

Abstract
Group B streptococci (GBS) are one of the leading causes of life-threatening illness in neonates. Proinflammatory responses to GBS mediated through host innate immune receptors play a critical role in the disease manifestation. However, the mechanisms involved in proinflammatory responses against GBS, and the contribution of signaling modulators involved in host immune defense, have not been fully elucidated. In the present study, we investigated the role of protein kinase D1 (PKD1) in the proinflammatory responses to GBS. We found that both live and antibiotic-killed GBS induce activation of PKD1 through a pathway that is dependent on the Toll-like receptor signaling adaptor MyD88 and its downstream kinase IRAK1, but independent of TRAF6. Our studies using pharmacological PKD inhibitors and PKD1-knockdown macrophages revealed that PKD1 is indispensable for GBS-mediated activation of MAPKs and NF-κB and subsequent expression of proinflammatory mediators. Furthermore, systemic administration of a PKD inhibitor protects D-galactosamine-sensitized mice from shock-mediated death caused by antibiotic-killed GBS. These findings imply that PKD1 plays a critical regulatory role in GBS-induced proinflammatory reactions and sepsis, and inhibition of PKD1 activation together with antibiotic treatment in GBS-infected neonates could be an effective way to control GBS diseases.
Keywords: Group B streptococci, Macrophages, Cytokines, Kinases, Inflammation, Toll-like receptor, MyD88, PKD1
INTRODUCTION
Group B streptococci (GBS) are capsulated Gram-positive bacteria that asymptomatically colonize the genitourinary and gastrointestinal tracts of approximately 15–20% of healthy adults (1). However, GBS disease can be lethal. It is one of the leading causes of life-threatening infection in neonates, young infants, and pregnant women (2, 3), and can cause of serious invasive infections in immunocompromised adults and elderly patients with severe underlying conditions (4–6). In pregnant women, GBS may cause preterm labor, premature rupture of membranes, urinary tract infections, chorioamnionitis, postpartum endometritis, postpartum wound infection, septic pelvic thrombophlebitis, endocarditis and sepsis (7). Pregnant women who are carriers of GBS have the potential to transmit GBS to their newborn infants, and GBS are one of the first pathogenic organisms that newborns may encounter at birth. Common manifestations of GBS diseases include pneumonia, septicemia, meningitis, bacteremia, and bone or joint infections, such as septic arthritis and osteomyelitis (8, 9). With the advent of intrapartum prophylaxis in 1999, the rate of early onset GBS infection has decreased, but it is still quite high in premature infants (10). Despite increased clinical awareness, prompt diagnosis and aggressive therapy, GBS are still the most common cause of early onset sepsis in neonates, especially premature infants (11). Even after the successful control of GBS infection using early and aggressive therapies with sensitive antibacterial agents (i.e., ampicillin and penicillin), permanent morbidity and mortality remain the frequent outcomes of GBS sepsis in the neonatal population, especially premature infants (12). Penicillin remains the drug of choice for these infections; however, many reports suggest that this conventional therapy may be less than optimal (13). Release of external and internal GBS contents due to rapid cell wall lysis by aggressive bactericidal activity of penicillin may be the causative factor for these outcomes.
Host defense against GBS infection in infants primarily relies on the innate immune system, as their adaptive immune system is not yet fully developed. GBS are recognized by the germline encoded pattern recognition receptors (PRRs), including Toll-like receptor (TLR) family members, present in host innate immune cells. For example, GBS cell wall components [capsular polysaccharide, peptidoglycan (PGN), lipoteichoic acid (LTA), and bacterial lipoprotein] and heat-labile soluble factor GBS-F are recognized by TLR2, TLR6 and co-receptor CD14 (14–20). Nucleic acids of GBS are recognized by TLRs 7, 9 and 13 (21–24). Other GBS components (including those released from heat-killed and antibiotic-killed GBS) may be recognized by other PRRs that have yet to be identified. This recognition of GBS by PRRs on/in the innate immune cells initiates a response that is characterized by local and systemic production of proinflammatory cytokines and mediators that limit the early proliferation and spread of GBS (20, 23, 25–29). However, an uncontrolled and exaggerated production of proinflammatory cytokines and mediators is often responsible for many of the manifestations of GBS diseases and can lead to detrimental outcomes in neonates and immunocompromised adults even after antibiotic treatment. Both heat-killed GBS and GBS-F induce neuronal apoptosis that contributes substantially to the serious morbidity associated with neonatal meningitis. This neuronal cell death is mediated by nitric oxide (NO) produced in microglia through a TLR2 and myeloid differentiation factor 88 (MyD88)-dependent pathway in response to GBS (18). In neonates with early onset GBS sepsis, plasma levels of proinflammatory cytokines are extremely elevated, and TNFα appears to have a major pathophysiologic role in mediating lethality (30–33). IL-1β, IL-6, and IL-18 have been demonstrated to play a pathophysiologic role in septic arthritis in neonates with late onset GBS infection and in immunocompromised adults with GBS infection (34, 35). GBS induces expression of these proinflammatory cytokines in macrophages through multiple TLRs (including TLR2, TLR6, TLR9, and yet to be identified TLRs) that are dependent for signal transduction through MyD88 (14, 17, 20). These findings indicate that under certain conditions of GBS infection, PRRs, especially MyD88-dependent TLRs such as TLR2, TLR6 and TLR9, contribute to an excessive and lethal cytokine storm, and that in addition to antibiotics treatment, inhibition of signal transduction of these molecules may improve the outcome of GBS diseases.
Previously, we reported that protein kinase D1 (PKD1), a serine/threonine kinase, is activated in an MyD88-dependent manner in innate immune cells in response to stimulation with various TLR ligands, IL-1β, and IL-18 in humans and mice, and plays a critical role in MyD88-mediated proinflammatory cytokine and chemokine production (36–38). These findings suggest a possibility that GBS may activate PKD1, and PKD1 may play a key regulatory role in GBS-induced proinflammatory responses. In the present study, we investigated whether live or antibiotic-killed GBS induce activation of PKD1 in an MyD88-dependent manner in macrophages, and whether PKD1 plays a biologic role in GBS-mediated proinflammatory responses in vitro and in vivo.
MATERIALS AND METHODS
Mice
C57BL/6 mice were obtained from The Frederick Cancer Research and Development Center, National Cancer Institute (Frederick, MD). MyD88 gene-deficient mice (MyD88−/−) and TLR9 gene-deficient mice (TLR9−/−) were provided by Dr. S. Akira (Osaka University, Osaka, Japan). MyD88−/− mice and TLR9−/− on a C57BL/6 background were bred at the University of Tennessee Health Science Center and were in at least the 9th generation backcross. All animal care and housing requirements set forth by the National Institutes of Health Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources were followed, and animal protocols were reviewed and approved by the University of Tennessee Animal Care and Use Committee.
Bacteria
A type Ia GBS isolated from a neonatal patient with sepsis (39) was used for the study. Serotyping was performed by the Centers for Disease Control and Prevention (Atlanta, GA). The bacteria were grown at 37°C in trypticase soy broth (Becton Dickinson and Co., Sparks, MD). Mid-log phase (OD600= 0.5) bacteria were washed three times in endotoxin-free phosphate buffered saline and the concentration was determined by colony counts. Final concentrations of 106 – 108 cfu/ml were used in experiments. Antibiotic-killed GBS were prepared by incubating GBS in the presence of 37.88 mg/1010 cfu/ml penicillin G for 6 h at 37°C. The concentration of antibiotic-killed GBS was given as the number of bacteria equivalent to cfu, determined immediately before antibiotic treatment (as determined on blood agar plate colony counting after overnight culture).
Isolation of murine peritoneal macrophages, cell lines, and culture conditions
Peritoneal macrophages were isolated as described (38). RAW264.7 cells (ATCC, Rockville, MD) were maintained in DMEM supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in a 5% CO2 humidified incubator. Human monocyte cell line THP1 cells (ATCC, Rockville, MD) were maintained in RPMI-1640 supplemented with 10% (v/v) heat-inactivated FBS, 2 mM L-glutamine, 25 mM HEPES, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in a 5% CO2 humidified incubator. For the induction of THP1 cell differentiation to macrophage, THP1 cells (5 × 105 cells/2 ml/well in 12-well plate) were incubated in the presence of PMA (20 ng/ml) for 72 h and then the medium was replaced with fresh medium without PMA. Twenty four hr later the cells were used for the designated experiments. Generation of specific gene knockdown RAW264.7 cells (Luciferase-shRNA, IRAK1-shRNA, TRAF6-shRNA, and PKD1-shRNA) and FLAG-tagged PKD-expressing RAW264.7 cells were previously described (37, 38, 40). For experiments with short-term culture (less than 2hr), cells were cultured in antibiotic-free DMEM supplemented with 10% heat-inactivated FBS and 2 mM L-glutamine. All culture reagents were purchased from Invitrogen (Gaithersburg, MD) and Sigma Chemical Co. (St. Louis, MO).
Reagents
Nuclease-resistant phosphorothioate oligodeoxynucleotides (S-ODN) 1826 (CpG DNA) were purchased from Coley Pharmaceutical Group (Kanata, ON, Canada) and further purified by ethanol precipitation. S-ODN had no detectable endotoxins by Limulus assay. The sequences of S-ODN used have been previously reported (41). Peptidoglycan (PGN) was purchased from Invivogen (San Diego, CA). PMA, ionomycin, and D-galactosamine (D-GalN) were purchased from Sigma Chemical Co. (St. Louis, MO). Gö6976, Gö6983, and CRT0066101 were purchased from Tocris (Minneapolis, MN). IRAK inhibitor 1 (IRAK4 inhibitor; 6-imidazo[1,2-a]pyridin-3-yl-N-piperidin-4-ylpyridin-2-amine) was purchased from APExBIO (Houston, TX).
In vivo experimental protocol
To investigate role of PKD on antibiotic-killed GBS-mediated proinflammatory responses, C57BL/6 mice were injected intraperitoneally (i.p.) with vehicle (7.6% v/v DMSO in PBS), PKC inhibitor Gö6983 (2.3 mg/kg) or a PKD inhibitor Gö6976 (2.3 mg/kg) 4 h and 1 h before the antibiotic-killed GBS (2 × 108 cfu/mouse) challenge. Two hr later, blood and spleen samples were obtained to prepare serum, cell extracts, and total RNA. To investigate role of TLR signaling modulators on GBS induced shock-mediated death of D-GalN-sensitized mice, mice (C57BL/6, MyD88−/−, or TLR9−/−) were challenged with the antibiotic-killed GBS (2 × 108 cfu/mouse) plus D-GalN (30 mg) by i.p. injection. In some experiments, C57BL/6 mice were injected i.p. with vehicle, Gö6983 or Gö6976 4 h and 1 h before and 2 h after the antibiotic-killed GBS plus D-GalN challenge. Fifteen mg of penicillin G was injected daily for the first 3 days to ensure complete killing of GBS. Viability of mice was observed up to 8 days.
Preparation of whole cell lysates and Western blot analysis
Whole cell lysates were prepared from RAW264.7 cells or whole spleen cells as previously described (42). To detect the presence or phosphorylation status of specific proteins in whole cell extracts, equal amounts of whole cell lysates were subjected to electrophoresis on a 10% polyacrylamide gel containing 0.1% SDS, and then Western blots were performed using specific antibodies, as previously described (42). All phospho-specific Abs were purchased from Cell Signaling (Beverly, MA). Antibodies specific for actin, PKD, IκBα or IκBβ were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
In vitro kinase assays
FLAG-tagged PKD-expressing RAW264.7 cells were stimulated with GBS. Each FLAG-tagged PKD protein in whole cell lysates was immunoprecipitated with anti-FLAG Ab. The resulting immune complexes were subjected to in vitro kinase assay using Syntide-2 (Sigma) as a PKD substrate, as previously described (43).
Cytokine-specific enzyme-linked immunosorbent assay (ELISA)
Levels of selected cytokines in culture supernatant or serum were analyzed by cytokine specific ELISA as described previously (44). All recombinant murine (rm) cytokines, antibodies specific for murine cytokines and recombinant human cytokines were purchased from BD Biosciences (San Diego, CA).
Preparation of DNA-free RNA and RT-PCR
DNA-free total RNA was isolated from RAW264.7 cells or spleens by using the RNeasy Mini Kit (Qiagen Inc., Valencia, CA) following the manufacturer’s protocol. To measure the relative amount of selected gene transcripts, isolated RNA (1 μg from each sample) were reverse transcribed with oligo(dT) primer using Superscript II reverse transcriptase (Moloney murine leukemia virus reverse transcriptase; Invitrogen). One tenth of the cDNA product was then amplified with gene specific primers. Twenty to forty cycles of PCR were conducted. PCR products were separated by 1% agarose gel electrophoresis and visualized. The sequences of RT-PCR primers for mouse genes are previously described (38, 45). The sequences of RT-PCR primers for human genes are: TNFα (F:5′ CCTGTAGCCCATGTTGTAGC3′, R:5′CAAAGTAGACCTGCCCAGAC3′), IL-6 (F:5′ACTCACCTCTTCAGAACGAA3′, R:5′CTCAAACTCCAAAAGACCAG3′), IL-8 (F:5′CACCCCAAATTTATCAAAGA3′, R:5′TCAAAAACTTCTCCACAACC3′), and β-actin (F:5′GTGGGCGCCCCAGGCACCA3′, R:5′CTCCTTAATGTCACGCACGATTTC3′). All primers were purchased from Integrated DNA Technologies, Inc. (Coralville, IA).
Flow cytometric analysis
To analyze cell surface expression of CD86, cells were stained with APC-conjugated rat anti–mouse CD86 or APC-conjugated isotype control. CD86 expression was analyzed with BD FACSAria II flow cytometer (BD Biosciences, San Diego, CA) and FlowJo flow cytometry data analysis software (FlowJo LLC, Ashland, OR). All Abs were purchased from BD Biosciences.
Statistical analysis
All experiments were repeated at least three times before analysis. Data are expressed as the mean ± S.D. of triplicates. Two-tailed Student’s t-test or two-tailed Fisher’s exact test was used to determine statistical significance. Kaplan-Meier survival curves were analyzed using XLSTAT (Addinsoft Co.) software, and statistical significance was determined using log rank test compared with antibiotic-killed GBS/D-GalN control. Statistical differences with p < 0.05, p < 0.005, and p < 0.001 are indicated as *, **, and #, respectively, and considered significant.
RESULTS
Live GBS and antibiotic-killed GBS induce activation of PKD1
We previously found that all TLR ligand, except TLR3 ligand, activate PKD1 in macrophages (37, 38). In addition, previous studies demonstrated that many of the biologic effects of GBS infection or GBS components (e.g., production of proinflammatory cytokines and NO) are mediated through MyD88-dependent signaling transduced from multiple TLRs, including TLR2/6, TLR9, and TLR13 (14, 15, 17–20, 23, 46). These findings led us to investigate whether GBS induce activation of PKD1 in macrophages. RAW264.7 cells (a murine macrophage cell line) and THP1 cells (a human macrophage cell line) were stimulated with either medium alone, PMA, live GBS, penicillin G-killed GBS (antibiotic-killed GBS), CpG DNA (TLR9 ligand) or PGN (TLR2 ligand). Phosphorylation of PKD, which is indicative of this protein’s activation, was detected by western blot. As shown in Figures 1A and 1B, both live GBS and antibiotic-killed GBS induced phosphorylation of PKD comparable to the TLR ligands CpG DNA or PGN. This indicates that both live and antibiotic-killed GBS induce activation of one or more PKD family members. To further identify the member(s) of the PKD family proteins activated by GBS, RAW264.7 cells stably expressing either empty vector, FLAG-tagged PKD1, FLAG-tagged PKD2, or FLAG-tagged PKD3 were stimulated with live GBS, antibiotic-killed GBS or PMA. As demonstrated in Figure 1C, both live and antibiotic-killed GBS induced kinase activity and phosphorylation of PKD1. However, neither live nor antibiotic-killed GBS induced kinase activity and phosphorylation of either PKD2 or PKD3. Of note, positive control PMA induced both kinase activity and phosphorylation of all three family members of PKD. This result indicates that the phosphorylated form of PKD detected after GBS stimulation in macrophages is PKD1.
Figure 1. Live GBS and antibiotic-killed GBS induce activation of PKD1.
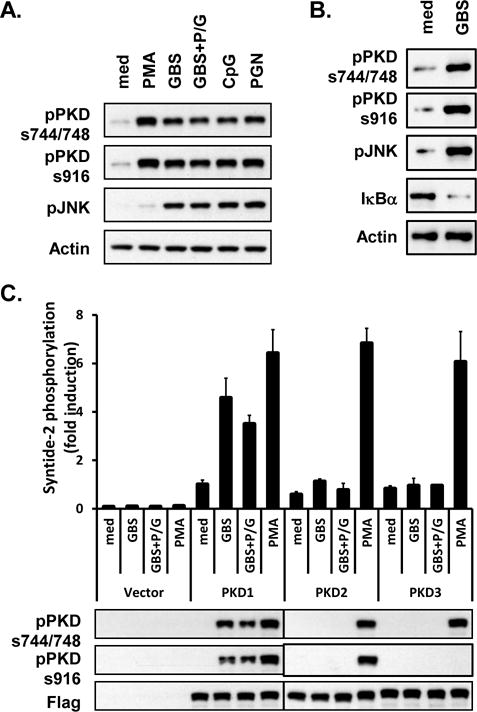
(A) RAW264.7 cells were stimulated with medium (med), PMA (10 ng/ml), live GBS (108 cfu/ml; GBS), antibiotic-killed GBS (108 cfu of GBS were treated with 1 mg of penicillin G for 6 h; GBS+P/G), CpG DNA (12 μg/ml), or PGN (1 μg/ml) for 45 min. (B) THP1 cells were stimulated with medium (med) or live GBS (1 × 108 cfu/ml; GBS) for 1 hr. Phosphorylation status of PKD (pPKDs744/748, pPKDs916) and JNK (pJNK), and protein levels of IκBα were detected by Western blot assay. Actin was used as a loading control. (C) RAW264.7 cells stably expressing empty vector, FLAG-tagged PKD1, FLAG-tagged PKD2, or FLAG-tagged PKD3 were stimulated with medium, live GBS or antibiotic-killed GBS for 45 min. Lysates were prepared and immunoprecipitated with anti-FLAG Ab. Kinase activity of PKD was analyzed in vitro using syntide-2 as a PKD substrate (Top). Expression and phosphorylation status of PKD were analyzed by immunoblotting with anti-FLAG or anti-phospho-PKD Abs (Bottom). All experiments were repeated at least three times with similar results.
GBS induce activation of PKD1 via a MyD88-dependent pathway
We next examined whether the activation of PKD1 induced by live and/or antibiotic-killed GBS that we observed in Figure 1 occurs in a MyD88-dependent manner. Primary peritoneal macrophages isolated from wild-type or MyD88 gene-deficient (MyD88−/−) mice were stimulated with either live or antibiotic-killed GBS, and phosphorylation of PKD was detected as an indication of PKD1 activation. As expected, both live and antibiotic-killed GBS, as well as PMA, CpG DNA, and PGN, induced phosphorylation of PKD1 in primary peritoneal macrophages isolated from wild-type mice (Fig. 2). In contrast, PKD1 activation in response to live GBS or antibiotic-killed GBS was completely abolished in primary peritoneal macrophages isolated from MyD88−/− mice. Ligands for MyD88-dependent TLRs (PGN and CpG DNA, ligands for TLR2 and TLR9 respectively) also failed to induce PKD1 activation in MyD88−/− macrophages, while PMA, which activates PKD1 through MyD88-independent pathway, induced activation of PKD in both wild-type and MyD88−/− macrophages at comparable level. These results provide direct evidence that GBS, as well as TLR ligands, induce activation of PKD1 through a MyD88-dependent signaling pathway.
Figure 2. Live GBS and antibiotic-killed GBS induce activation of PKD1 through an MyD88-dependent manner.
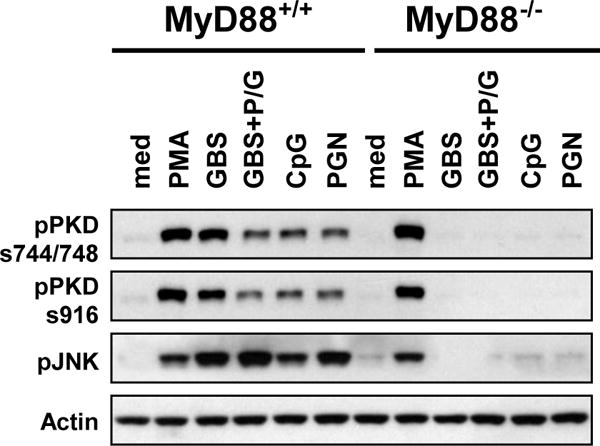
Peritoneal macrophages isolated from wild-type (WT) or MyD88−/− mice were stimulated with medium (med), PMA (10 ng/ml), CpG DNA (12 μg/ml), PGN (1 μg/ml), live GBS (108 cfu/ml), or antibiotic-killed GBS (GBS+P/G; 108 cfu of GBS were treated with 1 mg of penicillin G for 6 h) for 45 min. Activation status of PKD1 and JNK was detected by phospho-specific Western blot assay. Actin was used as a loading control. Data represent results obtained from three separate experiments.
Activation of PKD1 by live GBS or antibiotic-killed GBS requires IRAK1, but not TRAF6
We have previously demonstrated that PKD1 interacts with IRAK4, IRAK1, and TRAF6 upon TLR ligand stimulation (38) and activation of PKD1 by TLR ligands is dependent on IRAK4 and IRAK1 (37). Given that our current results indicate activation of PKD1 by live or antibiotic-killed GBS is dependent on MyD88 (Fig. 2), we further investigated whether PKD1 activation by live and/or antibiotic-killed GBS is also dependent on MyD88 downstream molecule IRAK1 and/or TRAF6. Control (luciferase-knockdown), IRAK1-knockdown, or TRAF6-knockdown macrophages were stimulated with either live or antibiotic-killed GBS. As shown in Figure 3, we found that both live GBS and antibiotic-killed GBS failed to induce activation of PKD1, as measured by phosphorylation of PKD, in IRAK1-knockdown macrophages. In contrast, PKD1 activation by these GBS was not inhibited in TRAF6-knockdown macrophages. Expression of TLRs, MyD88, and PKD1 was normal in IRAK1-knockdown macrophages and TRAF6-knockdown macrophages (37, 38). Activation of JNK (one of IRAK1- and TRAF6-downstream effectors in the MyD88-dependent TLR signaling) by GBS was completely inhibited in both IRAK1-knockdown macrophages and TRAF6-knockdown macrophages, validating effectiveness of the target gene knockdown. Of note, PMA-mediated PKD activation in IRAK1-knockdown or TRAF6-knockdown macrophages was comparable with that in the control luciferase-knockdown macrophages. These results demonstrate that IRAK1, but not TRAF6, is necessary for PKD1 activation by GBS.
Figure 3. Activation of PKD1 induced by GBS is dependent on IRAK1, but independent of TRAF6.
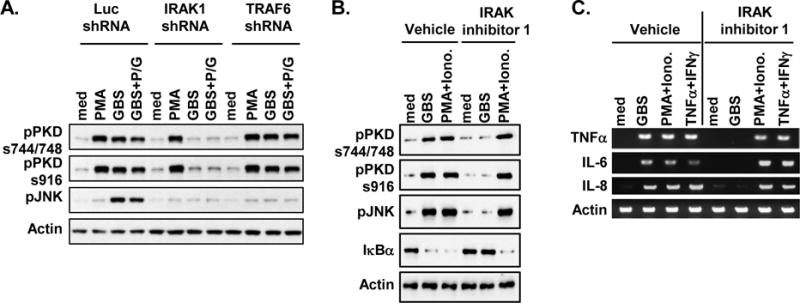
(A) Control luciferase-knockdown (Luc-shRNA), IRAK1-knockdown (IRAK1-shRNA) or TRAF6-knockdown (TRAF6-shRNA) RAW264.7 cells were stimulated with medium, PMA (10 ng/ml), live GBS (108 cfu/ml), or antibiotic-killed GBS (GBS+P/G; 108 cfu of GBS were treated with 1 mg of penicillin G for 6 h) for 45 min. (B, C) THP1 cells were pretreated with vehicle (0.5% v/v DMSO) or IRAK inhibitor 1 (5 μM) for 1 hr and then stimulated with medium, GBS (1 × 108 cfu/ml), or PMA (10 ng/ml) plus Ionomycin (10 ng/ml) for 1 hr (B) or 4 hr (C). Phosphorylation status of PKD (pPKDs744/748, pPKDs916) and JNK (pJNK), and protein levels of IκBα were detected by Western blot assay (A, B). Total RNA was isolated and mRNA levels of the indicated cytokines were analyzed by RT-PCR (C). Actin was used as a loading control. Data represent results obtained from three separate experiments.
To further verify that GBS also activate PKD1 via the same signaling pathway in human cells, we stimulated THP1 cells with GBS in the presence or absence of a specific pharmacological IRAK4 inhibitor, IRAK inhibitor 1, that inhibits kinase activity of IRAK4 (47, 48). As demonstrated in Figure 3B and 3C, GBS failed to activate PKD1 and its downstream effector JNK and NF-κB (judged by degradation of IκBα) in THP1 cells pretreated with IRAK inhibitor 1. In addition, GBS-induced expression of proinflammatory cytokines, TNFα, IL-6, and IL-8, was substantially inhibited in THP1 cells by IRAK inhibitor 1. Our results indicate that GBS induced PKD1 activation via an IRAK-dependent manner in human macrophages and kinase activity of IRAK4 is required for GBS-mediated PKD1 activation. These results also indicate that as in murine macrophages, GBS activate PKD1 through a TLR/MyD88-dependet pathway in human macrophages. Of note, IRAK inhibitor 1 did not suppress activation of PKD and JNK, degradation of IκBα, and expression of proinflammatory cytokines induced by PMA plus ionomycin or TNFα plus IFNγ in THP1 cells. These results show that IRAK inhibitor 1 is specific and not toxic to THP1 cells at the concentration we used in our study. Taken together, our results demonstrate that GBS induce activation of PKD1 through IRAK-dependent pathway, and that IRAK1, but not TRAF6, is necessary for PKD1 activation by GBS.
PKD1 plays an essential role in proinflammatory responses induced by GBS
Although a pharmacological inhibitor that specifically inhibits only PKD1 has yet to be developed, careful use of several pharmacological inhibitors can provide insight to the biologic role of PKD1. We previously found that Gö6976 selectively inhibits TLR ligand-mediated PKD1 activation in vitro and in vivo, while Gö6983 has no effect (36–38). In addition, a newly developed PKD inhibitor CRT0066101 has been demonstrated to selectively and effectively inhibit PKD1 and PKD2 in vitro and in vivo (49). To investigate the biologic role of GBS-activated PKD1, THP1 cells and RAW264.7 cells were pretreated with either vehicle, a PKD inhibitor (Gö6976 or CRT0066101) or a PKC inhibitor (Gö6983), followed by stimulation with GBS, after which activation of JNK, degradation of IκBα, and expression and production of the selected proinflammatory cytokines was measured by ELISA. Figure 4 shows that GBS induced activation of PKD1, JNK, and NF-κB, and expression and production of proinflammatory cytokines in THP1 cells and RAW264.7 cells. As demonstrated in Figure 4A and 4B, Gö6976 and CRT0066101 inhibited GBS-mediated activation of PKD1, JNK, and NF-κB, and expression of proinflammatory cytokines (TNFα, IL-6, and IL-8) in THP1 cells. In addition, production of proinflammatory cytokines (TNFα, IL-6, and IL-12) in response to GBS was significantly inhibited in the presence of PKD inhibitor Gö6976 (Fig. 4C–E) or CRT0066101 (Fig. 4F). In contrast, PKC inhibitor Gö6983 did not affect activation of PKD1, JNK, and NF-κB, and expression and production of those proinflammatory cytokines in response to GBS in both THP1 cells and RAW264.7 cells (Fig. 4A–E). Of note, TNFα plus IFNγ-induced proinflammatory cytokine expression was not affected by Gö6976 or CRT0066101 at the concentration used in THP1 cells (Fig. 4B). IFNγ-induced TNFα production was not affected by CRT0066101 at the concentration used in RAW264.7 cells (Fig. 4F). These results suggest that PKD1 plays a critical role in GBS-induced production of cytokines in both murine and human macrophages.
Figure 4. Effect of pharmacological PKD inhibitors on GBS-mediated activation of JNK and NF-κB and subsequent expression of proinflammatory cytokines in human and murine macrophages in vitro.

(A, B) THP1 cells were pretreated with vehicle (0.5% v/v DMSO), Gö6976 (1 μM) or Gö6983 (1 μM) or CRT0066101 (5 μM) for 1 hr and then stimulated with medium, GBS (1 × 108 cfu/ml) or TNFα (25 ng/ml) plus IFNγ (50 ng/ml) for 1 hr (A) or 4 hr (B). (A) Cell lysates were prepared. Phosphorylation status of PKD (pPKDs744/748, pPKDs916) and JNK (pJNK), and protein levels of IκBα were detected by Western blot assay. (B) Total RNA was isolated and mRNA levels of the indicated cytokines were analyzed by RT-PCR. Actin was used as a loading control. (C–F) RAW264.7 cells (1 × 106 cells/ml) were pretreated with vehicle (0.5% v/v DMSO), Gö6976 (0.66 μM) or Gö6983 (0.57 μM) for 30 min and then stimulated with medium or GBS (106 or 107 cfu/ml) for 24 hr in the complete DMEM supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin. (D) RAW264.7 cells (1 × 106 cells/ml) were pretreated with vehicle (0.5% v/v DMSO) or CRT0066101 (1 μM) for 30 min and then stimulated with medium, GBS (5 × 106 cfu/ml) or IFNγ (10 ng/ml) for 24 hr in the complete DMEM supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin. Levels of the selected cytokines in culture supernatants were analyzed by ELISA. Data represent the mean concentration (pg/ml) ± SD of quadruplicates. *, p < 0.05; **, p < 0.005; #, p < 0.0005. All experiments were repeated at least three times with similar results.
To further confirm the findings with pharmacological PKD inhibitors that PKD1 is required for GBS-mediated macrophage activation, we investigated GBS-mediated inflammatory response in PKD1-knockdown macrophages. Specificity and efficacy of PKD1-knockdown macrophages have previously been reported (37, 38). We confirmed this in Figure 5A, by demonstrating that expression of other PKD family members was comparable to that in control Luc-shRNA cells, while expression of PKD1 (at both mRNA and protein levels) was substantially suppressed in PKD1-knockdown macrophages. Control (luciferase-knockdown) or PKD1-knockdown macrophages were stimulated with GBS. As expected, GBS induced phosphorylation of PKD in control luciferase-knockdown macrophages (Fig. 5B). In contrast, GBS failed to induce phosphorylation of PKD in PKD1-knockdown macrophages. In addition, GBS-mediated activation of MAPK JNK and IKKα/β is substantially inhibited in PKD1-knockdown macrophages (Fig. 5B). GBS-mediated activation of MAPK p38 is also partially inhibited in PKD1-knockdown macrophages. Furthermore, production of cytokine (TNFα) and costimulatory factor CD86 in response to GBS was significantly inhibited in PKD1-knockdown macrophages compared to that in control luciferase-knockdown macrophages (Fig. 5C and 5D). Of note, levels of IFNγ-mediated TNFα production in PKD1-knockdown macrophages were comparable with those in control macrophages. Collectively, these results demonstrated that PKD1 plays an essential role in GBS-mediated activation of MAPKs and IKKα/β (indication of NF-κB activation), and subsequent production of cytokines and costimulatory factors.
Figure 5. Activation of MAPKs and IKKα/β and production of proinflammatory cytokines induced by GBS is dependent on PKD1 in vitro.

(A) Messenger RNA levels of PKD family members and protein levels of PKD1 in control luciferase-knockdown RAW264.7 cells (Luc-shRNA) and PKD1-knockdown RAW264.7 cells (PKD1-shRNA) were analyzed by RT-PCR and Western blot (WB) assay, respectively. GAPDH and Actin were used as loading controls. (B) (Left panel) Control luciferase-knockdown or PKD1-knockdown RAW264.7 cells were stimulated with medium, live GBS (5 × 107 cfu/ml) or IFNγ (10 ng/ml) for 45 min. Phosphorylation status or expression levels of the indicated proteins were detected by Western blot assay. Actin was used as a loading control. (Right panel) Quantitation of phosphor-p38 in Western blots by densitometry. The density of phosphor-p38 band in each sample was quantitated three times by densitometry and normalized to the density of the actin band in the same sample. Data represent the mean (fold induction from the normalized densitometric value of phosphor-p38 band of the unstimulated Luc-shRNA control sample) ± S.D. from two separate experiments. (C) Control luciferase-knockdown or PKD1-knockdown RAW264.7 cells (1 × 106 cells/ml) were stimulated with medium, GBS (5 × 106 cfu/ml) or IFNγ (10 ng/ml) for 24 hr in the complete DMEM supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin. Levels of TNFα in culture supernatants were analyzed by ELISA. Data represent the mean concentration (pg/ml) ± SD of triplicates. *, p < 0.05. (D) Control luciferase-knockdown or PKD1-knockdown RAW264.7 cells were stimulated with medium, GBS (108 cfu/ml) for 24 hr in the complete DMEM supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin. Surface expression levels of CD86 were detected by flow cytometric analysis. All experiments were repeated two to four times with similar results.
Inhibition of PKD1 activation in mice prior to exposure to antibiotic-killed GBS results in decreased expression of proinflammatory cytokines/chemokines and protects D-galactosamine (D-GalN)-sensitized mice from shock-mediated death
To investigate whether activation of signaling modulators and subsequent expression of cytokines and chemokines in response to antibiotic-killed GBS in vivo are also dependent on PKD1 activation, C57BL/6 mice were challenged with antibiotic-killed GBS in the presence or absence of a pharmacological PKD inhibitor Gö6976 or PKC inhibitor Gö6983. Administration of antibiotic-killed GBS to C57BL/6 mice induced activation of PKD1, MAPKs (JNK, p38, and ERK), degradation of IκBα and IκBβ (Fig. 6A and 6B), and expression of various inflammatory cytokines and chemokines (e.g., TNFα, IL-6, IL-10, IL-12p40, IP-10, MCP-1, CCL5, MIP-1β, MIP-2, LIX, and KC) (Fig. 6C and 6D) in spleen cells in vivo. In addition, serum levels of cytokines in these mice were increased following antibiotic-killed GBS challenge (Fig. 6E and 6F). In contrast, antibiotic-killed GBS-mediated activation of PKD1, JNK and ERK, and degradation of IκBα and IκBβ were substantially inhibited in the presence of a PKD inhibitor (Fig. 6A). Antibiotic-killed GBS-mediated activation of p38 was partially inhibited by Gö6976. As a consequence, expression and production of inflammatory cytokines and chemokines in response to antibiotic-killed GBS were significantly reduced in mice pretreated with PKD inhibitor Gö6976 (Fig. 6C and 6E). In agreement with in vitro response, antibiotic-killed GBS-mediated activation of those signaling modulators and expression and production of proinflammatory cytokines in vivo were not significantly affected by a PKC inhibitor Gö6983 (Fig. 6B, 6D, 6F). These results indicate that PKD1 plays an essential role in GBS-induced inflammatory responses in vivo.
Figure 6. Effects of PKD inhibitor Gö6976 on GBS-mediated activation of MAPKs and NF-κB and expression of cytokines and chemokines in vivo.
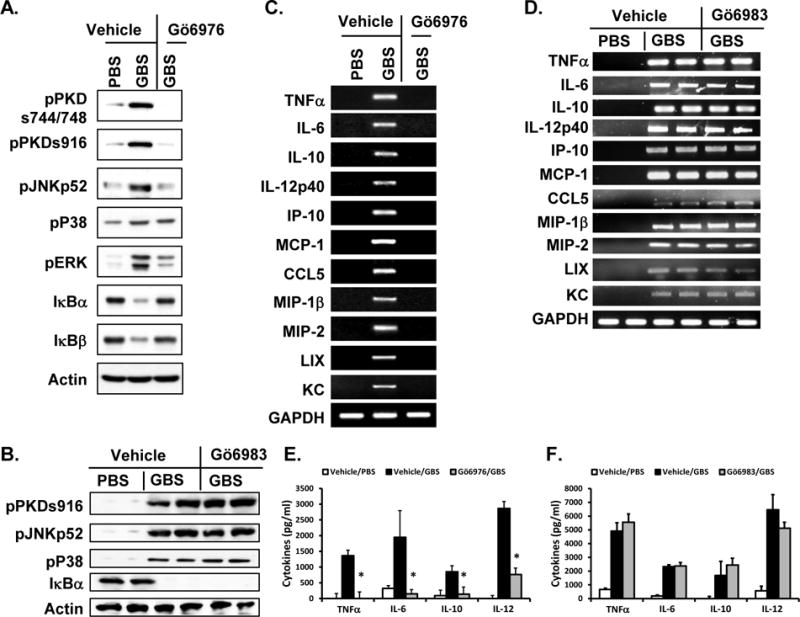
Four h and 1 h prior to antibiotic-killed GBS challenge, C57BL/6 mice (n=3/group for A, C, and E; n=2/group for B, D, and F) were injected i.p. with vehicle, Gö6976, or Gö6983. These mice were challenged with PBS or antibiotic-killed GBS (2 × 108 cfu/mouse) i.p. Two hr later, blood and spleens were harvested. (A, B) Whole spleen lysates were prepared and phosphorylation status or expression levels of the indicated proteins were detected by Western blot assay. Actin was used as a loading control. (C, D) Total RNA was isolated from spleen and mRNA levels of the indicated cytokines and chemokines were analyzed by RT-PCR. GAPDH was used as a loading control. (E, F) Levels of the selected cytokines in serum from each mouse were analyzed by ELISA. Data represent the mean serum cytokine concentration (pg/ml) ± SD of 3 different mice. *, p < 0.05. A, C, and E are representative results from two separate experiments and B, D, and F were done once.
We previously demonstrated that TLR ligands (e.g., LPS and CpG DNA) can induce shock-mediated death caused by overwhelming amounts of proinflammatory cytokines in C57BL/6 mice, and by TNFα in mice sensitized with D-GalN (44, 45, 50). We further investigated whether inhibition of PKD1 activation can prevent shock-mediated death of mice in both experimental models. First, we challenged mice with a lethal dose of E. coli DNA (TLR9 ligand) and LPS (TLR4 ligand) in the presence or absence of a PKD inhibitor Gö6976 or a PKC inhibitor Gö6983. As previously reported (44), 100% of mice died within 24 hr after the sequential challenge with E. coli DNA followed by LPS (Table 1). Treatment with a PKD inhibitor significantly protected mice from shock-mediated death (75% survival rate) induced after the sequential challenge with E. coli DNA followed by LPS. In contrast, PKC inhibitor failed to protect mice from shock-mediated death induced by the sequential challenge with E. coli DNA followed by LPS. Second, we investigated whether antibiotic-killed GBS can induce shock-mediated death in mice sensitized with D-GalN via a MyD88-dependent pathway and whether PKD1 play a critical role in this process. Mice (wild-type, MyD88−/−, or TLR9−/−) were challenged with antibiotic-killed GBS plus D-GalN and then viability of mice was observed over 8 days. We found that antibiotic-killed GBS induced a shock-mediated death of D-GalN-sensitized mice within 24 h after the challenge (Fig. 7). In contrast, antibiotic-killed GBS failed to induce a shock-mediated death of D-GalN-sensitized MyD88−/− mice (Fig. 7A). Deletion of TLR9 also significantly protected D-GalN-sensitized mice from GBS-induced death (Fig. 7A). To further investigate whether inhibition of PKD1 can protect mice from GBS-induced shock-mediated death in D-GalN-sensitized mice, mice pretreated with vehicle, Gö6976 or Gö6983 were challenged with antibiotic-killed GBS plus D-GalN and then viability of mice was observed over 8 days. Systemic administration of PKD inhibitor (Gö6976) significantly protected mice from shock-mediated death caused by lethal challenge with antibiotic-killed GBS and D-GalN (Fig. 7B). In contrast, systemic administration of PKC inhibitor (Gö6983) did not protect mice from shock-mediated death caused by lethal challenge with antibiotic-killed GBS and D-GalN (Fig. 7B). Taken together, these results indicate that TLR/MyD88-dependent PKD1 activation substantially contributes to the proinflammatory response induced by antibiotic-killed GBS. Our results also suggest that PKD1 can be an effective therapeutic target to improve the outcome of GBS diseases after antibiotic treatment.
Table I.
PKD1 play a critical role in shock-mediated death induced by TLR ligands.
| Pre-treatment | Challenge | Dead/Total |
|---|---|---|
| Vehicle | Saline | 0/4 * |
| Vehicle | EC DNA+LPS | 4/4 |
| Gő6976 | EC DNA+LPS | 1/4 |
| Gő6983 | EC DNA+LPS | 4/4 |
C57BL/6 mice (n=4/group) were injected i.p. with vehicle (5% v/v DMSO in PBS), a PKC inhibitor Gö6983 (2.3 mg/kg body weight) or a PKD inhibitor Gö6976 (2.3 mg/kg body weight) 4 h and 1 h before and 2 h after E. Coli DNA treatment. Mice received i.v. injection of E. Coli DNA (EC DNA; 300 μg) and then 4 h later challenged i.v. with a sublethal dose of LPS (100 pg). Viability of mice was observed at 24 h after the LPS challenge. Statistical significance was determined using Two-tailed Fisher’s exact test compared to the vehicle pretreated EC DNA plus LPS challenged control group.
p < 0.05.
Figure 7. MyD88 and PKD1 play a critical role in antibiotic-killed GBS-induced shock-mediated death of D-galactosamine (D-GalN)-sensitized mice.
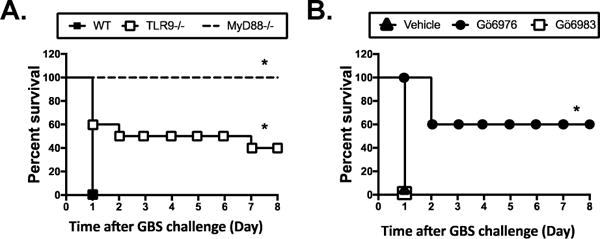
(A) Wilde-type (n=5), MyD88−/− (n=4), or TLR9−/− (n=10) mice were challenged i.p. with antibiotic-killed GBS (2 × 108 cfu) plus D-GalN (30 mg). (B) C57BL/6 mice were injected i.p. with vehicle (n=9), Gö6976 (n=5), or Gö6983 (n=5) at 4 h and 1 h before and 2 h after the GBS challenge and then challenged i.p. with the antibiotic-killed GBS (2 × 108 cfu) plus D-GalN (30 mg). Fifteen mg of penicillin G was injected daily for the first 3 days to ensure complete killing of GBS. Viability of mice was observed up to 8 days. *, p < 0.05.
DISCUSSION
Group B Streptococcus is a common pathogen that can cause sepsis and meningitis related illnesses in newborns. The clinical manifestation of diseases caused by GBS infection is related to the host-pathogen interaction and proinflammatory responses to GBS. The conventional treatment for GBS disease is intravenous administrations of antibiotics to destroy the bacteria. However, even after successful antibiotic treatment, an uncontrolled and exaggerated production of proinflammatory cytokines and mediators, triggered by the host reaction to GBS components released from dying and dead GBS, can lead to detrimental outcomes in neonates and immunocompromised adults. In line with this, we previously found that antibiotic killing of GBS leads to release of GBS components (that are not released by live GBS or heat-killed GBS, e.g., GBS genomic DNA) that are capable of inducing proinflammatory responses in macrophages in vitro (24). Multiple innate immune receptors, including TLRs, are involved in recognition of GBS components and signaling through MyD88-dependent and independent pathways contributes to the proinflammatory responses to GBS components (14–20, 23, 24, 46). A number of previous studies have focused on mechanisms whereby GBS induce various proinflammatory cytokines through TLRs, but not much is known about the role of an MyD88-dependent downstream signaling modulators. Over the last few years, it has become evident that a serine/threonine protein kinase PKD1 plays a significant regulatory role in the inflammatory process mediated by MyD88-dependent TLR and IL-1R family members (36–38, 40, 51, 52). However, it is currently unknown whether GBS induce activation of PKD1 and whether PKD1 contributes to proinflammatory responses to GBS. In the present study, we have demonstrated that GBS in the presence or absence of antibiotics activate PKD1, and PKD1 is one of the critical host signaling modulators for inflammatory reactions to GBS.
Activation of PKD1 by live or antibiotic-killed GBS appears to be mediated through interactions between GBS and TLRs. External and internal components of GBS have been reported to interact with TLRs 2, 6, 7, 9 and 13 (14, 15, 17, 19, 22, 23). Although it is not known whether TLR13 activates PKD1, all other TLRs that utilize MyD88 as a signaling adaptor activate PKD1 (37). In the current study we found that, as in the TLR signaling pathway (37, 38), GBS activates only PKD1 among three PKD family members, and fails to activate PKD1 in MyD88-defficient macrophages, IRAK4 kinase inhibitor-treated macrophages, and IRAK1-knockdown macrophages, while PKD1 activation by GBS is unaffected by silencing TRAF6 expression. Our findings indicate that PKD1 activation by GBS is also dependent on MyD88 and its downstream serine/threonine kinase IRAK1, but independent of TRAF6.
Our studies with pharmacological inhibitors and PKD1-knockdown macrophages demonstrate that PKD1 plays a substantial role in GBS-mediated activation of MAPKs and NF-κB, and proinflammatory gene expression, both in vitro and in vivo. Although our results suggest that Ca++-dependent conventional PKCs and PKCδ and ζ (which can be inhibited by Gö6983) may be dispensable (or less critical) for GBS-mediated activation of MAPKs and NF-κB and subsequent expression of proinflammatory mediators, we cannot completely rule out the possible involvement of other PKC isozymes. Among three MAPKs, JNK, ERK and p38, activation of p38 in response to GBS was least affected by PKD1-knockdown or by pharmacological PKD inhibitors. Compared to this, previous studies demonstrated that a PKD inhibitor Gö6976 significantly, if not completely, inhibits activation of p38 by TLR ligands, including LPS (TLR4 ligand), flagellin (TLR5 ligand) and CpG DNA (TLR9 ligand) (38, 51, 52). These indicate that in addition to TLR signaling pathway there are other signaling pathway(s) that might be activated by GBS and contribute to the p38 activation. In line with this, significant but partial inhibition of TNFα production in macrophages pre-treated with PKD inhibitors or PKD1-knockdown macrophages in response to GBS (especially to a high concentration of GBS) also indicate that there are other signaling pathways (e.g., TLR signaling pathway that leading to expression of type I IFNs and other non-TLR PRR signaling pathways activated by GBS) are involved in proinflammatory response to GBS. We previously found that PKD1 is not involved in TLR-mediated type I IFN expression and inflammasome signaling (37, 38).
As an indication of PKD activation, phosphorylation of two different sites (serine residues at a.a. 744 and 748 and serine residue at a.a. 916 in mouse PKD1) in PKD are often monitored using antibodies specific for the phosphorylated forms of PKD; pPKDs744/748-specific antibody detects phosphorylation of all PKD family proteins and pPKDs916-specific antibody detects phosphorylation of PKD1 and PKD2 (38). It has previously been demonstrated that activation of PKD is controlled by the phosphorylation of two activation loop sites, serine 744 and serine 748 residues (53). Phosphorylation of these serine 744 and serine 748 residues of PKD is absent from unstimulated cells but is induced by phorbol esters, bombesine, BCR ligation, or active PKCε or PKCη (54). Serine 916 is not trans-phosphorylated by an upstream kinase but is rather an autophosphorylation event that occurs following activation of PKD and the degree of serine 916 phosphorylation during lymphocyte activation and inhibition exactly correlated with the activation status of PKD (55). GBS induced phosphorylation of PKD1 at both sites and IRAK4 kinase activity was required for PKD1 phosphorylation at both sites, indicating that GBS components and TLR interaction leads to the activation of upstream kinase, presumably IRAK4, that in turn induces phosphorylation of PKD1 at serine 744/748. Although we demonstrated previously that PKD1 is one of components in TLR signaling receptor complex and that IRAK4 interacts with PKD1 upon TLR and its ligand binding (37, 38), it is still subject for further investigation whether PKD1 is a direct substrate for IRAK4 or there is another yet to be discovered kinase that bridging IRAK4 and PKD1 exist in GBS-mediated PKD1 activation pathway, as well as in the TLR signaling pathway.
PKD can be activated via PKC-dependent or -independent pathways (56, 57). In the case of the PKC-dependent pathway, the phosphorylation of serine residues within an activation loop (S744/748) of PKD is required (56). In contrast, phosphorylation of these serine residues is not required in the PKC-independent activation of PKD (57). Although GBS induces the phosphorylation of serine residues within the activation loop of PKD1 in macrophages, GBS-mediated PKD1 activation appears to be conventional PKC- and PKCδ and ζ-independent as judged by the failure of PKC inhibitor Gö6983 to inhibit activation of PKD1 by GBS. Of note, activation of PKDs by PMA, which occurs in a PKC-dependent manner, is suppressed similarly by either Gö6976 or Gö6983, indicating comparable effectiveness of Gö6976 and Gö6983 on inhibition of conventional PKC isoforms (36), which further support our finding. As mentioned previously, PKD family members are kinases whose activation is differently affected by Gö6976 and Gö6983 (38, 58). Although Gö6976 and CRT0066101 (PKD-specific inhibitor) inhibit activation of all three PKD family members, PKD1 may be the kinase whose activity was inhibited by these PKD inhibitors in macrophages stimulated with GBS, because GBS (both live and antibiotic-killed) activates only PKD1, not PKD2 or PKD3 and silencing PKD1 expression results in ablation of PKD phosphorylation in response to GBS.
Defects in PRR signaling pathway can lead to expansion and ineffective clearance of pathogenic invaders, whereas uncontrolled PRR signaling can lead to excess inflammation and tissue injury. Strong harmful proinflammatory responses of the host can be prolonged even after successful antibiotic killing of bacteria due to bacterial components released from dead bacteria. Different levels of proinflammatory cytokine responses to GBS killed by different class of antibiotics also support this concept (24). Therefore, in addition to antibiotic treatment, a targeted conjunctive therapy that reduces (or blocks) inflammatory responses induced by bacterial components released by dead bacteria can be a useful and desirable therapeutic approach for GBS diseases. In support of this, we found that PKD inhibitors significantly reduce proinflammatory cytokines and chemokine production in response to antibiotic-killed GBS both in vitro and in vivo. PKD inhibitor treatment protected mice from death by septic shock like syndrome caused by overwhelming cytokine storm induced by multiple TLR ligands. Furthermore, PKD inhibitor treatment significantly protected D-GalN-sensitized mice from TNFα-mediated death caused by antibiotic-killed GBS. In addition to early onset sepsis in neonates, serious complications of GBS diseases include meningitis and neuronal cell damage. Activation of microglia, the resident innate immune cells of the central nervous system, is detrimental to neurons and is involved in a number of neurodegenerative diseases. Previous studies demonstrated that GBS activates microglia to generate neurotoxic nitric oxide through a TLR2/MyD88-dependent pathway (18). Interestingly, TLR4 ligand LPS-mediated p38 activation and TNFα secretion in microglial cells are suppressed by a PKD inhibitor Gö6976 (52). Considering the indispensable role of PKD1 in TLR/MyD88-induced expression of proinflammatory cytokines and chemokines (36–38) and our new finding on the indispensable role of PKD1 in proinflammatory responses to both live and antibiotic-killed GBS, PKD1 is an attractive therapeutic target for a conjunctive therapy for GBS diseases, meningitis and neuronal cell damage.
In summary, we have demonstrated that both live GBS and antibiotic-killed GBS induce activation of PKD1 in macrophages through a MyD88- and IRAK1-dependent, but TRAF6-independent, mechanism. Activation of PKD1 is required for activation of MAPKs and transcription factor NF-κB and subsequent expression and production of proinflammatory mediators in response to both live GBS and antibiotic-killed GBS in macrophages in vitro and in vivo. Inhibition of PKD1 activation lead to suppressed inflammatory responses and significantly improved survival of D-GalN-sensitized mice lethally challenged with antibiotic-killed GBS. Collectively, our findings imply that PKD1 is one of the indispensable factors that play a regulatory role in GBS-induced proinflammatory reactions, and that inhibition of PKD1 activation together with antibiotic treatment in GBS-infected neonates could be an effective way to control GBS diseases.
Acknowledgments
We thank Dr. S. Akira (Osaka Univ., Osaka, Japan) for kindly providing TLR9−/− and MyD88−/− mice. We also thank Mrs. Elizabeth Meals for excellent technical assistance on experiments.
K.U., J.E.P. and Y.I.K. were supported by grants from Le Bonheur Children’s Hospital. A.K.Y. was supported by grants from NIH (AI053137, AR064723) and Arthritis Foundation (IRG 5942). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, Arthritis Foundation, the Children’s Foundation of Memphis, Le Bonheur Children’s Hospital, or University of Tennessee Health Science Center.
References
- 1.Manning SD, Neighbors K, Tallman PA, Gillespie B, Marrs CF, Borchardt SM, Baker CJ, Pearlman MD, Foxman B. Prevalence of group B streptococcus colonization and potential for transmission by casual contact in healthy young men and women. Clin Infect Dis. 2004;39:380–388. doi: 10.1086/422321. [DOI] [PubMed] [Google Scholar]
- 2.Krohn MA, Hillier SL, Baker CJ. Maternal peripartum complications associated with vaginal group B streptococci colonization. J Infect Dis. 1999;179:1410–1415. doi: 10.1086/314756. [DOI] [PubMed] [Google Scholar]
- 3.Schrag SJ, Zywicki S, Farley MM, Reingold AL, Harrison LH, Lefkowitz LB, Hadler JL, Danila R, Cieslak PR, Schuchat A. Group B streptococcal disease in the era of intrapartum antibiotic prophylaxis. N Engl J Med. 2000;342:15–20. doi: 10.1056/NEJM200001063420103. [DOI] [PubMed] [Google Scholar]
- 4.Farley MM. Group B streptococcal disease in nonpregnant adults. Clin Infect Dis. 2001;33:556–561. doi: 10.1086/322696. [DOI] [PubMed] [Google Scholar]
- 5.Munoz P, Llancaqueo A, Rodriguez-Creixems M, Pelaez T, Martin L, Bouza E. Group B streptococcus bacteremia in nonpregnant adults. Arch Intern Med. 1997;157:213–216. doi: 10.1001/archinte.1997.00440230087011. [DOI] [PubMed] [Google Scholar]
- 6.Perovic O, Crewe-Brown HH, Khoosal M, Karstaedt AS. Invasive group B streptococcal disease in nonpregnant adults. Eur J Clin Microbiol Infect Dis. 1999;18:362–364. doi: 10.1007/pl00015020. [DOI] [PubMed] [Google Scholar]
- 7.Keenan C. Prevention of neonatal group B streptococcal infection. Am Fam Physician. 1998;57:2713–2720. 2725. [PubMed] [Google Scholar]
- 8.Dan M. Neonatal septic arthritis. Isr J Med Sci. 1983;19:967–971. [PubMed] [Google Scholar]
- 9.Lai TK, Hingston J, Scheifele D. Streptococcal neonatal osteomyelitis. Am J Dis Child. 1980;134:711. doi: 10.1001/archpedi.1980.02130190077025. [DOI] [PubMed] [Google Scholar]
- 10.Schuchat A, Oxtoby M, Cochi S, Sikes RK, Hightower A, Plikaytis B, Broome CV. Population-based risk factors for neonatal group B streptococcal disease: results of a cohort study in metropolitan Atlanta. J Infect Dis. 1990;162:672–677. doi: 10.1093/infdis/162.3.672. [DOI] [PubMed] [Google Scholar]
- 11.Velaphi S, Siegel JD, Wendel GD, Jr, Cushion N, Eid WM, Sanchez PJ. Early-onset group B streptococcal infection after a combined maternal and neonatal group B streptococcal chemoprophylaxis strategy. Pediatrics. 2003;111:541–547. doi: 10.1542/peds.111.3.541. [DOI] [PubMed] [Google Scholar]
- 12.Weisman LE, Stoll BJ, Cruess DF, Hall RT, Merenstein GB, Hemming VG, Fischer GW. Early-onset group B streptococcal sepsis: a current assessment. J Pediatr. 1992;121:428–433. doi: 10.1016/s0022-3476(05)81801-3. [DOI] [PubMed] [Google Scholar]
- 13.Waterer GW. Monotherapy versus combination antimicrobial therapy for pneumococcal pneumonia. Curr Opin Infect Dis. 2005;18:157–163. doi: 10.1097/01.qco.0000160906.02308.3c. [DOI] [PubMed] [Google Scholar]
- 14.Draper DW, Bethea HN, He YW. Toll-like receptor 2-dependent and -independent activation of macrophages by group B streptococci. Immunol Lett. 2006;102:202–214. doi: 10.1016/j.imlet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, Theilacker C, Hubner J, Santos-Sierra S, Teti G, Golenbock DT, Poyart C, Trieu-Cuot P. Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J Immunol. 2008;180:6149–6158. doi: 10.4049/jimmunol.180.9.6149. [DOI] [PubMed] [Google Scholar]
- 16.Henneke P, Morath S, Uematsu S, Weichert S, Pfitzenmaier M, Takeuchi O, Muller A, Poyart C, Akira S, Berner R, Teti G, Geyer A, Hartung T, Trieu-Cuot P, Kasper DL, Golenbock DT. Role of lipoteichoic acid in the phagocyte response to group B streptococcus. J Immunol. 2005;174:6449–6455. doi: 10.4049/jimmunol.174.10.6449. [DOI] [PubMed] [Google Scholar]
- 17.Henneke P, Takeuchi O, van Strijp JA, Guttormsen HK, Smith JA, Schromm AB, Espevik TA, Akira S, Nizet V, Kasper DL, Golenbock DT. Novel engagement of CD14 and multiple toll-like receptors by group B streptococci. J Immunol. 2001;167:7069–7076. doi: 10.4049/jimmunol.167.12.7069. [DOI] [PubMed] [Google Scholar]
- 18.Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- 19.Lehnardt S, Wennekamp J, Freyer D, Liedtke C, Krueger C, Nitsch R, Bechmann I, Weber JR, Henneke P. TLR2 and caspase-8 are essential for group B Streptococcus-induced apoptosis in microglia. J Immunol. 2007;179:6134–6143. doi: 10.4049/jimmunol.179.9.6134. [DOI] [PubMed] [Google Scholar]
- 20.Mancuso G, Midiri A, Beninati C, Biondo C, Galbo R, Akira S, Henneke P, Golenbock D, Teti G. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J Immunol. 2004;172:6324–6329. doi: 10.4049/jimmunol.172.10.6324. [DOI] [PubMed] [Google Scholar]
- 21.Biondo C, Mancuso G, Beninati C, Iaria C, Romeo O, Cascio A, Teti G. The role of endosomal toll-like receptors in bacterial recognition. European review for medical and pharmacological sciences. 2012;16:1506–1512. [PubMed] [Google Scholar]
- 22.Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, Teti G, Beninati C. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol. 2009;10:587–594. doi: 10.1038/ni.1733. [DOI] [PubMed] [Google Scholar]
- 23.Signorino G, Mohammadi N, Patane F, Buscetta M, Venza M, Venza I, Mancuso G, Midiri A, Alexopoulou L, Teti G, Biondo C, Beninati C. Role of Toll-like receptor 13 in innate immune recognition of group B streptococci. Infect Immun. 2014;82:5013–5022. doi: 10.1128/IAI.02282-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talati AJ, Kim HJ, Kim YI, Yi AK, English BK. Role of bacterial DNA in macrophage activation by group B streptococci. Microbes Infect. 2008;10:1106–1113. doi: 10.1016/j.micinf.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Cusumano V, Genovese F, Mancuso G, Carbone M, Fera MT, Teti G. Interleukin-10 protects neonatal mice from lethal group B streptococcal infection. Infect Immun. 1996;64:2850–2852. doi: 10.1128/iai.64.7.2850-2852.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mancuso G, Cusumano V, Cook JA, Smith E, Squadrito F, Blandino G, Teti G. Efficacy of tumor necrosis factor alpha and eicosanoid inhibitors in experimental models of neonatal sepsis. FEMS Immunol Med Microbiol. 1994;9:49–54. doi: 10.1111/j.1574-695X.1994.tb00473.x. [DOI] [PubMed] [Google Scholar]
- 27.Mancuso G, Cusumano V, Genovese F, Gambuzza M, Beninati C, Teti G. Role of interleukin 12 in experimental neonatal sepsis caused by group B streptococci. Infect Immun. 1997;65:3731–3735. doi: 10.1128/iai.65.9.3731-3735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mancuso G, Tomasello F, Migliardo M, Delfino D, Cochran J, Cook JA, Teti G. Beneficial effects of interleukin-6 in neonatal mouse models of group B streptococcal disease. Infect Immun. 1994;62:4997–5002. doi: 10.1128/iai.62.11.4997-5002.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mancuso G, Tomasello F, von Hunolstein C, Orefici G, Teti G. Induction of tumor necrosis factor alpha by the group- and type-specific polysaccharides from type III group B streptococci. Infect Immun. 1994;62:2748–2753. doi: 10.1128/iai.62.7.2748-2753.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berner R, Niemeyer CM, Leititis JU, Funke A, Schwab C, Rau U, Richter K, Tawfeek MS, Clad A, Brandis M. Plasma levels and gene expression of granulocyte colony-stimulating factor, tumor necrosis factor-alpha, interleukin (IL)-1beta, IL-6, IL-8, and soluble intercellular adhesion molecule-1 in neonatal early onset sepsis. Pediatr Res. 1998;44:469–477. doi: 10.1203/00006450-199810000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Berner R, Schumacher RF, Bartelt S, Forster J, Brandis M. Predisposing conditions and pathogens in bacteremia in hospitalized children. Eur J Clin Microbiol Infect Dis. 1998;17:337–340. doi: 10.1007/BF01709456. [DOI] [PubMed] [Google Scholar]
- 32.Givner LB, Gray L, O’Shea TM. Antibodies to tumor necrosis factor-alpha: use as adjunctive therapy in established group B streptococcal disease in newborn rats. Pediatr Res. 1995;38:551–554. doi: 10.1203/00006450-199510000-00013. [DOI] [PubMed] [Google Scholar]
- 33.Teti G, Mancuso G, Tomasello F. Cytokine appearance and effects of anti-tumor necrosis factor alpha antibodies in a neonatal rat model of group B streptococcal infection. Infect Immun. 1993;61:227–235. doi: 10.1128/iai.61.1.227-235.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tissi L, McRae B, Ghayur T, von Hunolstein C, Orefici G, Bistoni F, Puliti M. Role of interleukin-18 in experimental group B streptococcal arthritis. Arthritis Rheum. 2004;50:2005–2013. doi: 10.1002/art.20014. [DOI] [PubMed] [Google Scholar]
- 35.Tissi L, Puliti M, Barluzzi R, Orefici G, von Hunolstein C, Bistoni F. Role of tumor necrosis factor alpha, interleukin-1beta, and interleukin-6 in a mouse model of group B streptococcal arthritis. Infect Immun. 1999;67:4545–4550. doi: 10.1128/iai.67.9.4545-4550.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim YI, Park JE, Brand DD, Fitzpatrick EA, Yi AK. Protein kinase D1 is essential for the proinflammatory response induced by hypersensitivity Pneumonitis-causing thermophilic actinomycetes saccharopolyspora rectivirgula. J Immunol. 2010;184:3145–3156. doi: 10.4049/jimmunol.0903718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JE, Kim Young In, Yi Ae Kyung. Protein kinase D1 is essential for MyD88-dependent TLR signaling pathway. Journal of Immunology. 2009;182:6316–6327. doi: 10.4049/jimmunol.0804239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park JE, Kim YI, Yi AK. Protein kinase D1: a new component in TLR9 signaling. J Immunol. 2008;181:2044–2055. doi: 10.4049/jimmunol.181.3.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brinkmann KC, Talati AJ, Akbari RE, Meals EA, English BK. Group B streptococci exposed to rifampin or clindamycin (versus ampicillin or cefotaxime) stimulate reduced production of inflammatory mediators by murine macrophages. Pediatr Res. 2005;57:419–423. doi: 10.1203/01.PDR.0000153946.97159.79. [DOI] [PubMed] [Google Scholar]
- 40.Kim YI, Park JE, Kwon KH, Hong CY, Yi AK. Interleukin-1 receptor-associated kinase 2- and protein kinase D1-dependent regulation of IRAK-monocyte expression by CpG DNA. PLoS One. 2012;7:e43970. doi: 10.1371/journal.pone.0043970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeo SJ, Gravis D, Yoon JG, Yi AK. Myeloid differentiation factor 88-dependent transcriptional regulation of cyclooxygenase-2 expression by CpG DNA: role of NF-kappaB and p38. J Biol Chem. 2003;278:22563–22573. doi: 10.1074/jbc.M302076200. [DOI] [PubMed] [Google Scholar]
- 42.Yi AK, Krieg AM. Rapid induction of mitogen-activated protein kinases by immune stimulatory CpG DNA. J Immunol. 1998;161:4493–4497. [PubMed] [Google Scholar]
- 43.Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem. 2003;278:154–163. doi: 10.1074/jbc.M208075200. [DOI] [PubMed] [Google Scholar]
- 44.Cowdery JS, Chace JH, Yi AK, Krieg AM. Bacterial DNA induces NK cells to produce IFN-gamma in vivo and increases the toxicity of lipopolysaccharides. J Immunol. 1996;156:4570–4575. [PubMed] [Google Scholar]
- 45.Kim YI, Park JE, Martinez-Hernandez A, Yi AK. CpG DNA prevents liver injury and shock-mediated death by modulating expression of interleukin-1 receptor-associated kinases. J Biol Chem. 2008;283:15258–15270. doi: 10.1074/jbc.M709549200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, Mayadas T, Nizet V, Akira S, Kasper DL, Golenbock DT. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol. 2002;169:3970–3977. doi: 10.4049/jimmunol.169.7.3970. [DOI] [PubMed] [Google Scholar]
- 47.Buckley GM, Ceska TA, Fraser JL, Gowers L, Groom CR, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, Sabin V. IRAK-4 inhibitors. Part II: a structure-based assessment of imidazo[1,2-a]pyridine binding. Bioorg Med Chem Lett. 2008;18:3291–3295. doi: 10.1016/j.bmcl.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 48.Marinho FV, Fahel JS, Scanga CA, Gomes MT, Guimaraes G, Carvalho GR, Morales SV, Bafica A, Oliveira SC. Lack of IL-1 Receptor-Associated Kinase-4 Leads to Defective Th1 Cell Responses and Renders Mice Susceptible to Mycobacterial Infection. J Immunol. 2016;197:1852–1863. doi: 10.4049/jimmunol.1502157. [DOI] [PubMed] [Google Scholar]
- 49.Harikumar KB, Kunnumakkara AB, Ochi N, Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R, Raynham T, Charles M, Bagherzadeh A, Foxton C, Boakes A, Farooq M, Maru D, Diagaradjane P, Matsuo Y, Sinnett-Smith J, Gelovani J, Krishnan S, Aggarwal BB, Rozengurt E, Ireson CR, Guha S. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Molecular cancer therapeutics. 2010;9:1136–1146. doi: 10.1158/1535-7163.MCT-09-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yi AK, Yoon H, Park JE, Kim BS, Kim HJ, Martinez-Hernandez A. CpG DNA-mediated induction of acute liver injury in D-galactosamine-sensitized mice: the mitochondrial apoptotic pathway-dependent death of hepatocytes. J Biol Chem. 2006;281:15001–15012. doi: 10.1074/jbc.M601337200. [DOI] [PubMed] [Google Scholar]
- 51.Ivison SM, Graham NR, Bernales CQ, Kifayet A, Ng N, Shobab LA, Steiner TS. Protein kinase D interaction with TLR5 is required for inflammatory signaling in response to bacterial flagellin. J Immunol. 2007;178:5735–5743. doi: 10.4049/jimmunol.178.9.5735. [DOI] [PubMed] [Google Scholar]
- 52.Jeohn GH, Cooper CL, Jang KJ, Liu B, Lee DS, Kim HC, Hong JS. Go6976 inhibits LPS-induced microglial TNFalpha release by suppressing p38 MAP kinase activation. Neuroscience. 2002;114:689–697. doi: 10.1016/s0306-4522(02)00356-1. [DOI] [PubMed] [Google Scholar]
- 53.Iglesias T, Rozengurt E. Protein kinase D activation by mutations within its pleckstrin homology domain. J Biol Chem. 1998;273:410–416. doi: 10.1074/jbc.273.1.410. [DOI] [PubMed] [Google Scholar]
- 54.Waldron RT, Rey O, Iglesias T, Tugal T, Cantrell D, Rozengurt E. Activation loop Ser744 and Ser748 in protein kinase D are transphosphorylated in vivo. J Biol Chem. 2001;276:32606–32615. doi: 10.1074/jbc.M101648200. [DOI] [PubMed] [Google Scholar]
- 55.Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem. 1999;274:26543–26549. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- 56.Cabrera-Poch N, Sanchez-Ruiloba L, Rodriguez-Martinez M, Iglesias T. Lipid raft disruption triggers protein kinase C and Src-dependent protein kinase D activation and Kidins220 phosphorylation in neuronal cells. J Biol Chem. 2004;279:28592–28602. doi: 10.1074/jbc.M312242200. [DOI] [PubMed] [Google Scholar]
- 57.Lemonnier J, Ghayor C, Guicheux J, Caverzasio J. Protein kinase C-independent activation of protein kinase D is involved in BMP-2-induced activation of stress mitogen-activated protein kinases JNK and p38 and osteoblastic cell differentiation. J Biol Chem. 2004;279:259–264. doi: 10.1074/jbc.M308665200. [DOI] [PubMed] [Google Scholar]
- 58.Chen Y, Khanna S, Goodyear CS, Park YB, Raz E, Thiel S, Gronwall C, Vas J, Boyle DL, Corr M, Kono DH, Silverman GJ. Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis. J Immunol. 2009;183:1346–1359. doi: 10.4049/jimmunol.0900948. [DOI] [PMC free article] [PubMed] [Google Scholar]