

Abstract
Mitochondria are double membrane-bound organelles mainly involved in supplying cellular energy, but also in signaling, cell differentiation and cell death. Mitochondria are implicated in carcinogenesis, and, as such, dozens of lethal signal transduction pathways converge on these organelles. Accordingly, mitochondria provide an alternative target for cancer management. In this study, F16, a drug targeting mitochondria, and chlorambucil (CBL), which is indicated for the treatment of selected human neoplastic diseases, were covalently linked, resulting in the synthesis of a multi-mitochondrial anticancer agent, FCBL. FCBL can associate with human serum albumin (HSA) to form a HSA-FCBL nanodrug, which selectively recognizes cancer, but not normal, cells. Systematic investigations show that FCBL partially accumulates in cancer cell mitochondria to depolarize mitochondrial membrane potential (MMP), increase reactive oxygen species (ROS) and attack mitochondrial DNA (mtDNA). With this synergistic effect on multiple mitochondrial components, the nanodrug can effectively kill cancer cells and overcome multiple drug resistance. Furthermore, based on its therapeutic window, HSA-FCBL exhibits clinically significant differential cytotoxicity between normal and malignant cells. Finally, while drug dosage and drug resistance typically limit first-line mono-chemotherapy, HSA-FCBL, with its ability to compromise mitochondrial membrane integrity and damage mtDNA, is expected to overcome those limitations to become an ideal candidate for the treatment of neoplastic disease.
Keywords: multi-mitochondrial antitumor agents, target cancer cell, delocalized lipophilic cation F16, human serum albumin nanodrug, chlorambucil
TOC image
A drug targeting mitochondria, F16, and chlorambucil (CBL), were covalently linked, resulting in the synthesis of a multi-mitochondrial anticancer agent, FCBL. FCBL can associate with human serum albumin (HSA) to form a HSA-FCBL nanodrug, which selectively recognizes cancer, but not normal, cells. Systematic investigations show that FCBL partially accumulates in cancer cell mitochondria to depolarize mitochondrial membrane potential, increase ROS and attack mtDNA. The nanodrug can effectively kill cancer cells and overcome multiple drug resistance.
INTRODUCTION
Based on their therapeutic efficacy, the primary choice for cancer management has been broad-spectrum DNA-damaging antineoplastic agents, such as anthracyclines, platinum-based agents and nitrogen mustard agents (1). These agents cause a wide range of lesions on nuclear DNA (nDNA) and consequently induce cell death. Unfortunately, they are also subject to dosage limits and multidrug resistance (MDR) after multiple treatment cycles, thus failing to provide long-term remission (2, 3). MDR can result from increased drug efflux and accelerated DNA repair by the nucleotide excision repair (NER) pathway (4, 5). Many elegant strategies have been developed to prevent the emergence of MDR and to fully regress tumors (6–8). One example involves a combination chemotherapy that utilizes the additive effect of individual antineoplastic agents on multiple bioactive molecules (9–12). However, since such combinations are typically based on empirical, not clinical, evidence, many have failed to significantly improve outcomes (13, 14). Therapeutic efficacy and dose-limiting cytotoxicity remain the main issues of combined therapies.
Mitochondria are implicated in carcinogenesis based on their key roles in cellular energy production and apoptosis (15, 16). Compared with their normal counterparts, cancer cell mitochondria can be structurally and functionally different, making cancer cells more susceptible to mitochondrial perturbations compared to normal cells. For example, the proximity of mtDNA to ROS production sites and the lack of an NER pathway make this genome vulnerable to oxidative damage or DNA-damaging agents (17, 18). Thus far, many agents have been developed against various mitochondria-associated substances, including the mitochondrial respiratory chain, the mitochondrial permeability transition pore complex, mtDNA, potassium channels on mitochondria, and various mitochondria-associated anti- and proapoptotic factors (19–22). These drugs can cause the permeabilization of mitochondria and induce cell death by the convergence of various lethal signal transduction pathways. Therefore, while combined therapies have effectively targeted mitochondria, few agents are able to act simultaneously on different mitochondrial components.
Therefore, in this study, delocalized lipophilic cation (DLC) F16, a drug targeting mitochondria, and FDA-approved nitrogen mustard chlorambucil (CBL), a DNA-damaging agent, were covalently linked, resulting in the synthesis of a multi-mitochondrial anticancer agent, FCBL. DLC F16 can accumulate in the mitochondrial matrix, resulting in depolarization of the mitochondrial membrane potential, release of cytochrome c, arrest of the cell cycle, and target cell apoptosis (23). Systematic investigations show that FCBL partially accumulates in cancer cell mitochondria to depolarize the mitochondrial membrane potential (MMP), increase the concentrations of reactive oxygen species (ROS) and attack mtDNA. With this synergistic effect on multiple mitochondrial components, the nanodrug can effectively kill cancer cells and overcome multiple drug resistance.
RESULTS
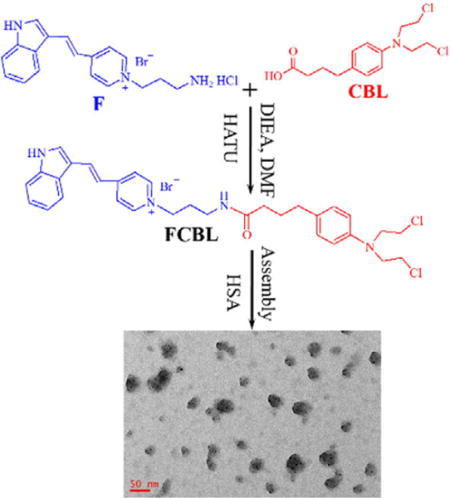
FCBL was synthesized by coupling a F16 derivative to CBL through HATU-mediated amide bond formation, as shown in Figure 1a. Details are described in the Experimental Methods and Figure S1. Purity was further characterized by HPLC (Figure S2a). The identity of the conjugate was confirmed by 1H NMR spectroscopy (SI, Methods), and its molecular weight was measured by mass spectrometry (MS) (Figure S2b). The synthesized FCBL was also characterized by UV−vis spectrophotometry (Figure 1b) and fluorescence spectroscopy (Figure 1c). These experimental results clearly demonstrate successful synthesis of FCBL. It is notable that FCBL emits green fluorescence with an emission peak wavelength of 538 nm using excitation with a 488-nm laser, thus providing a precision approach for investigating FCBL cellular uptake, intracellular distribution, and location.
Figure 1. Scheme of FCBL synthesis and its spectroscopic characterization.
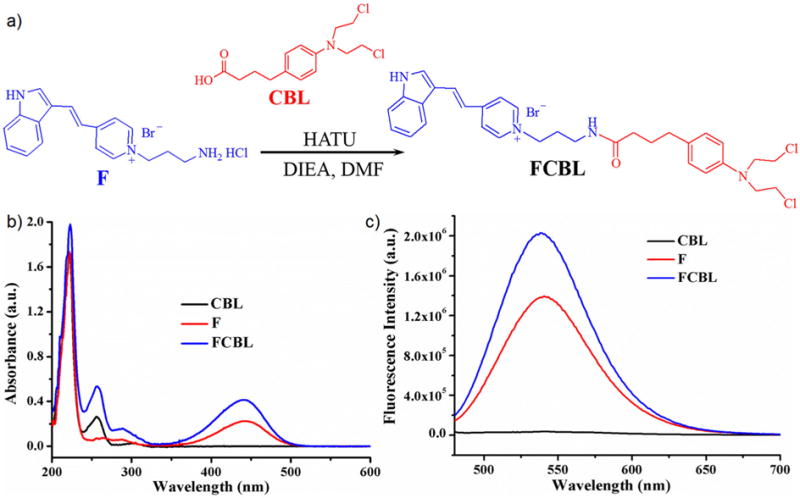
(a) FCBL was synthesized by coupling F16 derivative to CBL through HATU-mediated amide bond formation. (b) UV/Vis spectrum of FCBL, F and CBL dissolved in methanol. (c) Fluorescence emission spectra of FCBL, F and CBL dissolved in methanol. The excitation wavelength (λex) was 440 nm.
It is well known that CBL alkylates and crosslinks genomic DNA during all phases of the cell cycle, interfering with DNA replication and causing DNA damage. To study the effect of F16 derivative linkage on the alkylation and DNA-crosslinking activities of CBL, the alkylation ratios of FCBL and CBL on 4-(4-nitrobenzyl)pyridine were first tested. As shown in Figure S3, FCBL has better alkylation ability than CBL. The activity of FCBL and CBL on DNA plasmid samples was also studied by agarose gel electrophoresis after treatment with CBL (2 μM, 20 μM and 200 μM) or FCBL (2 μM, 20 μM and 50 μM) for 1 h. Figure 2 shows more DNA with high molecular weight in samples treated with FCBL compared to those treated with CBL. These results indicate that FCBL has better alkylation ability and DNA-crosslinking ability than unmodified CBL, which, in turn, means that the covalent coupling of F to CBL significantly improves the DNA-damaging ability of CBL. This improvement can be attributed to the substitution of the negatively charged carboxylic group of CBL for the positively charged F, making the resultant FCBL prone to associate with the negatively charged DNA.
Figure 2.
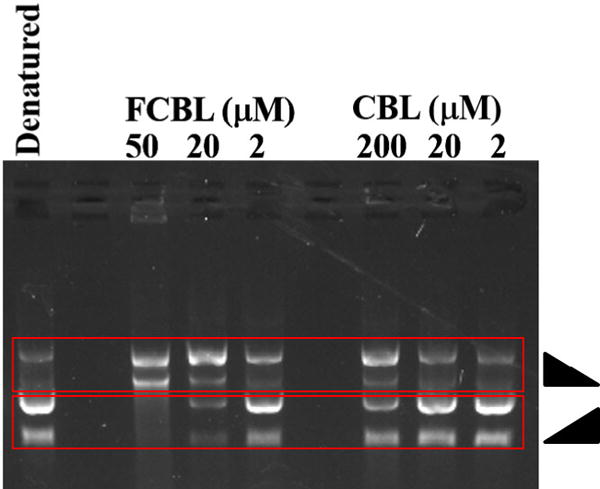
Analysis of crosslinking ability of FCBL and CBL on DNA plasmid samples.
Human serum albumin (HSA), which is the most abundant protein in blood, has been extensively used to bind and deliver hydrophobic molecules (24). Herein, we carefully studied the binding of FCBL to HSA by measuring the fluorescence signal of FCBL. Figure S4 demonstrates that FCBL exhibited a low fluorescence signal in H2O or Phosphate Buffered Saline (PBS). However, the fluorescence signal significantly increased upon the addition of HSA. This result indicates that FCBL can bind to HSA, even though FCBL can self-assemble into nanoparticles with a critical aggregation concentration value of 10 pM (Figure S5). Fluorescence increased when the ratio of FCBL to HSA increased from 1:4 to 5:1 (Figure S6a). However, obvious quenching of FCBL fluorescence was observed when the ratio of FCBL to HSA was further increased to 10:1 or 20:1. The binding of FCBL to HSA also led to an increase of HSA hydrodynamic diameter (Figure S6b). When the ratio of FCBL to HSA was less than 5:1, the size of HSA increased to about 140 nm. However, when the ratio further increased, HSA showed clear aggregation. Therefore, the molar ratio of FCBL to HSA (5:1) was chosen as the optimized formulation to prepare the HSA-FCBL nanodrug.
The as-prepared HSA-FCBL nanodrug was a canary yellow solution with an average hydrodynamic diameter of approximately 138 nm (Figure S7a). Transmission electron microscopy (TEM) indicated that the nanodrug consisted of spherical nanoparticles with an average size of approximately 45.5 nm (Figure S7b). This size is slightly smaller than that measured by DLS because of nanoparticle shrinkage during the process of drying in preparation for TEM. The HSA-FCBL nanodrug is very stable, and no obvious change in either size or polydispersity index (PDI) was observed during 12 days of storage (Figure S8). Leakage of FCBL from the nanodrug was tested, and results revealed negligible leakage of FCBL after 48-h dialysis (Figure S9). Formation of the HSA-FCBL nanodrug improved both the circulation time of the drug in blood and its accumulation in cancer cells via passive targeting. Importantly, HSA provides many active groups, such as NH2 and -COOH, for conjugation of ligands recognizing cancer cells, further increasing the accumulation of FCBL in cancer cells (24–26).
Selectivity of the HSA-FCBL nanodrug for cancer cells was studied by measuring its cellular uptake in the human cervical carcinoma HeLa cell line and the human normal liver LO2 cell line. Flow cytometric results showed that during 3-h incubation the HSA-FCBL easily accumulated in cancer HeLa cells, but only negligibly in normal cells (Figure 3a and Figure S10). Further quantitative analysis based on fluorescence spectroscopy showed that about 79% of FCBL could accumulate in HeLa cells and that only 21% of FCBL remained in cell medium when 11.29 μg of FCBL was applied to HeLa cells for 3 h (Table S1). These results indicate that HSA-FCBL has excellent cancer cell selectivity and easy intracellular accumulation. This selectivity can be explained by previous reports indicating the higher net negative charge on the surface of cancer cells when compared to that of normal cells (27).
Figure 3. Cancer cell-specific cellular uptake of HSA-FCBL and its intracellular distribution.
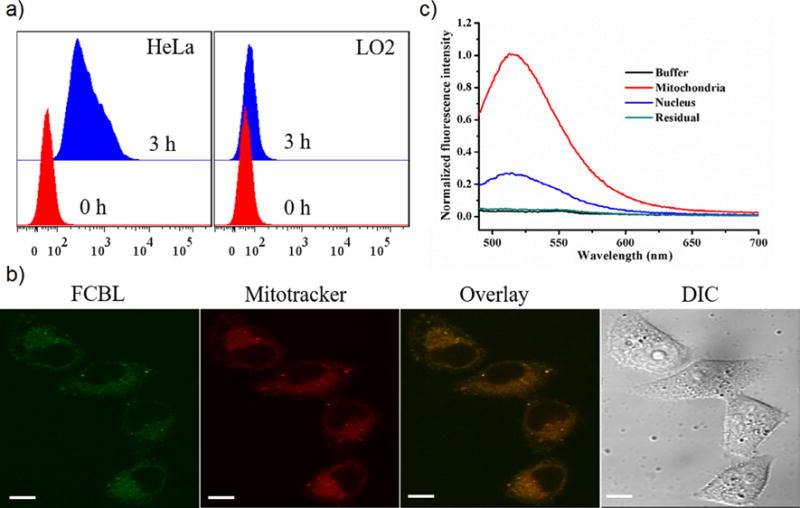
(a) Cellular uptake analysis of the HSA-FCBL in HeLa and LO2 cells after 3-h incubation. (b) Fluorescence analysis of FCBL in equivalent nuclear extracts, mitochondrial extracts and intracellular residual of HeLa cells. (c) CLSM analysis of HeLa cells incubated with HSA-FCBL after 3-h incubation. Mitochondria costained with MitoTracker® Deep Red FM. The scale bar was 10 μm.
Confocal laser scanning microscopy (CLSM) was used to analyze the intracellular distribution of FCBL in HeLa cells after 3-h incubation, followed by costaining with MitoTracker® Deep Red FM. Figure 3b shows that the green fluorescence of FCBL in HeLa cells exhibited an excellent overlay with the red fluorescence of MitoTracker, suggesting that F16 retained its mitochondria-targeting ability. To quantify the intracellular distribution of FCBL, the contents of FCBL in mitochondrial extracts and nuclear extracts were investigated by fluorescence spectroscopy analysis after incubation with FCBL. Figure 3c shows that mitochondrial extracts presented higher fluorescence signal of FCBL than equivalent nuclear extracts and other intracellular organelles. According to the standard curve, about 29% of intracellular FCBL was recovered from the mitochondria (Table S2). It is notable that most FCBL could still be recovered from the nuclear extracts, which accounted for about 60% of intracellular FCBL. At first glance, this may seem to contradict our previous observation that virtually no fluorescence signal was observed in the nucleus. However, the nucleus is larger in volume and size compared to mitochondria, resulting in lower average concentration of nuclear FCBL; therefore, the fluorescence signal in the nucleus could not be observed by CLSM.
Since the HSA-FCBL nanodrug could accumulate in both the mitochondria and nucleus, the effects of alkylation damage delivered by both FCBL and CBL on the two genomes were investigated by PCR amplification of DNA. We chose a 17.7 kb segment of nDNA at the b-globin gene and an 8.9 kb fragment of mtDNA for analysis after cells were treated with CBL or FCBL. As shown in Figure 4a, CBL primarily damaged nDNA with very few mitochondrial lesions, whereas FCBL caused a significant reduction of mitochondrial genome amplification with minimal effect on the nuclear genome. Thus, FCBL mainly damaged mtDNA, but not nDNA, although most intracellular FCBL accumulated in the nucleus.
Figure 4. FCBL activity on cancer cell mitochondria.
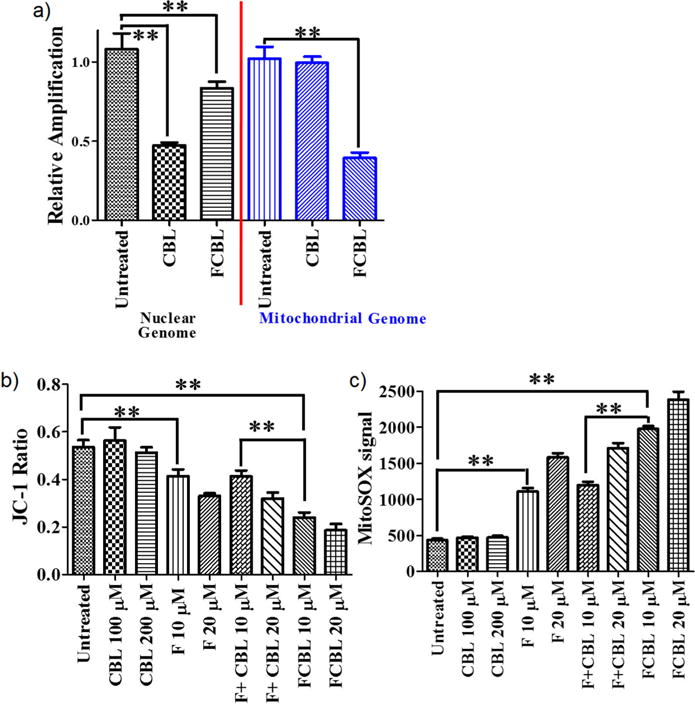
(a) FCBL and CBL induced DNA damage in the nuclear genome and mitochondrial genome. (b) The depolarizing effect of FCBL, F, CBL and F+CBL on MMP after 24-h incubation. (c) The effect of FCBL, F, CBL and the mixture of equivalent F and CBL, i.e., F+CBL, on ROS levels of mitochondria after 1.5-h incubation. Mean values plotted, n = 3, error bars equal SEM. Student’s two-tailed - t test was used to determine p values. ** indicates p value < 0.01.
According to Fantin et al. (23), F16 could compromise mitochondrial membrane integrity, subsequently reducing the depolarization of mitochondrial membrane potential (MMP) and increasing its ROS level, leading to target cell apoptosis. Therefore, we first investigated the effect of FCBL on MMP using the MMP dye JC-1. After cells were treated with F, CBL or the mixture of equivalent F and CBL (hereinafter termed as ‘F+CBL’), respectively, the red fluorescence signal (RFS) and the green fluorescence signal (GFS) of JC-1 in cells were recorded, and the JC-1 ratio of RFS to GFS was calculated. A decrease of JC-1 ratio is known to be proportional to decreased MMP. Thus, as shown in Figure 4b, FCBL significantly depolarized MMP compared to F, CBL, or F+CBL. FCBL also significantly increased reactive oxygen species (ROS) levels compared to F, CBL or F+CBL (Figure 4c). These data support the conclusion that FCBL has better activity on MMP and mtDNA lesions than F, CBL or F+CBL.
The cytotoxicity of the HSA-FCBL nanodrug was evaluated on three CBL-sensitive cancer cell lines (CCRF-CEM, U937 and MM.1S), four CBL-resistant cancer cell lines (MM.1R, K562, K562/ADR and HeLa), and two normal cell lines (LO2 and NIH3T3), followed by comparison to F, CBL, and F+CBL. Since synthesized F had cytotoxicity against HeLa cells and NIH3T3 similar to that of F16, F was used as control in the cytotoxicity analysis (Figure S11). Using the Annexin-V apoptosis assay kit (Figure 5a), FCBL was observed to induce apoptosis in both CBL-sensitive and CBL-resistant cell lines more effectively than F, CBL or F+CBL. For example, 48.2% of the cell population treated with FCBL was apoptotic for the CBL-resistant K562 cell line. However, after treatment with F, CBL and F+CBL, respectively, apoptosis was seen in only 8.20%, 11.24% and 23.10% of the cell population. To assess the antiproliferative ability of FCBL on these cell lines, MTT was employed, and the results are summarized in Table 1 and Figure S12. FCBL displayed little cytotoxicity (IC50 > 900 μM) on the two normal cell lines, similar to F or CBL. However, FCBL presented better cytotoxicity than F, CBL or F+CBL in all cancer cell lines tested. It is worth noting that FCBL can overcome deactivation of CBL and multidrug resistance in cancer cells. For example, the IC50 of FCBL (11.3 μM) for CBL-resistant K562 cells was about 36-fold lower than the IC50 CBL (416.8 μM), 7.8-fold lower than the IC50 of F (89.2 μM), and 6-fold lower than the IC50 of F+CBL (70.5 μM). Two factors can explain the enhanced activity of FCBL. First, FCBL accumulates in the nucleus and partially accumulates in the mitochondria, thus directing DNA-damaging activity to mtDNA, which lacks the NER pathway and is, therefore, susceptible to damage. Second, based on the additive effect of F and CBL, multiple components of mitochondrial structure and function are compromised, including damage to mtDNA, depolarization of MMP, and increased ROS levels, resulting in cancer cell apoptosis. According to previous reports (23, 28), mtDNA damage can increase the ROS level, further damaging mtDNA. Therefore, with this synergistic effect on multiple mitochondrial components, FCBL can effectively kill cancer cells and overcome multiple drug resistance.
Figure 5. Toxicity of FCBL in cell lines exhibiting apoptotic resistance and its activity in primary chronic lymphocytic leukemia (CLL) cells.
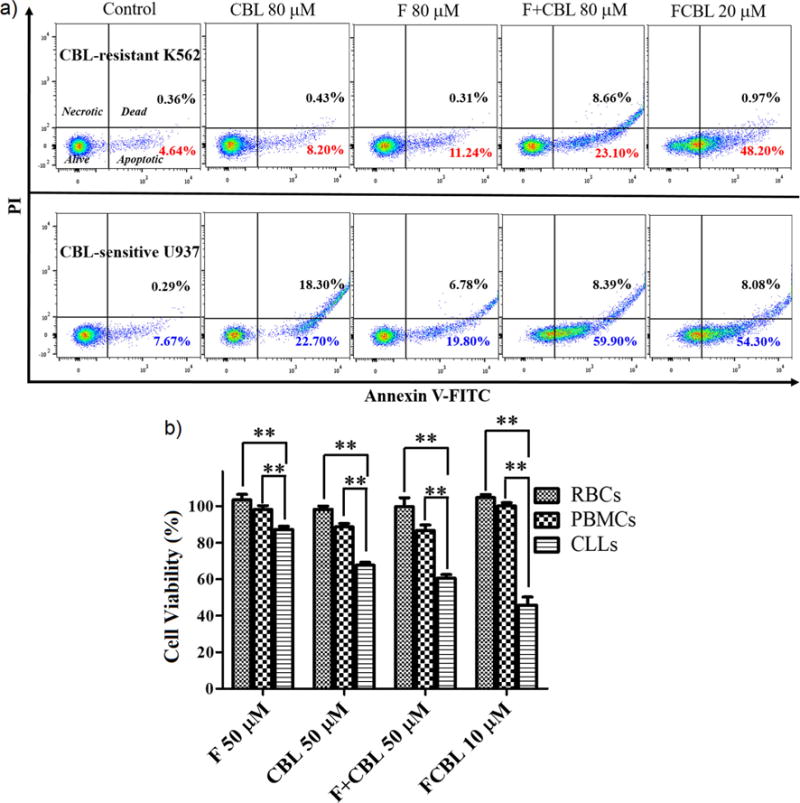
(a) Flow cytometry analysis of apoptosis of CBL-sensitive (U937) and CBL-resistant (K562) cells after 24-h incubation with F, CBL, the mixture of equivalent F and CBL, i.e., F+CBL, and FCBL, respectively. Lower left, A-FITC−/PI−, living cells; lower right, A−FITC+/PI−, apoptotic cells; upper right, A−FITC+/PI+, dead cells; upper left, A−FITC−/PI+, necrotic cells. Inserted numbers in the profiles indicate the percentage of cells present in that area. (b) Evaluation of HSA-FCBL nanodrug activity on red blood cells, peripheral blood mononuclear cells, and primary CLL cells. Mean values plotted, n > 5, error bars are SEM. Student’s two-tailed - t test was used to determine p values. ** indicate p value < 0.01.
Table 1.
Summary of IC50 values (μM, 24 h) in a panel of cell lines.
| Cell source | Cell line | CBL | F | CBL+F | FCBL |
|---|---|---|---|---|---|
| Human T cell lymphoblast-like cell | CCRF-CEM | 41.9 ± 5. 43 | 70.3 ± 6.78 | 32.7 ± 3.17 | 11.1 ± 1.97 |
| Human monocytic leukemia | U937 | 45.7 ± 3.71 | 66.8 ± 5.12 | 29.7 ± 3.86 | 10.3 ± 2.11 |
| Human multiple myeloma cell | MM.1S | 47.4 ± 5.08 | 63.3 ± 5.17 | 33.1 ± 4.16 | 12.1 ± 0.95 |
| Human multiple myeloma cell | MM.1R | 235.5 ± 16.88 | 66.7 ± 5.86 | 48.5 ± 3.99 | 13.5 ± 1.77 |
| Human chronic myeloid leukemia | K562 | 416.8 ± 27.88 | 89.2 ± 6.99 | 70.5 ± 6.37 | 11.3 ± 1.66 |
| Human chronic myeloid leukemia | K562/ADR | > 1000 ± 53.41 | 82.5 ± 7.31 | 65.4 ± 4.85 | 10.9 ± 1.74 |
| Human cervical cancer cell | HeLa | 320.7 ± 33.18 | 275.3 ± 20.78 | 159.5 ± 18.77 | 31.5 ± 3.57 |
| Human normal hepatoma cell | LO2 | > 600 ± 30.11 | > 900 ± 31.67 | > 600 ± 24.66 | > 900 ± 11.89 |
| Mouse embryonic fibroblasts cell | NIH3T3 | > 600 ± 17.66 | > 900 ± 20.19 | > 600 ± 16.47 | > 900 ± 22.15 |
Note: Cell survival was determined by MTT assays as described in Supporting Information. Data are mean ± SD of three independent experiments.
In their study on silver nanoparticles, Swanner, et al. (29) stated that the identification of differential sensitivity of cancer cells as compared to normal cells has the potential to reveal a therapeutic window for the use of silver nanoparticles (AgNPs) as a therapeutic agent for cancer therapy. Similarly, we herein evaluated whether HSA-FCBL would exhibit clinically significant differential cytotoxicity between normal and malignant cells. To accomplish this, we compared the activity of HSA-FCBL, F, CBL and F+CBL toward peripheral blood mononuclear cells (PBMCs) from normal healthy donors to patient-derived CLL cells. Figure 5b shows that treatment with 50 μM of CBL, F and F+CBL had moderate cytotoxicity and only killed 32.3%, 12.7% and 39.4% of CLL cells, respectively. Under the same conditions, CBL, F and F+CBL had clear side effects on PBMCs and killed 11%, 2% and 13.7% of cells, respectively. Importantly, however, treatment with 10 μM HSA-FCBL killed 54.2% of CLL cells, while, at the same time, showing little toxicity against normal PBMCs. Therefore, comparing CBL, F and F+CBL administered at 50 μM and HSA-FCBL administered at 10 μM, these results tell us that the concentration of drug needed to kill CLL cells is significantly lower than that needed to exert cytotoxicity against normal PBMCs, indicating that a clinically significant therapeutic windows does, indeed, exist for HSA-FCBL. Furthermore, HSA-FCBL showed nominal red blood cell hemolysis levels at the concentrations used in this study (Figure 5b), suggesting a lack of toxicity to these cells at concentrations where leukemia cells were ablated.
Conclusions
In summary, it is well known that mitochondria are susceptible to anticancer agents. Accordingly, we successfully developed an effective anticancer agent, FCBL, which targets mitochondria through covalent linkage of two existing drugs. Data from the study revealed that synthesized FCBL perfectly combines the activity of its two components, F16 derivative and CBL, and that it can specifically accumulate in cancer cell mitochondria, where HSA-FCBL simultaneously and synergistically acts on the integrity of mitochondrial membrane structure and mtDNA. Thus, with its synergistic effect on multiple mitochondrial components, FCBL can, when compared to F, CBL and F+CBL, effectively kill cancer cells and overcome multiple drug resistance. Furthermore, FCBL exhibited a clinically significant differential cytotoxicity between normal and malignant cells. Generally, our study has featured the preparation of a potent anticancer agent through covalent linkage of two drugs, each with relatively low therapeutic efficacy when administered separately, but producing a greater therapeutic efficacy when administered in a combined formulation. Moreover, while drug dosage and drug resistance typically limit first-line mono-chemotherapy, HSA-FCBL, with its ability to compromise mitochondrial membrane integrity and damage mtDNA, is expected to overcome those limitations and become an ideal candidate for the treatment of neoplastic disease. This is especially true because the CBL component of HSA-FCBL can be easily replaced with other DNA-damaging agents (e.g., doxorubicin and cisplatin) or photosensitizers (e.g., chlorine e6). Thus, our strategy is also universal for the rational development of new anticancer agents that work synergistically for strong cancer management regimes.
Supplementary Material
Acknowledgments
We thank the National Natural Science Foundation of China (NSFC 21521063, NSFC 21405041 and NSFC 21327009) and Science and Technology Project of Hunan Province (2015RS4019) and China Postdoctoral Science Foundation Funded Project (2016T90751, 2015M570679) for financial support. This work is also supported by the National Institutes of Health (GM079359). The authors thank Dr. Wenpei Liu for funding to support compound synthesis.
Footnotes
Supporting Information
Detail of FCBL synthesis; 1HNMR, UV−vis and fluorescence characterization of FCBL; preparation and characteristics of FCBL nanodrug; isolated DNA-crosslinking; in vitro cell cytotoxicity experimental procedures and cell apoptosis analysis data for this study. Supporting information for this article can be found under http://dx.doi.org/10.1002/cmdc.201600538.
Notes
The authors declare no competing financial interest.
References
- 1.Xu X, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci USA. 2013;110(46):18638–43. doi: 10.1073/pnas.1303958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92(16):1295–302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Iyer AK, Singh A, Ganta S, Amiji MM. Role of integrated cancer nanomedicine in overcoming drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1784–802. doi: 10.1016/j.addr.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci USA. 2008;105(45):17356–61. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu L, Chen T, Öçsoy I, Yasun E, Wu C, Zhu G, You M, Han D, Jiang J, Yu R, Tan W. A cell-targeted, size-photocontrollable, nuclear-uptake nanodrug delivery system for drug-resistant cancer therapy. Nano Lett. 2015;15(1):457–63. doi: 10.1021/nl503777s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng YR, Suntharalingam K, Johnstone TC, Yoo H, Lin W, Brooks JG, Lippard SJ. Pt(IV) prodrugs designed to bind non-covalently to human serum albumin for drug delivery. J Am Chem Soc. 2014;136(24):8790–8. doi: 10.1021/ja5038269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang P, Wang D, Su Y, Huang W, Zhou Y, Cui D, Zhu X, Yan D. Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug-drug conjugate for cancer therapy. J Am Chem Soc. 2014;136(33):11748–56. doi: 10.1021/ja505212y. [DOI] [PubMed] [Google Scholar]
- 9.te Velde EA, Vogten JM, Gebbink MF, van Gorp JM, Voest EE, Borel Rinkes IH. Enhanced antitumour efficacy by combining conventional chemotherapy with angiostatin or endostatin in a liver metastasis model. Br J Surg. 2002;89(10):1302–9. doi: 10.1046/j.1365-2168.2002.02183.x. [DOI] [PubMed] [Google Scholar]
- 10.Herskovic A, Martz K, al-Sarraf M, Leichman L, Brindle J, Vaitkevicius V, Cooper J, Byhardt R, Davis L, Emami B. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med. 1992;326(24):1593–8. doi: 10.1056/NEJM199206113262403. [DOI] [PubMed] [Google Scholar]
- 11.Citron ML, Berry DA, Cirrincione C, Hudis C, Winer EP, Gradishar WJ, Davidson NE, Martino S, Livingston R, Ingle JN, Perez EA, Carpenter J, Hurd D, Holland JF, Smith BL, Sartor CI, Leung EH, Abrams J, Schilsky RL, Muss HB, Norton L. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol. 2003;21(8):1431–9. doi: 10.1200/JCO.2003.09.081. [DOI] [PubMed] [Google Scholar]
- 12.van Hagen P, Hulshof MC, van Lanschot JJ, Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ, Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ, Cuesta MA, Blaisse RJ, Busch OR, ten Kate FJ, Creemers GJ, Punt CJ, Plukker JT, Verheul HM, Spillenaar Bilgen EJ, van Dekken H, van der Sangen MJ, Rozema T, Biermann K, Beukema JC, Piet AH, van Rij CM, Reinders JG, Tilanus HW, van der Gaast A, CROSS Group Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366(22):2074–84. doi: 10.1056/NEJMoa1112088. [DOI] [PubMed] [Google Scholar]
- 13.Henderson IC, Berry DA, Demetri GD, Cirrincione CT, Goldstein LJ, Martino S, Ingle JN, Cooper MR, Hayes DF, Tkaczuk KH, Fleming G, Holland JF, Duggan DB, Carpenter JT, Frei E, 3rd, Schilsky RL, Wood WC, Muss HB, Norton L. Improved outcomes from adding sequential Paclitaxel but not from escalating Doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. J Clin Oncol. 2003;21(6):976–83. doi: 10.1200/JCO.2003.02.063. [DOI] [PubMed] [Google Scholar]
- 14.Lu-Emerson C, Duda DG, Emblem KE, Taylor JW, Gerstner ER, Loeffler JS, Batchelor TT, Jain RK. Lessons from anti-vascular endothelial growth factor and anti-vascular endothelial growth factor receptor trials in patients with glioblastoma. J Clin Oncol. 2015;33(10):1197–213. doi: 10.1200/JCO.2014.55.9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 2008;18(4):165–73. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, Pesdar EA, Sobol M, Filimonenko A, Stuart S, Vondrusova M, Kluckova K, Sachaphibulkij K, Rohlena J, Hozak P, Truksa J, Eccles D, Haupt LM, Griffiths LR, Neuzil J, Berridge MV. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21(1):81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Marrache S, Pathak RK, Dhar S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc Natl Acad Sci USA. 2014;111(29):10444–9. doi: 10.1073/pnas.1405244111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–64. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 19.D’Souza GG, Wagle MA, Saxena V, Shah A. Approaches for targeting mitochondria in cancer therapy. Biochim Biophys Acta. 2011;1807(6):689–96. doi: 10.1016/j.bbabio.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 20.Lee C, Park HK, Jeong H, Lim J, Lee AJ, Cheon KY, Kim CS, Thomas AP, Bae B, Kim ND, Kim SH, Suh PG, Ryu JH, Kang BH. Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J Am Chem Soc. 2015;137(13):4358–67. doi: 10.1021/ja511893n. [DOI] [PubMed] [Google Scholar]
- 21.Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5(9):1032–8. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- 22.Wu JB, Lin TP, Gallagher JD, Kushal S, Chung LW, Zhau HE, Olenyuk BZ, Shih JC. Monoamine oxidase A inhibitor-near-infrared dye conjugate reduces prostate tumor growth. J Am Chem Soc. 2015;137(6):2366–74. doi: 10.1021/ja512613j. [DOI] [PubMed] [Google Scholar]
- 23.Fantin VR, Berardi MJ, Scorrano L, Korsmeyer SJ, Leder P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell. 2002;2(1):29–42. doi: 10.1016/s1535-6108(02)00082-x. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q, Liu Z. Albumin Carriers for Cancer Theranostics: A Conventional Platform with New Promise. Adv Mater. 2016 doi: 10.1002/adma.201600038. [DOI] [PubMed] [Google Scholar]
- 25.Tan W, Donovan MJ, Jiang J. Aptamers from cell-based selection for bioanalytical applications. Chem Rev. 2013;113(4):2842–62. doi: 10.1021/cr300468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc Natl Acad Sci USA. 2006;103(32):11838–43. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinthuvanich C, Veiga AS, Gupta K, Gaspar D, Blumenthal R, Schneider JP. Anticancer β-hairpin peptides: membrane-induced folding triggers activity. J Am Chem Soc. 2012;134(14):6210–7. doi: 10.1021/ja210569f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94(2):514–9. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swanner J, Mims J, Carroll DL, Akman SA, Furdui CM, Torti SV, Singh RN. Differential cytotoxic and radiosensitizing effects of silver nanoparticles on triple-negative breast cancer and non-triple-negative breast cells. Int J Nanomedicine. 2015;10:3937–53. doi: 10.2147/IJN.S80349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.