

Abstract
Detailed structural elucidation of protein glycosylation is a tedious process often involving several techniques. Glycomics and glycoproteomics approaches with mass spectrometry offer a rapid platform for glycan profiling but are limited by the inability to resolve isobaric species such as linkage and positional isomers. Recently, ion mobility spectrometry (IMS) has been shown to effectively resolve isobaric oligosaccharides, but the utility of IMS to obtain glycan structural information on a site-specific level with proteomic analyses has yet to be investigated. Here, we report that the addition of IMS to conventional glycoproteomics platforms adds additional information regarding glycan structure and is particularly useful for differentiation of sialic acid linkage isomers on both N- and O-linked glycopeptides. With further development IMS may hold the potential for rapid and complete structural elucidation of glycan chains at a site-specific level.
Graphical abstract
Glycosylation plays a role in nearly all aspects of biology including cell–cell communication, differentiation and development, modulating enzymatic activity, and immune regulation and is integral to many host–pathogen interactions.1,2 Glycan chains are attached to proteins either through the side chains of asparagine (N-linked) or serine and threonine residues (O-linked). All glycan chains are composed of relatively few types of monosaccharides, but the large number of possible branching patterns, linkage isomers, and other modifications (e.g., sulfation) results in an enormous diversity of structural variants with different biological effects. For example, sialic acids moieties can be linked α2-3 or α2-6 to the outer sugars of the nonreducing end, which alter host recognition of the influenza viruses.3 Despite their importance to clinical diagnostics and therapeutic development, our knowledge of glycosylation has been hindered by the structural complexity of carbohydrates, which create difficult problems for data analysis.4
Unlike DNA or proteins, for which technologies have enabled rapid sequencing, glycan sequencing remains a very involved and specialized task, sometimes requiring several orthogonal methods. Strategies for full glycan structural elucidation generally involve glycan removal either enzymatically (N-glycanase) or chemically (e.g., hydrazine) and either liquid chromatography or capillary electrophoresis to resolve each glycan structure.4–7 In many cases complex glycans need to be further analyzed with stepwise treatment using specific exoglycosidases for full structural elucidation. While such methods have been established for rapid parallel processing, one major limitation is the reliance on glycan removal from the protein. With proteins containing multiple glycosylation sites, this approach does not resolve differences among the glycan structures at the individual sites. Site-specific glycan characterization typically necessitates additional prefractionation to isolate each glycosylation site on a glycopeptide level. This additional step can be labor intensive and generally requires significantly more starting material.
Modern glycoproteomics with mass spectrometry offer a rapid platform for site-specific glycan profiling, but the information content is limited by the inability to resolve linkage- and positional isomers.8 Additional information on the glycan structures requires specific exoglycosidase treatments or derivatization schemes in conjunction with mass spectrometry.9–14 Ion mobility spectrometry (IMS) adds a dimension capable of resolving isobaric species by their gas-phase collision cross-section (CCS), which has proven useful at resolving oligosaccharides15–28 and databases for carbohydrate CCS values have recently been established.29 However, all of these studies have been limited to synthetic standards or glycan chains isolated from glycoproteins. Additionally, because of gas-phase rearrangements within certain types of sugars during typical collision-induced dissociation (CID), mass spectrometry based glycan structural characterization has largely focused on metal adducted glycan chains.30–32 Here we demonstrate how incorporation of IMS into glycoproteomics platforms can provide additional information regarding glycan structure on a site-specific level, particularly in regard to differentiation of sialylation linkages on N-linked and O-linked glycopeptides.
Results and Discussion
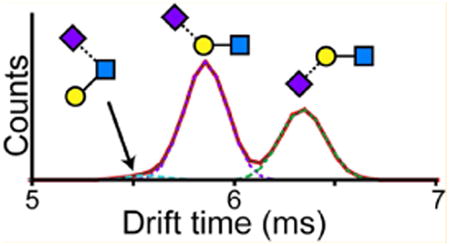
As a first assessment of the potential of IMS to resolve sialic acid isomers the arrival time distributions (ATD) of 3′ and 6′ sialyl-N-acetyllactosamine (SLN) were compared (Figure 1A). The two trisaccharides have identical structures with the exception of the linkage of the sialic acid (NeuAc) to the galactose (Gal), which is either linked α2-3 or α2-6. The ATD of the [M − H2O + H]+ species (657.24 Da), one of the abundant fragments that is observed during typical CID of an intact glycan chain, showed remarkable separation of the two linkage isomers. While 6′ SLN had a clear Gaussian peak at ∼6 ms, 3′ SLN showed a major peak at ∼6.4 ms with a minor peak near 6 ms. The fragment corresponding to the NeuAc-Gal species (m/z 454.17) was also examined, but was very poorly detected, presumably due to poor fragmentation efficiency at the Galβ1-4GlcNAc glycosidic bond and therefore was not used for further analysis.
Figure 1.
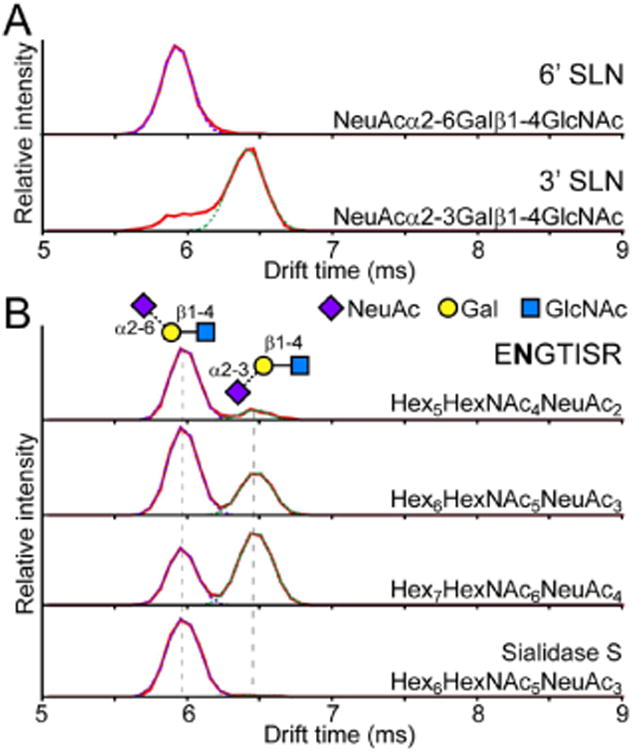
(A) Separation of α2-3 and α2-6 linked sialic acid by IMS. Arrival time distributions of the [M − H2O + H]+ fragment (m/z 657.24) of 3′ (top) and 6′ (bottom) sialyl-N-acetyllactosamine. The signal at ∼6 ms in the 3′ standard was not due to impurities (Figure S1) but most likely alternative gas phase conformations of the 3′ SLN standard. (B) Glycopeptide ENGTISR (residues 84–90) from a tryptic digest of α1AGP was isolated, subjected to collision induced dissociation, and the fragments were resolved by IMS. Differences in the ratio of α2-3 and α2-6 sialylation are evident between the biantennary bisialo (top), triantennary trisialo (2nd trace), and tetraantennary tetrasialo (3rd trace) glycoforms. Sialidase S treatment to specifically remove only α2-3 sialylation reveals an ATD showing purely the signal for the α2-6 linked species (bottom).
To test whether the additional peak in the 3′ SLN was due to some 6′ impurity, both isomers were analyzed by hydrophilic interaction chromatography (HILIC), which was able to resolve the two anomers of both 3′ and ′6 species (Figure S1A). From the ATDs, only one of the two anomers of the 3′ SLN shows a strong signal near 6 ms (Figure S1B). The presence of the second peak was not dependent on the collision energy used (Figure S2A). Structural rearrangements in the gas-phase have been reported to occur on several protonated glycan species even under mild CID conditions, albeit at relatively low levels.31–33 However, the CID spectra of both 3′ and 6′ SLN isomers showed only a trace peak (less than 0.1% base peak intensity) corresponding to a NeuAc-GlcNAc species (495.17 Da), that would arise from such rearrangements. The sodium adduct, which should not undergo any gas-phase rearrangements,32 also showed a second peak only in the 3′ SLN (Figure S2B), and its fragmentation pattern showed no evidence of any linkage rearrangements. A similar pattern was seen with the intact protonated ion, though for this case even the 6′ SLN showed two peaks, likely relating to the different anomers (Figure S2B). At present, we speculate that the minor peak in the 3′ SLN ATD is due to an alternate gas-phase conformation that is dependent on the anomeric configuration of the reducing sugar.
The same IMS separation was applied to resolve sialic acid linkage isomers on intact glycopeptides from a tryptic digest of human alpha-1-acid glycoprotein (α1AGP). Intact glycopeptides were resolved by LC, their precursor ions isolated in the quadrupole, fragmented by CID, and separated by IMS to resolve the glycan fragment isomers (LC–CID-IM-MS). Figure 1B shows the ATDs of the SLN fragment from one selected α1AGP glycopeptide with a biantennary, triantennary, or tetraantennary N-linked glycan chain. There is a clear signal for both the 3′ and 6′ isomers that matches the major peaks observed with the SLN standards. From the known structures of the N-linked glycans of α1AGP, the two peaks should correspond to NeuAcα2-3Galβ1-4GlcNAc and NeuAcα2-6Galβ1-4GlcNAc.34 To confirm these assignments, the tryptic digests were treated with Sialidase S to specifically cleave only α2-3 linked sialic acids. Accordingly, the ATD of the Sialidase S treated sample showed only the single peak at ∼6 ms (Figure 1B, bottom trace).
LC–CID-IM-MS was similarly performed on a tryptic digest of bovine Fetuin. The two peaks in the ATD for the SLN fragments matched those from α1AGP and the SLN standards (Figure 2A). An additional peak at ∼5.7 ms was evident when analyzing the fragments from the triantennary tetrasialo glycoform, which has been shown to contain a GlcNAc-linked sialic acid.35,36 After Sialidase S treatment the ∼6.5 ms peak disappeared, leaving only the 5.7 and 6 ms peaks. The tryptic digest was further treated with Sialidase A to remove all sialic acids. However, a signal for the triantennary glycoform bearing a single sialic acid remained, which has previously been observed to be resistant to sialidase treatment.35 The ATD of the SLN fragment from this glycoform showed a predominant peak at ∼5.7 ms, which therefore corresponds to the Galβ1-3(NeuAcα2-6)GlcNAc structure present on a fraction of triantennary N-linked glycans on Fetuin. Among the fragments from the tetrasialo glycoforms, there was also an abundant signal at m/z 495.2 and 948.34 that had a single peak in the ATD consistent with the fragments NeuAcα2-6GlcNAc and NeuAcα2-3Galβ1-3(NeuAcα2-6)GlcNAc, respectively (Figure 2B). These ions were not observed in the triantennary trisialo or bianternnary Fetuin glycopeptides or any of the α1AGP glycopeptides. Therefore, there is no detectable occurrence of gas-phase rearrangements between the sialic acid and the GlcNAc during CID of intact glycopeptides. Calibrated CCS values for all observed sialylated glycan fragments are reported in Table 1.
Figure 2.
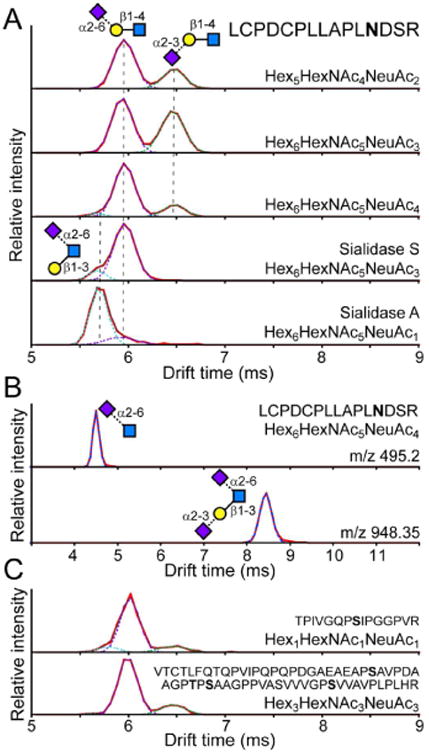
Patterns of sialylation linkage on glycopeptides from bovine Fetuin. (A) Glycopeptide 127-141 was isolated and fragments were resolved by IMS for the biantennary bisialo (1st trace), triantennary trisialo (2nd trace), and triantennary tetrasialo (3rd trace) glycoforms. After Sialidase S digestion, the predominant signal corresponds to the α2-6 linkage with an additional peak at ∼5.7 ms that was also faintly observed in the triantennary tetrasialo glycoform. ATD after Sialidase A digestion to remove all except certain GlcNAc-linked sialic acids (last trace). Glycan structures are indicated next to each peak. (B) ATD of the NeuAcα2-6GlcNAc fragment (m/z 495.2) and NeuAcα2-3Galβ1-3(NeuAcα2-6)GlcNAc fragment (m/z 948.35) from the triantennary tetrasialo glycoform of peptide 127-141 of Fetuin. (C) ATD of the SLN fragment obtained from isolation and fragmentation of Fetuin peptides 316-330 and 228-288 bearing sialylated O-linked glycans. Peptide sequence and glycan composition of the precursor ion is indicated within each panel.
Table 1. CCS Values for Sialic Acid Containing Glycan Fragment Ions.
| m/z | arrival time (ms) | CCS (Å2) | structure |
|---|---|---|---|
| 292.10 | 2.88 | 162.3 ± 0.3 | NeuAc |
| 657.24 | 5.93 | 236.9 ± 0.4 | NeuAcα2-6Galβ1-4GlcNAc |
| 657.24 | 6.41 | 247.2 ± 0.6 | NeuAcα2-3Galβ1-4GlcNAc |
| 657.24 | 5.67 | 231.0 ± 0.6 | Galβ1-3(NeuAcα2-6)GlcNAc |
| 495.18 | 4.47 | 204.0 ± 0.7 | NeuAcα2-6GlcNAc |
| 948.33 | 8.40 | 283.0 ± 0.9 | NeuAcα2-3Galβ1-3(NeuAcα2-6)GlcNAc |
In the course of examining the various glycoforms of each glycopeptide, we observed a trend in which the higher degree of branching correlated with an increase in the content of α2-3 linked sialylation. Such correlations have been previously observed during comprehensive NMR structural elucidation of Fetuin N-linked glycans.35 However, with the ability to resolve variations in sialylation patterns on a site-specific level we can see that not all sites share identical patterns (Figure S3). For example, the biantennary bisialo glycopeptide 142-169 shows 41.3% α2-3 while glycopeptide 54-85 with the same glycoform contains only 23.2% (Table S1). These types of differences are also observed among the different biantennary glycopeptides in α1AGP, with the content of α2-3 linked sialylation ranging from 4.3 to 23.6%. As a complementary way to monitor relative sialylation levels, we examined the signal intensity of each glycoform before and after sialidase S treatment as a qualitative assessment of the degree of α2-3 sialylation. Overall, the trends are consistent with the IMS results in that biantennary glycans have the highest content of 6′ sialylation and tetraantennary glycans have the most 3′ (Figure S4). While several studies have examined the distributions of Fetuin and α1AGP glycoforms along with relative amounts of sialylation linkages,35,37 to our knowledge this is the first example of such a quantitative examination of sialylation at a site-specific level.
The Fetuin digest also provided a sample to test whether IMS can be used to obtain structural information for glycopeptides bearing O-linked glycans. We examined peptides 316-330 bearing a single glycosylation site, and 228-288 which bears 4 well-characterized glycosylation sites.38 The most abundant glycoforms with resolved precursor ions were examined by IMS. Though the overall signal was relatively poor the ATDs of the sialyl-N-acetyllactosamine fragments could still be monitored (Figure 2C). With both peptides there were three species with the predominant peak at 6 ms and minor peaks at 6.3 and 5.6 ms. The most prevalent O-linked glycan structure on Fetuin is α2-3 sialylated with galactose linked β1-3 to a N-acetylgalactosamine (NeuAcα2-3Galβ1-3GalNAc).39 Two other glycoforms have α2-6 sialylation on the GalNAc: NeuAcα2-3Galβ1-3(NeuAcα2-6)GalNAc and Galβ1-3(NeuAcα2-6)GalNAc); the later of which might correspond to the peak at 5.6 ms, as seen with the Galβ1-3(NeuAcα2-6)GlcNAc) on the Fetuin N-linked glycopeptides. Additionally there is also a minor glycoform containing a second N-acetyllactosamine chain attached to the GalNAc (NeuAcα2-3Galβ1-3(NeuAcα2-3Galβ1-4GlcNAcβ1-6)-GalNAc).40 This hexasaccharide can fragment to yield NeuAcα2-3Galβ1-4GlcNAc that would be consistent with the peak at 6.3 ms, as observed with the N-linked glycopeptides. Further work will be necessary for definitively establishing the drift times of structures relevant to O-linked glycosylation, but for now this data demonstrates the utility of IMS for resolving different structures of O-linked glycans.
To assess the overall quantitative accuracy of sialylation by IMS an additional LC–MS run was performed with a high cone voltage to induce fragmentation of all precursor ions and observe the combined ATD from all glycopeptides (Figure 3). This signal should be dominated by the N-linked glycans, as the Fetuin O-linked glycopeptide precursor ions were of relatively weak intensity. From the combined ATD the overall ratios of sialic acid linkage isomers observed on Fetuin is approximately 38.4% α2-3, 59.4% α2-6, and 2.2% attributed to sialylation at the GlcNAc. These overall ratios are consistent with previous quantitative measurements of sialylation of Fetuin that range from 38:62 up to 51:49 in α2-3:α2-6 sialylation, possibly reflecting differences in the methods used for glycan isolation, detection, and variations in sample preparations.35,37 Additionally, the overall content of sialylation at GlcNAc is similar to that observed by nuclear magnetic resonance (∼2.6%).35
Figure 3.
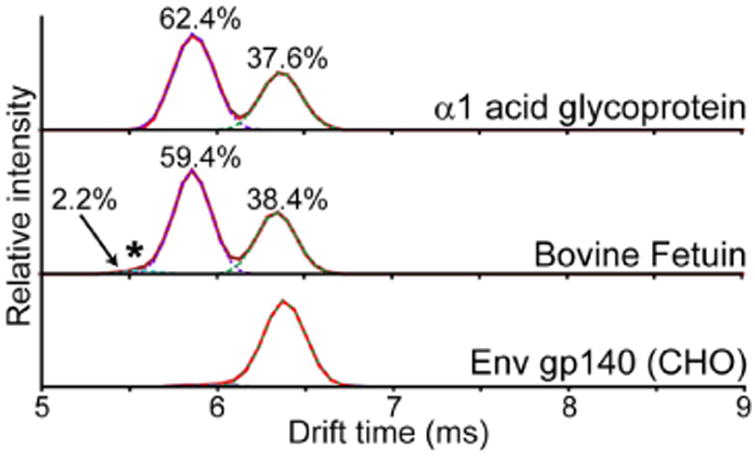
Untargeted ATDs of glycoprotein digests. Digests of α1AGP (top), Fetuin (middle), and Env gp140 from CHO cells (bottom) were analyzed by LC–IMS using a high cone voltage to induce glycan fragmentation in all eluting glycopeptides. The combined ATD for the sialyl-N-acteyllactosamine fragment (m/z 657.24) for the entire LC run is shown. Gaussian fits for quantitation are shown in dashed lines. Relative percentages of different sialic acid linkages are indicated above each peak, including the GlcNAc-linked sialic acid on Fetuin (*).
As an additional control we examined the sialylation pattern of a HIV-1 Env gp140 glycoprotein purified from Chinese Hamster Ovary (CHO) cells. gp140 trimers contain 28 N-linked glycosylation sites on each of the three subunits, some of which bear biantennary monosialylated glycan structures.41 Expression in CHO cells produces proteins with only α2-3 linked sialylation as CHO cells lack α2-6 sialyltransferase.42 A tryptic digest of gp140 was analyzed using the same high cone voltage approach as described for α1AGP and Fetuin. The resulting combined ATD showed only a single peak at 6.3 ms (Figure 3, lower panel). Examining the individual patterns for each glycopeptide in such a complex glycoprotein was beyond the scope of this study. However, this data sufficiently demonstrates that the underlying cause for the multiple peaks observed in the ATD of the 3′ SLN standard (Figure 1A) is not confounding the analysis of sialyl-N-acetyllactosamine fragments from intact glycopeptides.
Lastly we examined the effect of the collision energy (CE) on the ATD profile of the glycan fragments. The m/z 657.24 peak was observed even with the lowest CE (4 V), became more abundant as the voltage was increased, and eventually diminished above 40 V, likely due to further fragmentation of the SLN fragment (Figure S5A). Even at the highest CE the ATD for m/z 657.24 fit well to two Gaussian distributions, unlike that observed in the 3′ SLN standard (Figures S1B and S5C). The relative intensities of the 3′ and 6′ SLN peaks are consistent at lower CE, but deviate above 20 V, with a drop in the observed 3′ SLN content (Figure S5B). The offsets at higher CE may be due to further fragmentation if one of the sialylation isomers is more susceptible to secondary fragmentation. Overall, this indicates that a lower CE is favorable for a more accurate quantitative measure of the sialylation linkage by IMS.
Conclusion
A number of studies have demonstrated the power of IMS to resolve linkage and positional isomers in synthetic oligosaccharides and differentiate intact glycan chains. However, the utility of IMS to obtain glycan information on a site-specific level during glycoproteomics analyses is only now becoming appreciated. Here, we report that the addition of IMS to conventional glycoproteomics platforms adds the ability to differentiate sialic acid linkage isomers on both N- and O-linked glycopeptides. During the revision stage of this article, another group reported a very similar strategy for differentiating sialylation linkage by IMS,43 further illustrating the utility of this approach. With further optimization of separation conditions for closely related glycans present on biologically relevant glycan structures, IMS has the potential to extract a higher level of information from glycoproteomics. Such a tool can elucidate fine glycan structural information on a site-specific level with minimal sample requirements and without additional sample preparation steps beyond current proteomic protocols.
Methods
Preparation of Glycopeptides
In total, 4 mg of bovine Fetuin and human α1AGP (Sigma-Aldrich, St. Louis, MO) were resuspended in 100 μL of 8 M urea, 100 mM Tris pH 8.0, 50 mM dithiothreitol (DTT) and heated at 85 °C for 30 min. Cysteines were alkylated by the addition of 100 mM iodoacetamide (IAM) and incubation for 1 h in the dark, followed by addition of 50 mM DTT to quench remaining IAM. The samples were diluted 16-fold in 20 mM Tris pH 8.0 and proteins were digested with TPCK-treated trypsin (Sigma-Aldrich) at a 1:50 ratio of trypsin–substrate overnight at 37 °C and subsequently quenched with 1 mM PMSF. A volume of 100 μL of the digest was treated with 10 mU Sialidase A (Arthrobacter ureafaciens, Prozyme, Hayward, CA) at 37 °C for 4 h. A second 100 μL portion was acidified to pH 6.0 with sodium acetate and treated with 10 mU Sialidase S (Streptococcus pneumoniae, Prozyme) at 37 °C for 4 h.
HIV-1 Env gp140 was expressed in CHO cells and purified as described previously.44 A volume of 50 μL of Env gp140 (0.4 mg/mL in PBS pH 7.4) was reduced with 20 mM DTT at 85 °C for 20 min, acidified to pH 2.5 with 0.5% formic acid, and digested with 3 μg of pepsin (Worthington Biochemicals, MA) at 37 °C for 15 min. Samples were neutralized with 10 μL 1 M sodium acetate for a final pH of 5.0 to stop digestion.
LC–IMS-MS
Tryptic digests (2 μg per injection) of glycoproteins were resolved with a Waters Aquity HPLC over a 1 mm × 100 mm 1.7 μm BEH C18 column (Waters, Milford, MA) with a linear gradient of 3% to 37% B over 18 min at a flow rate of 70 μL/min (A, 0.1% formic acid; B, 0.1% formic acid, 100% acetonitrile). Eluting peptides were analyzed online by electrospray with a Synapt G2-Si mass spectrometer (Waters). Source and desolvation temperatures were 100 and 250 °C, and the capillary and cone voltages were set to 2.5 kV and 40 V. Initial runs were collected in a data-dependent fashion to select ions with a quadrupole resolution of 7 and perform MS/MS on precursor ions from 800 up to 2000 Da with a charge state of at least 2. Glycopeptides were identified by using fragmentation data and exact mass to assess the glycan composition and the peptide sequence.45
Targeted glycopeptide runs were subsequently performed by running identical injections in a data-independent manner with fragmentation in the trap prior to the IMS cell (premobility MS/MS). Glycopeptides were selected by retention times and narrow m/z windows (2 Da) for quadrupole selections. Two second acquisitions for each MS/MS were collected with the collisions energy ramped from 30 to 50 V. The IMS cell pressure was ∼2.85 mbar with a N2 gas flow of 90 mL/min and the traveling wave height and velocity were set at 40 V and 650 m/s, respectively. Six additional injections and LC runs were performed with a fixed trap CE value of 0, 5, 10, 20, 35, and 50 V. For untargeted IMS analysis of glycan fragments from all glycopeptides, an identical LC–IMS-MS run was performed with a cone voltage of 120 V. For CCS calibration, IMS-MS was collected for 5 minutes before and after the series of LC runs using a 0.01 mg/mL polyalanine ladder (Waters) in 0.1% formic acid infused at 40 μL/min with identical IMS and MS settings.
3′ and 6′ sialyl-N-acetyllactosamines (SLNs) were purchased from Prozyme (Hayward, CA). SLNs were resuspended in 0.1% formic acid for a final concentration of 10 μM and infused at 40 μL/min into a Waters Synapt GS-Si mass spectrometer with settings as described above. For HILIC separation, 4 μg of either 3′ or 6′ SLN was injected over a GlycoSep 1 mm × 50 mm XBridge Glycan BEH amide column (Waters) in 90% acetonitrile. A linear gradient of 95 to 65% B over 20 min was used to elute SLNs at 150 μL/min (A, 0.1% formic acid, B, 0.1% formic acid, 100% acetonitrile). The flow was coupled to a Synapt G2-Si, and spectra were collected over a mass range of 300 to 1200 every 2 s with ion mobility engaged as described above.
Data Analysis
Data were analyzed in MassLynx 4.1 and DriftScope v2.8 (Waters). ATDs for sialylated glycan fragments (see Table 1) with a mass window of 0.05 Da were exported and copied into Excel (Microsoft) for fitting Gaussian distributions with custom macros. The corrected drift times of the 1+ ions for Ala3 up to Ala14 relative to their known collision cross section (CCS) values was used to calculate CCS values for all glycan fragments as described previously.46,47
Supplementary Material
Acknowledgments
We wish to thank Andras Guttman, Kelly Hines, and Libin Xu for insightful discussions and assistance with data interpretation. HIV-1 Env gp140 purified from CHO cells was a kind gift from Al Cupo and John P. Moore. This work was supported by NIH grants R01-GM099989 (K.K.L.) as well as the Bill and Melinda Gates Foundation Global Vaccine Accelerator Program (GV-VAP) grant OPP1033102 (K.K.L.).
Footnotes
ASSOCIATED CONTENT
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.6b00265.
Content of sialic acid linkages in α1AGP and Fetuin glycopeptides; HILIC traces of purification of 3′ and 6′ sialyl-N-acetyllactosamine; ATDs of SLN at different collision energies, the protonated, and the sodiated species; ATD of sialyl-N-acetyllactosamine fragments from α1AGP and Fetuin glycopeptides; extracted ion chromatograms of glycopeptides after treatment with sialidases; and effect of the collision energy (CE) on the observed sialylation linkage by IMS (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.Varki A, Cummings R, Esko J, Freeze H, Stanley P, Bertozzi C, Hart GW, Etzler M. Essentials of Glycobiology. 2nd. Cold Spring Harbor Laboratory Press; Plainview, NY: 2009. [PubMed] [Google Scholar]
- 2.Brooks SA. Mol Biotechnol. 2009;43:76–88. doi: 10.1007/s12033-009-9184-6. [DOI] [PubMed] [Google Scholar]
- 3.Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Nature. 2006;440:435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 4.Marino K, Bones J, Kattla JJ, Rudd PM. Nat Chem Biol. 2010;6:713–723. doi: 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- 5.Guttman A. Nature. 1996;380:461–462. doi: 10.1038/380461a0. [DOI] [PubMed] [Google Scholar]
- 6.Royle L, Dwek RA, Rudd PM. Current Protocols in Protein Science. 2006;43:12.16.11–12.16.45. doi: 10.1002/0471140864.ps1206s43. [DOI] [PubMed] [Google Scholar]
- 7.Campbell MP, Royle L, Rudd PM. Methods Mol Biol. 2015;1273:17–28. doi: 10.1007/978-1-4939-2343-4_2. [DOI] [PubMed] [Google Scholar]
- 8.Medzihradszky KF, Kaasik K, Chalkley RJ. Mol Cell Proteomics. 2015;14:2103–2110. doi: 10.1074/mcp.M115.050393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leavell MD, Leary JA. J Am Soc Mass Spectrom. 2001;12:528–536. doi: 10.1016/S1044-0305(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 10.Wheeler SF, Harvey DJ. Anal Chem. 2000;72:5027–5039. doi: 10.1021/ac000436x. [DOI] [PubMed] [Google Scholar]
- 11.Wheeler SF, Domann P, Harvey DJ. Rapid Commun Mass Spectrom. 2009;23:303–312. doi: 10.1002/rcm.3867. [DOI] [PubMed] [Google Scholar]
- 12.Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M. Anal Chem. 2014;86:5784–5793. doi: 10.1021/ac500335t. [DOI] [PubMed] [Google Scholar]
- 13.de Haan N, Reiding KR, Haberger M, Reusch D, Falck D, Wuhrer M. Anal Chem. 2015;87:8284–8291. doi: 10.1021/acs.analchem.5b02426. [DOI] [PubMed] [Google Scholar]
- 14.Kolarich D, Weber A, Turecek PL, Schwarz HP, Altmann F. Proteomics. 2006;6:3369–3380. doi: 10.1002/pmic.200500751. [DOI] [PubMed] [Google Scholar]
- 15.Gabryelski W, Froese KL. J Am Soc Mass Spectrom. 2003;14:265–277. doi: 10.1016/S1044-0305(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 16.Clowers BH, Dwivedi P, Steiner WE, Hill HH, Jr, Bendiak B. J Am Soc Mass Spectrom. 2005;16:660–669. doi: 10.1016/j.jasms.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Dwivedi P, Bendiak B, Clowers BH, Hill HH., Jr J Am Soc Mass Spectrom. 2007;18:1163–1175. doi: 10.1016/j.jasms.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Isailovic D, Kurulugama RT, Plasencia MD, Stokes ST, Kyselova Z, Goldman R, Mechref Y, Novotny MV, Clemmer DE. J Proteome Res. 2008;7:1109–1117. doi: 10.1021/pr700702r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plasencia MD, Isailovic D, Merenbloom SI, Mechref Y, Novotny MV, Clemmer DE. J Am Soc Mass Spectrom. 2008;19:1706–1715. doi: 10.1016/j.jasms.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamagaki T, Sato A. Anal Sci. 2009;25:985–988. doi: 10.2116/analsci.25.985. [DOI] [PubMed] [Google Scholar]
- 21.Zhu M, Bendiak B, Clowers B, Hill HH., Jr Anal Bioanal Chem. 2009;394:1853–1867. doi: 10.1007/s00216-009-2865-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams JP, Grabenauer M, Holland RJ, Carpenter CJ, Wormald MR, Giles K, Harvey DJ, Bateman RH, Scrivens JH, Bowers MT. Int J Mass Spectrom. 2010;298:119–127. [Google Scholar]
- 23.Fenn LS, McLean JA. Phys Chem Chem Phys. 2011;13:2196–2205. doi: 10.1039/c0cp01414a. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Bendiak B, Siems WF, Gang DR, Hill HH., Jr Anal Chem. 2013;85:2760–2769. doi: 10.1021/ac303273z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pagel K, Harvey DJ. Anal Chem. 2013;85:5138–5145. doi: 10.1021/ac400403d. [DOI] [PubMed] [Google Scholar]
- 26.Both P, Green AP, Gray CJ, Sardzik R, Voglmeir J, Fontana C, Austeri M, Rejzek M, Richardson D, Field RA, Widmalm G, Flitsch SL, Eyers CE. Nat Chem. 2013;6:65–74. doi: 10.1038/nchem.1817. [DOI] [PubMed] [Google Scholar]
- 27.Hoffmann W, Hofmann J, Pagel K. J Am Soc Mass Spectrom. 2014;25:471–479. doi: 10.1007/s13361-013-0780-0. [DOI] [PubMed] [Google Scholar]
- 28.Hofmann J, Hahm HS, Seeberger PH, Pagel K. Nature. 2015;526:241–244. doi: 10.1038/nature15388. [DOI] [PubMed] [Google Scholar]
- 29.Struwe WB, Pagel K, Benesch JL, Harvey DJ, Campbell MP. Glycoconjugate J. 2015 doi: 10.1007/s10719-015-9613-7. [DOI] [PubMed] [Google Scholar]
- 30.Wuhrer M, Koeleman CA, Hokke CH, Deelder AM. Rapid Commun Mass Spectrom. 2006;20:1747–1754. doi: 10.1002/rcm.2509. [DOI] [PubMed] [Google Scholar]
- 31.Wuhrer M, Koeleman CA, Deelder AM. Anal Chem. 2009;81:4422–4432. doi: 10.1021/ac900278q. [DOI] [PubMed] [Google Scholar]
- 32.Leymarie N, Zaia J. Anal Chem. 2012;84:3040–3048. doi: 10.1021/ac3000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harvey DJ, Mattu TS, Wormald MR, Royle L, Dwek RA, Rudd PM. Anal Chem. 2002;74:734–740. doi: 10.1021/ac0109321. [DOI] [PubMed] [Google Scholar]
- 34.Yoshima H, Matsumoto A, Mizuochi T, Kawasaki T, Kobata A. J Biol Chem. 1981;256:8476–8484. [PubMed] [Google Scholar]
- 35.Green ED, Adelt G, Baenziger JU, Wilson S, Van Halbeek H. J Biol Chem. 1988;263:18253–18268. [PubMed] [Google Scholar]
- 36.Takasaki S, Kobata A. Biochemistry. 1986;25:5709–5715. doi: 10.1021/bi00367a054. [DOI] [PubMed] [Google Scholar]
- 37.Cointe D, Leroy Y, Chirat F. Carbohydr Res. 1998;311:51–59. doi: 10.1016/s0008-6215(98)00196-7. [DOI] [PubMed] [Google Scholar]
- 38.Windwarder M, Altmann F. J Proteomics. 2014;108:258–268. doi: 10.1016/j.jprot.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 39.Merry AH, Neville DC, Royle L, Matthews B, Harvey DJ, Dwek RA, Rudd PM. Anal Biochem. 2002;304:91–99. doi: 10.1006/abio.2002.5620. [DOI] [PubMed] [Google Scholar]
- 40.Edge AS, Spiro RG. J Biol Chem. 1987;262:16135–16141. [PubMed] [Google Scholar]
- 41.Behrens AJ, Vasiljevic S, Pritchard LK, Harvey DJ, Andev RS, Krumm SA, Struwe WB, Cupo A, Kumar A, Zitzmann N, Seabright GE, Kramer HB, Spencer DI, Royle L, Lee JH, Klasse PJ, Burton DR, Wilson IA, Ward AB, Sanders RW, Moore JP, Doores KJ, Crispin M. Cell Rep. 2016;14:2695–2706. doi: 10.1016/j.celrep.2016.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee EU, Roth J, Paulson JC. J Biol Chem. 1989;264:13848–13855. [PubMed] [Google Scholar]
- 43.Hinneburg H, Hofmann J, Struwe WB, Thader A, Altmann F, Varon Silva D, Seeberger PH, Pagel K, Kolarich D. Chem Commun (Cambridge, U K) 2016;52:4381–4384. doi: 10.1039/c6cc01114d. [DOI] [PubMed] [Google Scholar]
- 44.Chung NP, Matthews K, Kim HJ, Ketas TJ, Golabek M, de Los Reyes K, Korzun J, Yasmeen A, Sanders RW, Klasse PJ, Wilson IA, Ward AB, Marozsan AJ, Moore JP, Cupo A. Retrovirology. 2014;11:33. doi: 10.1186/1742-4690-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medzihradszky KF. Methods Enzymol. 2005;405:116–138. doi: 10.1016/S0076-6879(05)05006-8. [DOI] [PubMed] [Google Scholar]
- 46.Bush MF, Campuzano ID, Robinson CV. Anal Chem. 2012;84:7124–7130. doi: 10.1021/ac3014498. [DOI] [PubMed] [Google Scholar]
- 47.Forsythe JG, Petrov AS, Walker CA, Allen SJ, Pellissier JS, Bush MF, Hud NV, Fernandez FM. Analyst. 2015;140:6853–6861. doi: 10.1039/c5an00946d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.