x. [DOI] [PubMed] [Google Scholar]
- 23.Patti GJ, Kim SJ, Yu T-Y, Dietrich E, Tanaka KSE, Parr TR, Jr, Far AR, Schaefer J. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J. Mol. Biol. 2009;392:1178–1191. doi: 10.1016/j.jmb.2009.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SJ, Singh M, Sharif S, Schaefer J. Cross-link formation and peptidoglycan assemby in the FemA mutant of Staphylococcus aureus. Biochemistry. 2014;53:4755–4757. doi: 10.1021/bi4016742. [DOI] [PMC free article] [PubMed] [Google Scholar]