

Abstract
Background
Nitric oxide (NO), a highly versatile signaling molecule, exerts a broad range of regulatory influences in the cardiovascular system that extends from vasodilation to myocardial contractility, angiogenesis, inflammation, and energy metabolism. Considerable attention has been paid to deciphering the mechanisms for such diversity in signaling. S-nitrosylation of cysteine thiols is a major signaling pathway through which NO exerts its actions. An emerging concept of NO pathophysiology is that the interplay between NO and reactive oxygen species (ROS), the nitroso/redox balance, is an important regulator of cardiovascular homeostasis.
Scope Of Review
ROS react with NO, limit its bioavailability, and compete with NO for binding to the same thiol in effector molecules. The interplay between NO and ROS appears to be tightly regulated and spatially confined based on the co-localization of specific NO synthase (NOS) isoforms and oxidative enzymes in unique subcellular compartments. NOS isoforms are also in close contact with denitrosylases, leading to crucial regulation of S-nitrosylation.
Major Conclusions
Nitroso/redox balance is an emerging regulatory pathway for multiple cells and tissues, including the cardiovascular system. Studies using relevant knockout models, isoform specific NOS inhibitors, and both in vitro and in vivo methods have provided novel insights into NO- and ROS-based signaling interactions responsible for numerous cardiovascular disorders.
General Significance
An integrated view of the role of nitroso/redox balance in cardiovascular pathophysiology has significant therapeutic implications. This is highlighted by human studies where pharmacologic manipulation of oxidative and nitrosative pathways exerted salutary effects in patients with advanced heart failure. This article is part of a Special Issue entitled Regulation of Cellular Processes by S-nitrosylation.
Keywords: Nitric oxide, Nitrosative and oxidative stress, Excitation, contraction coupling, Adrenergic contractility, Angiogenesis, Inflammation
1. Introduction
Nitric oxide (NO) is a labile free radical gas that functions as a highly versatile and ubiquitous signaling molecule exerting a broad range of regulatory influences in the cardiovascular and renal systems, ranging from control of systemic and microvascular tone to platelet function, myocardial contractility, calcium (Ca2+) cycling, vascular inflammatory and growth responses, renal sodium excretion, and cellular energy metabolism [1–7]. Much attention has been paid to deciphering the mechanisms underlying such diversity in signaling.
In addition to cGMP-dependent signaling [8], S-nitrosylation of specific cysteine thiol residues or metal centers is a major signaling pathway through which NO modifies protein activity and thereby exerts its widespread and diverse effects [9,10]. An emerging concept of NO pathophysiology is that the interplay between NO and reactive oxygen species (ROS), the nitroso/redox balance, is an important regulator of cardiovascular homeostasis [11,12]. ROS readily react with NO and limit its bioavailability and also compete with NO for binding to the same sites in effector molecules. Accumulating evidence shows that nitrosative stress, an impairment in NO signaling caused by increased amounts of reactive nitrogen species (RNS), is caused by or associated with a disturbance in the cellular redox state. RNS of biological significance include NO, low and high molecular weight S-nitrosothiols (SNO), and peroxynitrite. This review addresses the role of S-nitrosylation in cardiovascular cell function and the significant interplay between oxidative stress and S-nitrosylation-based signaling in cardiovascular health and disease.
2. NO based signaling
2.1. Sources of NO
NO is produced from the amino acid l-arginine by the enzymatic action of NO synthases (NOS) or by the breakdown of nitrite or other compounds [13–16]. NOS generated NO is under complex, tight control to dictate specificity of its signaling and to limit toxicity to other cellular components, due to its potent chemical reactivity and high diffusibility. There are three major NOS isoforms in mammalian systems: Neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and endothelial NOS (eNOS or NOS3), each of which oxidizes the terminal guanidino nitrogen of l-arginine to form NO and the amino acid l-citrulline. They share a common basic structural organization and requirement for substrates (arginine and NADPH) and cofactors (tetrahydrobiopterin, heme, calmodulin, FAD, and FMN) for enzymatic activity. The binding of calmodulin is triggered by transient elevations in intracellular Ca2+ levels and serves as an allosteric modulator of the three NOS isoforms. NOS1 is expressed in neural tissues, myocardium, skeletal muscle, and macula densa as well as other renal tubule segments and is a Ca2+/calmodulin-dependent enzyme that is also subject to transcriptional and other post-translational controls [16–19]. Transcription of NOS2 is induced in nearly all tissues in response to cytokines, endotoxin, or other proinflammatory stimuli. NOS2 is less responsive to intracellular Ca2+ transients owing to tight calmodulin binding at ambient intracellular Ca2+levels [20]. NOS3 is expressed in the endothelium and myocardium and plays a central role in the regulation of systemic blood pressure, cardiovascular remodeling, myocardial contractility, and angiogenesis [16,19,21,22]. NOS3 is subject to rapid regulation by calcium Ca2+/calmodulin as well as a variety of transcriptional, post-transcriptional, and post-translational controls. A major structural difference between NOS1 and NOS3 is that the NOS1α isoform has an N-terminal PDZ domain that is crucial in regulation of its spatial localization and protein–protein interactions [23]. Furthermore, we have shown that NOS1 and NOS3 reside in precise subcellular organelles in cardiac myocytes and interact with oxidative enzymes in a spatially confined manner, as discussed in more detail later [19,24–26].
2.2. cGMP pathway
NO activates soluble guanylate cyclase (sGC), a heterodimer with an α subunit and a β subunit,to generate the second messenger 3′,5′-cyclic guanosine monophosphate (cGMP) [8]. Activation of this enzyme is mediated by the binding of NO to the heme moiety of sGC to form the nitrosyl-heme adduct of sGC [27]. As a result, the heme iron is shifted out of the plane of the porphyrine ring configuration, initiating binding of GTP and the formation of cGMP. Cyclic GMP activates cGMP-dependent protein kinase (PKG), which in turn phosphorylates a number of proteins involved in vascular smooth muscle relaxation, the proliferative process, adhesion molecule expression, and platelet aggregation [28]. cGMP signaling is terminated by the action of the cyclic nucleotide-hydrolyzing phosphodiesterases (PDE). PDE5 is spatially localized within cells in proximity to NOS. In the case of the cardiac myocyte, PDE5 is found at the cell membrane associated with caveolae [29]. cGMP mediated signaling has been extensively reviewed elsewhere and is not the focus of this review (Fig. 1A) [8].
Fig. 1.
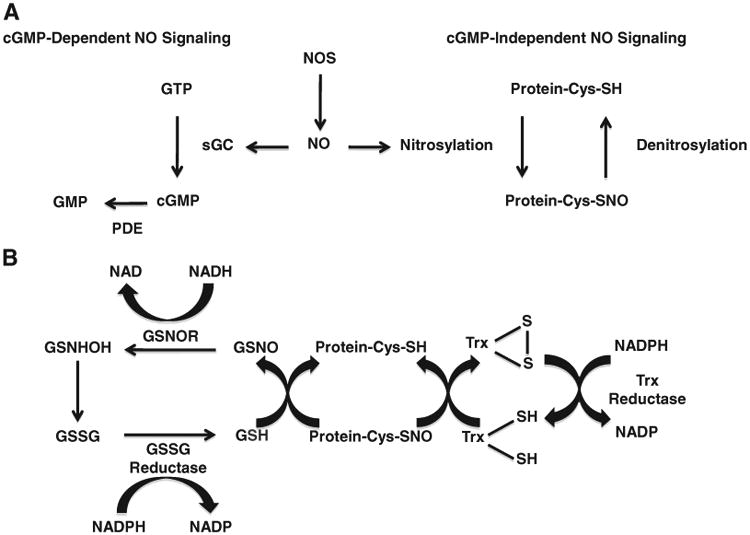
Panel A shows cGMP-dependent and independent nitric oxide (NO) signaling pathways. NO synthase (NOS) produces NO, which activates soluble guanylate cyclase (sGC) to generate the second messenger 3′,5′-cyclic guanosine monophosphate (cGMP). cGMP signaling is terminated by cyclic nucleotide-hydrolyzing phosphodiesterases (PDE). NO also exerts its actions independently of the cGMP pathway by the nitrosylation of sulfhydryl groups on proteins. Panel B shows denitrosylation pathways through the S-nitrosoglutathione (GSNO) reductase (GSNOR) and thioredoxin (Trx) systems. GSNOR uses NADH to reduce GSNO into glutathione S-hydroxysulfenamide (GSNHOH), which in turn is converted to oxidized glutathione (GSSG). Reduction of GSSG by glutathione reductase terminates the denitrosylation reaction. Trx mediates the denitrosylation of S-nitrosylated proteins. Following denitrosylation, the Trx system uses Trx reductase and NADPH to regenerate reduced Trx. Adapted from Lima, B. et al., Circulation Research 2010;106:633–646.
Of note, S-nitrosylation has been shown to modulate cGMP levels by inhibiting sGC [30] and to inhibit NOS3 [31] and NOS3 regulating proteins, including heat shock protein 90 and Akt [32,33]. PKG has regulatory thiols that may also be susceptible to S-nitrosylation. In addition, S-nitrosylation has been shown to activate arginase and inhibit dimethylarginine dimethylaminohydrolase, which leads to decreased NOS substrate levels and increased levels of methylarginine NOS inhibitors, respectively [34,35].
2.3. S-nitrosylation
Besides the cGMP pathway, NO exerts its actions by the nitrosylation of sulfhydryl groups on proteins and small molecules (Fig. 1A) [36,37]. S-nitrosylation is a ubiquitous post-translational modification that regulates diverse biologic processes [9,38,39]. Nitrosylation of specific cysteine thiol residues or metal centers is a reversible covalent modification that modulates protein activity. Cysteines susceptible to nitrosylation tend to be located between an acidic and a basic amino acid and a consensus motif predictive of sites of S-nitrosylation has been described [37,39].
Protein S-nitrosylation occurs by transnitrosylation from low-molecular weight SNO, such as S-nitrosoglutathione (GSNO) or S-nitrosocysteine, by transition metal catalyzed addition of NO, or by endogenous NO-mediated nitrosylating agents such as dinitrogen trioxide (N2O3), which is formed by the autoxidation of NO and is particularly increased within the hydrophobic interior of biological membranes [39–41]. There is evidence that the redox state and ultrastructural accessibility of cysteine residues determines whether a particular thiol in a protein is S-nitrosylated [42,43]. Increases in cellular ROS and RNS production lead to decreases in the intracellular reduced glutathione (GSH) pool, thereby attenuating GSH-mediated trans- or denitrosylation and stabilizing SNO formation [44]. The stability of SNO is favored by low ambient oxygen, whereas with increasing oxygen S-thiolation is promoted [45,46]. The intermediate thiyl radical may be involved in the decomposition of SNO [47].
As evidenced from studies in the myocardium [2], the sub-cellular localization of NOS determines the local concentration of NO and appears to becritical in the formation of SNO. For example, sarcolemmal NOS3 inhibits the L-type Ca2+ channel (LTCC) [48] via S-nitrosylation, whereas NOS1 colocalizes with the sarcoplasmic reticulum (SR) Ca2+-release channel (ryanodine receptor, RYR) and stimulates its activity via S-nitrosylation (Fig. 2) [49,50]. The role of S-nitrosylation in regulating cardiac calcium signaling is discussed in greater detail later.
Fig. 2.
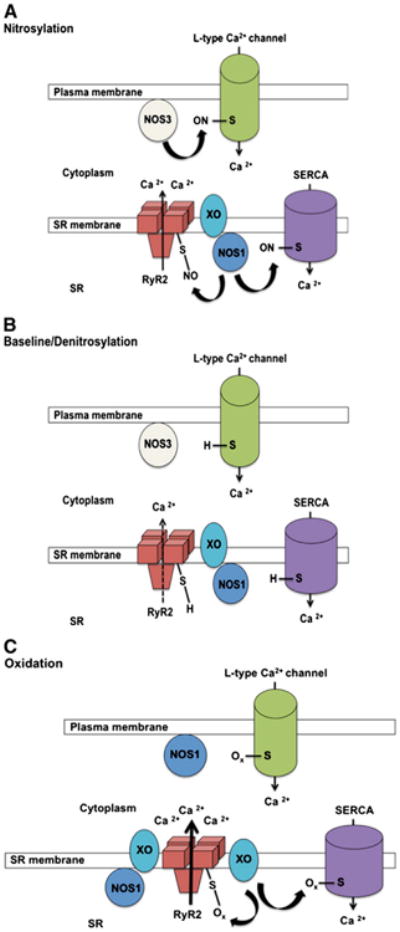
Nitroso-redox regulationof cardiac excitation–contraction coupling. Depicted Is NOS isoform-specific signaling in subcellular compartments as well as potential mechanisms for S-nitrosylation and oxidation regulation of the ryanodine receptor/Ca2+-release channel (RyR2). The specificity of NOS localization is critical for the subcellular generation of NO because it provides a localized signal for protein S-nitrosylation (Panels A and B). NOS3 is localized primarily to the plasma membrane and t tubules. NOS1 colocalizes with the tetrameric RyR2 in the sarcoplasmic reticulum (SR) but translocates to the plasma membrane under conditions of stress (Panel C), including myocardial infarction and heart failure. RyR2 is closely associated with the plasma membrane L-type Ca2+ channel (LTCC), facilitating Ca2+-mediated Ca2+ release from the SR. Panels A and B illustrate the physiological and specific S-nitrosylation and denitrosylation that occur on a millisecond time scale and therefore participate in the regulation of contraction in systole and relaxation in diastole. Panel C shows oxidation of cysteine thiols on RyR2, which leads to irreversible channel activation with maladaptive loss of regulatory control. Oxidation may occurat thiols that are nitrosylated or at other sites, which could change permissiveness to S-nitrosylation via allosteric effects. Reactive oxygen species (ROS) leading to cysteine oxidation are derived from multiple sources, including xanthine oxidase (XO). In heart failure, the excess of ROS derived from xanthine oxidase (XO) impairs the normal nitrosylation of cysteines, which become oxidized. In this condition, the activity of RyR2 increases, leading to diastolic leak that reduces the Ca2+ content of the SR. The LTCC and SR Ca2+ ATPase (SERCA) are similarly regulated by redox mechanisms at cysteine thiols.
2.4. Denitrosylation
S-nitrosylation-based signaling is regulated not only by NO production, but also by enzymatic degradation (Fig. 1B). A recent series of reports demonstrates that GSNO reductase (GSNOR) is a key regulator of S-nitrosylation and is important in protecting cells from nitrosative stress, the mechanisms of which have been recently reviewed [9,51]. GSNOR has widespread activity in cells, using NADH to reduce GSNO into glutathione S-hydroxysulfenamide (GSNHOH), which in turn is converted to oxidized glutathione (GSSG) [52]. Reduction of GSSG by glutathione reductase terminates the denitro-sylation reaction (Fig. 1). Although GSNOR selectively metabolizes GSNO, by depleting the pool of GSNO it shifts the equilibrium and therefore limits levels of S-nitrosylated proteins. A knockout mouse (GSNOR−/−) has been generated and manifests increased levels of S-nitrosylated proteins despite similar production of NO, suggesting that S-nitrosylation is tightly regulated both at the level of formation and decomposition of SNO bonds [53]. We and others have shown that GSNOR−/− mice have increased mortality following endotoxic challenge and manifest hypotension under anesthesia, whereas they exhibit decreased cardiac infarct size and remodeling and increased cardiac function and survival after myocardial infarction [53,54]. These findings support the notion that hypo- and hyper-nitrosylation of specific protein targets correlate with pathophysiology.
Another major enzymatic system mediating intracellular protein denitrosylation is the thioredoxin system [55]. Unlike GSNOR which specifically denitrosylates GSNO, the cytoplasmic and mitochondrial thioredoxins mediate the denitrosylation of multiple S-nitrosylated proteins in a stimulus coupled, substrate specific, and spatially restricted manner [51,55,56]. Following denitrosylation, the thioredoxin system uses thioredoxin reductase and NADPH to regenerate reduced thioredoxin (Fig. 1B). This enzymatic system has been shown to reverse the S-nitrosylation-induced reduction of NOS activity and inhibition of protein kinase C and thioredoxin is itself regulated by S-nitrosylation of multiple residues [57–59]. Other enzymatic systems reported to function as denitrosylases include carbonyl reductase 1 [60], Cu, Zn-superoxide dismutase [61], and xanthine oxidoreductase [62]. All of these denitrosylases are coupled to cellular antioxidant redox systems, highlighting the importance of the cellular redox state in regulating the level of protein S-nitrosylation and thereby cellular signaling.
3. Myocardial cellular processes regulated by S-nitrosylation
NO has both autocrine and paracrine activities within the heart [63], and influences myocardial contractility [25,64,65], ventricular relaxation [66,67], and mitochondrial respiration [68–71]. At a biochemical level, these effects are due to interactions with Ca2+ handling proteins, including the L-type Ca2+ channel (LTCC), ryanodine receptor/Ca2+-release channel (RYR2), and SR Ca2+ ATPase (SERCA) [9,49,72], the contractile myofilaments [73], and respiratory complexes [74], respectively. Extensive study by multiple groups has led to a view of isoform-specific NO signaling in precise subcellular compartments (Fig. 2)[24,25]. NOS knockout mice have been a valuable tool for deciphering these signaling pathways, and overexpression or reconstitution strategies have continued to support isoform specific activity [19,26,50,75,76].
3.1. Spatial localization of NO production
In the cardiac myocyte, NO activity is predominantly determined by its site of production, which in turn is controlled by spatial localization of the NOS enzymes (Fig. 2) [24,25]. NOS3 is localized primarily to caveolae of the sarcolemma and t tubules, where its function is regulated by interaction with caveolin-3 and is linked to multiple cell surface receptors, including muscarinic, β-adrenergic, and bradykinin receptors [65]. Activation of NOS3 results in negative inotropy and chronotropy, an effect that is enhanced by β-adrenergic activation, suggesting that NO acts as a negative feedback mechanism over contractile reserve [77]. NOS1 has been localized to the SR, where it influences Ca2+ cycling and thereby exerts positive inotropic effects in the heart.Of note, immunoprecipitation studies have demonstrated that NOS1 binds to RyR2 [25,49,50,72,78]. There is also evidence of a mitochondrial NOS expressed in the inner mitochondrial membrane or matrix [79], although this remains controversial [80,81]. Another source of mitochondrial NO may be NOS1 associated with the SR, since the SR membrane has been shown to be attached to the outer mitochondrial membrane [82]. Importantly, this specificity of NOS localization is critical for the subcellular organelle generation of NO because it provides a localized signal for protein S-nitrosylation (Fig. 2).
3.2. Alteration in subcellular localization following tissue injury
NOS enzyme activity and subcellular localization are altered after myocardial infarction and in heart failure (Fig. 2C) [83–85]. In this regard, it has been shown that NOS1 expression is not only increased post-myocardial infarction, but NOS1 is shifted in localization within the cell to the level of the sarcolemma with interactions with caveolin-3, in contrast to its location at the SR in mice without myocardial infarction. Furthermore, NOS1 accounted for the majority of the NO produced within the cell, as NOS3 expression and activity decreased post-myocardial infarction. This translocation of NOS1 to the sarcolemma may explain the observation that NOS1 signaling inhibits myocardial β-adrenergic contractility after myocardial infarction [86]. This likely represents an adaptive response since others and we have shown that NOS1−/− mice exhibit increased mortality after myocardial infarction (Fig. 3) [87,88]. Indeed, the cardioprotective effect of NOS1 post-myocardial infarction was associated with increased S-nitrosylation of the LTCC leading to decreased Ca2+ influx [89]. In turn, the reduced Ca2+ entry within the cardiac myocyte prevented Ca2+ overload-induced injury. NOS1 has also been shown to protect against the development of myocardial remodeling after infarction [87,88]. In summary, based on their specific spatial localization within the cardiac myocyte, NOS1 and NOS3 play a specific and unique role in cardiac biology and pathophysiology (24).
Fig. 3.
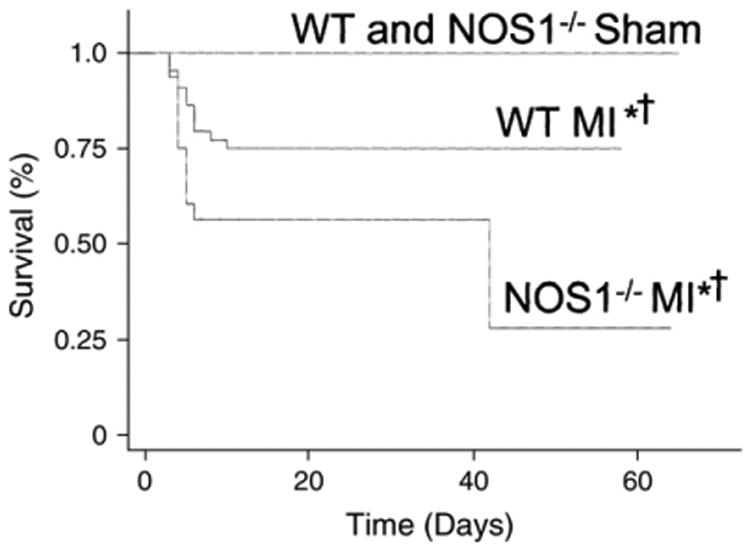
Survival curves after myocardial infarction (MI) in wild type (WT) and NOS1−/−-mice followed up for 60 days. Post-MI survival was significantly reduced in NOS1−/− vs. WT mice (*P=0.036). Both MI groups had reduced survival in relation to their respective sham-operated controls (†P=0.014 between WT subgroups and P=0.0001 between NOS1−/− subgroups). There was no significant difference in survival after sham operation between WT and NOS1−/− mice.
Reprinted with permission from Saraiva, R. M. et al. Circulation 2005;112:3415–3422.
3.3. Calcium signaling
Depolarization of the cardiac myocyte plasma membrane triggers a cascade of events leading to a rapid increase in cytosolic Ca2+ and resulting in muscle contraction, termed excitation–contraction coupling [2]. S-nitrosylation modulates the function of ion channels that regulate excitation–contraction coupling and therefore normal systolic and diastolic function [9,11].
In the normal heart, intracellular Ca2+ concentration ([Ca2+]i) is tightly regulated throughout the cardiac cycle. In excitation–contraction coupling, a small amount of Ca2+ enters through the LTCC during membrane depolarization. This influx triggers massive Ca2+ release from the SR,mainly trough theCa2+release channel RyR2. Thiselevated [Ca2+]i causes the binding of Ca2+ to troponin C in the myofilaments, which activates the contraction. Relaxation is initiated when Ca2+ is transported out of the cytosol, which is achieved by SERCA in the SR and the sarcolemmal sodium–calcium exchanger (NCX).
Defects in intracellular Ca2+ handling, such as reductions in the systolic [Ca2+]i, increase in diastolic [Ca2+]i, and impairments in diastolic Ca2+ re-uptake, have an important role in the depressed contractility and cardiac reserve observed in heart failure [90]. The down-regulation of SERCA and up-regulation of NCX that occurs in heart failure act in concert to shift Ca2+ out of the cell and reduce SR Ca2+ content, leaving less Ca2+ available for contraction. Beside the changes in SERCA and NCX expression, there is evidence for diastolic Ca2+ leak in heart failure [90]. Redox modifications of RyR2 (S-nitrosylation, oxidation) in the setting of heart failure play an important role in the activity of the channel [91–95]. Due to its high number of cysteines (89 cysteine residues per subunit), it is susceptible to S-nitrosylation, S-glutathionylation, and disulphide oxidation [49,96]. A few of these cysteines can be rapidly oxidized by ROS and RNS, including H2O2, , NO, and GSNO [42,96–98].An increased oxidation of the RyR2 channel has been demonstrated in animal models of heart failure [92,94,95]. This was restored by β-adrenergic-blockers and antioxidants, associated with improved cardiac function, although it was not investigated whether the change in free thiols was due to increased S-nitrosylation, glutathionylation, or further oxidation (disulphide bonds, sulphenic or sulphonic acids). Our group has shown that with a low concentration of NO donor, the increase in S-nitrosylation of the channel is associated with an increase in contractility and that loss of S-nitrosylation decreases inotropic responsiveness [50,99–101]. This highlights the importance of determining the redox state of RyR2 in disease.
ROS and RNS species play a significant pathophysiological role in heart failure. The cardiac RyR2 has low-level basal SNO. S-nitrosylation of additional cysteines leads to further activation of the channel [49]. This is a highly reversible modification that can occur on a time scale proportionate to excitation–contraction coupling. On the other hand, oxidation of cysteine residues on RyR2 leads to irreversible activation of the channel, leading to SR leak and resulting in SR Ca2+ depletion (Fig. 2C). We have shown that cardiac RyR2 is hyponitrosylated in heart failure, due to nitroso-redox imbalance (Fig. 4) [50,101]. We evaluated excitation–contraction coupling and nitroso-redox balance in spontaneously hypertensive-heart failure (SHHF) rats with dilated cardiomyopathy and age matched Wistar–Kyoto rats. SHHF cardiomyocytes were characterized by depressed contractility, increased diastolic Ca2+ leak, hyponitrosylation of RyR2, and enhanced xanthine oxidase (XO)-derived superoxide [101]. Global S-nitrosylation was decreased in failing hearts compared to non-failing. XO inhibition restored global and RyR2 nitrosylation and reversed the diastolic SR Ca2+ leak, improving Ca2+ handling and contractility. Together these findings demonstrate that nitroso-redox imbalance causes RyR2 oxidation, hyponitrosylation, and SR Ca2+ leak, a hallmark of cardiac dysfunction. The reversal of this phenotype by inhibition of XO has important pathophysiological and therapeutic implications [102,103].
Fig. 4.

Decreased S-nitrosylation of ryanodine receptor/Ca2+-release channel (RyR2) in isolated cardiomyocytes from spontaneously hypertensive-heart failure (SHHF) rats. Confocal microscopy showing red staining for RyR2 (left panels), green staining for nitrosylated cysteines (middle panels) and the combination of both (merge), right panels. The upper panels show a sequence of images of cardiac myocytes from a non-failing heart (Wistar–Kyoto rat). The lower panels show the same sequence for myocytes from a failing heart (SHHF rat). Note the degree of colocalization of RyR2 with the S-nitrosocysteines (Cys-NO) in the non-failing heart, which is lost in the failing myocardium.
XO is an important source of ROS in the cardiovascular system and our studies have provided significant new insights into the mechanism of cardiac XO signaling and its interaction with NOS1. We and others have demonstrated that NOS1 deficiency increases mortality, remodeling, and ventricular arrhythmia after myocardial infarction, associated with increased XO activity and decreased S-nitrosylation of Ca2+ handling proteins, while specific myocardial NOS1 overexpression has been shown to protect from remodeling by preserving Ca2+ cycling components [26,87,88,104,105]. Increased expression of NOS1 in the caveolae is associated with increased S-nitrosylation of the LTCC leading to decreased Ca2+ current. In turn, the reduced Ca2+ influx within the cardiac myocyte prevented Ca2+overload-induced injury [69,89,106]. This is a protective mechanism, since it has been recently shown that increasing Ca2+ influx through the LTCC channel after myocardial infarction prevents depressed myocyte contractility but increases the risk of ischemic injury, precipitates sudden death, and exacerbates depressed cardiac pump function [107].
We have shown that XO inhibition preserves the expression of components of Ca2+ handling, such as SERCA, phosphorylated phospholamban (PLB) and NCX [108]. In this regard, it has been described that NOS1 influences PLB phosphorylation, probably through phosphatase activity [66]. Interestingly, it has been proposed that physiologically, this NOS1-dependent effect on PLB phosphorylation is mediated by peroxynitrite, formed from the concerted action of NOS1 and XO [109]. More recently, phospholamban has been shown to be S-nitrosylated in the heart, leading to activation of SERCA [67]. In NOS1 deficient mice, after myocardial infarction, we have shown that XO inhibition prevented remodeling and contractility by decreasing ROS/RNS damage and preserving the components of Ca2+ cycling [26].
NO activity has been shown by some [106,110], but not all [42,111] investigators to inhibit SR Ca2+ uptake. S-nitrosylation reactions have the potential to modify SERCA via thiol reactions [106,111–113]. Previous studies have shown that thiol-oxidation reduces Ca2+ pump activity. To the extent that nitrosative stress and ROS compete for the same thiol [39,114], it is possible that reversible S-nitrosylation of SERCA may also modulate pump activity to coordinate with S-nitrosylation regulation of the RYR2 [50]. It is also conceivable that oxidant signaling may directly regulate cross-bridge cycling kinetics, thereby modulating the efficiency of contraction [115,116]. In so far as modulation of proteins via thiol S-nitrosylation and oxidation is a general phenomenon, many potential proteins involvedincardiac Ca2+ signaling have the potential to be influenced by NOS, XO, and other oxidase signaling [11,72]. Whether these proteins are modulated reversibly so as to preserve physiologic signaling or irreversibly so as to cause toxicity is determined by nitroso/redox balance.
3.4. Voltage-gated potassium and sodium channel function
Cardiac voltage-gated potassium (K+) and sodium (Na+) channels, important in the regulation of the cardiac action potential, have been shown to be subject to regulation by S-nitrosylation [72]. Voltage-gated K+ channels determine the resting membrane potential and the duration of the cardiac action potential. The delayed rectifier K+ current is one of the major components that determines the timing of repolarization of cardiac myocytes and consists of a rapidly activating (IKr) and a slowly activating component (IKs). It has been reported that S-nitrosylation of cysteine 445 in the pore-forming subunit KCNQ1 increases IKs in a NOS3 dependent manner [117–119]. This activation resulted in shortening of the action potential duration. S-nitrosylation of a cysteine in the Kir2.1 channel protein has also been shown to shorten the action potential by increasing the inward-rectifying K+ current [120].
In cardiomyocytes, the voltage-gated Na+channels are responsible for fast depolarization. The main Na+ channel expressed in the mammalian myocardium is encoded by the gene SCN5A. Although Na+channels typically inactivate very quickly, in cardiomyocytes a late current is observed that has been shown to be dependent on S-nitrosylation coupled to NOS activity [121]. The Na+ channel is rich in cysteines that could also be subject to oxidation under oxidative stress conditions [122,123]. Of note, a mutation in α-syntrophin associated with a form of the long QT syndrome results in aberrant S-nitrosylation of the Na+ channel [124]. α-syntrophin, a dystrophin-associated protein, normally serves as scaffold protein for NOS1 and the plasma membrane CA2+-ATPase, an interaction that results in inhibition of NO production [125]. The α-syntrophin mutation results in a disruption of the CA2+-ATPase-NOS1 complex and favors interaction of NOS1 with the Na+ channel, promoting S-nitrosylation and increased late Na+ currents [124]. Although the impact of this process on the action potential duration was not investigated, it has been shown by others that mutations of the Na+ channel that lead to similar increased late currents prolong the duration of the action potential [126].
3.5. β-adrenergic receptor signaling
In the heart, NOS3 is activated by coupling to numerous receptors, including the β-adrenergic, muscarinic, and bradykinin receptors. The prototypic mode of activation appears to occur via agonist-stimulated increase in Ca2+, leading to Ca2+/calmodulin activation of NOS3. NOS3 is localized to caveolae of the sarcolemma and t-tubules, where the scaffolding protein caveolin-3 inactivates it until displaced by Ca2+/ calmodulin [65,127]. NOS3 can also be activated directly by Akt phosphorylation without intracellular increases in Ca2+[128,129].
NO exerts negative inotropic effects, which are more marked when contractility is stimulated by either β-adrenergic activation or heart rate, a finding reminiscent of the phenomenon of “accentuated antagonism” that is observed with vagal nerve stimulation [130]. The observation that NO inhibition of contractility is more apparent during β-adrenergic stimulation has led to the proposal that NO serves as a negative feedback mechanism over contractile reserve, a notion supported by the finding that adrenergic agonists directly stimulate NO production [77]. We have shown that the β3-adrenergic receptor, present in myocardium [131], is linked to NO production [132]. Studies performed using NOS−/− mice recapitulate central features of NO biology. For example, NOS3−/− mice have an enhanced β-adrenergic inotropic response [133], and some [134], but not all [135], patch clamp experiments demonstrate a requirement for NOS3 in cholinergic inhibition of intracellular Ca2+.
Studies have shown that NOS3-induced S-nitrosylation of β-arrestin 2, dynamin, and G protein coupled receptor kinase (GRK2) regulates agonist-induced β2-adrenergic receptor trafficking by promoting receptor internalization, promoting endocytosis, and decreasing receptor phosphorylation and desensitization, respectively [136–139]. β-arrestin 2 serves as a scaffold that functionally colocalizes NOS3 and β2-adrenergic receptor. Isoproterenol stimulation results in activation of NOS3 and S-nitrosylation of β-arrestin 2, which promotes its dissociation from NOS3 and its association with clathrin heavy chain/β-adaptin. This facilitates routing of the β2-adrenergic receptor into the clathrin-based endocytotic pathway, and β-arrestin 2 is subsequently denitrosylated. Inhibition of GRK2 by isoproterenol induced S-nitrosylation suppresses β2 adrenergic receptor phosphorylation, β-arrestin 2 recruitment, and receptor desensitization and downregulation. On the other hand, desensitization is enhanced by inhibiting NO production. NOS3 also mediates S-nitrosylation of dynamin, which promotes clathrin-dependent endocytosis and internalization of the β2 adrenergic receptor.
3.6. Mitochondrial function
Many studies indicate that mitochondrial NO regulates energy metabolism. In studies measuring muscle O2 consumption, NO donors and agonists suppress tissue O2 consumption in a fashion that could be attenuated by NOS inhibitors [140]. Myocardial O2 consumption is also physiologically inhibited by NO in a manner that improves mechanical efficiency [68]. It has been demonstrated in both anesthetized and conscious animals, that myocardial oxygen consumption (MVO2) increases in response to NOS inhibition [68,141,142]. Thus, NO regulates not only the major energy consuming process of the heart, contraction, but also mitochondrial energy production. Collectively, these studies suggest that NO promotes mechanoenergetic coupling and thereby enhances myocardial mechanical efficiency.
There is evidence that S-nitrosylation of proteins is involved in the regulation of mitochondrial energetics [69]. S-nitrosylation inhibits the activity of Complex 1 and F1F0ATPase, thereby attenuating ROS generation and reducing ATP consumption, respectively, during ischemia–reperfusion [69]. Cytochrome c oxidase activity is also inhibited by S-nitrosylation leading to decreased oxygen consumption [70]. S-nitrosylation of creatine kinase inhibits its activity and suppresses contractility under stress [71]. On the other hand, the activity of α-KGDH is increased by S-nitrosylation, which may prevent oxidative inactivation upon ischemia–reperfusion [69].
4. Vascular cellular processes regulated by S-nitrosylation
4.1. Vasodilation
In addition to being the largest reservoir of oxygen (O2), hemoglobin is a major NO donor that vasodilates blood vessels in response to low oxygen tension, thereby matching perfusion with tissue O2 demand, a process termed hypoxic vasodilation [143]. Hemoglobin is a tetramer of 2 alpha and 2 beta chains that exhibit cooperative binding of O2 and exists in one of 2 structural states, R (relaxed, high O2 affinity) and T (tense, low O2 affinity). NO is carried both by binding to hemes in a manner similar to O2 and by S-nitrosylation of Cys93 of the β subunit. S-nitrosohemoglobin (SNO-Hb) has been shown to mediate hypoxic vasodilation [120]. SNO-Hb formation is favored in the oxygenated R structure, whereas in hypoxia or low Ph the T structure releases NO and S-nitrosothiols to the surrounding tissues with resultant vasodilation. There is evidence that the coronary vasodilator nitroglycerin improves myocardial perfusion by utilizing SNO-Hb mediated O2 delivery in concert with NO release [121].
A recent clinical study has shown that inorganic nitrate capsules or a dietary nitrate load results in dose-dependent increases in plasma nitrite concentration via bioconversion in vivo [15]. This bioactive nitrite, after reduction to NO, causes dose-dependent decreases in blood pressure and prevents ischemia–reperfusion-induced endothe-lial dysfunction in healthy volunteers. Nitrite, within the realm of physiological concentrations, vasodilates both the arterial and venous sides of the forearm circulation and systemic nitrite application decreases blood pressure in humans [13,14]. It is thought that these effects of nitrite are mainly because of its reduction to NO within the blood vessel wall and within the red blood cell. Of note, nitrite infusions have been shown to be associated with rapid formation of iron-nitrosylated hemoglobin and, to a lesser extent, S-nitroso-hemoglobin [13]. Therefore, red blood cells play key role in autoregulation of blood flow and disturbances in nitroso-redox balance may underlie vascular dysfunction in a variety of disease states, including heart failure [122], pulmonary hypertension [123], sickle cell disease [144], and diabetic cardiovascular disease [145].
4.2. Angiogenesis
Vascular endothelial growth factor (VEGF), a major promoter of angiogenesis, stimulates NOS3 production of NO [146,147]. NOS3−/− mice are deficient in VEGF responsiveness, supporting the important role of NO in angiogenesis [148]. Accumulating evidence suggests that S-nitrosylation mediates the pro-angiogenic effects of NO. VEGF has been shown to induce S-nitrosylation of mitogen activated protein kinase phosphatase 7 (MKP7), which facilitates endothelial cell migration [149]. In addition, S-nitrosylation-mediated activation of dynamin, a regulator of endocytosis, promotes endothelial cell survival and angiogenesis [138]. In vascular diseases associated with aging [150] and diabetes mellitus [151], alterations in endothelial cell protein S-nitrosylation have also been reported.
Hypoxia stimulates angiogenesis via the transcription factor hypoxia-inducible factor (HIF), which increases VEGF expression [152]. SNO donors have been shown to exert an effect similar to hypoxia, leading to increased HIF nuclear expression and S-nitrosylation-mediated HIF stabilization [153–156]. Furthermore, studies in normoxic GSNOR−/− mice demonstrated constitutively S-nitrosylated HIF with increased binding to the VEGF gene [54]. These mice also manifested cardioprotection after myocardial infarction, associated with increased myocardial capillary density. Collectively, these studies support the notion that NO promotes angiogenesis via protein S-nitrosylation.
4.3. Inflammation and apoptosis
Extensive evidence has demonstrated that NO exerts anti-inflammatory effects in the vasculature. NO donors decrease, whereas NOS inhibitors increase, leukocyte-endothelial adherence [6,157]. Studies in mice lacking specific NOS isoforms have further supported this notion [158] [159]. S-nitrosylation has been shown to be the NO-based signaling mechanism regulating endothelial protein trafficking and suppression of nuclear factor κB (NFκB)-dependent expression of proinflammatory cytokines and adhesion molecules [160]. In regard to protein trafficking in endothelial cells, S-nitrosylation of N-ethylmaleimide sensitive factor suppresses exocytosis of granules (i.e. Weibel–Palade bodies) and thereby externalization of the adhesion molecule P-selectin [161]. This inhibits leukocyte rolling and thus vascular inflammation. A similar mechanism is operative in platelets, reducing activation, adhesion, aggregation, and thrombosis [162]. Studies have demonstrated inhibitory S-nitrosylation of both NFκB and its activating enzyme complex, inhibitoryκB kinase [163,164]. Thus, the S-nitrosylation mediated anti-inflammatory actions of NO are relevant to a wide range of cardiovascular disease processes, including atherosclerosis, sepsis, and autoimmune disorders.
The antiapoptotic effects of NO have been shown to be mediated, at least in part, by S-nitrosylation of caspase-3 [165–167]. A cysteine residue on the active site of caspase-3 is S-nitrosylated thereby inhibiting its proapoptotic effects. A recent study has reported that thioredoxin-mediated denitrosylation is the mechanism by which caspase-3 undergoes stimulus-coupled activation [55]. To the extent that these mechanisms are operative in endothelial cells, the balance between S-nitrosylation and denitrosylation may play a pivotal role in endothelial cell survival.
5. Renal cellular processes regulated by S-nitrosylation
NO plays a key role in regulating the capability of the kidneys to excrete sodium, an important determinant of arterial blood pressure [5,168]. Studies evaluating the intrarenal effects of NO donors and NOS inhibitors indicated that NO serves as a diuretic and natriuretic agent [5]. Experimental evidence from proximal tubule and cortical collecting duct cells and isolated proximal tubule and collecting duct segments demonstrated that this effect of NO is mediated by direct inhibition of epithelial transport mechanisms [4]. NO inhibits the sodium/hydrogen (Na+/H+) antiporter on the luminal membrane of the proximal tubule and attenuates the sodium/potassium (Na+/K+) ATPase activity on the basolateral membrane of the proximal tubule and collecting duct segments [169]. However, the signaling mechanisms mediating the inhibitory effects of NO on renal epithelial ion channels have not been fully elucidated. There is also evidence suggesting that the effects of intrarenal NOS inhibitors and NO donors on tubular reabsorptive function are mediated indirectly by the associated changes in peritubular hemodynamicsor interstitial pressure [5,170].
NO has been shown to be abundantly produced in the renal medulla. Renal medullary cells adapt to the hyperosmotic interstitial environment by increased expression of osmoprotective genes, which is driven by the transcriptional activator, tonicity-responsive enhancer I.H. Schulman, J.M. Hare / Biochimica et binding protein (TonEBP) [171]. A recent study addressed the effect of NO on the expression of osmoprotective genes and TonEBP activation in Madin–Darby Canine Kidney (MDCK) epithelial cells. NO donors blunted tonicity-induced up-regulation of TonEBP target genes. 8-bromo-cGMP and peroxynitrite failed to reproduce the inhibitory effect of NO, indicating that NO acts directly on TonEBP. S-nitrosylation of TonEBP was found to correlate with reduced DNA binding and transcriptional activity. Thus, this study demonstrated a novel SNO-mediated inhibitory effect on TonEBP, a mechanism relevant for regulation of osmoprotective genes in the renal medulla.
Ecto-5′-nucleotidase (5′-ribonucleoside phosphohydrolase, 5′-NU) is a membrane-bound glycoprotein that hydrolyzes extracellular nucleotides into membrane-permeable nucleosides [172]. In the kidney, 5′-NU is expressed mainly in plasma membranes of proximal tubular cells and, to a lesser extent, in glomerular mesangial cells, interstitial fibroblasts, and intercalated cells of the collecting tubule. It has been shown that NO inhibits 5′-NU activity in a cGMP- and protein synthesis-independent manner, most likely through S-nitrosylation of the enzyme [172]. The inhibition of 5′-NU activity by NO affected renal proximal phosphate reabsorption.
In glomerular mesangial cells, NO can modulate cell migration, cell proliferation, and the expression of extracellular matrix (ECM) proteins, degrading proteases, and intrinsic protease inhibitors [173]. The regulatory effects of NO on the expression pattern of cytokine-inducible genes that contribute to ECM homeostasis are considered to be a critical step in the development and progression of fibrotic processes within the kidney [174]. The role of protein S-nitrosylation in mediating the effects of NO on mesangial cells has not been extensively studied. However, using the biotin-switch method combined with two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and peptide mass fingerprinting, 31 novel protein targets of S-nitrosylation in NO-treated and cytokine-activated murine mesangial cells have been identified, including signaling proteins, receptors and membrane proteins, cytoskeletal and cell matrix proteins, and cytoplasmic proteins [175]. More recently, PARP-1, a trans-activator of the NOS2 promoter in mesangial cells, has been shown to be a target of NO-mediated S-nitrosylation [176]. This modification limits its DNA binding activity and ability to trans-activate the NOS2 promoter. This NO-mediated negative feedback regulation of PARP-1 binding and action at the NOS2 promoter represents an endogenous mechanism to limit excessive NO generation in pathological states. Further research is warranted to elucidate the pathophysiological role of S-nitrosylation and denitrosylation signaling pathways in the kidney.
6. Summary
NO plays a central role in cardiovascular physiology. Protein S-nitrosylation, a reversible, thiol-based, and redox-sensitive post-translational modification, has emerged as a crucial and ubiquitous NO-based signal. The regulatory effects of S-nitrosylation involve altering protein structure and function by modifying specific thiols and shielding modified thiols from further irreversible modification under oxidative/nitrosative stress. In addition, the spatial localization of NO and SNO signaling, the level of protein S-nitrosylation, and the interaction with other signaling pathways determines whether the overall effect of S-nitrosylation is protective or detrimental. Future research on the mechanisms of S-nitrosylation and denitrosylation and interactions with ROS-based signaling pathways may help identify potential therapeutic targets in cardiovascular diseases.
Acknowledgments
This work was supported by grants from the NHLBI (RO1 HL65455 and RO1 HL094849) to J.M. Hare. Dr. Hare is also supported by NIH grants: P20 HL101443, RO1 HL084275, RO1 HL107110, and U54 HL081028.
Abbreviations
- [Ca2+]i
Intracellular calcium concentration
- cGMP
3′,5′-cyclic guanosine monophosphate
- ECM
Extracellular matrix
- GRK2
G protein coupled receptor kinase
- GSH
Glutathione
- GSNHOH
Glutathione S-hydroxysulfenamide
- GSNO
S-nitrosoglutathione
- GSNOR
S-nitrosoglutathione reductase
- GSSG
Oxidized glutathione
- HIF
hypoxia inducible factor
- K+
potassium
- LTCC
L-type Ca2+ channel
- MKP7
mitogen activated protein kinase phosphatase 7
- MVO2
Myocardial oxygen consumption
- Na+
sodium
- NCX
Sodium-calcium exchanger
- NFkB
nuclear factor kB
- NO
Nitric oxide
- N2O3
dinitrogen trioxide
- NOS
Nitric oxide synthases
- NSF
N-ethylmaleimide sensitive factor
- 5′-NU
Ecto-5′-nucleotidase
- O2
Oxygen
- PDE
Phosphodiesterase
- PKG
cGMP-dependent protein kinase
- PLB
Phospholamban
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- RYR2
Ryanodine receptor/Ca2+-release channel
- SERCA
Sarcoplasmic reticulum Ca2+ ATPase
- sGC
Soluble guanylate cyclase
- SHHF
Spontaneously hypertensive-heart failure
- SNO
S-nitrosothiols
- SNO-Hb
S-nitrosohemoglobin
- SR
Sarcoplasmic reticulum
- TonEBP
Tonicity-responsive enhancer binding protein
- VEGF
Vascular endothelial growth factor
- XO
Xanthine oxidase
Footnotes
This article is part of a Special Issue entitled Regulation of Cellular Processes by S-nitrosylation.
Conflict of interest statement: All the authors declared no conflict of interest that could influence this work.
References
- 1.Gewaltig MT, Kojda G. Vasoprotection by nitric oxide: mechanisms and therapeutic potential. Cardiovasc Res. 2002;55:250–260. doi: 10.1016/s0008-6363(02)00327-9. [DOI] [PubMed] [Google Scholar]
- 2.Hare JM. Nitric oxide and excitation–contraction coupling. J Mol Cell Cardiol. 2003;35:719–729. doi: 10.1016/s0022-2828(03)00143-3. [DOI] [PubMed] [Google Scholar]
- 3.Freedman JE, Ting B, Hankin B, Loscalzo J, Keaney JF, Jr, Vita JA. Impaired platelet production of nitric oxide predicts presence of acute coronary syndromes. Circulation. 1998;98:1481–1486. doi: 10.1161/01.cir.98.15.1481. [DOI] [PubMed] [Google Scholar]
- 4.Stoos BA, Garvin JL. Actions of nitric oxide on renal epithelial transport. Clin Exp Pharmacol Physiol. 1997;24:591–594. doi: 10.1111/j.1440-1681.1997.tb02097.x. [DOI] [PubMed] [Google Scholar]
- 5.Majid DS, Navar LG. Nitric oxide in the control of renal hemodynamics and excretory function. Am J Hypertens. 2001;14:74S–82S. doi: 10.1016/s0895-7061(01)02073-8. [DOI] [PubMed] [Google Scholar]
- 6.Tomita H, Egashira K, Kubo-Inoue M, Usui M, Koyanagi M, Shimokawa H, Takeya M, Yoshimura T, Takeshita A. Inhibition of NO synthesis induces inflammatory changes and monocyte chemoattractant protein-1 expression in rat hearts and vessels. Arterioscler Thromb Vasc Biol. 1998;18:1456–1464. doi: 10.1161/01.atv.18.9.1456. [DOI] [PubMed] [Google Scholar]
- 7.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 8.Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- 9.Lima B, Forrester MT, Hess DT, Stamler JS. S-Nitrosylation in Cardiovascular Signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmet JM, Hare JM. Nitroso-redox interactions in the cardiovascular system. Circulation. 2006;114:1531–1544. doi: 10.1161/CIRCULATIONAHA.105.605519. [DOI] [PubMed] [Google Scholar]
- 12.Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, III, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 14.Dejam A, Hunter CJ, Tremonti C, Pluta RM, Hon YY, Grimes G, Partovi K, Pelletier MM, Oldfield EH, Cannon RO, III, Schechter AN, Gladwin MT. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116:1821–1831. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- 15.Kapil V, Milsom AB, Okorie M, Maleki-Toyserkani S, Akram F, Rehman F, Arghandawi S, Pearl V, Benjamin N, Loukogeorgakis S, MacAllister R, Hobbs AJ, Webb AJ, Ahluwalia A. Inorganic nitrate supplementation lowers blood pressure in humans: role for nitrite-derived NO. Hypertension. 2010;56:274–281. doi: 10.1161/HYPERTENSIONAHA.110.153536. [DOI] [PubMed] [Google Scholar]
- 16.Kone BC, Kuncewicz T, Zhang W, Yu ZY. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am J Physiol Renal Physiol. 2003;285:F178–F190. doi: 10.1152/ajprenal.00048.2003. [DOI] [PubMed] [Google Scholar]
- 17.Wilcox CS, Welch WJ, Murad F, Gross SS, Taylor G, Levi R, Schmidt HH. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc Natl Acad Sci U S A. 1992;89:11993–11997. doi: 10.1073/pnas.89.24.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boissel JP, Schwarz PM, Forstermann U. Neuronal-type NO synthase: transcript diversity and expressional regulation. Nitric Oxide. 1998;2:337–349. doi: 10.1006/niox.1998.0189. [DOI] [PubMed] [Google Scholar]
- 19.Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM. Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res. 2003;92:1322–1329. doi: 10.1161/01.RES.0000078171.52542.9E. [DOI] [PubMed] [Google Scholar]
- 20.Abu-Soud HM, Loftus M, Stuehr DJ. Subunit dissociation and unfolding of macrophage NO synthase: relationship between enzyme structure, prosthetic group binding, and catalytic function. Biochemistry. 1995;34:11167–11175. doi: 10.1021/bi00035a023. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Wallerath T, Forstermann U. Physiological mechanisms regulating the expression of endothelial-type NO synthase. Nitric Oxide. 2002;7:132–147. doi: 10.1016/s1089-8603(02)00127-1. [DOI] [PubMed] [Google Scholar]
- 22.Schulman IH, Zhou MS, Raij L. Interaction between nitric oxide and angiotensin II in the endothelium: role in atherosclerosis and hypertension. J Hypertens Suppl. 2006;24:S45–S50. doi: 10.1097/01.hjh.0000220406.46246.f2. [DOI] [PubMed] [Google Scholar]
- 23.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 24.Hare JM. Spatial confinement of isoforms of cardiac nitric-oxide synthase: unravelling the complexities of nitric oxide's cardiobiology. Lancet. 2004;363:1338–1339. doi: 10.1016/S0140-6736(04)16083-2. [DOI] [PubMed] [Google Scholar]
- 25.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 26.Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, Li D, Berkowitz DE, Hare JM. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation–contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–15948. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ignarro LJ, Cirino G, Casini A, Napoli C. Nitric oxide as a signaling molecule in the vascular system: an overview. J Cardiovasc Pharmacol. 1999;34:879–886. doi: 10.1097/00005344-199912000-00016. [DOI] [PubMed] [Google Scholar]
- 28.Massberg S, Sausbier M, Klatt P, Bauer M, Pfeifer A, Siess W, Fassler R, Ruth P, Krombach F, Hofmann F. Increased adhesion and aggregation of platelets lacking cyclic guanosine 3′,5′-monophosphate kinase I. J Exp Med. 1999;189:1255–1264. doi: 10.1084/jem.189.8.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, Paolocci N, Tomaselli GF, Hare JM, Kass DA. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001;15:1718–1726. doi: 10.1096/fj.00-0538com. [DOI] [PubMed] [Google Scholar]
- 30.Sayed N, Kim DD, Fioramonti X, Iwahashi T, Duran WN, Beuve A. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res. 2008;103:606–614. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ravi K, Brennan LA, Levic S, Ross PA, Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc Natl Acad Sci U S A. 2004;101:2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Ruiz A, Villanueva L, Gonzalez de Orduna C, Lopez-Ferrer D, Higueras MA, Tarin C, Rodriguez-Crespo I, Vazquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci U S A. 2005;102:8525–8530. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280:7511–7518. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 34.Santhanam L, Lim HK, Miriel V, Brown T, Patel M, Balanson S, Ryoo S, Anderson M, Irani K, Khanday F, Di Costanzo L, Nyhan D, Hare JM, Christianson DW, Rivers R, Shoukas A, Berkowitz DE. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res. 2007;101:692–702. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]
- 35.Leiper J, Murray-Rust J, McDonald N, Vallance P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc Natl Acad Sci U S A. 2002;99:13527–13532. doi: 10.1073/pnas.212269799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S) NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Murphy E. Protein S-Nitrosylation and Cardioprotection. Circ Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 40.Hogg N. The biochemistry and physiology of S-nitrosothiols. Annu Rev Pharmacol Toxicol. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Miller MJ, Joshi MS, Thomas DD, Lancaster JR., Jr Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc Natl Acad Sci U S A. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- 43.Galkin A, Moncada S. S-nitrosation of mitochondrial complex I depends on its structural conformation. J Biol Chem. 2007;282:37448–37453. doi: 10.1074/jbc.M707543200. [DOI] [PubMed] [Google Scholar]
- 44.Das DK, Maulik N. Preconditioning potentiates redox signaling and converts death signal into survival signal. Arch Biochem Biophys. 2003;420:305–311. doi: 10.1016/j.abb.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 45.Foster MW, Liu L, Zeng M, Hess DT, Stamler JS. A genetic analysis of nitrosative stress. Biochemistry. 2009;48:792–799. doi: 10.1021/bi801813n. [DOI] [PubMed] [Google Scholar]
- 46.Koshiishi I, Takajo T, Tsuchida K. Regulation of S-thiolation and S-nitrosylation in the thiol/nitric oxide system by radical scavengers. Nitric Oxide. 2007;16:356–361. doi: 10.1016/j.niox.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 47.Steffen M, Sarkela TM, Gybina AA, Steele TW, Trasseth NJ, Kuehl D, Giulivi C. Metabolism of S-nitrosoglutathione in intact mitochondria. Biochem J. 2001;356:395–402. doi: 10.1042/0264-6021:3560395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 50.Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A. 2007;104:20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 52.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 53.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 54.Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanghani PC, Davis WI, Fears SL, Green SL, Zhai L, Tang Y, Martin E, Bryan NS, Sanghani SP. Kinetic and cellular characterization of novel inhibitors of S-nitrosoglutathione reductase. J Biol Chem. 2009;284:24354–24362. doi: 10.1074/jbc.M109.019919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patel JM, Zhang J, Block ER. Nitric oxide-induced inhibition of lung endothelial cell nitric oxide synthase via interaction with allosteric thiols: role of thioredoxin in regulation of catalytic activity. Am J Respir Cell Mol Biol. 1996;15:410–419. doi: 10.1165/ajrcmb.15.3.8810647. [DOI] [PubMed] [Google Scholar]
- 58.Kahlos K, Zhang J, Block ER, Patel JM. Thioredoxin restores nitric oxide-induced inhibition of protein kinase C activity in lung endothelial cells. Mol Cell Biochem. 2003;254:47–54. doi: 10.1023/a:1027380828645. [DOI] [PubMed] [Google Scholar]
- 59.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin dependon S-nitrosylation at cysteine 69. Nat Cell Biol. 2002;4:743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 60.Bateman RL, Rauh D, Tavshanjian B, Shokat KM. Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J Biol Chem. 2008;283:35756–35762. doi: 10.1074/jbc.M807125200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okado-Matsumoto A, Fridovich I. Putative denitrosylase activity of Cu, Zn-superoxide dismutase. Free Radic Biol Med. 2007;43:830–836. doi: 10.1016/j.freeradbiomed.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 62.Trujillo M, Alvarez MN, Peluffo G, Freeman BA, Radi R. Xanthine oxidase-mediated decomposition of S-nitrosothiols. J Biol Chem. 1998;273:7828–7834. doi: 10.1074/jbc.273.14.7828. [DOI] [PubMed] [Google Scholar]
- 63.Hare JM, Stamler JS. NOS: modulator, not mediator of cardiac performance. Nat Med. 1999;5:273–274. doi: 10.1038/6486. [DOI] [PubMed] [Google Scholar]
- 64.Hare JM, Loh E, Creager MA, Colucci WS. Nitric oxide inhibits the positive inotropic response to beta-adrenergic stimulation in humans with left ventricular dysfunction. Circulation. 1995;92:2198–2203. doi: 10.1161/01.cir.92.8.2198. [DOI] [PubMed] [Google Scholar]
- 65.Hare JM, Lofthouse RA, Juang GJ, Colman L, Ricker KM, Kim B, Senzaki H, Cao S, Tunin RS, Kass DA. Contribution of caveolin protein abundance to augmented nitric oxide signaling in conscious dogs with pacing-induced heart failure. Circ Res. 2000;86:1085–1092. doi: 10.1161/01.res.86.10.1085. [DOI] [PubMed] [Google Scholar]
- 66.Zhang YH, Zhang MH, Sears CE, Emanuel K, Redwood C, El-Armouche A, Kranias EG, Casadei B. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ Res. 2008;102:242–249. doi: 10.1161/CIRCRESAHA.107.164798. [DOI] [PubMed] [Google Scholar]
- 67.Garofalo F, Parisella ML, Amelio D, Tota B, Imbrogno S. Phospholamban S-nitrosylation modulates Starling response in fish heart. Proceedings of the Royal Society B Biological Sciences. 2009;276:4043–4052. doi: 10.1098/rspb.2009.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Recchia FA, McConnell PI, Bernstein RD, Vogel TR, Xu X, Hintze TH. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res. 1998;83:969–979. doi: 10.1161/01.res.83.10.969. [DOI] [PubMed] [Google Scholar]
- 69.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 70.Zhang J, Jin B, Li L, Block ER, Patel JM. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol. 2005;288:C840–C849. doi: 10.1152/ajpcell.00325.2004. [DOI] [PubMed] [Google Scholar]
- 71.Arstall MA, Bailey C, Gross WL, Bak M, Balligand JL, Kelly RA. Reversible S-nitrosation of creatine kinase by nitric oxide in adult rat ventricular myocytes. J Mol Cell Cardiol. 1998;30:979–988. doi: 10.1006/jmcc.1998.0662. [DOI] [PubMed] [Google Scholar]
- 72.Gonzalez DR, Treuer A, Sun QA, Stamler JS, Hare JM. S-Nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:188–195. doi: 10.1097/FJC.0b013e3181b72c9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Canton M, Menazza S, Sheeran FL, Polverino de Laureto P, Di Lisa F, Pepe S. Oxidation of myofibrillar proteins in human heart failure. J Am Coll Cardiol. 2011;57:300–309. doi: 10.1016/j.jacc.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 74.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barouch LA, Cappola TP, Harrison RW, Crone JK, Rodriguez ER, Burnett AL, Hare JM. Combined loss of neuronal and endothelial nitric oxide synthase causes premature mortality and age-related hypertrophic cardiac remodeling in mice. J Mol Cell Cardiol. 2003;35:637–644. doi: 10.1016/s0022-2828(03)00079-8. [DOI] [PubMed] [Google Scholar]
- 76.Champion HC, Georgakopoulos D, Takimoto E, Isoda T, Wang Y, Kass DA. Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circ Res. 2004;94:657–663. doi: 10.1161/01.RES.0000119323.79644.20. [DOI] [PubMed] [Google Scholar]
- 77.Kanai AJ, Mesaros S, Finkel MS, Oddis CV, Birder LA, Malinski T. Beta-adrenergic regulation of constitutive nitric oxide synthase in cardiac myocytes. Am J Physiol. 1997;273:C1371–C1377. doi: 10.1152/ajpcell.1997.273.4.C1371. [DOI] [PubMed] [Google Scholar]
- 78.Sun J, Yamaguchi N, Xu L, Eu JP, Stamler JS, Meissner G. Regulation of the cardiac muscle ryanodine receptor by O(2) tension and S-nitrosoglutathione. Biochemistry. 2008;47:13985–13990. doi: 10.1021/bi8012627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc Natl Acad Sci U S A. 2001;98:14126–14131. doi: 10.1073/pnas.241380298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Venkatakrishnan P, Nakayasu ES, Almeida IC, Miller RT. Absence of nitric-oxide synthase in sequentially purified rat liver mitochondria. J Biol Chem. 2009;284:19843–19855. doi: 10.1074/jbc.M109.003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lacza Z, Pankotai E, Busija DW. Mitochondrial nitric oxide synthase: current concepts and controversies. Front Biosci. 2009;14:4436–4443. doi: 10.2741/3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hayashi T, Martone ME, Yu Z, Thor A, Doi M, Holst MJ, Ellisman MH, Hoshijima M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci. 2009;122:1005–1013. doi: 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Damy T, Ratajczak P, Robidel E, Bendall JK, Oliviero P, Boczkowski J, Ebrahimian T, Marotte F, Samuel JL, Heymes C. Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. FASEB J. 2003;17:1934–1936. doi: 10.1096/fj.02-1208fje. [DOI] [PubMed] [Google Scholar]
- 84.Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363:1365–1367. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 85.Takimoto Y, Aoyama T, Tanaka K, Keyamura R, Yui Y, Sasayama S. Augmented expression of neuronal nitric oxide synthase in the atria parasympathetically decreases heart rate during acute myocardial infarction in rats. Circulation. 2002;105:490–496. doi: 10.1161/hc0402.102662. [DOI] [PubMed] [Google Scholar]
- 86.Bendall JK, Damy T, Ratajczak P, Loyer X, Monceau V, Marty I, Milliez P, Robidel E, Marotte F, Samuel JL, Heymes C. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in beta-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation. 2004;110:2368–2375. doi: 10.1161/01.CIR.0000145160.04084.AC. [DOI] [PubMed] [Google Scholar]
- 87.Saraiva RM, Minhas KM, Raju SV, Barouch LA, Pitz E, Schuleri KH, Vandegaer K, Li D, Hare JM. Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction: role of nitrosoredox equilibrium. Circulation. 2005;112:3415–3422. doi: 10.1161/CIRCULATIONAHA.105.557892. [DOI] [PubMed] [Google Scholar]
- 88.Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation. 2005;112:3729–3737. doi: 10.1161/CIRCULATIONAHA.105.539437. [DOI] [PubMed] [Google Scholar]
- 89.Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 90.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 91.Cohen RI, Wilson D, Liu SF. Nitric oxide modifies the sarcoplasmic reticular calcium release channel in endotoxemia by both guanosine-3′,5′ (cyclic) phosphate-dependent and independent pathways. Crit Care Med. 2006;34:173–181. doi: 10.1097/01.ccm.0000194722.12260.f9. [DOI] [PubMed] [Google Scholar]
- 92.Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49:1722–1732. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 93.Sanchez G, Escobar M, Pedrozo Z, Macho P, Domenech R, Hartel S, Hidalgo C, Donoso P. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res. 2008;77:380–386. doi: 10.1093/cvr/cvm011. [DOI] [PubMed] [Google Scholar]
- 94.Sanchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005;39:982–991. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 95.Deng J, Wang G, Huang Q, Yan Y, Li K, Tan W, Jin C, Wang Y, Liu J. Oxidative stress-induced leaky sarcoplasmic reticulum underlying acute heart failure in severe burn trauma. Free Radic Biol Med. 2008;44:375–385. doi: 10.1016/j.freeradbiomed.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 96.Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- 97.Marengo JJ, Hidalgo C, Bull R. Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys J. 1998;74:1263–1277. doi: 10.1016/S0006-3495(98)77840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun J, Xu L, Eu JP, Stamler JS, Meissner G. Nitric oxide, NOC-12, and S-nitrosoglutathione modulate the skeletal muscle calcium release channel/rya-nodine receptor by different mechanisms. An allosteric function for O2 in S-nitrosylation of the channel. J Biol Chem. 2003;278:8184–8189. doi: 10.1074/jbc.M211940200. [DOI] [PubMed] [Google Scholar]
- 99.Gonzalez DR, Fernandez IC, Ordenes PP, Treuer AV, Eller G, Boric MP. Differential role of S-nitrosylation and the NO–cGMP–PKG pathway in cardiac contractility. Nitric Oxide. 2008;18:157–167. doi: 10.1016/j.niox.2007.09.086. [DOI] [PubMed] [Google Scholar]
- 100.Paolocci N, Ekelund UE, Isoda T, Ozaki M, Vandegaer K, Georgakopoulos D, Harrison RW, Kass DA, Hare JM. cGMP-independent inotropic effects of nitric oxide and peroxynitrite donors: potential role for nitrosylation. Am J Physiol Heart Circ Physiol. 2000;279:H1982–H1988. doi: 10.1152/ajpheart.2000.279.4.H1982. [DOI] [PubMed] [Google Scholar]
- 101.Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J Biol Chem. 2010;285:28938–28945. doi: 10.1074/jbc.M110.154948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hare JM, Mangal B, Brown J, Fisher C, Jr, Freudenberger R, Colucci WS, Mann DL, Liu P, Givertz MM, Schwarz RP. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol. 2008;51:2301–2309. doi: 10.1016/j.jacc.2008.01.068. [DOI] [PubMed] [Google Scholar]
- 103.Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kinugawa S, Huang H, Wang Z, Kaminski PM, Wolin MS, Hintze TH. A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ Res. 2005;96:355–362. doi: 10.1161/01.RES.0000155331.09458.A7. [DOI] [PubMed] [Google Scholar]
- 105.Loyer X, Gomez AM, Milliez P, Fernandez-Velasco M, Vangheluwe P, Vinet L, Charue D, Vaudin E, Zhang W, Sainte-Marie Y, Robidel E, Marty I, Mayer B, Jaisser F, Mercadier JJ, Richard S, Shah AM, Benitah JP, Samuel JL, Heymes C. Cardiomyocyte overexpression of neuronal nitric oxide synthase delays transition toward heart failure in response to pressure overload by preserving calcium cycling. Circulation. 2008;117:3187–3198. doi: 10.1161/CIRCULATIONAHA.107.741702. [DOI] [PubMed] [Google Scholar]
- 106.Burger DE, Lu X, Lei M, Xiang FL, Hammoud L, Jiang M, Wang H, Jones DL, Sims SM, Feng Q. Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation. 2009;120:1345–1354. doi: 10.1161/CIRCULATIONAHA.108.846402. [DOI] [PubMed] [Google Scholar]
- 107.Zhang H, Chen X, Gao E, MacDonnell SM, Wang W, Kolpakov M, Nakayama H, Zhang X, Jaleel N, Harris DM, Li Y, Tang M, Berretta R, Leri A, Kajstura J, Sabri A, Koch WJ, Molkentin JD, Houser SR. Increasing cardiac contractility after myocardial infarction exacerbates cardiac injury and pump dysfunction. Circ Res. 2010;107:800–809. doi: 10.1161/CIRCRESAHA.110.219220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saliaris AP, Amado LC, Minhas KM, Schuleri KH, Lehrke S, St John M, Fitton T, Barreiro C, Berry C, Zheng M, Kozielski K, Eneboe V, Brawn J, Hare JM. Chronic allopurinol administration ameliorates maladaptive alterations in Ca2+ cycling proteins and beta-adrenergic hyporesponsiveness in heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H1328–H1335. doi: 10.1152/ajpheart.00461.2006. [DOI] [PubMed] [Google Scholar]
- 109.Wang H, Kohr MJ, Traynham CJ, Wheeler DG, Janssen PM, Ziolo MT. Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. Am J Physiol Cell Physiol. 2008;294:C1566–C1575. doi: 10.1152/ajpcell.00367.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 112.Wawrzynow A, Collins JH. Chemical modification of the Ca(2+) -ATPase of rabbit skeletal muscle sarcoplasmic reticulum: identification of sites labeled with aryl isothiocyanates and thiol-directed conformational probes. Biochim Biophys Acta. 1993;1203:60–70. doi: 10.1016/0167-4838(93)90036-q. [DOI] [PubMed] [Google Scholar]
- 113.Eley DW, Eley JM, Korecky B, Fliss H. Impairment of cardiac contractility and sarcoplasmic reticulum Ca2+ ATPase activity by hypochlorous acid: reversal by dithiothreitol. Can J Physiol Pharmacol. 1991;69:1677–1685. doi: 10.1139/y91-249. [DOI] [PubMed] [Google Scholar]
- 114.Stamler JS, Hausladen A. Oxidative modifications in nitrosative stress. Nat Struct Biol. 1998;5:247–249. doi: 10.1038/nsb0498-247. [DOI] [PubMed] [Google Scholar]
- 115.Duncan JG, Ravi R, Stull LB, Murphy AM. Chronic xanthine oxidase inhibition prevents myofibrillar protein oxidation and preserves cardiac function in a transgenic mouse model of cardiomyopathy. Am J Physiol Heart Circ Physiol. 2005;289:H1512–H1518. doi: 10.1152/ajpheart.00168.2005. [DOI] [PubMed] [Google Scholar]
- 116.Kogler H, Fraser H, McCune S, Altschuld R, Marban E. Disproportionate enhancement of myocardial contractility by the xanthine oxidase inhibitor oxypurinol in failing rat myocardium. Cardiovasc Res. 2003;59:582–592. doi: 10.1016/s0008-6363(03)00512-1. [DOI] [PubMed] [Google Scholar]
- 117.Bai CX, Takahashi K, Masumiya H, Sawanobori T, Furukawa T. Nitric oxide-dependent modulation of the delayed rectifier K+current and the L-type Ca2+ current by ginsenoside Re, an ingredient of Panax ginseng, in guinea-pig cardiomyocytes. Br J Pharmacol. 2004;142:567–575. doi: 10.1038/sj.bjp.0705814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bai CX, Namekata I, Kurokawa J, Tanaka H, Shigenobu K, Furukawa T. Role of nitric oxideinCa2+ sensitivity of the slowly activating delayed rectifierK+current in cardiac myocytes. Circ Res. 2005;96:64–72. doi: 10.1161/01.RES.0000151846.19788.E0. [DOI] [PubMed] [Google Scholar]
- 119.Asada K, Kurokawa J, Furukawa T. Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel. J Biol Chem. 2009;284:6014–6020. doi: 10.1074/jbc.M807158200. [DOI] [PubMed] [Google Scholar]
- 120.Gomez R, Caballero R, Barana A, Amoros I, Calvo E, Lopez JA, Klein H, Vaquero M, Osuna L, Atienza F, Almendral J, Pinto A, Tamargo J, Delpon E. Nitric oxide increases cardiac IK1 by nitrosylation of cysteine 76 of Kir2.1 channels. Circ Res. 2009;105:383–392. doi: 10.1161/CIRCRESAHA.109.197558. [DOI] [PubMed] [Google Scholar]
- 121.Ahern GP, Hsu SF, Klyachko VA, Jackson MB. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem. 2000;275:28810–28815. doi: 10.1074/jbc.M003090200. [DOI] [PubMed] [Google Scholar]
- 122.Evans JR, Bielefeldt K. Regulation of sodium currents through oxidation and reduction of thiol residues. Neuroscience. 2000;101:229–236. doi: 10.1016/s0306-4522(00)00367-5. [DOI] [PubMed] [Google Scholar]
- 123.Kurata Y, Hisatome I, Tsuboi M, Uenishi H, Zhang G, Oyaizu M, Sato R, Imanishi S. Effect of sulfhydryl oxidoreduction on permeability of cardiac tetrodotoxin-insensitive sodium channel. Life Sci. 1998;63:1023–1035. doi: 10.1016/s0024-3205(98)00364-6. [DOI] [PubMed] [Google Scholar]
- 124.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Williams JC, Armesilla AL, Mohamed TM, Hagarty CL, McIntyre FH, Schomburg S, Zaki AO, Oceandy D, Cartwright EJ, Buch MH, Emerson M, Neyses L. The sarcolemmal calcium pump, alpha-1 syntrophin, and neuronal nitric-oxide synthase are parts of a macromolecular protein complex. J Biol Chem. 2006;281:23341–23348. doi: 10.1074/jbc.M513341200. [DOI] [PubMed] [Google Scholar]
- 126.George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Feron O, Saldana F, Michel JB, Michel T. The endothelial nitric-oxide synthase-caveolin regulatory cycle. J Biol Chem. 1998;273:3125–3128. doi: 10.1074/jbc.273.6.3125. [DOI] [PubMed] [Google Scholar]
- 128.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 130.Henning RJ, Khalil IR, Levy MN. Vagal stimulation attenuates sympathetic enhancement of left ventricular function. Am J Physiol. 1990;258:H1470–H1475. doi: 10.1152/ajpheart.1990.258.5.H1470. [DOI] [PubMed] [Google Scholar]
- 131.Gauthier C, Leblais V, Kobzik L, Trochu JN, Khandoudi N, Bril A, Balligand JL, Le Marec H. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest. 1998;102:1377–1384. doi: 10.1172/JCI2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Varghese P, Harrison RW, Lofthouse RA, Georgakopoulos D, Berkowitz DE, Hare JM. beta(3) -adrenoceptor deficiency blocks nitric oxide-dependent inhibition of myocardial contractility. J Clin Invest. 2000;106:697–703. doi: 10.1172/JCI9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gyurko R, Kuhlencordt P, Fishman MC, Huang PL. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am J Physiol Heart Circ Physiol. 2000;278:H971–H981. doi: 10.1152/ajpheart.2000.278.3.H971. [DOI] [PubMed] [Google Scholar]
- 134.Han X, Kubota I, Feron O, Opel DJ, Arstall MA, Zhao YY, Huang P, Fishman MC, Michel T, Kelly RA. Muscarinic cholinergic regulation of cardiac myocyte ICa-L is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:6510–6515. doi: 10.1073/pnas.95.11.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vandecasteele G, Eschenhagen T, Scholz H, Stein B, Verde I, Fischmeister R. Muscarinic and beta-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nat Med. 1999;5:331–334. doi: 10.1038/6553. [DOI] [PubMed] [Google Scholar]
- 136.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci U S A. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 139.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 140.Xie YW, Shen W, Zhao G, Xu X, Wolin MS, Hintze TH. Role of endothelium-derived nitric oxide in the modulation of canine myocardial mitochondrial respiration in vitro. Implications for the development of heart failure. Circ Res. 1996;79:381–387. doi: 10.1161/01.res.79.3.381. [DOI] [PubMed] [Google Scholar]
- 141.Saavedra WF, Paolocci N, St John ME, Skaf MW, Stewart GC, Xie JS, Harrison RW, Zeichner J, Mudrick D, Marban E, Kass DA, Hare JM. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res. 2002;90:297–304. doi: 10.1161/hh0302.104531. [DOI] [PubMed] [Google Scholar]
- 142.Suto N, Mikuniya A, Okubo T, Hanada H, Shinozaki N, Okumura K. Nitric oxide modulates cardiac contractility and oxygen consumption without changing contractile efficiency. Am J Physiol. 1998;275:H41–H49. doi: 10.1152/ajpheart.1998.275.1.H41. [DOI] [PubMed] [Google Scholar]
- 143.Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- 144.Pawloski JR, Hess DT, Stamler JS. Impaired vasodilation by red blood cells in sickle cell disease. Proc Natl Acad Sci U S A. 2005;102:2531–2536. doi: 10.1073/pnas.0409876102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Padron J, Peiro C, Cercas E, Llergo JL, Sanchez-Ferrer CF. Enhancement of S-nitrosylation in glycosylated hemoglobin. Biochem Biophys Res Commun. 2000;271:217–221. doi: 10.1006/bbrc.2000.2617. [DOI] [PubMed] [Google Scholar]
- 146.Kroll J, Waltenberger J. VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR) Biochem Biophys Res Commun. 1998;252:743–746. doi: 10.1006/bbrc.1998.9719. [DOI] [PubMed] [Google Scholar]
- 147.Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ahmad S, Hewett PW, Wang P, Al-Ani B, Cudmore M, Fujisawa T, Haigh JJ, le Noble F, Wang L, Mukhopadhyay D, Ahmed A. Direct evidence for endothelial vascular endothelial growth factor receptor-1 function in nitric oxide-mediated angiogenesis. Circ Res. 2006;99:715–722. doi: 10.1161/01.RES.0000243989.46006.b9. [DOI] [PubMed] [Google Scholar]
- 149.Pi X, Wu Y, Ferguson JE, III, Portbury AL, Patterson C. SDF-1alpha stimulates JNK3 activity via eNOS-dependent nitrosylation of MKP7 to enhance endothelial migration. Proc Natl Acad Sci U S A. 2009;106:5675–5680. doi: 10.1073/pnas.0809568106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hoffmann J, Haendeler J, Aicher A, Rossig L, Vasa M, Zeiher AM, Dimmeler S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res. 2001;89:709–715. doi: 10.1161/hh2001.097796. [DOI] [PubMed] [Google Scholar]
- 151.Wadham C, Parker A, Wang L, Xia P. High glucose attenuates protein S-nitrosylation in endothelial cells: role of oxidative stress. Diabetes. 2007;56:2715–2721. doi: 10.2337/db06-1294. [DOI] [PubMed] [Google Scholar]
- 152.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 153.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Palmer LA, Doctor A, Chhabra P, Sheram ML, Laubach VE, Karlinsey MZ, Forbes MS, Macdonald T, Gaston B. S-nitrosothiols signal hypoxia-mimetic vascular pathology. J Clin Invest. 2007;117:2592–2601. doi: 10.1172/JCI29444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, Vujaskovic Z, Dewhirst MW, Li CY. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. 2007;26:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sumbayev VV, Budde A, Zhou J, Brune B. HIF-1 alpha protein as a target for S-nitrosation. FEBS Lett. 2003;535:106–112. doi: 10.1016/s0014-5793(02)03887-5. [DOI] [PubMed] [Google Scholar]
- 157.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lefer DJ, Jones SP, Girod WG, Baines A, Grisham MB, Cockrell AS, Huang PL, Scalia R. Leukocyte–endothelial cell interactions in nitric oxide synthase-deficient mice. Am J Physiol. 1999;276:H1943–H1950. doi: 10.1152/ajpheart.1999.276.6.H1943. [DOI] [PubMed] [Google Scholar]
- 159.Chen J, Kuhlencordt PJ, Astern J, Gyurko R, Huang PL. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation. 2001;104:2391–2394. doi: 10.1161/hc4501.099729. [DOI] [PubMed] [Google Scholar]
- 160.Lowenstein CJ. Nitric oxide regulation of protein trafficking in the cardiovascular system. Cardiovasc Res. 2007;75:240–246. doi: 10.1016/j.cardiores.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O'Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–150. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Morrell CN, Matsushita K, Chiles K, Scharpf RB, Yamakuchi M, Mason RJ, Bergmeier W, Mankowski JL, Baldwin WM, III, Faraday N, Lowenstein CJ. Regulation of platelet granule exocytosis by S-nitrosylation. Proc Natl Acad Sci U S A. 2005;102:3782–3787. doi: 10.1073/pnas.0408310102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Marshall HE, Stamler JS. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 164.Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci U S A. 2004;101:8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibitionofinterleukin-1beta-converting enzyme (ICE) -likeand cysteine protease protein (CPP) -32-like proteases. J Exp Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 167.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 168.Zhou MS, Schulman IH, Raij L. Nitric oxide, angiotensin II, and hypertension. Semin Nephrol. 2004;24:366–378. doi: 10.1016/j.semnephrol.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 169.Gabbai FB, Blantz RC. Role of nitric oxide in renal hemodynamics. Semin Nephrol. 1999;19:242–250. [PubMed] [Google Scholar]
- 170.Majid DS, Said KE, Omoro SA, Navar LG. Nitric oxide dependency of arterial pressure-induced changes in renal interstitial hydrostatic pressure in dogs. Circ Res. 2001;88:347–351. doi: 10.1161/01.res.88.3.347. [DOI] [PubMed] [Google Scholar]
- 171.Neuhofer W, Fraek ML, Beck FX. Nitric oxide decreases expression of osmoprotective genes via direct inhibition of TonEBP transcriptional activity. Pflugers Arch. 2009;457:831–843. doi: 10.1007/s00424-008-0540-3. [DOI] [PubMed] [Google Scholar]
- 172.Siegfried GR, Amiel C, Friedlander GR. Inhibition of ecto-5'-nucleotidase by nitric oxide donors. J Biol Chem. 1996;271:4659–4664. doi: 10.1074/jbc.271.9.4659. [DOI] [PubMed] [Google Scholar]
- 173.Akool ES, Doller A, Muller R, Gutwein P, Xin C, Huwiler A, Pfeilschifter J, Eberhardt W. Nitric oxide induces TIMP-1 expression by activating the transforming growth factor β-smad signaling pathway. J Biol Chem. 2005;280:39403–39416. doi: 10.1074/jbc.M504140200. [DOI] [PubMed] [Google Scholar]
- 174.Pfeilschifter J, Beck KF, Eberhardt W, Huwiler A. Changing gears in the course of glomerulonephritis by shifting superoxide to nitric oxide-dominated chemistry. Kidney Int. 2002;61:809–815. doi: 10.1046/j.1523-1755.2002.00225.x. [DOI] [PubMed] [Google Scholar]
- 175.Kuncewicz T, Sheta EA, Goldknopf IL, Kone BC. Proteomic analysis of S-nitrosylated proteins in mesangial cells. Mol Cell Proteomics. 2003;2:156–163. doi: 10.1074/mcp.M300003-MCP200. [DOI] [PubMed] [Google Scholar]
- 176.Yu Z, Kuncewicz T, Dubinsky WP, Kone BC. Nitric oxide-dependent negative feedback of PARP-1 trans-activation of the inducible nitric-oxide synthase gene. J Biol Chem. 2006;281:9101–9109. doi: 10.1074/jbc.M511049200. [DOI] [PubMed] [Google Scholar]