

Abstract
The combination of anti-VEGF blockade (bevacizumab) with immune checkpoint anti–CTLA-4 blockade (ipilimumab) in a phase I study showed tumor endothelial activation and immune cell infiltration that were associated with favorable clinical outcomes in patients with metastatic melanoma. To identify potential immune targets responsible for these observations, post-treatment plasma from long-term responding patients were used to screen human protein arrays. We reported that ipilimumab plus bevacizumab therapy elicited humoral immune responses to galectin-1 (Gal-1) which exhibits pro-tumor, pro-angiogenesis, and immunosuppressive activities in 37.2% of treated patients. Gal-1 antibodies purified from post-treatment plasma suppressed the binding of Gal-1 to CD45, a T-cell surface receptor that transduces apoptotic signals upon binding to extracellular Gal-1. Antibody responses to Gal-1 were found more frequently in the group of patients with therapeutic responses and correlated with improved overall survival. In contrast, another subgroup of treated patients had increased circulating Gal-1 protein instead, and they had reduced overall survival. Our findings suggest that humoral immunity to Gal-1 may contribute to the efficacy of anti-VEGF and anti–CTLA-4 combination therapy. Gal-1 may offer an additional therapeutic target linking anti-angiogenesis and immune checkpoint blockade.
Keywords: Immune therapy, melanoma, galectin-1, antibody responses to galectin-1, Anti-VEGF
Introduction
CTLA-4 ligation on activated T cells down regulates T-cell responses, acting as a brake to T-cell activation (1). Clinical studies have shown that ipilimumab, a fully human monoclonal antibody (mAb) that blocks CTLA-4 activity, improves overall survival in patients with metastatic melanoma (2,3). Emerging evidence has suggested potential synergies between anti-angiogenesis agents and immune checkpoint blockade (4–10). In a phase I trial of ipilimumab plus bevacizumab (Ipi-Bev) in patients with metastatic melanoma (11), the response rate was approximately 20%, and the disease control rate was 67.4% in 46 treated patients. Treatment was associated with enhanced intratumoral endothelial activation, infiltration of lymphocytes, particularly CD8+ T cells. Most patients controlled their disease and a subset experienced long-term survival. The mechanisms responsible for the effects of this combination therapy warrant additional investigation.
CTLA-4 is important for suppression of B-cell responses and antibody production by regulating T follicular helper (TFH) cells, T follicular regulatory (TFR) cells, and T regulatory cells (Tregs) (12,13). Consistent with this, we have reported that Ipi-Bev elicited humoral immune responses in melanoma patients (11,14,15). One current goal in cancer immunotherapy is to better understand the importance of immune responses to specific targets for both cellular and humoral immunity. One method to do this is to analyze serologic responses in patients experiencing clinical benefits from treatment. Previous findings demonstrated that anti–CLTA-4 blockade increased IgG that is specific for certain targets in melanoma patients (10,16,17). To develop IgG responses to specific antigen, class switching is coordinated by cellular responses from helper CD4+ T cells. Targets that have been identified in such serologic screens also had functional activity (10,16,17). Based on these findings, we hypothesized that humoral immune responses may detect key targets involved in maintaining long-term clinical benefits in patients treated with immune checkpoint blockade. To test this hypothesis, we screened human protein arrays with plasma from patients responding to Ipi-Bev treatment and identified robust humoral immune responses to galectin-1, -3, and -9 (11).
Galectin-1 (Gal-1; LGALS1) is significantly upregulated and secreted in many tumors (18). High levels of Gal-1 are associated with tumor aggressiveness, metastasis, and poor survival for many cancers (18–22). Extracellular Gal-1 promotes tumor growth and progression by facilitating tumor cell proliferation, invasion/metastasis, angiogenesis, and immune escape (18,19,23–27). Depletion of Gal-1 in melanoma cells impeded tumor growth and improved survival in murine models (28), highlighting its potential significance for melanoma. Given Gal-1’s protumoral, proangiogenic, and immunosuppressive roles, we sought to further characterize humoral immune reaction to Gal-1 in melanoma patients receiving Ipi-Bev, Ipi alone, and PD-1 blockade in the current study. Humoral immune responses to angiogenic and immune regulatory factors such as Gal-1 provide an avenue to better understand the importance of these responses in -relation to outcomes and potential functional significance.
Materials and Methods
Patients
Patients with metastatic melanoma enrolled in the phase I Ipi-Bev trial (NCT00790010) and treated with ipilimumab alone or with PD-1 blockade (nivolumab; pembrolizumab) have been described previously (11,15). Informed consent was obtained from all the patients involved in this study after the nature and possible consequences of the studies were explained.
Collection of patient plasma
Blood samples were collected from patients at Dana-Farber/Harvard Cancer Center (Boston, MA) through Institutional Review Board (IRB) approved protocols. Blood samples were collected in Vacutainer tubes containing heparin. They were diluted with equal volume of RPMI1640 and subjected to Ficoll density gradient separation of PBMCs. The supernatant above the PBMC layer was collected and used as plasma. Aliquots of the plasma samples were stored at ≤ −20 °C.
Detection of Gal-1 antibody in patient plasma samples
ProtoArray Human Protein Microarray V5 (Life Technologies, Grand Island, NY) was used to screen antibodies in the posttreatment plasma samples from three long-term responders to Ipi-Bev to identify potential reactive antigens as guided by the manufacturer (11). Briefly, protein arrays were blocked in synthetic blocking solution (Life Technologies) for 1 hour and then incubated with plasma samples diluted in the blocking solution (1:500) overnight at 4°C. The arrays were washed and detected with Alexa Fluor® 647 goat anti-human IgG (Life Technologies). The arrays were scanned and image data were acquired using a Gene Pix scanner. Image data were analyzed using the ProtoArray® Prospector data analysis software (Life Technologies). Potential antibody targets were identified using Z factor cutoff of 0.4 as recommended by the manufacturer. Gal-1 was identified as a potential target by this screening (11). The presence of Gal-1 antibodies in the plasma samples was further confirmed by immunoblot analysis and ELISA using recombinant human Gal-1 (R&D Systems, Minneapolis, MN). Immunoblot analyses of Gal-1 antibodies with plasma samples have been previously described (11). Gal-1 antibodies in plasma were quantitatively assessed with recombinant human Gal-1 and a His tag with 8 His residues using the ELISA protocol previously described (15). Plasma samples were diluted 1:500 to 1:2,000 fold.
Affinity purification of Gal-1 antibodies from patient plasma
Recombinant human Gal-1 (6 μg) was coupled to activated NHS (N-hydroxysuccinimide) magnetic beads (40 μl) per the manufacturer (Thermo Scientific, Waltham, MA). Plasma samples (400 μl) were diluted with PBS (800 μl) and incubated with Gal-1-coupled beads with rotation at 4°C overnight. The beads were isolated with a magnet and washed with PBS five times. Antibodies bound to the beads were eluted with 0.1 M glycine (pH 2.5) and neutralized with 1/10 volume of 1 M Tris-Cl (pH 9.0). The antibodies were concentrated using an Amicon Ultra filter and stored in PBS supplemented with 0.02% BSA at 4°C. IgG content was determined by ELISA against normal human IgG (Life Technologies).
Preparation of biotinylated His-Avi-SUMO tagged Gal-1
The Expresso® Biotin Cloning & Expression System (Lucigen, Middleton, WI) was used to produce biotinylated Gal-1 with His, Avi, and SUMO tags at the N-terminus (HAS-Gal-1). Primer design and PCR amplification to incorporate His, Avi, and SUMO tags into Gal-1 cDNA were performed according to the instructions provided by the manufacturer. The Gal-1 cDNA was amplified from total RNA isolated from HUVEC using the following primers: 5′-CGCGAACAGATTGGAGGTgcttgtggtctggtcgccagcaac-3′ (sense) and 5′-GTGGCGGCCGCTCTATTAGtcaaaggccacacatttgatctt-3′ (antisense). The resultant PCR fragment was mixed with pAviTag N-His Vector and used to transform BIOTIN XCell F’ Chemically Competent Cells (Lucigen). The insertion of HAS-Gal-1 sequence was confirmed by DNA sequencing. For HAS-Gal-1 expression, instructions provided by the manufacturer (Lucigen) were followed. Briefly, a single colony was inoculated in LB with kanamycin (30 μg/ml) and 0.5% glucose overnight. Bacterial culture was diluted with LB (1:100) and incubated with shaking until OD600 reached 0.8. Rhamnose, arabinose, and biotin were added to the culture at final concentrations of 0.2%, 0.01%, and 50 μM respectively and incubated overnight with shaking to induce the expression of biotinylated HAS-Gal-1. Cells were collected and suspended in PBS and subjected to sonication. The lysate was supplemented with 1% Triton X-100 and 500 mM NaCl and incubated for 1 hour with shaking on ice, followed by centrifugation at 10,000 rpm for 10 min. HAS-Gal-1 in the supernatant was purified using HisPur Ni-NTA resin (Thermo Scientific) following the instructions. HAS-Gal-1 was eluted in PBS plus 250 mM Imidazole and dialyzed against PBS. The sample was supplemented with 8 mM DTT and stored in aliquots at −20 °C. Protein identity and biotinylation was confirmed by immunoblot analysis and ELISA using Gal-1 antibody (R&D Systems, Minneapolis, MN) and streptavidin-HRP respectively.
Binding of Gal-1 to CD45
Recombinant human CD45 (R&D Systems) was coated onto 96-well plates (25 ng/well) at 4°C overnight. The coated wells were blocked with 2% BSA in PBS for 1 hour at RT. HAS-Gal-1(0.1 or 0.5 μg/ml) in TBST plus 0.1% BSA was added to each well coated with CD45 and incubated for 1 hour at RT. The plates were washed with PBST and incubated with streptavidin-HRP diluted in PBST with 1% BSA for 1 hour at RT. The plates were thoroughly washed with PBST. Substrate TBM (Sigma, St Louis, MO) was added and incubated for an appropriate period of time. The reaction was stopped with 1 N HCl. OD450 and OD570 were read in a microplate reader. In some experiments, HAS-Gal-1 was incubated with equal amounts of a commercial anti-Gal-1 antibody (R&D Systems), control antibody (R&D Systems), Gal-1 antibodies purified from patient plasma, or normal human IgG (Life Technologies) for 2 hours at 4 °C before addition to the coated CD45.
Measurement of circulating Gal-1
Gal-1 concentrations in pretreatment and posttreatment (5–10 weeks after treatment initiation) plasma samples from Ipi-Bev-treated patients were determined using the previously described ELISA method(29). Gal-1 in plasma samples from ipilimumab-treated patients was measured using Magnetic Luminex Screening Assay kits (R&D Systems) per manufacturer’s instructions.
Immunohistochemistry for Gal-1
Pretreatment and posttreatment (approximately 12 weeks after initiation of therapy) tumor tissues were collected, fixed in 10% neutral buffered formalin, dehydrated, and embedded in paraffin. Five-micrometer thick sections were cut, deparaffinized, rehydrated, and heated in a steamer for 30 minutes for antigen retrieval in citrate buffer pH 6.0 (Invitrogen, Carlsbad, CA). After cooling, sections were incubated with peroxidase blocker (DAKO, Carpinteria, CA) for 5 min and serum-free protein blocker (DAKO) for 20 minutes. Slides were then incubated at RT for one hour with primary monoclonal antibody against Gal-1(30) diluted (1:15,000) in Da Vinci green diluent (Biocare Medical, Concord, CA). For the secondary antibody, Envision anti-mouse HRP-labeled polymer (DAKO) was applied to the sections and stained for 30 min. The slides were visualized with diaminobenzidine (DAKO), washed in distilled water, hematoxylin counterstained, dehydrated, and mounted. Positive and negative controls were included in each panel of staining for all markers. Immunoreactivity for Gal-1 was detected in tumor cells and endothelial cells of small blood vessels. All slides were evaluated and scored semiquantitatively by a pathologist (X.L.) blinded to clinical data. The intensity (0, negative; 1, weak; 2, moderate; 3, intense) and the percentage of staining (0–100%) were assessed. H-Scores were based on the results of staining intensity multiplied by staining percentage. The scores of tumor cells and endothelial cells were recorded separately and added to obtain a final score, except when the positive staining in both tumor cells and immune cells made them difficult to distinguish. All scoring was performed twice. Samples were also reviewed by a second pathologist (S.R.) blinded to the initial review.
Statistical analysis
Two analytic techniques were used to investigate the relationship between the fold change of Gal-1 antibodies and survival. The first technique employed an 18-week conditional landmark analysis, which corresponded to the maximum number of weeks between samples. Patients who were alive at 18 weeks were classified according to whether or not they achieved a meaningful fold change and followed forward in time. This reduced the sample size from 43 to 42 patients. The second technique was based on the extended Cox model with time-dependent covariates. All patients were initially classified as not having a meaningful fold change for Gal-1 antibodies in circulation. At the time the fold change (post/pre ratio) increased to at least 1.5, a division that divides the distribution of fold change values into the lower 2/3 and upper 1/3, the patient was classified as having achieved a meaningful change. With this approach, differences in the hazard of death for patients who achieved a meaningful change, compared with those who had not yet or never did, were estimated. This analysis used the full sample size of 43 patients. To investigate if an increase in Gal-1 antibody in the circulation was associated with clinical response, 27 patients who were assessed for antibody changes on or before the date that response was first recorded were analyzed. The median time between pre- and post-antibody assessments was 8 weeks (range: 4 to 14 weeks). Clinical responses were determined using RECIST criteria and dichotomized into complete responses/partial responses (CR/PR) or stable disease/progressive disease (SD/PD). In the dataset, 7 patients had CR/PR and 20 patients had SD/PD. The relationship between response and antibody change and the comparisons of antibody responses to Gal-1 between Ipi-Bev, ipilimumab, and PD-1 blockade treatments were assessed using Fisher’s exact test. P values are two-sided. The association of fold change of circulating Gal-1 with survival was assessed based on fold change of 1.2. This division point was determined by the method of Contal-O’Quigley(31) and also corresponds to a division of the distribution of fold change values into the lower 2/3 and upper 1/3. The differences in binding of Gal-1 to CD45 were evaluated using an unpaired, two-tailed t test. P < 0.05 was considered statistically significant for all comparisons.
Results
Ipilimumab plus bevacizumab elicited humoral immune responses to Gal-1
To identify potential targets of humoral immune responses, posttreatment plasma samples from three responding patients (P12, P13, and P27; Supplementary Table S1) of Ipi-Bev- and one pembrolizumab-treated patient (P152) were used to screen protein arrays. Proteins with a Z-factor of 0.4 or greater were considered potential targets, per manufacturer’s guidance. Gal-1 was identified in protein array screens with plasma of P12 (the Ipi-Bev patient who achieved complete response) and P152 (durable partial response) having Z-factors of 0.49 and 0.71, respectively (Supplementary Fig. S1). To confirm Ipi-Bev-induced antibody responses, Gal-1 antibodies in the pretreatment and posttreatment plasma samples of five patients with varying outcomes [complete response (CR), partial response (PR), stable disease (SD) or progressive disease (PD)] were first assessed by immunoblot analysis and ELISA. Gal-1 antibody increases were detected in four patients by both immunoblot analysis (Fig. 1A) and ELISA (Fig. 1B).
Figure 1.
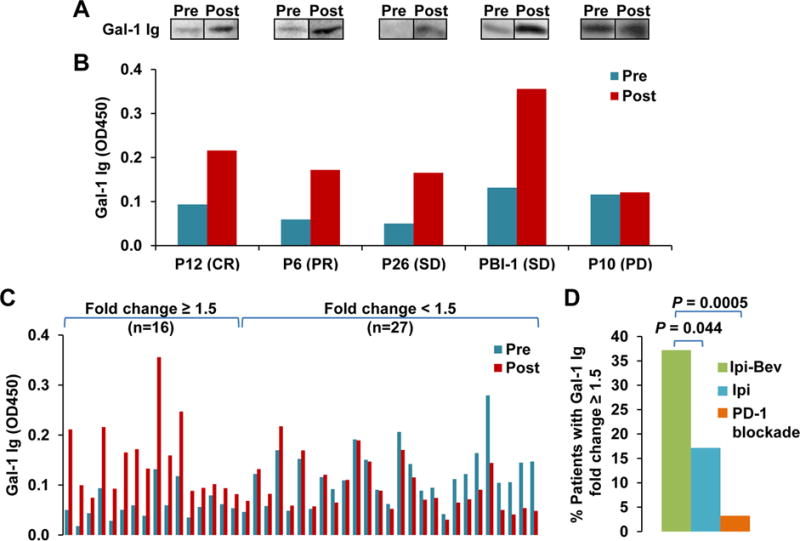
Ipilimumab plus bevacizumab (Ipi-Bev) elicited antibody responses to Gal-1 in patients with metastatic melanoma. A and B, Detection of Gal-1 antibodies in pretreatment and posttreatment plasma samples of representative patients by immunoblot analyses (A) and ELISA (B). Clinical responses of the patients are also indicated (CR, complete response; PR, partial response; SD, stable disease; and PD, progressive disease). C, Gal-1 antibody titers in the pretreatment and posttreatment plasma samples of 43 Ipi-Bev-treated patients measured by ELISA. Patients were grouped by Gal-1 antibody fold change ≥ 1.5 or < 1.5. The numbers of patients in each group are indicated. D, Proportions of patients treated with Ipi-Bev (n = 43), ipilimumab (n = 35), and PD-1 blockade (n = 31) with Gal-1 antibody fold change ≥ 1.5. Statistically significant differences are noted between treatment groups with Ipi-Bev patients having the greatest incidence of Gal-1 antibody fold change ≥ 1.5 (P = 0.044 to ipilimumab alone; P = 0.0005 to PD-1 blockade).
To further investigate the potential importance of Gal-1 immune responses, Gal-1 antibodies in pretreatment and posttreatment plasma samples from 43 Ipi-Bev-treated patients were assessed by ELISA. Sample collection and fold changes for Gal-1 antibodies are summarized in Table S2. Median follow-up time (based on the inverted Kaplan-Meier censor) was 204 weeks (95% CI: 131 to 225 weeks) and there were 29 deaths (67%) reported. An increase in Gal-1 antibody titer by 50% (i.e. fold change = 1.5) was considered significant and used as the cut-off point. This division point corresponds to a division of the distribution of fold change values into the lower 2/3 and upper 1/3. Based on this pre-determined cut-off, 16 (37.2%) of the patients displayed increases in Gal-1 antibody titers as a function of treatment (Fig. 1C).
Given the initial success with CTLA-4 mAb-based treatments in patients and the now broad application of anti–PD-1 therapies for multiple cancers, a logical next question of investigation is to ask whether the immune regulation seen with anti–CTLA-4 may also be relevant to anti–PD-1. PD-1 is a clinically active immune check point known to be involved in regulation of cellular, as well as humoral, immune response. CTLA-4 and PD-1 have both similar and different effects on the development and effector functions of TFH cells and TFR cells (12,13,32,33). Therefore, it is reasonable to ask whether humoral immune targets identified with CTLA-4 blockade would also have relevance in anti–PD-1 therapy. To address whether ipilimumab monotherapy and PD-1 blockade also enhanced antibody responses to Gal-1, Gal-1 antibodies were measured in pretreatment and posttreatment plasma samples collected from 35 ipilimumab-treated and 31 PD-1 blockade-treated patients with metastatic melanoma. These patients were on different trials and not combined with anti-VEGF, whereas a small part of the PD-1 blockade-treated patients received prior ipilimumab treatment. Six (17.1%) of the ipilimumab-treated and one (3.2%) of the PD-1 blockade-treated patients displayed an increase in Gal-1 antibody concentration by 50% or more respectively (Fig. 1D; Supplementary Fig. S2A and B). These findings suggest that Gal-1 antibody responses may occur less frequently in patients receiving ipilimumab or PD-1 blockade than the Ipi-Bev combination therapy.
Humoral responses to Gal-1 associated with favorable clinical outcomes to Ipi-Bev therapy
We next examined if enhanced humoral immune responses to Gal-1 were associated with clinical outcomes to Ipi-Bev therapy. Among the 16 patients with increased antibody titers to Gal-1 as a function of treatment, 5 (31.3%) had CR/PR, 8 (50.0%) had SD, and 3 (18.8%) experienced PD (Fig. 2A and B). An increase in antibody response to Gal-1 occurred in 62.5% (5 out of 8) of CR/PR patients, 36.4% (8 out of 22) of SD patients, and 23.1% (3 out of 13) of PD patients (Fig. 2C and Table S3). Patients with Gal-1 antibody fold change ≥ 1.5 appeared to have a higher clinical response rate than those with fold change < 1.5 (41.7 % vs. 13.3%), although it did not reach statistical significance presumably due to small number of patients who were assessed for antibody changes on or before the date that response was first recorded (n = 27; Table S4). The median survival of the patients with Gal-1 antibody fold change < 1.5 was 70 weeks (95% CI, 47–81 weeks), whereas that of patients with fold change ≥ 1.5 was undefined because > 50% of the patients were still alive at the time of the analysis (Fig. 2D). Patients who achieved a Gal-1 antibody fold change of at least 1.5 had a hazard of death that was reduced by 73% compared with patients who had not yet or never achieved a fold change ≥ 1.5 [Hazard ratio: (yes vs. no) 0.27 (95% CI, 0.11–0.67); Wald P = 0.005]. These observations suggested that humoral immune responses to Gal-1 were associated with clinical outcomes to Ipi-Bev therapy.
Figure 2.
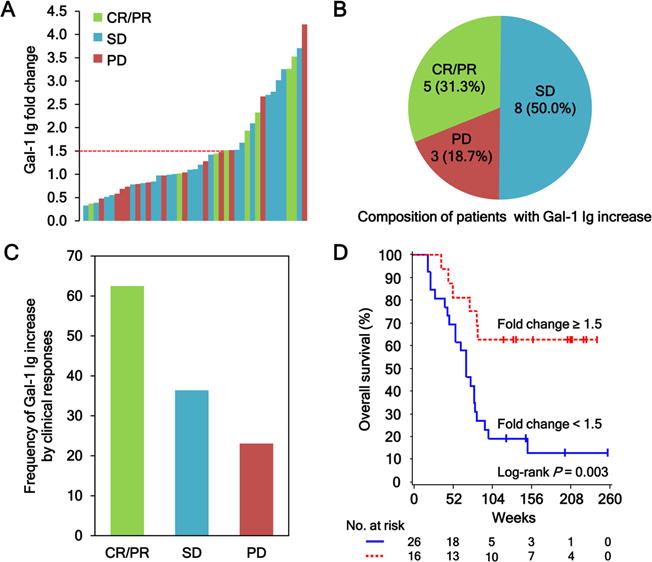
Antibody responses to Gal-1 correlate with clinical outcomes in metastatic melanoma patients treated with Ipi-Bev. A, Patients were plotted based on their Gal-1 antibody fold changes. Each bar represents a patient and the color of the bar indicates clinical response of the patient (CR, complete response; PR, partial response; SD, stable disease; and PD, progressive disease). There were 8 CR/PR (1 CR and 7 PR), 22 SD, and 13 PD patients. Gal-1 antibody titer was considered as increased when fold change ≥ 1.5. B, Composition of the 16 patients with a Gal-1 antibody increase. Numbers (and %) of CR/PR, SD and PD patients with Gal-1 antibody fold changes ≥ 1.5 are shown. C, Frequencies of Gal-1 antibody increase by clinical responses. D, Kaplan-Meier survival curves of patients based on Gal-1 antibody fold change ≥ 1.5 or < 1.5 (P = 0.0031). The median survival of the patients with Gal-1 antibody fold change < 1.5 was 70 weeks (95% CI, 47–81), whereas that of patients with fold change ≥ 1.5 was unreached.
Longitudinal analysis of Gal-1 antibody responses
In order to better understand Gal-1 humoral immune responses following Ipi-Bev treatment, time courses of Gal-1 antibody titers as a function of treatment were conducted in five patients who experienced durable clinical benefit from therapy (P5, P6, P12, P13, and P26, Table S1). All five patients displayed an increase in Gal-1 antibodies following initial treatment of Ipi-Bev (Supplementary Fig. S3A–E). Somewhat varying dynamic patterns of Gal-1 antibodies were observed later in the course of treatment: Gal-1 antibody concentrations either declined slightly after initial increase and remained at that level (Supplementary Fig. S3A and B) or the antibody titers waxed and waned over time (Supplementary Fig. S3C–E).
Circulating Gal-1 antibodies are functional in neutralizing Gal-1
Given the development of high titer antibodies to Gal-1 in a number of treated patients receiving long-term clinical benefit, we next determine if these Gal-1 antibodies were functional and could block the biological activities of Gal-1. The binding of Gal-1 to CD45 on T cells has been reported to transduce apoptotic signals (19,34–37). Therefore, we wanted to test if this binding could be blocked by circulating Gal-1 antibodies in patients. Gal-1 protein was expressed in a fusion form (designated as HAS-Gal-1) with His, Avi, and SUMO tags at its N-terminus in bacterial cells (Supplementary Fig. S4A and B). The Avi tag contained the biotinylation consensus sequence and was biotinylated (Supplementary Fig. S4C). Gal-1 antibodies were isolated from the posttreatment plasma sample collected from a long-term SD patient (PBI-12, Table S1) who displayed a high titer antibody response to Gal-1 (Supplementary Fig. S5A and B). Successful enrichment of Gal-1 antibodies was confirmed by their depletion from the plasma sample and their recognition of Gal-1 and HAS-Gal-1 (Supplementary Fig. S5C and D). In contrast to the enriched Gal-1 antibodies, normal human IgG did not recognize Gal-1 or HAS-Gal-1 (Supplementary Fig. S5D). The binding of HAS-Gal-1 to coated CD45 was demonstrated by ELISA (Fig. 3 and Supplementary Fig. S6A). This binding was blocked by a commercial Gal-1-specific polyclonal antibody, but not a control antibody (Fig. 3 and Supplementary Fig. S6A), indicating that this binding was mediated via interaction between CD45 and the Gal-1 sequence, and not the tags of HAS-Gal-1. This binding was also blocked by β-lactose but not sucrose (Fig. 3 and Supplementary Fig. S6B), confirming it is β-galactoside dependent. The binding of HAS-Gal-1 to CD45 was significantly inhibited by the enriched Gal-1 antibodies from the treated patient, while normal human IgG had little effect (Fig. 3 and Supplementary Fig. S6C). These findings indicate that Gal-1 antibodies induced by Ipi-Bev therapy can functionally block galactoside-dependent Gal-1 binding.
Figure 3.
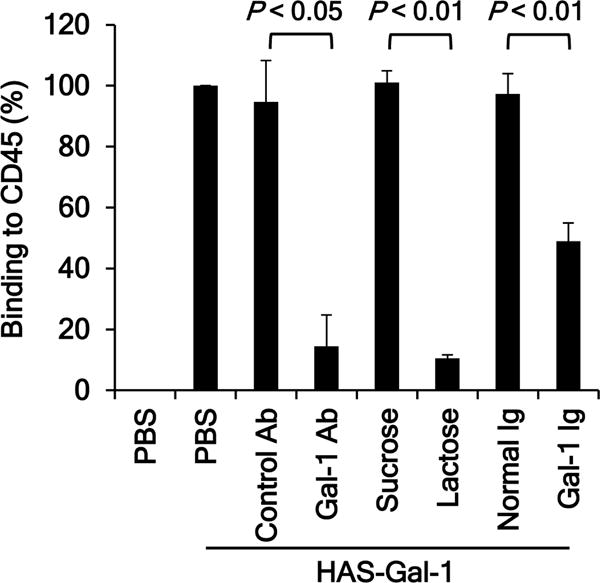
Anti-Gal-1 antibodies isolated from an Ipi-Bev-treated patient abrogate Gal-1 binding to CD45. Anti-Gal-1 antibodies were affinity purified from plasma. Biotinylated HAS-Gal-1 (250 ng/ml) was incubated with a commercial anti-Gal-1 polyclonal antibody (Gal-1 Ab) or control antibody (10 μg/ml), enriched endogenous Gal-1 antibodies (Gal-1 Ig) or normal human IgG (1.98 μg/ml) prior to incubation with coated CD45. The binding of HAS-Gal-1 to CD45 was detected with streptavidin-HRP. Sucrose and lactose were added to the reaction at 5 mM. Data are presented as Mean ± SD of 3 experiments.
Association of increased circulating Gal-1 with reduced overall survival
To further address if endogenous Gal-1 antibodies may be clinically relevant, we examined Gal-1 expression in melanoma tumors and in the circulation before and after Ipi-Bev treatment initiation. Gal-1 expression was analyzed in paired pretreatment and posttreatment tumor biopsies of 8 patients receiving Ipi-Bev including 3 PR, 3 SD, and 2 PD (Table S5). Gal-1 expression in melanoma cells and tumor associated endothelial cells (TEC) before treatment varied from being undetectable to highly expressed (Fig. 4A–F, Supplementary Fig. S7, and Table S5). Complex patterns of Gal-1 expression in response to Ipi-Bev were observed: some of the patients displayed increased Gal-1 expression in the posttreatment tumor cells and endothelia (Fig. 4A and B; Table S5) or in endothelia but not in tumor cells (Fig. 4C and D; Table S5), whereas the others showed decreased Gal-1 expression in tumor and endothelial cells (Fig. 4E and F; Table S5). An association between Gal-1 expression in the tumors and clinical responses of these patients was not observed, presumably due to small sample size.
Figure 4.
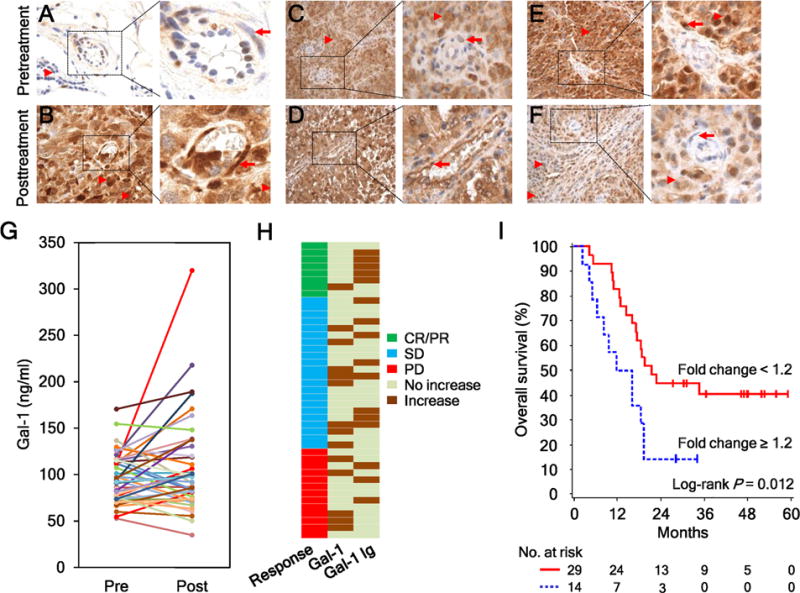
Ipi-Bev therapy altered Gal-1 expression in tumors and in the circulation. A and B, Patient P27. Gal-1 expression was hardly detected in the pretreatment tumors (A) but was intensive in tumor cells and tumor associated endothelial cells (TEC) posttreatment (B). Representative TEC and tumor cells are indicated with arrows and arrow heads respectively. C and D, Patient P20. Gal-1 expression was highly expressed in tumor cells but not TEC pretreatment (C). Strong Gal-1 expression was observed in tumor cells and TEC posttreatment (D). E and F, Patient P28. Gal-1 was highly expressed in tumor cells and TEC pretreatment (E). Reduced and no Gal-1 expression was detected in tumor cells and TEC posttreatment (F). G, Pretreatment and posttreatment circulating Gal-1 concentrations of the patients (n = 43). H, Different subsets of patients had increased circulating Gal-1 and antibody responses to Gal-1 in response to Ipi-Bev. Patients are grouped by clinical responses. I, Kaplan-Meier survival curves of patients based on circulating Gal-1 fold changes as a result of Ipi-Bev treatment. Increases in circulating Gal-1 were significantly associated with shortened overall survival (P = 0.012). The median survival of the patients with circulating Gal-1fold change ≥ 1.2 was 13.9 months (95% CI, 5–19), while that of patients with fold change < 1.2 was 21.6 months (95% CI, 16 to ∞).
Ipi-Bev also altered circulating Gal-1 concentrations (Fig. 4G) and was associated with increased circulating Gal-1 in a subset of patients that included 1 (12.5%), 8 (36.4%), and 5 (38.5%) of CR/PR, SD, and PD patients, respectively, (Fig. 4H). Of note, this subgroup of patients had limited (2 out of 14) overlap with those displayed increased Gal-1 antibody titers (Fig. 4H) and had significantly shortened overall survival [median survival: 13.9 (95% CI, 5–19) vs. 21.6 (95% CI, 16 to ∞) months, P = 0.0118; Fig. 4I]. Similar to Ipi-Bev treated patients (Supplementary Fig. S8A), pretreatment Gal-1concentrations in the serum were not associated with survival (Supplementary Fig. S8B), whereas circulating Gal-1 increases were associated with reduced survival in patients treated with ipilimumab alone (Supplementary Fig. S9).
Discussion
Gal-1 is known to have protumoral, proangiogenic, and immunosuppressive activities in preclinical studies (18). We show here that a subgroup of melanoma patients increased circulating Gal-1 in response to Ipi-Bev and this increase was associated with reduced survival, whereas a different subset of patients demonstrated enhanced antibody responses to Gal-1 that correlated with better clinical outcomes. Our findings suggest that circulating Gal-1 and circulating Gal-1 antibodies may be relevant to clinical benefit of Ipi-Bev therapy, providing a possible biomarker for immune therapy, but also serving as a therapeutic target alone or in combination with checkpoint blockade.
The extracellular functions of Gal-1 require the β-galactoside-dependent binding to numerous ligands on the surface of tumor cells, tumor-associated endothelial cells, or activated T cells(18,19). Specifically, binding of Gal-1 to T cells and induction of T-cell apoptosis by Gal-1 are β-galactoside dependent and can be inhibited by lactose (14, 18, 30). Our in vitro studies demonstrated the neutralizing activity of patient-derived Gal-1 antibodies on β-galactoside-dependent binding of Gal-1 to CD45. These findings suggest that the humoral responses to Gal-1 arising from the combined Ipi-Bev treatment could be providing functional blocking of the β-galactoside-dependent activities of Gal-1 and may contribute to favorable antitumor effects of Ipi-Bev.
Gal-1 plays a key role in tumor immune evasion by inducing apoptosis of effector cytotoxic T cells(19,26,34,37–41). Antitumor effector T cells express large amounts of Gal-1 ligands that transmit proapoptotic signals and inhibit the production of antitumor cytokine IFNγ upon binding to Gal-1(19,34,35,38,40,42). Blocking tumor-derived Gal-1 with a neutralizing antibody reduces tumor growth, enhances tumor rejection, and is accompanied by increases in tumor-specific cytotoxic T lymphocytes and IFNγ production(25,27). It is plausible that an increase in functional Gal-1 antibodies resulting from Ipi-Bev treatment could block Gal-1-mediated T-cell apoptosis and thus enhance antitumor immunity. Increased infiltration of CD8+ T cells has been observed in the posttreatment tumors of Ipi-Bev patients(11). A functional antibody response to Gal-1 may contribute to this via multiple mechanisms, including enhancement of survival of the activated lymphocytes in the tumor microenvironment.
Gal-1 is known to stimulate tumor angiogenesis and promote metastasis(18,21,23,28,43,44). Gal-1 is upregulated and secreted from tumors by anti-VEGF treatment as well as hypoxia, and promotes VEGF-independent angiogenesis, thereby providing a compensatory mechanism for the growth of tumors resistant to anti-VEGF therapy(27). Mice treated with a neutralizing Gal-1 antibody exhibited decreased tumor angiogenesis and growth of anti-VEGF–resistant tumors(27). A functional antibody response to Gal-1 might prevent or reduce Gal-1–mediated VEGF-independent angiogenesis in the settings of Ipi-Bev therapy.
Our analyses revealed dynamic changes of Gal-1 in the circulation of treated patients and heterogeneous expression in tumors in response to Ipi-Bev therapy. Ipi-Bev induced an early increase in circulating Gal-1 in a subgroup of patients with limited antibody responses to Gal-1 and this early increase was associated with reduced overall survival. These observations are in agreement with previously reported roles of Gal-1. A number of factors, including cytokines (such as TNFα, IL1β, and IFNγ) and activation of endothelial cells, have been reported to modulate Gal-1 expression(22,45). Significant proportions of Ipi-Bev-treated patients have profound increases in circulating TNFα, IL1β, and IFNγ, and prominent activation of endothelia in their tumors(11,14). Anti-VEGF treatment with bevacizumab upregulates Gal-1 expression and secretion from tumors in animal models of pancreatic cancer(27). This in itself may contribute to the immune recognition of Gal-1 via the addition of bevacizumab to ipilimumab, resulting in a higher incidence of increased antibody titers to Gal-1. In addition, significantly more patients treated with Ipi-Bev developed the highest titers of antibodies to Gal-1, compared to patients treated with Ipi alone. This suggests a mechanism of immune recognition involving the addition of bevacizumab in the setting of immune checkpoint blockade that may relate to the recognized functional relationships between Gal-1 and VEGF. It is also worthy to note that PD-1 blockade induced the least immunity to Gal-1. This suggests that anti–CLTA-4 and anti–PD-1 may have distinct influences of humoral immunity, at least to Gal-1, which may be attributed to different roles of CTLA-4 and PD-1 in the effector function of TFR cells and antibody production (12,13,32,33).
In summary, our findings demonstrated that increases in circulating Gal-1 were associated with poor clinical outcomes, whereas immune checkpoint treatment-related antibody responses to Gal-1 were functional and correlated with favorable clinical outcomes. These findings provide useful mechanistic insights into the antitumor and potential synergistic effects of combining immune checkpoint blockade with anti-angiogenesis agents, and further establish Gal-1 as a therapeutic target.
Supplementary Material
Acknowledgments
NIH CA143832 (F.S.H.), the Melanoma Research Alliance (F.S.H.), the Sharon Crowley Martin Memorial Fund for Melanoma Research (F.S.H.) and the Malcolm and Emily Mac Naught Fund for Melanoma Research (F.S.H.) at Dana-Farber Cancer Institute, Genentech/Roche, and Bristol-Myers Squibb.
Abbreviations
- CR
complete response
- Gal-1
galectin-1
- HAS-Gal-1
galectin-1 with His, Avi, and SUMO tags
- Ipi-Bev
ipilimumab plus bevacizumab
- NHS
n-hydroxysuccinimide
- mAb
monoclonal antibody
- PD
progressive disease
- PR
partial response
- SD
stable disease
- TEC
tumor associated endothelial cells
- THF
T follicular helper cells
- TFR
T follicular regulatory cells
- Tregs
T regulatory cells
Footnotes
F.S.H has served as a consultant to Genentech and has received research support to institution from Bristol-Myers Squibb. Patent application antibody responses to Gal-1 as biomarker is pending.
References
- 1.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 4.Yuan J, Zhou J, Dong Z, Tandon S, Kuk D, Panageas KS, et al. Pretreatment serum VEGF is associated with clinical response and overall survival in advanced melanoma patients treated with ipilimumab. Cancer Immunol Res. 2014;2:127–32. doi: 10.1158/2326-6066.CIR-13-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunol Res. 2001;23:263–72. doi: 10.1385/IR:23:2-3:263. [DOI] [PubMed] [Google Scholar]
- 6.Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol. 1998;160:1224–32. [PubMed] [Google Scholar]
- 7.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–51. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J. 2015;29:227–38. doi: 10.1096/fj.14-250985. [DOI] [PubMed] [Google Scholar]
- 10.Schoenfeld J, Jinushi M, Nakazaki Y, Wiener D, Park J, Soiffer R, et al. Active immunotherapy induces antibody responses that target tumor angiogenesis. Cancer Res. 2010;70:10150–60. doi: 10.1158/0008-5472.CAN-10-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2:632–42. doi: 10.1158/2326-6066.CIR-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014;41:1026–39. doi: 10.1016/j.immuni.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sage PT, Sharpe AH. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol. 2015;36:410–8. doi: 10.1016/j.it.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu X, Giobbie-Hurder A, Liao X, Lawrence D, McDermott D, Zhou J, et al. VEGF Neutralization Plus CTLA-4 Blockade Alters Soluble and Cellular Factors Associated with Enhancing Lymphocyte Infiltration and Humoral Recognition in Melanoma. Cancer Immunol Res. 2016;4:858–68. doi: 10.1158/2326-6066.CIR-16-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Giobbie-Hurder A, Liao X, Connelly C, Connolly EM, Li J, et al. Angiopoietin-2 as a Biomarker and Target for Immune Checkpoint Therapy. Cancer Immunol Res. 2017;5:17–28. doi: 10.1158/2326-6066.CIR-16-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A. 2003;100:4712–7. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinushi M, Hodi FS, Dranoff G. Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc Natl Acad Sci U S A. 2006;103:9190–5. doi: 10.1073/pnas.0603503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Astorgues-Xerri L, Riveiro ME, Tijeras-Raballand A, Serova M, Neuzillet C, Albert S, et al. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat Rev. 2014;40:307–19. doi: 10.1016/j.ctrv.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16:137R–57R. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- 20.Braeuer RR, Shoshan E, Kamiya T, Bar-Eli M. The sweet and bitter sides of galectins in melanoma progression. Pigment Cell Melanoma Res. 2012;25:592–601. doi: 10.1111/j.1755-148X.2012.01026.x. [DOI] [PubMed] [Google Scholar]
- 21.Thijssen VL, Hulsmans S, Griffioen AW. The galectin profile of the endothelium: altered expression and localization in activated and tumor endothelial cells. Am J Pathol. 2008;172:545–53. doi: 10.2353/ajpath.2008.070938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Croci DO, Salatino M, Rubinstein N, Cerliani JP, Cavallin LE, Leung HJ, et al. Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. J Exp Med. 2012;209:1985–2000. doi: 10.1084/jem.20111665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thijssen VL, Postel R, Brandwijk RJ, Dings RP, Nesmelova I, Satijn S, et al. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc Natl Acad Sci U S A. 2006;103:15975–80. doi: 10.1073/pnas.0603883103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He J, Baum LG. Presentation of galectin-1 by extracellular matrix triggers T cell death. J Biol Chem. 2004;279:4705–12. doi: 10.1074/jbc.M311183200. [DOI] [PubMed] [Google Scholar]
- 25.Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5:241–51. doi: 10.1016/s1535-6108(04)00024-8. [DOI] [PubMed] [Google Scholar]
- 26.Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon S, Mendez-Huergo SP, et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res. 2013;73:1107–17. doi: 10.1158/0008-5472.CAN-12-2418. [DOI] [PubMed] [Google Scholar]
- 27.Croci DO, Cerliani JP, Dalotto-Moreno T, Mendez-Huergo SP, Mascanfroni ID, Dergan-Dylon S, et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell. 2014;156:744–58. doi: 10.1016/j.cell.2014.01.043. [DOI] [PubMed] [Google Scholar]
- 28.Mathieu V, de Lassalle EM, Toelen J, Mohr T, Bellahcene A, Van Goietsenoven G, et al. Galectin-1 in melanoma biology and related neo-angiogenesis processes. J Invest Dermatol. 2012;132:2245–54. doi: 10.1038/jid.2012.142. [DOI] [PubMed] [Google Scholar]
- 29.Ouyang J, Plutschow A, Pogge von Strandmann E, Reiners KS, Ponader S, Rabinovich GA, et al. Galectin-1 serum levels reflect tumor burden and adverse clinical features in classical Hodgkin lymphoma. Blood. 2013;121:3431–3. doi: 10.1182/blood-2012-12-474569. [DOI] [PubMed] [Google Scholar]
- 30.Ouyang J, Juszczynski P, Rodig SJ, Green MR, O’Donnell E, Currie T, et al. Viral induction and targeted inhibition of galectin-1 in EBV+ posttransplant lymphoproliferative disorders. Blood. 2011;117:4315–22. doi: 10.1182/blood-2010-11-320481. [DOI] [PubMed] [Google Scholar]
- 31.Contal COQJ. An application of changepoint methods in studying the effect of age on survival in breast cancer. Computational Statistics and Data Analysis. 1999;30:253–70. [Google Scholar]
- 32.Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol. 2013;14:152–61. doi: 10.1038/ni.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev. 2016;271:246–59. doi: 10.1111/imr.12411. [DOI] [PubMed] [Google Scholar]
- 34.Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–9. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 35.Pace KE, Lee C, Stewart PL, Baum LG. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J Immunol. 1999;163:3801–11. [PubMed] [Google Scholar]
- 36.Nguyen JT, Evans DP, Galvan M, Pace KE, Leitenberg D, Bui TN, et al. CD45 modulates galectin-1-induced T cell death: regulation by expression of core 2 O-glycans. J Immunol. 2001;167:5697–707. doi: 10.4049/jimmunol.167.10.5697. [DOI] [PubMed] [Google Scholar]
- 37.Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, et al. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006;176:778–89. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- 38.Perillo NL, Uittenbogaart CH, Nguyen JT, Baum LG. Galectin-1, an endogenous lectin produced by thymic epithelial cells, induces apoptosis of human thymocytes. J Exp Med. 1997;185:1851–8. doi: 10.1084/jem.185.10.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood. 2007;109:2058–65. doi: 10.1182/blood-2006-04-016451. [DOI] [PubMed] [Google Scholar]
- 40.Rabinovich GA, Ilarregui JM. Conveying glycan information into T-cell homeostatic programs: a challenging role for galectin-1 in inflammatory and tumor microenvironments. Immunol Rev. 2009;230:144–59. doi: 10.1111/j.1600-065X.2009.00787.x. [DOI] [PubMed] [Google Scholar]
- 41.Cedeno-Laurent F, Opperman MJ, Barthel SR, Hays D, Schatton T, Zhan Q, et al. Metabolic inhibition of galectin-1-binding carbohydrates accentuates antitumor immunity. J Invest Dermatol. 2012;132:410–20. doi: 10.1038/jid.2011.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–34. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 43.Hsieh SH, Ying NW, Wu MH, Chiang WF, Hsu CL, Wong TY, et al. Galectin-1, a novel ligand of neuropilin-1, activates VEGFR-2 signaling and modulates the migration of vascular endothelial cells. Oncogene. 2008;27:3746–53. doi: 10.1038/sj.onc.1211029. [DOI] [PubMed] [Google Scholar]
- 44.Laderach DJ, Gentilini LD, Giribaldi L, Delgado VC, Nugnes L, Croci DO, et al. A unique galectin signature in human prostate cancer progression suggests galectin-1 as a key target for treatment of advanced disease. Cancer Res. 2013;73:86–96. doi: 10.1158/0008-5472.CAN-12-1260. [DOI] [PubMed] [Google Scholar]
- 45.Thijssen VL, Poirier F, Baum LG, Griffioen AW. Galectins in the tumor endothelium: opportunities for combined cancer therapy. Blood. 2007;110:2819–27. doi: 10.1182/blood-2007-03-077792. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.