

Abstract
Aim
Primary congenital hypothyroidism is frequently seen endocrine disorder and one of the preventable cause of mental retardation. Aim of study was to evaluate the frequency of permanent/transient hypothyrodism, and to detect underlying reason to identfy any marker which carries potential to discriminate permanent/transient form.
Material and Methods
Forty eight cases older than 3 years of age, diagnosed as primary congenital hypothyroidism and started thyroxin therapy in newborn-period, and followed up between January 2007–June 2013 were included in the study. Thyroid hormon levels were evaluated and thyroid ultrasonography was performed in cases who are at the end of their 3 years of age, after 6 weeks of thyroxine free period. Thyroid sintigraphy was performed if serum thyroid-stimulating hormone was high (≥ 5 mIU/mL) and perchlorate discharge test was performed if uptake was normal or increased on sintigraphy. Cases with thyroid-stimulating hormone levels ≥ 5 mIU/mL were defined as permanent primary congenital hypothyroidism group and as transient primary congenital hypothyroidism group with normal thyroid hormones during 6 months.
Results
The mean age was 3.8±0.7 years. Mean diagnosis age was 16.6±6.5 days and 14 cases (29.2%) were diagnosed by screening program of Ministry of Health. There were 23 cases (14F, 9M) in permanent primary congenital hypothyroidism group and 12 (52.2%) of them were dysgenesis (8 hypoplasia, 4 ectopia), and 11 (47.8%) dyshormonogenesis. In transient primary congenital hypothyroidism group, there were 25 cases (17M, 8F). The mean thyroid-stimulating hormone levels at diagnosis were similar in two groups. The mean thyroxin dose in permanent primary congenital hypothyroidism group was significantly higher than transient group at the time of thyroxin cessation (2.1±0.7, 1.5±0.5 mg/kg/d, respectively, p=0.004). Thyroxin dose ≥1.6 mcg/kg/d was 72% sensitive and 69.6% specific for predicting permenant primary congenital hypothyroidism.
Conclusions
Transient primary congenital hypothyroidism is more frequent than expected and found often in males in the primary congenital hypothyroidism cases, started thyroxin therapy in neonatal period. While fT4, thyroid-stimulating hormone, Tg levels at diagnosis do not predict transient/permenant primary congenital hypothyroidism, thyroxin dose before the therapy cessation at the age of 3 may make the distinction between transient/permenant primary congenital hypothyroidism.
Keywords: Primary congenital hypothyroidism, etiology, thyroxin dose
Introduction
Primary congenital hypothyroidism (PCH) is one of the most common causes of preventable mental retardation and occurs in 1/2000–1/4000 births in newborn babies (1–3). Thyroid hormones are essential for development and function of the brain because they are involved in neuron formation and migration, formation of axons and dendrites, myelinization, development of synapses, and regulation of specific neurotransmitters in the fetal and postnatal periods (4). Therefore, PCH causes permanent mental retardation if the diagnosis is delayed and treatment is not administered (1, 2).
When primary congenital hypothyroidism is evaluated in terms of the underlying cause and the disease period, it is divided into two subgroups as permanent and transient PCH. Permanent PCH occurs as a result of a developmental defect of the thyroid gland (thyroid dysgenesis) or thyroid hormone production defect (dyshormogenesis), and thyroid hormone replacement treatment is administered lifelong (3). The most common cause of PCH is thyroid dysgenesis (70–80%), and this is followed by dyshormogenesis (20–30%) (2, 3). Transient primary congenital hypothyroidism is a condition characterized by normalization of thyroid hormone deficiency, which is noted at birth, within a few months, and years. The causes of transient PCH include iodine deficiency, prenatal-perinatal iodine load, antibodies inhibiting maternal thyroid-stimulating hormone (TSH) receptors that pass the placenta, exposure of the mother to radioiodine or antithyroid drugs, and transient organogenesis defects (THOX2 or DUOXA2 heterozygous mutations) (1–3, 5). Although studies have reported different prevalences for transient PCH, it is about 30–50% (5, 6). According to the general opinion, subjects with transient PCH should be treated until the age of 3 years, though there are cases where treatment can be terminated earlier because transient thyroid hormone failure is in question (2). The rate of subjects diagnosed based on clinical findings is 5% because the diagnostic clinical findings in primary congenital hypothyroidism generally appear after the sixth postnatal week (2, 7, 8). PCH should be diagnosed and thyroxine replacement treatment should be initiated in the first two weeks to prevent mental retardation (1, 9). Therefore, screening programs were initiated throughout the world with the objective of early diagnosis (2, 10–12). In our country, PCH has been included in the screening program by the Ministry of Health since December 2006 (13). Investigations directed to the underlying cause are postponed until the age of three years when central nervous system myelinization is completed to a great extent, because treatment is usually initiated without investigations directed to the etiologic cause in cases of primary congenital hypothyroidism (1, 2). In cases of primary congenital hypothyroidism, thyroxine treatment is discontinued for 4–6 weeks after the age of three years and a diagnosis of permanent or transient PCH is made by detecting the underlying cause with investigations directed to the etiology. Thus, treatment is terminated confidently in subjects with a diagnosis of transient PCH and hereditary characteristics and prognosis of the subjects with a diagnosis of permanent PCH can be enlightened (5, 6, 14). By means of this differential diagnosis, unnecessary long-term follow-up and treatment is prevented in children with transient PCH and an appropriate way is specified for the regulation and monitoring of treatment in patients with permanent PCH.
In this study, we aimed to determine the prevalence of permanent and transient PCH in subjects with a diagnosis of PCH were under follow up in our clinic, to determine the etiology in children with permanent PCH, and specify laboratory findings that might be predictive markers for permanent/transient PCH.
Material and Methods
Fourty-eight subjects who had recently passed three years of age who were under follow up in Trakya University, Faculty of Medicine, Division of Pediatric Endocrinology Outpatient Clinic between January 2007 and June 2013, who were diagnosed as having PCH in the neonatal period, and who started to receive thyroxine treatment were included in the study. Information related with the current ages, ages at the time of diagnosis, sexes, presence of consanguineous marriage, method of diagnosis (in peripheral blood with neonatal screening test or incidentally), free thryoxine (fT4), TSH levels at the time of diagnosis, and initial thyroxine doses were obtained retrospectively from hospital files and recorded. In the subjects in whom thyroxine treatment was discontinued for six weeks at the age of three years, sT4, TSH, thyroglobulin (Tg) and thyroglobulin antibody (anti-Tg) levels were measured at the end of this period. Thyroid ultrasonography (USG) was performed in all subjects. Thyroid scintigraphy was performed in subjects in whom the TSH level was high (≥5 mIU/mL). Perchlorate discharge test was performed in subjects in whom scintigraphy revealed normal or increased iodine uptake. Subjects who had increased TSH level (≥5 mIU/mL) at the end of the six-week period during which treatment was discontinued were considered to have permanent PCH, and subjects who had normal fT4 and TSH (<5 mIU/mL) levels were considered as having transient PCH.
Serum fT4, TSH, Tg and AntiTg levels were measured using a Beckman Coulter Unicel DxI 800 (2004, USA) device with radioimmunoassasy kits. Thyroid USG was performed using an Esaote MyLab60 XVision device and thyroid volumes were calculated based on the study of Kurtoğlu et al. (15). Thyroid scintigraphy was performed using a Medical Imaging Electronics Orbiter Digi 37 with technetium 99 pertechnetate (Tc99). A perchlorate discharge test was performed using a Biodex Atolamb 950 spectrometer and a ≥20% reduction in I-131 uptake in the second hour after sodium perchlorate administration was considered positive (14).
Statistical analysis
Mean, standard deviation, percentage, median and quertile values were calculated as descriptive statistics related with the data. The compatibility of the quantitative variables to normal distribution was tested using the one sample Kolmogorov-Smirnov test. Comparisons between the groups were performed using the Mann-Whitney U test because the variables were not distributed normally. Pearson’s Chi-square test was used in the comparison of the categorical data between the groups. In the differentiation of permanent and transient PCH, ROC analysis was used to determine the cut-off value of thyroxine dose at the time of treatment discontinuation as a prediction criterion and sensitivity and specificity values were calculated for this cutoff value. The power of the study was calculated to as 94.2% with post-hoc power analysis based on the thyroxine dose at the time of treatment discontinuation with an area under the curve (AUC) value of 0.745, with a magrin of error of α=0.05. IBM SPSS (IBM Statistical Package for the Social Sciences; Armonk, NY, USA) 20.0, Medcalc and NCSS statistical package programs were used in the analyses of the data. A p value of <0.05 was considered statistically significant.
Ethics committee approval was obtained for the study (Protocol number: TÜTF-TÜBAPK 2012–151, date: 28.06.2012).
Results
Twenty-eight (54.2%) of 48 subjects included in the study were male, 22 (45.8%) were female, and the female/male ratio was 1:1.1. The mean age at the time of the study was 3.8±0.7 years. The mean age at the time of diagnosis was 16.6±6.5 days. Twenty-two subjects’ (45.8%) PCH was diagnosed in the first 14 days of life, and 26 (54.2%) were diagnosed as having PCH between the 15th and 30th days. The diagnosis was made using the Ministry of Health Screening Program in 14 subjects (29.2%). Twenty-six (54.2%) subjects’ PCH was diagnosed when peripheral blood samples were obtained during investigations performed because of prolonged jaundice. Eight subjects’ (16.6%) PCH was diagnosed incidentally. None of the subjects had a family history of consanguineous marriage. At the time of diagnosis, the mean TSH level was 43.1±43.6 mIU/mL and the mean fT4 level was found as 0.98±0.32 ng/dL. Six weeks after treatment was discontinued, the mean TSH level was found as 23±56.7 mIU/mL and the mean fT4 level was 0.97±0.27 ng/dL. Anti-Tg was negative in all subjects and the mean Tg level was 47.6±46.4 ng/mL (Table 1). The mean thyroxine dosage was 8±2.5 mcg/kg/day at the time of treatment initiation and 1.83±0.72 mcg/kg/day at the time of treatment discontinuation. Twenty-three subjects (47.9%) whose TSH level was >5 mIU/mL after the 6-week period without treatment were diagnosed as having permanent PCH, and 25 subjects (52.1%) whose TSH level was <5 mIU/mL for six months in the follow-up were diagnosed as having transient PCH.
Table 1.
Clinical and laboratory findings subjects with primary congenital hypothyroidism
| Clinical and laboratory characteristics | Subjects with PCH (n=48) | |
|---|---|---|
| Mean±SD | Median (quartile) | |
| Age at the time of the study (current) (years±SD) | 3.8±0.7 | 3.6 (3.3–4.1) |
| Gender (n, %) | ||
| Male | 26 (54.2) | - |
| Female | 22 (45.8) | - |
| Age at the time of diagnosis (n, %) | ||
| 0–14 days | 22 (45.8) | - |
| 15–30 days | 26 (54.2) | - |
| Way of diagnosis (n, %) | ||
| Screening test | 14 (29.2) | - |
| Peripheral blood (for investigating jaundice) | 26 (54.2) | - |
| Incidental | 8 (16.6) | - |
| At the time of diagnosis | ||
| TSH (mIU/mL) | 43.1±43.6 | 25.1 (17.1–62.9) |
| sT4 (ng/dL) | 0.98±0.32 | 1 (0.7–1.1) |
| Initial treatment dosage (mgc/kg/day) | 1.83±0.72 | 1.5 (1.2–2.2) |
| LT4 dose at the time of treatment discontinuation (mcg/kg/day) | 1,83±0,72 | 1,5 ( 1,2–2,2) |
| After discontinuation of treatment (sixth week) | ||
| TSH (mIU/mL) | 23±56.7 | 5.8 (3.1–15.7) |
| sT4 (ng/dL) | 0.97±0.27 | 1 (0.8–1.1) |
| Tg (ng/mL) | 47.6±46.4 | 34 (26.4–52.1) |
| Permanent PCH (n, %) | 23 (47.9) | |
| Dysgenesis | 12 (52.2) | |
| Hypoplasia | 8 (34.8) | |
| Ectopy | 4 (17.4) | |
| Dyshormogenesis | 11 (47.8) | |
| Transient PCH (n, %) | 25 (52,1) | |
| Etiology | unknown | |
PCH: primary congenital hypothyroidism; fT4: free thyroxine; Tg: thyroglobulin; TSH: thyroid-stimulating hormone
In the permanent PCH group, nine (39.1%) of 29 subjects were male, 14 (60.9%) were female, and the female/male ratio was 1.5. The mean age at the time of the study was 3.6±0.5 years. The mean age at the time of diagnosis was 16.5±7.1 days, and 10 subjects (43.5%) were diagnosed as having PCH in the first 14 days of life and treatment was initiated. The diagnosis was made in the Ministry of Health Screening Program in nine subjects (39.2%). Eleven (47.8%) subjects’ PCH was diagnosed when peripheral blood samples were obtained during investigations performed because of prolonged jaundice. Three (13%) subjects’ PCH was diagnosed incidentally. Twelve (5.2%) of the subjects were diagnosed as having dysgenesis (eight hypoplasia, four ectopia), and eleven (47.8%) were diagnosed as having dyshormogenesis. The general characteristics of the subjects with permanent PCH are shown in Table 2.
Table 2.
Comparison of the clinical and laboratory characteristics between the subjects with transient and permanent primary congenital hypothyroidism
| Permanent PCH (n=23; 47.8%) | Transient PCH (n=25; 52.1%) | ||||
|---|---|---|---|---|---|
| Clinical and laboratory characteristics | Mean±SD | Median (quartile) | Mean±SD | Median (quartile) | p |
| Current age | 3.6±0.5 | 3.5 3.5 (3.4–3.7) | 4±0.7 | 3.7 (3.3–4.8) | 0.192 |
| Sex | |||||
| Male | 9 (39.1) | - | 17 (68)a | - | 0.045a |
| Female | 14 (60.9) | - | 8 (32) | - | |
| Age at the time of diagnosis | |||||
| 0–14 days | 10 (43.5) | - | 12 (48) | - | 0.753 |
| 15–30 days | 13 (56.5) | - | 13 (52) | - | |
| Way of diagnosis | |||||
| Screening test | 9 (39.2) | - | 5 (20) | - | |
| Peripheral blood (for investigating jaundice ) | 11 (47.8) | - | 15 (60) | - | 0.336 |
| Inicidental | 3 (13) | - | 5 (20) | - | |
| Familial history | |||||
| Consanguineous marriage | 0 (0) | - | 0 (0) | - | |
| At the time of diagnosis | |||||
| TSH (mIU/mL) | 47.6±34.1 | 38.7 (16.3–73) | 38.9±51.2 | 19.6 (17.2–30.3) | 0.170 |
| sT4 (ng/dL) | 0.8±0.3 | 0.9 (0.6–1.1) | 1±0.2 | 1.1 (0.9–1.1) | 0.057 |
| After treatment discontinuation | |||||
| TSH (mIU/mL) | 43.2±76.8 | 20 (11.4–21.6) | 3.7±1.1 | 3.3 (2.8–4.2) | <0.001 |
| sT4 (ng/dL | 0.8±0.3 | 1 (0.7–1) | 1±0.1 | 1 (0.9–1.1) | 0.190 |
| Tg (ng/mL) | 60.4±63.6 | 39.3 (27.4–86.7) | 35.8±14.6 | 33.6 (23.7–48) | 0.415 |
| Thyroxine dosage used (mcg/kg/day) | |||||
| At the time of diagnosis | 8.4±2.7 | 8.3 (6.6–10) | 7.7±2.2 | 7.4 (6.4–8.4) | 0.409 |
| At the time of treatment discontinuation | 2.1±0.7 | 2.1 (1.5–2.7) | 1.5±0.5 | 1.4 (1.1–1.8) | 0.004b |
The rate of male sex is significantly higher in group 2 (p=0.045)
Threshold value 1.6 mcg/kg/day (sensitivity 69.6%, specificity 72%)
PCH: primary congenital hypothyroidism; fT4: free thyroxine; Tg: thyroglobulin; TSH: thyroid-stimulating hormone
In the transient PCH group, 17 (68%) of 25 subjects were male, eight (32%) were female, and the female/male ratio was 1:2.1 (the number of male subjects was significantly higher; p=0.045). The mean age at the time of the study was 4±0.7 years and the mean age at the time of diagnosis was 16.6±6 days. Twelve (48%) subjects’ PCH was diagnosed in the first 14 days of life. Fifteen (60%) subjects’ PCH was diagnosed when peripheral blood samples were obtained during investigations performed because of prolonged jaundice. Five (20%) subjects’ PCH was diagnosed in the Ministry of Health Screening Program, and five (20%) diagnoses were made incidentally (Table 2).
No significant differences were found between the groups in terms of age at the time of the study, age at the time of diagnosis, the way of diagnosis, and TSH and fT4 levels at the time of diagnosis (p>0.05). In addition, the thyroxine dosage used at the time of treatment discontinuation was significantly higher in group 1 compared with group 2 (p<0.05) (Table 2).
The optimal threshold value for thyroxine dosage at the time of treatment discontinuation as a preditvie marker in the differentiation of permanent and transient PCH was found as >1.6 mcg/kg/day with ROC analysis. The AUC was 0.745, which was statistically significant (p<0.001). This threshold value had a diagnostic sensitivity of 69.6% and a specificity of 72.0% (Figure 1).
Figure 1.
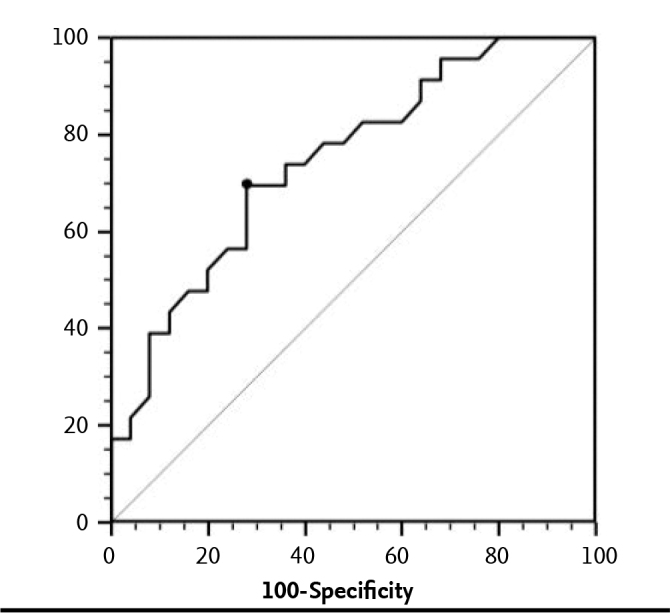
ROC analysis results of the optimal thyroxine dose threshold value at the time of treatment discontinuation as a prediction criterion in differentiation of permanent and transient PCH
Discussion
The diagnosis of PCH should be made in the first two weeks after birth and treatment should be initiated to prevent mental retardation and motor dysfunction (muscle weakness, poor motor coordination, spastic diplegia, ataxia) (1, 2, 4, 9, 16, 17). Another significant point in primary congenital hypothyroidism, in addition to early diagnosis, is specification of the underlying cause such that overtreatment of subjects with transient PCH and insufficient treatment in subjects with permanent PCH is prevented. The prevalence of PCH is 1/2000–4000 in neonatal screening programs (1–3). The female/male ratio is 1.1–2:1 (3, 8, 9, 17–20). In Turkey, the prevalence of PCH was found as 1/650 as a result of screening programs performed between 2008 and 2010 and the female/male ratio was similar (17).
In our study, the female/male ratio (1:1.1) was found different compared with ratios reported in the literature, but similar to the ratio reported in the study conducted by Ünüvar et al. in the Egean region from our country. It was thought that the similarity in sex distribution might be related with limited the number of subjects or a finding reflecting a characteristic of the Western regions of Turkey. In the literature, it has been reported that transient PCH occurs more commonly in the male sex (16, 18, 21). In our study, the number of male subjects was higher and the female/male ratio was 1:2.1 in the transient PCH group, similar to the literature.
The age at the time of diagnosis has decreased markedly in primary congenital hypothyroidism with neonatal screening programs, which were initiated because of the importance of early diagnosis and treatment. The mean age at the time of diagnosis in various studies was 11–12 days. In our country, the mean age at the time of diagnosis was found 19.2 days in 2008, whereas it decreased to 15.7 days in 2010 (17, 19, 20). The most common clinical finding in cases of primary congenital hypothyroidism is prolonged jaundice (2, 16). In our study, the mean age at the time of diagnosis (16.6 days) was at least 4–5 days older compared with the literature and one day older compared with our national neonatal screening program 2010 data. This older age at the time of diagnosis may be related with the fact that the rate of diagnosis by way of the screening program was low. Only 29.2% of our subjects were diagnosed by way of the screening program, the majority (54.2%) were diagnosed as having PCH during investigations performed because of prolonged jaundice.
Although the prevalence of permanent and transient PCH varies in different studies, an increase in transient PCH has been observed with the widespread use of screening programs. In the literature, the prevalence of transient PCH has been reported to range between 40.2% and 54% in studies related with PCH (5, 6, 16, 18). In a study that showed the neonatal hypothyroidism screening results of 2008–2010 in our country, the prevalence of PCH was reported as 1/650, but the prevalence of permanent and transient PCH was not specified (17). In our country, Ünüvar et al. (16) found the prevalence of transient PCH as 54%. In our study, the prevalence of transient PCH (52.1%) was similar to that in the study of Ünüvar et al. (16), but higher compared with rates reported in other studies in the literature. The cause of transient PCH may be maternal iodine deficiency, iodine load in the mother/baby or passage of TSH-blocking antibodies to the fetus because of maternal autoimmune thyroiditis (1–3, 5). In a study that reported the iodine status in our country, iodine levels were found as normal in the region of Trakya (22). Considering this fact, the reason of the high prevalence of transient PCH in our study may be iodine load rather than iodine deficiency. Currently, use of iodine solutions is still continuing both in umbilical care of babies and maternal postoperative care after cesarean section and normal vaginal deliveries (23). However, it is not possible to state that there was iodine load or iodine deficieny because we could not measure urine iodine levels of the babies and mothers at the time of diagnosis. In addition, it is not possible to make an interpretation in terms of presence of maternal autoimmune thyroiditis as an underlying cause, because thyroid autoantiboodies were not measured in the babies and mothers. Studies have reported the prevalence of permanent hypothyroidism as 46–70% in subjects with PCH (5, 6, 16, 18). The most common cause of permanent PCH (50–85%) is thyroid dysgenesis. Dyshormogenesis has been reported with a rate of 20–30% (5, 16, 18). Consanguineous marriage is important because dyshormogenesis may be inherited autosomal recessively (24). In the study of Tamam et al. (6), dyshormogenesis was found in 44% of subjects with permanent PCH. In our study, the prevalence of permanent PCH (47.9%) was similar to the literature data. Thyroid dysgenesis was found in 52.2% of the subjects and dyshormogenesis was found in 47.8%. Although the number of subjects with dyshormogenesis was higher than expected, it was compatible with the study of Tamam et al. (6). The absence of consanguineous marriage in all patients with PCH with dyshormogenesis in our study suggests that factors other than Mendelian inheritance should be investigated in the etiology. In addition, it should also be determined as to whether regional differences in the prevalence of PCH cases with a diagnosis of dyshormogenesis are present in our country.
Differentiation of permanent and transient primary congenital hypothyroidism is important in terms of prevention of unnecessary treatment in transient cases and providing appropriate treatment for permanent cases. However, etiologic investigations are mostly delayed until after the age of three years because some investigations directed to the etiology (e.g., thyroid scintigraphy) are time consuming and it is mandatory to initiate treatment early to prevent mental retardation. However, crtieria that could predict a diagnosis of permanent or transient PCH have been investigated (5, 6, 16, 18, 25). A few studies conducted in this area found different results as clinical and laboratory prediction criteria. Although it was reported that the fT4 level at the time of diagnosis could be a criterion in the differentiation of transient and permanent PCH in one study, other studies concluded that fT4 level was not determinative (6, 16, 25). In the literature, studies have reported that TSH level may be a criterion in predicting transient and permanent PCH, whereas others concluded that it could not be used as a criterion (5, 16, 18). In our study, we found that fT4 and THS levels at the time of diagnosis were not a predictive criterion to be used in the differential diagnosis of permanent and transient PCH. When the thyroxine dosage was evaluated as a predictive criterion in the study of Hashemipour et al. for the differential diagnosis of permanent and transient PCH, it was reported the thyroxine dosage given at the time of treatment initiation could be used as a criterion (18). In contrast, Delvecchio et al. (25) found that the thyroxine dosagee at the time of treatment initiation was not different in the permanent and transient PCH groups. In our study, the thyroxine dose at the time of treatment initiation was similar in the permanent and transient PCH groups, in accordance with the literature. When we evaluated if the thyroxine dose used at the time of treatment discontinuation could be used as a criterion for differential diagnosis, however, it was found that the subjects with permanent PCH received significantly higher thyroxine doses compared with subjects with transient PCH. Ünüvar et al. (16) found that the thyroxine dosage used during treatment discontinuation might be a predictive criterion, similar to the conclusion of our study. In the study conducted by Nair et al. (5), it was found that the thyroxine dose was significantly different between subjects with permanent and transient PCH, but this difference was eliminated when the dose values were adjusted by body weight and height. In our study, the thyroxine dosage threshold value at the time of treatment discontinuation, which was found as a predictive criterion for differential diagnosis of permanent and transeint PCH, was found as 1.6 mcg/kg/day. When this threshold value was considered, the sesitivity for prediction of permanent or transient PCH was found as 69.6% and the specificity was 72%. Although the sensitivity and specificity are low, we think that it would be appropriate to evaluate this value with larger scale studies because specification of a criterion to predict permanent or transient PCH is important, especially in terms of making an early decision for treatment discontinuation in subjects with transient PCH.
In conclusion, the high rate of transient PCH in this study, in which the number of subjects was limited, suggested that timely investigations directed to etiology in subjects who are being followed up for PCH are important in terms of preventing unneccessary treatment. It may be necessary to determine if the high male/female ratio in patients with PCH, and especially in transient PCH, is specific for our country. In addition, thyroid hormone levels at the time of diagnosis are not significant in the prediction of permanent and transient PCH, but the thyroxine dosage at the time of treatment discontinuation at the age of three years may be used as a predictive criterion. Large-scale studies should be conducted to evaluate this result.
Acknowledgements
We thank to Prof. Necdet Süt for statistichal analysis.
Footnotes
Ethics Committee Approval: Ethics committee approval was received for this study from the ethics committee of Trakya University School of Medicine (06.28.2012, TÜTF-TÜBAPK 2012-151).
Informed Consent: Written informed consent was not obtained from the parents of the patients because the study was retrospective designed.
Peer- review: Externally peer- reviewed.
Author Contributions: Concept - F.T., D.B.; Design - F.T., D.B., E.D., N.T.; Supervision - F.T.; Funding - F.T., D.B.; Materials - D.B.; Data Collection and /or Processing - D.B., E.D., F.T.; Analysis and /or interpretation - D.B., F.T.; Literature Review – D.B., E.D.; Writer - D.B.; Critical Review - F.T.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.LaFranchi SH. Approach to the diagnosis and treatment of neonatal hypothyroidism. J Clin Endocrinol Metab. 2011;96:2959–67. doi: 10.1210/jc.2011-1175. https://doi.org/10.1210/jc.2011-1175. [DOI] [PubMed] [Google Scholar]
- 2.Kurtoğlu S, Akın MA. Konjenital hipotiroidizm. In: Kurtoğlu S, editor. Yenidoğan dönemi endokrin hastalıkları. İstanbul: Nobel Matbaacılık; pp. 449–73. 2011.syf. [Google Scholar]
- 3.Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis. 2010;5:17. doi: 10.1186/1750-1172-5-17. https://doi.org/10.1186/1750-1172-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher DA, Grueters A. Thyroid disorders in childhood and adolecence. In: Sperling MA, editor. Pediatric endocrinology. 3rd ed. Philadelphia: Saunders Elsevier; 2008. pp. 227–53. [Google Scholar]
- 5.Nair PS, Sobhakumar S, Kailas L. Diagnostic re-evaluation of children with congenital hypothyroidism. Indian Pediatr. 2010;47:757–60. doi: 10.1007/s13312-010-0115-1. https://doi.org/10.1007/s13312-010-0115-1. [DOI] [PubMed] [Google Scholar]
- 6.Tamam M, Adalet I, Bakir B, et al. Diagnostic spectrum of congenital hypothyroidism in Turkish children. Pediatr Int. 2009;51:464–8. doi: 10.1111/j.1442-200X.2008.02790.x. https://doi.org/10.1111/j.1442-200X.2008.02790.x. [DOI] [PubMed] [Google Scholar]
- 7.Van Vliet G. Neonatal hypothyroidism: Treatment and outcome. Thyroid. 1999;9:79–84. doi: 10.1089/thy.1999.9.79. https://doi.org/10.1089/thy.1999.9.79. [DOI] [PubMed] [Google Scholar]
- 8.LaFranchi S. Congenital hypothyroidism: etiologies, diagnosis, and management. Thyroid. 1999;9:735–43. doi: 10.1089/thy.1999.9.735. https://doi.org/10.1089/thy.1999.9.735. [DOI] [PubMed] [Google Scholar]
- 9.Agrawal P, Philip R, Saran S, et al. Congenital hypothyroidism. Indian J Endocrinol Metab. 2015;19:221–7. doi: 10.4103/2230-8210.131748. https://doi.org/10.4103/2230-8210.131748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dussault JH, Coulombe P, Laberge C, Guyda H, Khoury K. Preliminary report on a mass screening program for neonatal hypothyroidism. J Pediatr. 1975;86:670–4. doi: 10.1016/s0022-3476(75)80349-0. https://doi.org/10.1016/S0022-3476(75)80349-0. [DOI] [PubMed] [Google Scholar]
- 11.Klett M, Schönberg D. Neonatal screening for hypothyroidism in the Federal Republic of Germany. Dtsch Med Wochenschr. 1981;106:6–12. doi: 10.1055/s-2008-1070246. https://doi.org/10.1055/s-2008-1070246. [DOI] [PubMed] [Google Scholar]
- 12.Fisher DA, Dussault JH, Folley TP, Jr, et al. Screening for congenital hypothyroidism: result of screening one million North American infants. J Pediatr. 1979;94:700–5. doi: 10.1016/s0022-3476(79)80133-x. https://doi.org/10.1016/S0022-3476(79)80133-X. [DOI] [PubMed] [Google Scholar]
- 13.Yordam M, Calikoglu AS, Hatun S, et al. Screening for congenital hypothyroidim in Turkey. Eur J Pediatr. 1995;154:614–6. doi: 10.1007/BF02079061. https://doi.org/10.1007/BF02079061. [DOI] [PubMed] [Google Scholar]
- 14.Beltrão CB, Juliano AG, Chammas MC, Watanabe T, Sapienza MT, Mauri S. Etiology of congenital hypothyroidism using thyroglobulin and ultrasound combination. Endocr J. 2010;57:587–93. doi: 10.1507/endocrj.k10e-009. https://doi.org/10.1507/endocrj.K10E-009. [DOI] [PubMed] [Google Scholar]
- 15.Kurtoglu S, Covut IE, Kendirci M, Uzum K, Durak AC, Kırış A. Normal thyroid volume of children in Turkey: Pilot study in Kayseri province. IDD Newsletter. 1995;11:41–2. [Google Scholar]
- 16.Ünüvar T, Demir K, Abacı A, Büyükgebiz A, Böber E. The role of initial clinical and laboratory findings in infants with hyperthytropinemia to predict transient or permanent hypothyroidism. J Clin Res Endocrinol. 2013;5:170–3. doi: 10.4274/Jcrpe.931. https://doi.org/10.4274/Jcrpe.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dilli D, Özbaş S, Acıcan D, et al. Establishment and development of a national newborn screening programme for congenital hypothyroidism in Turkey. J Clin Res Pediatr Endocrinol. 2013;5:73–9. doi: 10.4274/Jcrpe.929. https://doi.org/10.4274/Jcrpe.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashemipour M, Hovsepian S, Kelishadi R, et al. Permenant and transient congenital hypothyroidism in Isfahan-Iran. J Med Screen. 2009;16:11–6. doi: 10.1258/jms.2009.008090. https://doi.org/10.1258/jms.2009.008090. [DOI] [PubMed] [Google Scholar]
- 19.Perry RJ, Maroo S, Maclennan AC, Jones JH, Donaldson MDC. Combined ultrasound and isotope scanning is more informative in the diagnosis of congenital hypothyroidism than single scanning. Arch Dis Child. 2006;91:972–6. doi: 10.1136/adc.2006.096776. https://doi.org/10.1136/adc.2006.096776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanukoğlu A, Perlman K, Shamis I, Brnjac L, Rovet J, Daneman D. Relationship of etiology to treatment in congenital hypothyroidism. J Clin Endocrinol Metab. 2001;86:186–91. doi: 10.1210/jcem.86.1.7124. https://doi.org/10.1210/jcem.86.1.7124. [DOI] [PubMed] [Google Scholar]
- 21.Yang R, Zhu Z, Zhou X, Zhao Z. Treatment and follow-up of children with transient congenital hypothyroidism. JZUS-B. 2005;6:1206–9. doi: 10.1631/jzus.2005.B1206. https://doi.org/10.1631/jzus.2005.B1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erdoğan MF. The Iodine Status of Turkey: Where Were We? Where are We Now? Türkiye Klinikleri J Endocrin Special Topics. 2008;1:8–13. [Google Scholar]
- 23.Keskin M. İyot yüklenmesi ve etkileri. In: Kurtoğlu S, Bayram F, editors. Her yönüyle iyot. Kayseri: M Grup Matbaacılık; pp. 94–7. 2016.syf. [Google Scholar]
- 24.Grasberger H, Retetoff S. Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr Opin Pediatr. 2011;23:421–8. doi: 10.1097/MOP.0b013e32834726a4. https://doi.org/10.1097/MOP.0b013e32834726a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delvecchio M, Faienza MF, Acquafredda A, Zecchino C, Peruzzi S, Cavallo L. Longitudinal assessment of levo-thyroxine therapy for congenital hypothyroidism: relationship with aetiology, bone maturation and biochemical features. Horm Res. 2007;68:105–12. doi: 10.1159/000100373. https://doi.org/10.1159/000100373. [DOI] [PubMed] [Google Scholar]