

Abstract
Acute myeloid leukemia (AML) comprises a heterogeneous group of leukemias frequently defined by recurrent cytogenetic abnormalities, including rearrangements involving subunits of the core-binding factor (CBF) transcriptional complex. To better understand the genomic landscape of CBF-AMLs, we analyzed both pediatric (n=87) and adult (n=78) samples, including cases with RUNX1-RUNX1T1 (n=85) or CBFB-MYH11 (n=80) rearrangements, by whole-genome or whole-exome sequencing. In addition to previously reported somatic mutations in the Ras signaling pathway, we identified recurrent stabilizing mutations in CCND2, suggesting a recurrent and previously unappreciated cooperating pathway in CBF-AML. Outside of signaling alterations, RUNX1-RUNX1T1 and CBFB-MYH11 AMLs demonstrated a remarkably different spectrum of cooperating mutations as RUNX1-RUNX1T1 cases harbored recurrent somatic mutations in DHX15 and ZBTB7A, as well as an enrichment of somatic mutations in epigenetic regulators, including ASXL2, and in components of the cohesin complex. This detailed analysis provides insights into the pathogenesis and development of CBF-AML, while highlighting dramatic differences in the landscape of cooperating mutations between these related AML subtypes.
CBF-AMLs, which account for approximately 30% of pediatric and 15% of adult AMLs, contain chromosomal translocations or inversions that target the transcription factors RUNX1 (AML1) or CBFB. Normally RUNX1 and CBFB heterodimerize to bind DNA and recruit lineage defining transcription factors to regulate hematopoietic differentiation1. The fusion products found in CBF-AMLs [RUNX1-RUNX1T1/t(8;21)(q22;q22) or CBFB-MYH11/inv(16)(p13.1q22) or t(16;16)(p13.1;q22)] block myeloid differentiation but are not sufficient to induce leukemia, as leukemia development in murine models of both fusions requires secondary mutations, including those in the Ras pathway2–5.
While initial studies focused on validating known mutations, especially those in the Ras pathway6–17, and recent profiling studies focused on limited gene sets18–20 or had limited sample numbers21–23, we sought to comprehensively understand genetic variations contributing to CBF-AML development by analyzing 165 cases by whole-genome sequencing (WGS, n=17) or whole-exome sequencing (WES, n=148) (Table 1 and Supplemental Table 1). In this cohort, the age distribution of patients with RUNX1-RUNX1T1 (n=85) or CBFB-MYH11 (n=80) rearrangements were comparable, as were the outcomes (Supplemental Figure 1). An average of 9.86 ± 6.16 (± s.d.) somatic mutations with functional consequences were identified per case, with more mutations in RUNX1-RUNX1T1 cases (11.86 ± 6.40 vs. 7.74 ± 5.13 for CBFB-MYH11, p <0.0001) and adult cases (12.56 ± 6.55 vs. 7.44 ± 4.62 for pediatric, p <0.0001) (Supplemental Figure 2 and Supplemental Tables 2–7), while the spectrum of mutations for both CBF-AMLs subtypes was similar to the previously reported signature for AML24 (Supplemental Figure 3). Subclonal variants were identified in all CBF-AMLs analyzed by WGS followed by capture validation at > 1200× coverage, consistent with other AMLs25,26 (Supplemental Figure 4). Few copy number alterations (CNAs) or additional structural variations (SVs) (Supplemental Figure 5 and Supplemental Tables 8 and 9) were present. Consistent with previous reports14,27,28, 66.1% of the cases harbored activating mutations in NRAS, KIT, FLT3, KRAS, PTPN11 and/or loss-of-function mutations in NF1 (Figure 1); cumulatively these mutations were not associated with outcome (Supplemental Figure 6). NRAS was the most frequently mutated gene in CBF-AMLs, yet was not associated with outcome (p=0.638). Not only were NRAS mutations more common in CBFB-MYH11 AMLs (p=0.032), but the spectrum of mutations was also different as codon 61 mutations were more common in CBFB-MYH11 cases (p=0.0023, Supplemental Figure 7). Additionally, there was an enrichment of KIT exon 17 mutations (p=0.0051) in the RUNX1-RUNX1T1 cohort13,15, which were associated with an inferior outcome (p=0.0097, See Supplementary Note and Supplemental Figures 6, 8, and 9).
Table 1.
Characteristics CBF-AML cohort
| Subtype | Total | Pediatric | Adult | WGS | WES | RNAseq |
|---|---|---|---|---|---|---|
|
|
||||||
| RUNX1-RUNX1T1 | 85 | 44 | 41 | 7 | 78 | 27 |
| CBFB-MYH11 | 80 | 43 | 37 | 10 | 70 | 9 |
|
|
||||||
| Total | 165 | 87 | 78 | 17 | 148 | 36 |
WES, whole exome sequencing; WGS, whole genome sequencing
Figure 1. Mutational landscape of CBF-AML.
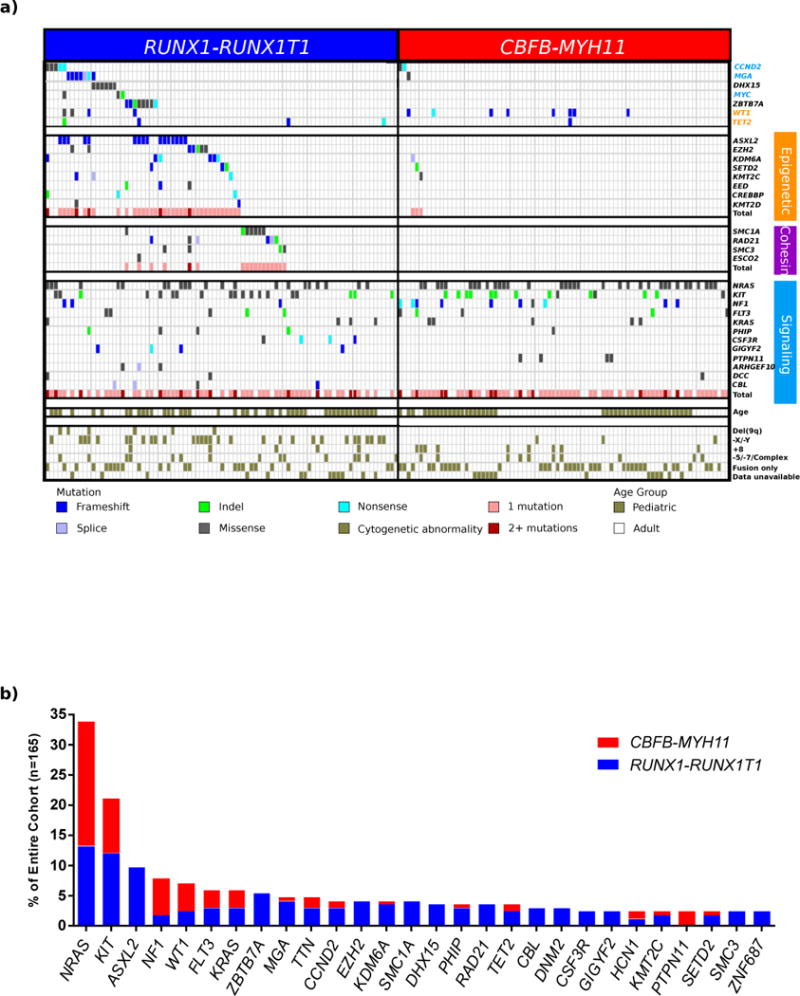
(a) Mutational data for 165 CBF-AML cases sequenced either by whole genome (n=17) or whole exome sequencing (n=148). Signaling, epigenetic, and cohesin genes are grouped into functional groups. Cytogenetic abnormalities and patient age group (adult or pediatric) are shown along the bottom of the figure. Mutations in both epigenetic (p=4.3E–10) and cohesin (p=2.2E–16) genes are significantly enriched in RUNX1-RUNX1T1 AML (Fisher’s Exact test). (b) The frequency of recurrently mutated genes (n>3) separated by CBF-AML fusion type is shown. Of the 10 FLT3 mutations, 4 are internal tandem duplications (ITD), 5 are located in the tyrosine kinase domain, and 1 is classified as neither ITD nor TKD.
MGA, a negative regulator of MYC signaling29, was also recurrently mutated in CBF-AMLs (Figure 2). The MGA mutations in RUNX1-RUNX1T1 AMLs are all predicted to be deleterious (including the splice site variant) and would eliminate the helix-loop-helix domain and binding of MGA to MAX29 (Figure 2). Similar loss-of-function mutations in MGA have also recently identifed in lung cancer, chronic lymphocytic leukemia, ETV6-RUNX1 acute lymphoblastic leukemia, NK/T cell lymphomas and in a single case of RUNX1-RUNX1T1 AML in the AML TCGA study30–34. We also found three cases with MYC mutations and seven cases (five RUNX1-RUNX1T1 and two CBFB-MYH11) with mutations in cyclin D2 (CCND2), a known downstream target of MYC35. The CCND2 mutations (Figure 2 and Supplemental Figure 10) all surround a conserved phosphorylation site (Thr280) that regulates ubiquitination of Lys270 by FBXL236 and degradation by the proteosome37. Germline and somatic mutations in this region of CCND2 have been identified in patients with Megalencephaly-Polymicrogyria-Polydactyly-Hydrocephalus (MPPH) syndrome38, but have been uncommon in myeloid malignancies25,39. Much like the germline mutations in MPPH, the CCND2 mutations identified here led to increased stability of CCND2 (Figure 2 and Supplemental Figure 10). Collectively, these data suggest that stabilization of CCND2 is a previously unidentified and potentially targetable mutation in CBF-AML leukemias.
Figure 2. Recurrent mutations in CCND2 and MGA.
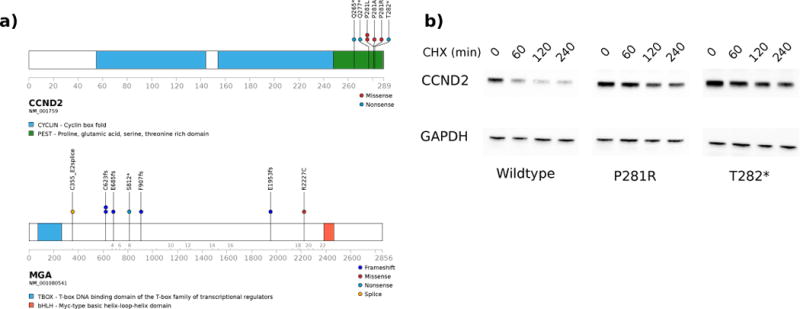
Domain structure and the localization of mutations are shown for (a) CCND2 and MGA. (b) Representative western blot (of three independent experiments) of HEK293T cells expressing wild-type, P281R, or T282* CCND2 treated with cyclohexamide (CHX) and harvested at the indicated time points. Data show the expected increase in the levels of mutant CCND2 protein. GAPDH serves as a loading control.
Recurrent p. R222G mutations in DHX15, a member of the DExD/H-box family of RNA helicases whose yeast homolog (Prp43) participates in spliceosome function and ribosomal biogenesis40–43, were also identified exclusively in the RUNX1-RUNX1T1 cohort. Interestingly, ribosomal biogenesis is also affected by RUNX1 deficiency44. The R222G missense mutation lies in the back of the RecA1 domain along the translocation site and, therefore, is unlikey to affect its ATPase activity or RNA binding ability (Supplemental Figure 11). We used RNAi-mediated knockdown of DHX15 to mimic a complete loss of function and observed differential expression of splicing genes and genes involved in ribosomal biogenessis (Supplemental Tables 10–12) as well as an increase in the number of alternative splicing events upon loss of DHX15 protein. A similar increase in alternative splicing events was obtained when the R222G mutant was overexpressed in vitro. These data led us to examine proteins with differential binding capabilities between the wildtype and mutant DHX15 protein via mass spectroscopy and co-immunoprecipitation (Supplemental Table 13). We found that TFIP11, a component of the splicing machinery, exhibits reduced binding to the DHX15 R222G mutant (Figure 3). While common in RUNX1-RUNX1T1 cases, to date this mutation has not been seen in other myeloid neoplasms; surprisingly other myeloid neoplasms harbor mutations in other spliceosome components (e.g. SF3B1, U2AF1)45, which were not observed in CBF-AMLs.
Figure 3.
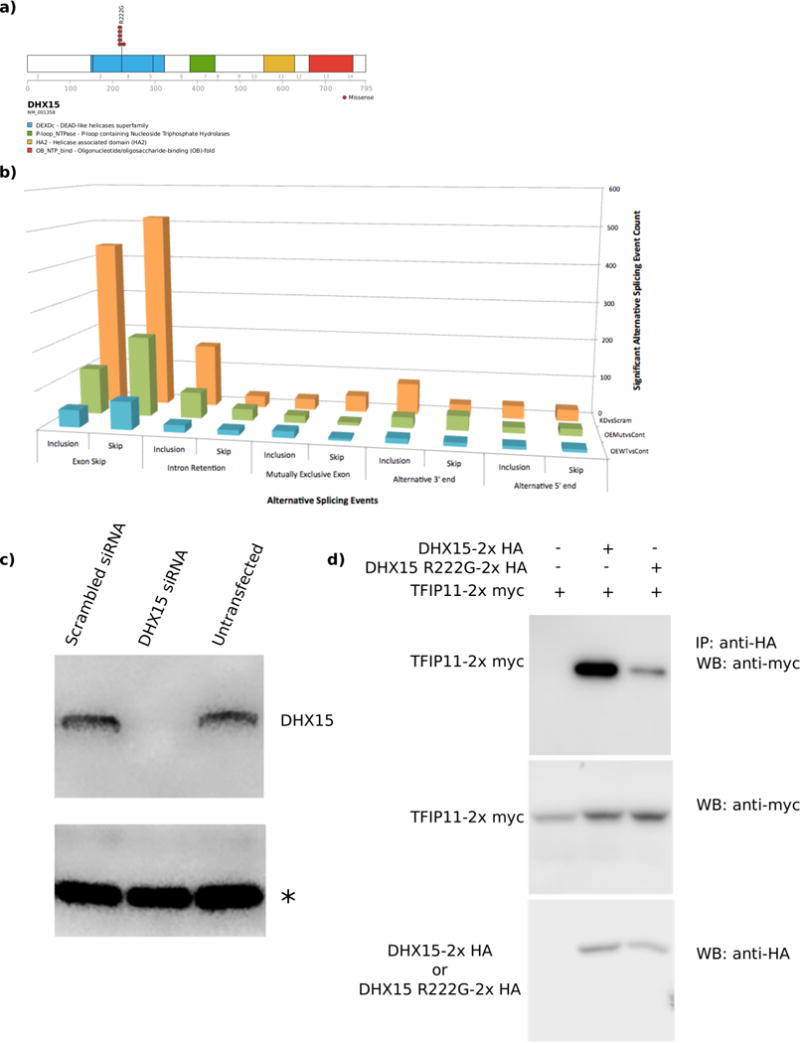
DHX15 is recurrently mutated in RUNX1-RUNX1T1 AML. (a) Domain structure and the localization of mutations for DHX15. (b) Increased numbers of alternative splicing events were observed upon DHX15 knockdown (red bars) or overexpression of the R222G mutant (green bars) compared to overexpression of wildtype DHX15 (blue bars). (c) siRNA mediated knockdown of DHX15 leads to an enrichment of differentially regulated genes associated with splicing and ribosomal biogenesis. (d) Western blot showing the effectiveness of the DHX15 knockdown. Equal amounts of protein were loaded for each sample. An asterisk indicates a non-specific band also used as a loading control. (e) Co-immunoprecipitation of TFIP11 with DHX15 demonstrates reduced binding of TFIP11 to the R222G mutant form of DHX15.
A striking and statistically significant enrichment for putative loss-of-function mutations in chromatin modifying genes (ASXL2, EZH2, KDM6A, EED, SETD2, KMT2D, KMT2C, and CREBBP) was observed in the RUNX1-RUNX1T1 cases (44% vs. 4% in CBFB-MYH11 cases, p=4.3E–10, Figure 1, Supplemental Figure 12). Consistent with previous reports, frameshift mutations in ASXL2 were common and found exclusively in RUNX1-RUNX1T1 CBF-AMLs46,47. These mutations were identified in both pediatric and adult cases, and were not associated with outcome (Supplemental Figure 13). These truncating mutations would eliminate the PHD protein-protein interacting domain, and are similar to mutations in ASXL1 that inhibit myeloid differentiation and induce a myelodysplastic syndrome (MDS)-like disease in mice48,49. Whether ASXL2 modulates PRC2 function, like ASXL1, however, is currently unknown48. In vitro studies using two separate ASXL2 mutations suggested that ASXL2 may play a role in myeloid differentiation (Supplemental Figure 14). We did not identify mutations in ASXL1 in the de novo leukemias, despite adequate sequence coverage in our WES cohort (Supplemental Figure 15). Many of the other mutated epigenetic regulators, such as KMT2C50,51, KDM6A52, and CREBBP53, functionally interact in complexes that alter enhancer states54, suggesting that enhancer dysregulation may be an important cooperating event with RUNX1-RUNX1T1. As such, recent data generated from Lowe and colleagues observed that disruption of Kmt2c in a RUNX1-RUNX1T1 model was associated with resistant disease55. Interestingly, of the 4 patients with KMT2C mutations in our cohort, 3 relapsed in <12 months and the 4th patient had residual disease after one course of remission induction therapy.
Mutations in the cohesin complex (n=17) were exclusively identified in RUNX1-RUNX1T1 AMLs (Supplemental Figure 16). SMC1A, SMC3 and RAD21 encode members of the complex responsible for sister chromatid cohesin during mitosis and post-replicative DNA repair56. Mutations in these genes have been reported in AML19,57,58 and germline mutations are some of the underlying causes of the cohesinopathies56,59. The seventeen cases with cohesin mutations lack evidence of aneuploidy or an increase rate of DNA mutations (Supplemental Tables 1 and 2) and mutation status has no effect on outcome (Supplemental Figure 17). We speculate that these mutations, similar to those in the epigenetic regulators mentioned above, alter transcriptional programs by dysregulating enhancer function50. We also identified recurrent mutations in ZBTB7A/Pokemon (Supplemental Figure 18). The putative loss-of-function mutations identified here are consistent with recent reports of ZBTB7A acting as a tumor suppressor in RUNX1-RUNX1T1 AML22,60
Transcriptome analysis was performed on the 36 (27 RUNX1-RUNX1T1 and 9 CBFB-MYH11) cases for which material was available. Differential expression analysis generated data consistent with previous array-based studies61,62 (Supplemental Figure 19 and Supplemental Tables 14–16), yet mutation specific differences could not be definitively determined due to our relatively small sample size. We were able to detect the alternatively spliced 9A isoform of RUNX1T1 in every RUNX1-RUNX1T1 case, ranging in abundance from 13% to 64% (Supplemental Figure 20)63.
Relapse material was characterized by WES for 8 samples in the cohort, demonstrating dynamic patterns of clonal evolution during disease progression including loss, retention or gain of somatic mutations and copy number alterations (Figure 4A, Supplemental Figure 21 and Supplemental Tables 17 and 18). For example, multiclonal KIT mutations were present at diagnosis in one case, with expansion of only one subclonal population at relapse (Figure 4B), whereas another case retained a KRAS mutation while losing a KIT mutation at relapse.
Figure 4. Mutant allele frequency (MAF) at diagnosis and relapse in CBF-AMLs.
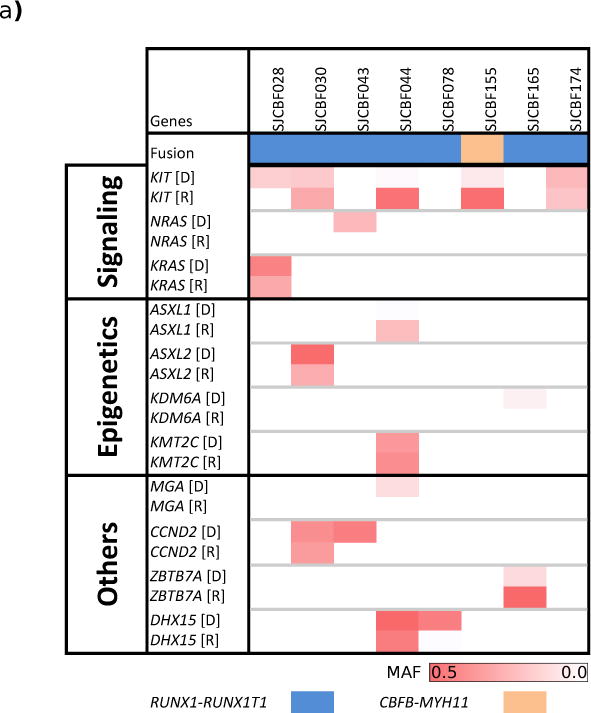
a) The mutant allele frequencies of mutations in the indicated genes are shown for the eight samples with relapse material available. D – diagnosis, R – relapse. Darker color indicates higher MAF.
Despite sharing a common molecular alteration involving a component of the core binding factor transcription complex, AMLs with RUNX1-RUNX1T1 and CBFB-MYH11 alterations have a remarkably different spectrum of cooperating mutations (Supplemental Figure 22). Signaling mutations are common in both subtypes, yet RUNX1-RUNX1T1 cases have different types of signaling alterations that include CCND2. Mutations involving epigenetic regulators and the cohesin complex, as well as mutations in DHX15 and ZBTB7A, are also frequently observed in RUNX1-RUNX1T1 cases. While suggestive of a functional role in RUNX1-RUNX1T1 leukemogenesis, future studies will be required to evaluate their role in CBF-AML pathogenesis.
METHODS
Subject cohorts and sample detail
Tumor and germline samples from pediatric CBF-AMLs from the St. Jude Children’s Research Hospital tissue resource core facility and adult CBF-AMLs from the University Hospital of Ulm were obtained with informed consent using a protocol approved by the St. Jude Children’s Research Hospital institutional review board. Detailed clinicopathological and sequencing information are provided in Supplementary Tables 1–9. The study cohort comprised 165 leukemias (85 RUNX1-RUNX1T1 and 80 CBFB-MYH11 with matched germline samples) in 2 cohorts: a cohort for whole genome sequencing (n=17; 7 RUNX1-RUNX1T1 and 10 CBFB-MYH11, all pediatric cases) and a cohort for whole exome sequencing (n=148, 78 RUNX1-RUNX1T1 and 70 CBFB-MYH11). Matched germline DNA was acquired at remission from either bone marrow or peripheral blood or was flow sorted from leukemic samples.
Whole-genome and whole-exome sequencing and analysis
Whole-genome and whole-exome sequencing were performed as previously described64,65. Paired-end sequencing was performed using Illumina HiSeq platform with a 100-bp read length. Whole-genome sequencing mapping, coverage and quality assessment, SNV and indel detection, tier annotation for sequence mutations and prediction of the deleterious effects of missense mutations have been described previously64,65. Mapping statistics and coverage for each tumor is summarized in Supplemental Tables 3 and 4. SNVs discovered by whole genome sequencing were classified as tier 1, tier 2, tier 3 or tier 4 as previously described64,65. All tier 1–3 SNVs were validated by a custom capture platform, as well as all coding indels and SVs (Supplemental Table 5). The overall validation rate was 83%, with a median validation rate of 87% per sample. All recurrent SNVs (Supplemental Table 6) in the whole-exome sequencing cohort were validated by either amplicon sequencing using the MiSeq platform or Sanger sequencing, as previously described64,65. Briefly, PCR primers were designed to flank the putative variant using Primer3. Amplicon sizes ranged from approximately 400bp to 600b. PCR was performed using AmpliTaq Gold 360 master mix (Applied BioSystems), 400 nM of each primer (IDT) and 20 ng of repli-G whole genome amplified DNA (QIAGEN) in a 25 ul reaction volume. Thermo cycling was performed using the following parameters: 95°C for 10 min., 95°C for 30 sec., 65°C for 30 sec., 72°C for 1 min. for 35 cycles, 72°C for 7 min. All amplicons were quality checked on a 2% agarose E-gel (Invitrogen). Pooled amplicons were used to create DNA libraries using the Nextera XT kit (Illumina) following the manufacturer’s instructions. Libraries were normalized and sequenced on an Illumina MiSeq using a 2 × 150 paired-end version 2 sequencing kit. Sanger Sequencing was also performed on a subset of amplicons. Primer pairs and SNV targets used in validation are included in Supplemental Table 18. All validated SNVs are summarized in Supplemental Table 7. CNAs that were identified by WGS data were analyzed by evaluating the difference in read depth between each tumor and its matching normal (germline) sample using CONSERTING66 (Supplemental Table 8). Other CNAs were detected by Affymetrix SNP6.0 arrays, as previously described28,29. Structural variations in whole genome sequencing data were analyzed using CREST and were annotated as previously described64,67(Supplemental Table 9). To assess tumor contamination of the germline sample we evaluated the existence of SVs supporting the RUNX1-RUNX1T1 and CBFB-MYH11 fusions in germline samples using CREST67. The level of tumor in normal contamination is calculated as number of reads supporting fusion / number of total reads and was under the limit of detection in each sample (Supplemental Table 3).
Transcriptome sequencing
For library construction, 2–5 μg of total RNA was extracted from tumor samples by using Qiagen RNeasy Mini kits according to the manufacturer’s instructions. RNA concentration was measured by using a NanoDrop 100 Spectrophotometer (Thermo Scientific). RNA integrity was measured by using an Agilent Technologies 2100 Bioanalyzer Lab-on-a-chip system. Total RNA was treated with DNAse I (Invitrogen) and enriched for poly A containing mRNA using oligo dT beads (Dynabeads, Invitrogen). The cDNA synthesis used random hexamers and the Superscript Double-Stranded cDNA Synthesis kit (Invitrogen). Paired-end reads from mRNA-seq were aligned to the following 4 database files by using a Burrows-Wheeler Aligner (0.5.5): (i) human NCBI Build 37 reference sequence, (ii) RefSeq, (iii) a sequence file representing all possible combinations of non-sequential pairs in RefSeq exons, and (iv) AceView flat file downloaded from UCSC, representing transcripts constructed from human expressed sequence tag (EST). The final BAM (compressed binary version of the Sequence Alignment/Map [SAM] format) file was constructed by selecting the best alignment in the four databases.
CNA detection using WES data
Samtools68 mpileup command was used to generate an mpileup file from matched normal and tumor BAM files with duplicates removed. VarScan269 was then used to take the mpileup file to call somatic CNAs after adjusting for normal/tumor sample read coverage depth and GC content. Circular Binary Segmentation algorithm70 implemented in the DNAcopy R package was used to identify the candidate CNAs for each sample. B-allele frequency info for all high quality dbSNPs heterozygous in the germline sample was also used to assess allele imbalance.
Accession numbers
Genomic data have been deposited at the European Genome-phenome Archive (EGA), which is hosted by the European Bioinformatics Institute (EBI), under EGAS00001000349.
Lineage-negative enrichment and differentiation assays of murine hematopoietic cells
Experiments were approved by the St. Jude Children’s Research Hospital Institutional Animal Care and Use Committee. Briefly, bone marrow cells were harvested from 8-wk old C57BL/6 mice. Lineage-negative cells were purified by magnetic separation and cultured for 48hr in IMDM/20% FBS supplemented with penicillin-streptomycin, L-glutamine, recombinant mouse IL-3 (10 ng/ml), IL-6 (20 ng/ml), and SCF (50 ng/ml) (Peprotech). Cells were infected on RetroNectin-coated plates for 48hr (Takara Bio Inc.) with MSCV-IRES-mCherry retrovirus expressing wild-type or mutant Flag-tagged ASXL2. Transduced mCherry+ cells were obtained by fluorescence-activated cell sorting. For flow analysis, cells in culture were stained with anti-mouse-Gr-1 (RB6-8C5), anti-mouse-Mac1 (M1/70), anti-mouse-cKit (2B8), and anti-mouse-Sca1 (E13-161.7) (BD Biosciences) on day 14 post transduction. The data was analyzed using FlowJo software.
Mutagenesis
pCMV6 constructs containing wild-type human CCND2 and DHX15 were obtained from Origene. CCND2 mutations (P281R and T282*) and DHX15 (R222G) were introduced using the Quickchange II XL kit (Agilent) per manufacturer’s instructions using the primer pairs shown below:
|
| |
| T282*_F | 5′-atcccgcacgtctcaaggggtgctggcttg-3′ |
|
| |
| T282*_R | 5′-caagccagcaccccttgagacgtgcgggat-3′ |
|
| |
| P281R_F | 5′-ccgcacgtctgtacgggtgctggcttg-3′ |
|
| |
| P281R_R | 5′-caagccagcacccgtacagacgtgcgg-3′ |
|
| |
| R222G_F | 5′-ctactgcagtcttcaaatccaatggagtaaccaacttcc-3′ |
|
| |
Transient transfection of CCND2 mutants
HEK293T (ATCC) cell lines were transiently transfected using FuGene HD (Promega) as per the manufacturer’s instructions. Cells were incubated for 24 hr, and then subjected to 50 μg/mL cyclohexamide and harvested at the indicated time points in NP-40 lysis buffer (150 mM sodium chloride, 1% NP-40 and 50 mM Tris, pH 8.0) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (ThermoScientific). Western blotting of 293T cells expressing CCND2 constructs was performed using 10 μg of protein lysates prepared using NuPAGE LDS buffer (Life Technology) electrophoresed through 4–12% NuPAGE Bis-TRIS gels under reducing conditions. After transferring to PVDF membrane (iBlot, Life Technology), membranes were probed with total CCND2 (Abcam, ab3085) and GAPDH (Santa Cruz Biotechnology, 6C5) antibodies using the iBind system (Life Technology). Quantification of western blots was completed using the ImageJ software.
Modeling of DHX15
The structure of PRP43 bound to ADP was obtained from the Protein Data Bank71 (PDB: 2XAU72). Mutations and models were generated using Pymol73.
DHX15 knockdown, overexpression, and mass spectrometry
HEK293T cells were transfected with a pool of siRNAs targeting DHX15 (Supplemental Table 19) using Lipofectamine RNAiMax (ThermoFisher Scientific) according to manufacturer instructions. Cells were transfected again after 24 hours and harvested at day 4. For overexpression experiments, HEK293T cells were transfected with pCMV6-DHX15 or pCMV6-DHX15 R222G. RNA was extracted after 48 hours with Trizol and libraries prepared as described above. Pull down experiments for mass spectrometry were done using the Pierce c-Myc Tag IP/Co-IP Kit (Thermo Scientific). The MS analysis was performed according to the optimized platform as previously reported74. Proteins in the gel band were in-gel digested by trypsin. Resulting peptides were loaded on a nanoscale reverse-phase column, and eluted by a gradient (~30 min). Eluted peptides were detected by an inline LTQ Orbitrap Elite mass spectrometer (Thermo Scientific). Acquired data were searched against the protein database using the Sequest algorithm and filtered to reduce the protein false discovery rate to below 1%. For binary comparison, statistical analysis was also performed based on the G-test. FDR was derived according to biological replicates and null hypothesis75.
Western blots and co-immunoprecipitations
Co-immunoprecipitations (CO-IP) and immunoblots (WB) used standard techniques as described previously76. Briefly, mouse embryonic fibroblasts were transduced to stably coexpress human TFIP11, 2× myc tagged with either wild type human DHX15, 2× HA tagged or R222G mutant, 2× HA tagged. Cells were lysed in NP-40 buffer (0.5% NP-40, 20 mM Tris, pH 7.5, 137 mM NaCl, 2 mM EDTA and 10% glycerol, 100 ug/mL RNase A). Agarose-conjugated anti-HA (Roche) or anti-myc (Cell Signaling Technology) antibodies were used for CO-IP. Horse radish peroxidase (HRP)-conjugated ani- HA (Roche) or anti-myc (Cell Signaling Technology) antibodies were used for WB.
Gene set enrichment analysis
Array data from Ross et al62 was used for Limma77 analysis to create a list of differentially expressed genes between cases of RUNX1-RUNX1T1 and CBFB-MYH11 CBF leukemia (Supplemental Table 16). The resulting gene list was truncated at a false discovery rate of 0.001 and the remaining genes were used to create a gene set that represented the gene signature of each fusion. The transcriptome data was then used for gene set enrichment analysis (GSEA)78.
Mutational spectrum analysis
We used the published computational framework for mutation signature identification24,79. The algorithm deciphers the minimal set of mutational signatures that optimally explains the proportion of each mutation type found in each catalogue and then estimates the contribution of each signature to each catalogue. We used somatic mutations called from the whole exome sequencing diagnostic samples of RUNX1-RUNX1T1 (70 samples, 1595 mutations) and CBFB-MYH11 (66 samples, 1024 mutations).
Statistical Methods
Enrichment in the number of cases harboring mutations in epigenetic and cohesin genes in the RUNX1-RUNX1T1 vs. CBFB-MYH11 cohorts was calculated using the Fisher’s Exact test. Differences in the expression level of CCND2 were calculated using the Student’s t-test. The association of a mutation with event free survival (EFS) or overall survival (OS) was performed using Cox proportional hazard model; survival curves were also compared by log rank test. Event-free survival (EFS) was defined as the time elapsed from on study date to induction failure, withdrawal, relapse, secondary malignancy, or death, with those living and event-free at last follow-up censored. Overall survival (OS) was defined as the time elapsed from on study date to death with those still living at last follow-up considered censored. Outcome data was available for 159 out of 165 CBF-AML patients.
Supplementary Material
Acknowledgments
We thank all the patients and their parents from the St. Jude Children’s Research Hospital (USA) and the adult patients and their families from the German-Austrian AML Study Group (AMLSG). We thank the Tissue Resources Laboratory, the Flow Cytometry and Cell Sorting Core and the Clinical Applications of Core Technology Laboratories of the Hartwell Center for Bioinformatics and Biotechnology of St. Jude Children’s Research Hospital. This work was funded by the St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project, the American Lebanese and Syrian Associated Charities of St. Jude Children’s Research Hospital and by a grant from the US National Institutes of Health (P30 CA021765). C.G.M. is a Pew Scholar in Biomedical Sciences and a St. Baldrick’s Scholar. J.M.K. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. Supported in part by grants 01GI9981 and 01KG0605 from the German Bundesministerium für Bildung und Forschung (BMBF), grant 111911 from the Deutsche Krebshilfe, and grant DO 704/3-1 from the Deutsche Forschungsgemeinschaft (DFG); KD, HD, and LB are supported by the Collaborative Research Center SFB 1074 funded by the DFG; LB is a Heisenberg Professor of the DFG (BU 1339/8-1).
Footnotes
Contributions
Z.J.F., A.L.G, J.C., I.R., J-R.C., A.K.A., J.D., L.D., Z.C., R.H., and E.P. contributed to the design of the study and conducted experiments. X.C., Y.L., G.S., J.M., M.P.W., G.W., M.E., M.R., C.Q., Y.L., J.W., E.H., H.M., K.B., B.V., D.Y., J.N., J.E., S.S., R.S.F., L.L.F., L.D, E.R.M., R.K.W. and J.Z. contributed to the preparation and analysis of the sequencing data. X.C. and S.B.P. provided statistical support. P.P., R.F.S., L.B, H.D., K.D., C-H.P., and J.E.R. provided clinical samples and data. Z.J.F., A.L.G. and J.M.K. wrote the manuscript. R.K.W., T.A.G., C.G.M., L.B., J.Z., J.M.K., and J.R.D contributed to the design and oversaw the study.
References
- 1.Downing JR. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. Br J Haematol. 1999;106:296–308. doi: 10.1046/j.1365-2141.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 2.Castilla LH, et al. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia. Nat Genet. 1999;23:144–6. doi: 10.1038/13776. [DOI] [PubMed] [Google Scholar]
- 3.Higuchi M, et al. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell. 2002;1:63–74. doi: 10.1016/s1535-6108(02)00016-8. [DOI] [PubMed] [Google Scholar]
- 4.Yuan Y, et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc Natl Acad Sci U S A. 2001;98:10398–403. doi: 10.1073/pnas.171321298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuber J, et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009;23:877–89. doi: 10.1101/gad.1771409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. Implications of NRAS mutations in AML: a study of 2502 patients. Blood. 2006;107:3847–53. doi: 10.1182/blood-2005-08-3522. [DOI] [PubMed] [Google Scholar]
- 7.Kuchenbauer F, et al. Identification of additional cytogenetic and molecular genetic abnormalities in acute myeloid leukaemia with t(8;21)/AML1-ETO. Br J Haematol. 2006;134:616–9. doi: 10.1111/j.1365-2141.2006.06229.x. [DOI] [PubMed] [Google Scholar]
- 8.Valk PJ, et al. Second hit mutations in the RTK/RAS signaling pathway in acute myeloid leukemia with inv(16) Haematologica. 2004;89:106. [PubMed] [Google Scholar]
- 9.Schessl C, et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest. 2005;115:2159–68. doi: 10.1172/JCI24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nanri T, Matsuno N, Kawakita T, Mitsuya H, Asou N. Imatinib mesylate for refractory acute myeloblastic leukemia harboring inv(16) and a C-KIT exon 8 mutation. Leukemia. 2005;19:1673–5. doi: 10.1038/sj.leu.2403889. [DOI] [PubMed] [Google Scholar]
- 11.Nanri T, et al. Mutations in the receptor tyrosine kinase pathway are associated with clinical outcome in patients with acute myeloblastic leukemia harboring t(8;21)(q22;q22) Leukemia. 2005;19:1361–6. doi: 10.1038/sj.leu.2403803. [DOI] [PubMed] [Google Scholar]
- 12.Bowen DT, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood. 2005;106:2113–9. doi: 10.1182/blood-2005-03-0867. [DOI] [PubMed] [Google Scholar]
- 13.Care RS, et al. Incidence and prognosis of c-KIT and FLT3 mutations in core binding factor (CBF) acute myeloid leukaemias. Br J Haematol. 2003;121:775–7. doi: 10.1046/j.1365-2141.2003.04362.x. [DOI] [PubMed] [Google Scholar]
- 14.Goemans BF, et al. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia. 2005;19:1536–42. doi: 10.1038/sj.leu.2403870. [DOI] [PubMed] [Google Scholar]
- 15.Boissel N, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML) Leukemia. 2006;20:965–70. doi: 10.1038/sj.leu.2404188. [DOI] [PubMed] [Google Scholar]
- 16.Paschka P, et al. Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): a study of the German-Austrian AML Study Group (AMLSG) Blood. 2013;121:170–7. doi: 10.1182/blood-2012-05-431486. [DOI] [PubMed] [Google Scholar]
- 17.Kuhn MW, et al. High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood. 2012;119:e67–75. doi: 10.1182/blood-2011-09-380444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He J, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–14. doi: 10.1182/blood-2015-08-664649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duployez N, et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood. 2016;127:2451–9. doi: 10.1182/blood-2015-12-688705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olsson L, et al. The genetic landscape of paediatric de novo acute myeloid leukaemia as defined by single nucleotide polymorphism array and exon sequencing of 100 candidate genes. Br J Haematol. 2016 doi: 10.1111/bjh.14056. [DOI] [PubMed] [Google Scholar]
- 21.Farrar JE, et al. Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res. 2016;76:2197–205. doi: 10.1158/0008-5472.CAN-15-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavallee VP, et al. RNA-sequencing analysis of core binding factor AML identifies recurrent ZBTB7A mutations and defines RUNX1-CBFA2T3 fusion signature. Blood. 2016;127:2498–501. doi: 10.1182/blood-2016-03-703868. [DOI] [PubMed] [Google Scholar]
- 23.Sood R, et al. Somatic mutational landscape of AML with inv(16) or t(8;21) identifies patterns of clonal evolution in relapse leukemia. Leukemia. 2016;30:501–4. doi: 10.1038/leu.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krauth MT, et al. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: frequency and impact on clinical outcome. Leukemia. 2014;28:1449–58. doi: 10.1038/leu.2014.4. [DOI] [PubMed] [Google Scholar]
- 28.Haferlach C, et al. AML with CBFB-MYH11 rearrangement demonstrate RAS pathway alterations in 92% of all cases including a high frequency of NF1 deletions. Leukemia. 2010;24:1065–9. doi: 10.1038/leu.2010.22. [DOI] [PubMed] [Google Scholar]
- 29.Hurlin PJ, Steingrimsson E, Copeland NG, Jenkins NA, Eisenman RN. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. The EMBO journal. 1999;18:7019–28. doi: 10.1093/emboj/18.24.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research, N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papaemmanuil E, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46:116–25. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edelmann J, et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood. 2012;120:4783–94. doi: 10.1182/blood-2012-04-423517. [DOI] [PubMed] [Google Scholar]
- 34.Landau DA, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–30. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouchard C, et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18:5321–33. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen BB, et al. F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood. 2012;119:3132–41. doi: 10.1182/blood-2011-06-358911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kida A, Kakihana K, Kotani S, Kurosu T, Miura O. Glycogen synthase kinase-3beta and p38 phosphorylate cyclin D2 on Thr280 to trigger its ubiquitin/proteasome-dependent degradation in hematopoietic cells. Oncogene. 2007;26:6630–40. doi: 10.1038/sj.onc.1210490. [DOI] [PubMed] [Google Scholar]
- 38.Mirzaa GM, et al. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nature genetics. 2014;46:510–5. doi: 10.1038/ng.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dolnik A, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012;120:e83–92. doi: 10.1182/blood-2011-12-401471. [DOI] [PubMed] [Google Scholar]
- 40.Arenas JE, Abelson JN. Prp43: An RNA helicase-like factor involved in spliceosome disassembly. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11798–802. doi: 10.1073/pnas.94.22.11798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Combs DJ, Nagel RJ, Ares M, Jr, Stevens SW. Prp43p is a DEAH-box spliceosome disassembly factor essential for ribosome biogenesis. Molecular and cellular biology. 2006;26:523–34. doi: 10.1128/MCB.26.2.523-534.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin A, Schneider S, Schwer B. Prp43 is an essential RNA-dependent ATPase required for release of lariat-intron from the spliceosome. The Journal of biological chemistry. 2002;277:17743–50. doi: 10.1074/jbc.M200762200. [DOI] [PubMed] [Google Scholar]
- 43.Mayas RM, Maita H, Semlow DR, Staley JP. Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10020–5. doi: 10.1073/pnas.0906022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai X, et al. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell. 2015;17:165–77. doi: 10.1016/j.stem.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 46.Huether R, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nature communications. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Micol JB, et al. Frequent ASXL2 mutations in acute myeloid leukemia patients with t(8;21)/RUNX1-RUNX1T1 chromosomal translocations. Blood. 2014 doi: 10.1182/blood-2014-04-571018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdel-Wahab O, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. The Journal of experimental medicine. 2013;210:2641–59. doi: 10.1084/jem.20131141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inoue D, et al. Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. The Journal of clinical investigation. 2013;123:4627–40. doi: 10.1172/JCI70739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herz HM, et al. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012;26:2604–20. doi: 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu D, et al. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013;33:4745–54. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee MG, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–50. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 53.Mansour MR, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–7. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tie F, Banerjee R, Conrad PA, Scacheri PC, Harte PJ. Histone demethylase UTX and chromatin remodeler BRM bind directly to CBP and modulate acetylation of histone H3 lysine 27. Mol Cell Biol. 2012;32:2323–34. doi: 10.1128/MCB.06392-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen C, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25:652–65. doi: 10.1016/j.ccr.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bose T, Gerton JL. Cohesinopathies, gene expression, and chromatin organization. The Journal of cell biology. 2010;189:201–10. doi: 10.1083/jcb.200912129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kon A, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nature genetics. 2013;45:1232–7. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
- 58.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–78. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deardorff MA, et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet. 2007;80:485–94. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hartmann L, et al. ZBTB7A mutations in acute myeloid leukaemia with t(8;21) translocation. Nat Commun. 2016;7:11733. doi: 10.1038/ncomms11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bullinger L, et al. Gene-expression profiling identifies distinct subclasses of core binding factor acute myeloid leukemia. Blood. 2007;110:1291–300. doi: 10.1182/blood-2006-10-049783. [DOI] [PubMed] [Google Scholar]
- 62.Ross ME, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–87. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- 63.Yan M, et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12:945–9. doi: 10.1038/nm1443. [DOI] [PubMed] [Google Scholar]
- 64.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–34. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen X, et al. CONSERTING: integrating copy-number analysis with structural-variation detection. Nat Methods. 2015;12:527–30. doi: 10.1038/nmeth.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011;8:652–4. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–72. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 71.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walbott H, et al. Prp43p contains a processive helicase structural architecture with a specific regulatory domain. EMBO J. 2010;29:2194–204. doi: 10.1038/emboj.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schrodinger, LLC. The PyMOL Molecular Graphics System. 2010. (Version 1.3r1). [Google Scholar]
- 74.Xu P, Duong DM, Peng J. Systematical optimization of reverse-phase chromatography for shotgun proteomics. J Proteome Res. 2009;8:3944–50. doi: 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou JY, et al. Improved LC-MS/MS spectral counting statistics by recovering low-scoring spectra matched to confidently identified peptide sequences. J Proteome Res. 2010;9:5698–704. doi: 10.1021/pr100508p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chao JR, et al. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature. 2008;452:98–102. doi: 10.1038/nature06604. [DOI] [PubMed] [Google Scholar]
- 77.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 79.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–59. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.