

Abstract
Hypertension is a major health problem with great consequences for public health. Despite its role as the primary cause of significant morbidity and mortality associated with cardiovascular disease, the pathogenesis of essential hypertension remains largely unknown. The central nervous system in general, and the hypothalamus in particular, are intricately involved in the development and maintenance of hypertension. Over the last several decades, the understanding of the brain’s role in the development of hypertension has dramatically increased. This brief review is to summarize the neural mechanisms of hypertension with a focus on neuroendocrine and neurotransmitter involvement, highlighting recent findings which suggest that hypothalamic inflammation disrupts key signaling pathways to affect the central control of blood pressure, and therefore suggesting future development of interventional strategies that exploit recent findings pertaining to the hypothalamic control of blood pressure as well as the inflammatory-sympathetic mechanisms involved in hypertension.
Keywords: central nervous system, hypothalamus, hypertension, inflammation
Introduction
Hypertension is characterized by a chronic elevation in arterial pressure and is a major risk factor for many common causes of morbidity and mortality including stroke, myocardial infarction, congestive heart failure, and end-stage renal disease in many segments of the population (1). In the United States alone, high blood pressure affects an estimated 65 million individuals (2, 3) and contributes to the deaths of as many as 360,000 Americans every year. Globally, hypertension is the biggest contributor to disease burden and mortality in the world, accounting for 9.4 million deaths each year (4). Over the next decade, the global prevalence of hypertension is predicted to increase by 60% (5), despite advancements in awareness, antihypertensive therapy, and control of high blood pressure (6). For this reason, preventive strategies for those at risk and methods to both identify the undiagnosed and manage uncontrolled hypertension are urgently needed. Resolving these issues requires a deeper understanding of the physiology of blood-pressure regulation, the genetic traits that contribute to hypertensive phenotypes, and the identity of environmental factors that confer risk in susceptible individuals. Pertinently, attempts to study the pathogenic mechanisms of hypertension increasingly point to alterations in central nervous system (CNS) regulation of arterial pressure as a critical modulating factor (7). Many of these functional changes are concentrated in the hypothalamus (8), an area of the brain consisting of several nuclei that acts as the interface between the nervous and endocrine systems. The hypothalamus plays a crucial role in coordinating and integrating the activity of neural networks that control central blood pressure (9, 10). The intent of this brief review is to highlight recent findings that implicate the nervous system and the hypothalamus in particular in the pathogenesis and maintenance of hypertension. Particular emphasis is placed on recent findings that point to hypothalamic inflammation as a potential driver of pathogenic hypertension and therefore likely to inform new translational advances in the field.
Brief overview on pathophysiology of blood pressure regulation
Hypertension is broadly categorized as primary or secondary depending on the underlying pathogenic mechanism (11). Primary or essential hypertension represents the majority of cases, typically arising in middle or old age as a result of the interaction between non-specific genetic and environmental factors. A genetic link is supported by high heritability of blood pressures, elevated sibling recurrence-risk ratio, and higher concordance of blood pressures among monozygotic twins in comparison to dizygotic twins (12). Although rare mendelian hypertensive phenotypes are associated with mutations in a single gene (13–17), the genetic risk seems to be more commonly derived from variations in at least 65 distinct loci affecting blood pressure, each of modest effect size (18–22). Progression from a normotensive to hypertensive phenotype among genetically-predisposed individuals is likely to be influenced by a combination of environmental, behavioral and dietary factors. Common determinants of primary hypertension include aging, obesity, insulin resistance and excessive intake of salt, calories, and alcohol (11). Other potential risk factors that have garnered attention in recent years include sedentary lifestyle, stress, depression, low potassium intake, low calcium intake, intrauterine programming and early life events. In contrast to essential hypertension, secondary hypertension affects far fewer patients, develops at an earlier age, and is linked to an identifiable cause such as renal or endocrine disorder and oral contraceptive use. Notwithstanding the insights into the multi-factorial nature of hypertension, the precise cellular and molecular mechanisms that influence physiology to raise blood pressure remain poorly understood.
Unraveling the etiology of hypertension requires consideration of different systems that contribute to short-term blood pressure control. These include the well-characterized interactions between the vasculature, the kidney, and the central and sympathetic nervous systems (SNS), mediated by various, often shared, receptors and ligands. Mechanisms that maintain normotensive arterial pressure include baroreceptors that sense acute changes in blood vessel pressure and decrease or increase sympathetic nervous system (SNS) activity; activation of the renin-angiotensin system (RAS) due to a fall in renal perfusion pressure; adrenergic receptors (or adrenoceptors) that bind catecholamines and increase heart rate; factors produced by endothelial cells that cause vasodilation (e.g. nitric oxide) or vasoconstriction (e.g. endothelin); secretion of natriuretic peptides in response to increased pressure; and the kinin-kallikrein system, which influences vascular tone and renal salt handling (Figure 1). Many of these systems function autonomously to locally regulate blood flow (via alterations in cardiac output and blood volume), resistance (via arterial contraction and relaxation) and ultimately blood pressure.
Figure 1. Systemic responses to blood pressure change.
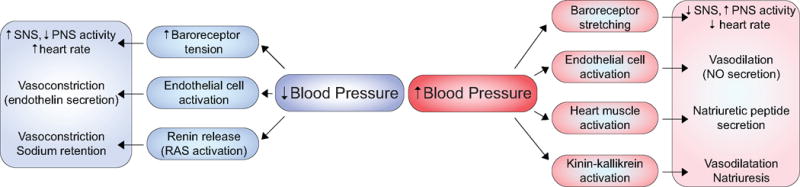
The body responds to changes in blood pressure by activating multiple homeostatic mechanisms. In response to decreased blood pressure, baroreceptors immediately sense decreased tension and signal for increased SNS outflow and decreased PNS outflow, effectively increasing heart rate. Concurrently, endothelial cells secrete endothelin, which constricts blood vessels. Renin released from juxtaglomerular cells of the kidney activates the RAS. In response to increased blood pressure, baroreceptors detect stretching and signal increased PNS outflow and decreased SNS outflow, effectively decreasing heart rate. Endothelial cells secrete nitric oxide, which dilates blood vessels. Cardiac muscle secretes natriuretic peptides in conjunction with activation of the kinin-kallikrein system to promote natriuresis and vasodilatory effects.
At the same time, the nervous system integrates signals from peripheral organs and helps to coordinate homeostatic responses (23, 24). The contributions of central pathways are perhaps best exemplified in the pathophysiological hallmarks of “neurogenic” essential hypertension. This form of hypertension is due to autonomic nervous system abnormalities originating in the afferent arm (e.g. baroreceptors, chemoreceptors and renal afferents) or in the central circuitry without a primary vascular or renal defect (10). Studies in animals – in which the contribution of causal factors and pathways underlying hypertension can be studied in a more systematic manner – reveal that circumventricular organs (CVOs), the hypothalamus, and the brain stem are critical regulatory regions. Some of the earliest studies in experimental models found that damage to the brain stem or the afferent components of the baroreceptor reflex pathway that terminate within the nucleus tractus solitarius (NTS) produce short-term (25) and long-term elevations in arterial pressure (26–28). Mechanistically, the increased arterial pressure is caused by increased regional vascular resistance (29) as a result of enhanced sympathetic tone (30) that is normally suppressed by inhibitory baroreceptor input. Thus, activation of carotid baroreceptors (31) or chemical stimulation of the NTS with adrenaline, noradrenaline and dopamine (32) decreases arterial pressure and heart rate. These early studies were instrumental in documenting neural mechanisms that could lead to enhanced central sympathetic outflow in hypertension.
Central modulation of blood pressure also involves the RAS. As mentioned above, it is well studied that peripheral RAS activation controls fluid and electrolyte balance when renal blood flow is reduced. However, components of the RAS (i.e. renin, angiotensinogen, angiotensin, angiotensin converting enzyme, angiotensin II, and angiotensin II receptor subtypes) are also found in the brain (33) and compelling evidence suggests the RAS can contribute to hypertension by modulating cardiovascular effects through the CNS (34, 35). In particular, angiotensin II (Ang II) stimulates the organum vasculosum and the subfornical organ, CVOs surrounding the anterior part of the third ventricle (36). Both sites are highly vascularized and lack a blood-brain barrier (BBB) making them responsive to both locally-produced (37) and circulating Ang II (38). Indeed, high levels of circulating Ang II induce the development of hypertension, which is mediated by increased production of reactive oxygen species (ROS) in the subfornical organ (39, 40). Most of the known actions of Ang II are mediated by angiotensin II type 1 (AT1) receptors. Their activation in hypertension is likely to have an effect on multiple brain structures in the network that controls SNS outflow including the paraventricular nucleus (PVN) in the hypothalamus, the median preoptic nucleus, and the rostral ventrolateral medulla in the brain stem (24, 41–43). Consistent with this point, several studies suggest that oxidative stress in the rostral ventrolateral medulla is a potent factor in the dysregulation of sympathetic outflow that accompanies the spontaneous development of hypertension (44–46). Ang II derived from the brain’s RAS (as opposed to circulating Ang II) is likely to play similar roles in the development of hypertension (47), but the factors that regulate this pathway’s activity remain unknown.
Salt sensitivity is one such factor that is likely to affect this pathway. High salt intake acutely reduces circulating renin-angiotensin activity and aldosterone concentration (48, 49). A third of essential hypertension patients with high-sodium consumption have lower plasma renin activity (50, 51), but are responsive to RAS inhibitors (52). One hypothesis to reconcile this apparent discrepancy is that the brain’s RAS response to salt intake may differ from the body’s RAS response (50, 53). High sodium intake in rats leads to a sustained increase in renin gene expression in the hypothalamus, despite reduced renal renin expression (54). In line with this evidence, angiotensin-converting enzyme (ACE) and AT1 expression in the hypothalamus and brain stem are elevated in salt-sensitive hypertension, particularly following activation of sodium channels in the brain (55). Blocking these sodium channels reduces blood pressure and SNS hyperactivity induced by hypertonic saline loading in the brain (56, 57). These results suggest high sodium loading increases brain RAS activity locally, which in turn increases sympathetic outflow to promote hypertension.
Central hypertensive regulation is also tightly coordinated by mineralocorticoid receptor (MR) expression and ligand responsiveness. Upon ligand binding, neuronal MRs enter the nucleus, forming dimers that complex with transcription factors to activate or repress target gene expression that culminates in increased SNS activity (58–60). Importantly, they have similar affinity to physiologic mineralocorticoids (aldosterone) and glucocorticoids (cortisol and corticosterone). However, aldosterone-targeted cells express both the MR and localized 11-β-hydroxysteroid dehydrogenase 2 (11βHSD2), an enzyme which converts cortisol and corticosterone into inactive metabolites; this increases relative aldosterone concentrations in close proximity to MRs (61). 11βHSD2 utilizes NAD+ as a cofactor, producing NADH and depleting MR-proximal concentrations of NAD+ (62). In the brain, 11βHSD2 is expressed at low levels apart from aldosterone-target neurons in the BBB-deficient zone of the NTS that influences sodium appetite (62–64). With sodium intake, these neurons quiesce resulting in decreased sodium appetite (64). However, NADH generated from 11βHSD2 activity limits the transcriptional activity of glucocorticoid-bound MRs. In the absence of 11βHSD2, increased NAD+ is thought to change the conformation of glucocorticoid-bound MR allowing it to have similar transcriptional activity to aldosterone-bound MR (62). Oxidative stress mimics this NADH-depleted state by redox imbalance, impairing normal MR function and activating glucocorticoid-bound MRs (65). Additionally, plasma levels of glucocorticoids are 1000 fold (total) or 100 fold (free) higher than that of aldosterone, and brain levels of these hormones have been shown to be similar (61, 62). Thus, neuronal MRs are bound and activated by basal glucocorticoids in normal physiological conditions.
MRs also work in congruence with AT1 receptors in the brain to drive SNS activity and subsequent hypertensive drive in the presence of excess mineralocorticoid (66). Both MR and the AT1 receptor in the subfornical organ increase Ang II-induced ROS production in the PVN and rostral ventrolateral medulla (67). Ang II activates NADPH oxidase to drive ROS production (68, 69), potentiating SNS hyperactivity (70). ACE mRNA and AT1 receptor mRNA are upregulated by aldosterone in hypothalamic tissue, further increasing Ang II production and subsequent ROS formation to drive hypertension (71).
Interestingly, the glucocorticoid receptor (GR) has only 1/10 the affinity to glucocorticoids that the MR does. Despite their widespread expression in the brain, GRs are thought to only be occupied with ligand during stress or the zenith of the circadian cycle for this reason (62, 72). The highest concentration of MRs is localized to the hippocampus, and activated GRs help to regulate the MR-mediated non-genomic stress response (73, 74) as well as hippocampal explicit memory formation (75). Imbalance between MR and GR function and expression contributes simultaneously to psychopathological disorders such as anxiety and PTSD and loss of cognitive function by dysregulating the hypothalamic-pituitary-adrenal axis (HPA axis) (75, 76). Increased GR increases HPA axis activity, while decreased GR decreases HPA axis activity. Due to the diurnal levels of glucocorticoids, the MR regulates HPA axis activity basally, and the GR during stressed conditions (77). In the hippocampus, MR and GR signal to inhibit or stimulate respectively secretion of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) from the PVN of the hypothalamus (78). Taken together, these studies demonstrate the complexity of action and signaling of glucocorticoids, mineralocorticoids, and their respective receptors in the nervous system.
Hypothalamic mechanisms of hypertension
Regulation of vasopressin secretion in hypertension
Accumulating evidence implicates increased AVP signaling in the pathogenesis of hypertension. AVP is produced by magnocellular neurons in the PVN and supraoptic nucleus (SON) of the hypothalamus and stimulates water reabsorption in the kidney to help maintain blood pressure. The concentration of circulating AVP is normally too low to have a measureable effect on blood pressure, but the AVP neuronal activity is dysregulated (79) fairly early in the development of hypertension (80). This effect on AVP neurons may be attributable, at least in part, to reduced inhibitory GABAergic input from baroreceptors in response to high salt intake (81). More recent findings suggest that such impairments in inhibitory signaling are mediated by brain-derived neurotrophic factor leading to increased excitability of hypothalamic AVP-secreting neurons. This in turn drives excess AVP release, which elevates arterial pressure (82, 83). Indeed, increased AVP expression is critical in the maintenance of hypertension in several experimental models involving RAS hyperactivity (79, 84–86). Although the precise mechanisms by which excess AVP secretion drives high blood pressure remain an ongoing topic of discussion, several pathways may be implicated including sympathoexcitation via V1a receptors in the PVN (87), brain RAS hyperactivity via V2 receptors (88), and peripheral vasoconstriction via V1 receptors (Figure 2) (82).
Figure 2. Hypothalamic mechanisms of hypertension.
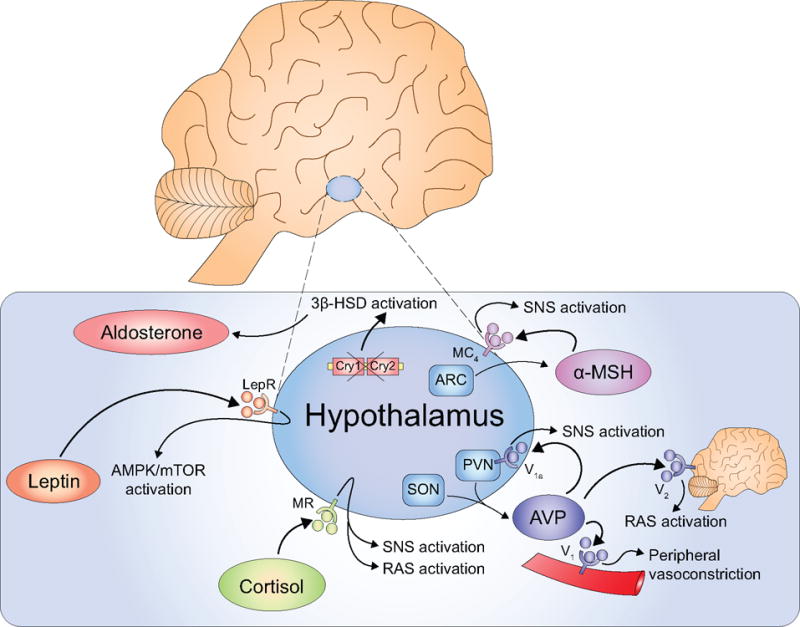
The hypothalamus activates the SNS and other pathways contributing to the pathogenesis of hypertension. Dysregulated AVP neurons in the SON and PVN produce excess AVP, which activates hypothalamic V1a, brain V2, and peripheral V1a receptors, thus activating the SNS, RAS, or endothelial cells, respectively. Circulating cortisol activates MRs in the hypothalamus to simultaneous stimulate the SNS and RAS. Leptin binds to the LepR to activate AMPK and the SNS. The ARC produces α-MSH, which binds to the MC4 in the hypothalamus to increase SNS outflow. Dysregulated clock gene expression promotes aldosterone production leading to salt-sensitive hypertension.
Hypertensive effect of steroid hormones in hypothalamus
Although the mechanisms underlying the centrally-mediated hypertensive responses to aldosterone have been well studied (89), the central effects of glucocorticoids are less understood. For example, intracerebroventricular injection of hydrocortisol increases SNS activity and induces hypertensive responses that are reversible with pretreatment using an Ang II antagonist or ACE inhibitor (90). As discussed previously, under normal conditions, cortisol can be converted to inactive metabolites by 11βHSD2 before acting on MRs (50, 91). While MRs are expressed in the hypothalamus, 11βHSD2 is barely detectable (92, 93), suggesting that circulating cortisol can act on the hypothalamus directly through the third ventricle, to increase sympathetic activity and blood pressure. Recent findings suggest that MR stimulation by cortisol may also modulate RAS activity downstream (91, 94). Thus, hypothalamic MRs sit at a delicate interface between glucocorticoid and mineralocorticoid stimulation.
Leptin-induced SNS activity in hypertension
Leptin is a hormone produced by adipose cells that helps to regulate energy balance by inhibiting hunger. Leptin levels are increased in obese humans (95) and have been shown to drive hypertension in rats (96). In addition, peripheral administration of an anti-leptin antibody decreases blood pressure and SNS activity in obese mice fed with a high fat diet (97). Although the leptin receptor (LepR) is expressed in multiple sites in the brain, leptin’s effects on SNS activity prominently involve the ventromedial hypothalamus, arcuate nucleus (ARC), and dorsomedial areas in the hypothalamus (97, 98). LepR deletion in the ARC attenuates leptin-induced increases in renal sympathetic discharge and resolves increased arterial pressure in diet-induced obese mice (99). In fact, ablation of LepR specifically in proopiomelanocortin (POMC) neurons, a major type of neuron in the ARC, can effectively reduce blood pressure (100). Recent evidence suggests that leptin-evoked increases in SNS activity are mediated by intracellular AMP-activated protein kinase (101) and mammalian target of rapamycin (mTORC1) signaling pathways (102), thus offering potential therapeutic targets to treat obesity-associated hypertension in the future.
Melanocortin receptor 4 (MC4) signaling in hypertension
MC4 is a member of the G-protein coupled receptor family and is activated by alpha-melanocyte-stimulating hormone (α-MSH). POMC neurons in the ARC send projections to the PVN and lateral hypothalamus where they release α-MSH. Thus, MC4 expression in POMC neurons is a critical component in the melanocortin system’s actions on feeding behavior, regulation of metabolism, SNS activation, and blood pressure homeostasis (103). Microinjection of a MC4 agonist into the PVN increases renal SNS activity and blood pressure in rats (104), while pharmacological blockade of MC4 in the PVN attenuates lumbar sympathetic nerve activity due to hyperinsulinemia (105). Intracerebroventricular injection of an MC4 antagonist markedly reduces blood pressure in spontaneously hypertensive rats in an SNS-dependent manner, irrespective of body weight fluctuations (106). Renal SNS activity due to central leptin and insulin administration on can be attenuated and abolished in heterozygous and homozygous MC4 knockout mice, respectively (107). Taken together, MC4 in POMC neurons plays a key role in several forms of hypertension.
Hypertension caused by circadian rhythm in the hypothalamus
It is well-known that cardiovascular functions, including blood pressure, show diurnal oscillation. Incidences of life-threatening cardiovascular events, such as stroke and acute myocardial infraction, also display a diurnal pattern, with increased incidence during the morning (108). The suprachiasmatic nucleus of the hypothalamus is the “central clock” that regulates physiological functions through the autonomic nervous system and humoral mediators. Clock genes are expressed in a circadian manner in the SCN; circadian variations associated with blood pressure are related to modifications in clock gene-regulated endogenous sleep-wake rhythms (109). Indeed, acute changes in blood pressure brought on by morning or sleep surge can modify cardiovascular risk (110). The underlying mechanism is hypothesized to involve multiple components including increased circulating blood volume due to salt sensitivity, excessive salt intake, autonomic nervous dysfunction, abnormal clock genes, and/or altered secretion of melatonin (109). The generation and maintenance of circadian rhythms involves two flavoproteins: cryptochrome-1 (Cry1) and cryptochrome-2 (Cry2) (111). Cry1 and Cry2-deficient mice are prone to salt sensitive hypertension due to increased activity of the adrenocortical aldosterone-producing enzyme, 3 beta-hydroxyl-steroid dehydrogenase (111). Recent studies also suggest that melatonin has multiple beneficial effects on the cardiovascular system; melatonin administration at bedtime reduces blood pressure in hypertensive patients (112). Thus, it is possible that alterations in circadian rhythm may affect melatonin levels resulting in autonomic nervous dysfunction, increased aldosterone, and subsequent hypertension.
Hypothalamic inflammatory mechanisms of hypertension
As detailed above, the hypothalamus acts as the central regulator of energy homeostasis – it senses metabolic cues and in turn modulates neurohormonal and neurotransmitter systems via endocrine signaling, inflammatory signaling, and neuronal plasticity (113–115). POMC and neuropeptide Y/agouti-related peptide (AGRP) neurons are the two major cell types in the mediobasal hypothalamus that play a vital role in energy balance. They reciprocally regulate energy homeostasis via anorexigenic and orexigenic effects, respectively. In addition, both POMC and AGRP neurons are regulated by leptin in opposite manners to affect energy homeostasis via negative and positive energy balance (116–119). Mounting evidence from experimental and clinical studies unequivocally has shown overnutrition is an important environmental factor capable of promoting neuroinflammation (120, 121). Obesity-associated hypertension is associated with the activation of pro-inflammatory signaling pathways (122) that promote the development of metabolic syndromes in several tissues (123–127). Metabolic inflammation chronically and negatively impacts neuronal regulatory functions including leptin and insulin signaling. This results in altered regulations, including central leptin and insulin resistance, that can drive increased blood pressure and energy imbalance (97, 128). Over activity of the hypothalamic IKKβ/NF-κB pathway has been recently shown to be as a critical modulator of hypothalamic inflammation (Figure 3). In particular, IKKβ/NF-κB driven hypothalamic inflammation induces blood pressure imbalance and insulin resistance in an obesity-independent manner (129–133). This inflammation seems to originate from the network of neurons, astrocytes and microglia, representing a new perspective on central inflammatory metabolic disorders (134, 135). The following describes the hypothalamic mechanisms of hypertension from a few bases that have been consistently connected with hypothalamic inflammation.
Figure 3. Pro-inflammatory hypertensive signalling in the hypothalamus.
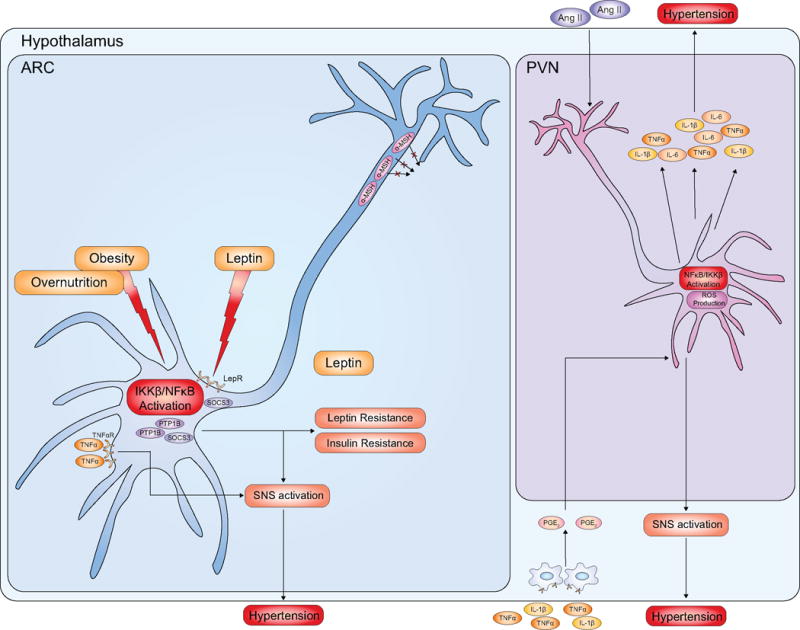
In response to overnutrition states, pro-inflammatory signalling including IKKβ/NFκB is activated in certain hypothalamic neurons such as POMC neurons in the ARC. NFκB activation triggers a variety of molecular reactions, such as increased levels of SOCS3 and of PTP1b, contributing to SNS activation and subsequent increased blood pressure. In addition, POMC neurons bind TNFα, which further stimulates SNS activation. Also, TNFα and IL-1β activate perivascular macrophages that produce prostaglandin E2, which signals through the PVN to activate the SNS and subsequent hypertension. Central RAS activation and Ang II production stimulates IKKβ/NFκB activation and ROS production in PVN neurons. Subsequent release of pro-inflammatory cytokines further contributes to ROS production, mitochondrial dysfunction, neuroinflammation and sustained increase in blood pressure leading to pathological hypertension.
Hypothalamic cytokines in hypertension
Cytokines orchestrate all phases of the immune response and function in highly complex networks to maintain homeostasis. A dynamic balance between pro and anti-inflammatory cytokines is required, and this contributes to changes in CNS physiology that promote hypertension. Circulating pro-inflammatory cytokines can pass through leaky blood vessels in CVOs or in areas where the BBB is disrupted. Alternatively, neuroactive cytokines including tumor necrosis factor-alpha (TNF-α) and interleukin-1β (IL-1β) can increase the activity of cyclooxygenase-2 in perivascular macrophages to generate prostaglandin E2. This chain of events results in increased discharge from PVN neurons, which regulate adrenocorticotropic hormone release, sympathetic outflow, and ultimately blood pressure elevation (136–138). Increased expression of pro-inflammatory cytokines in the hypothalamus is also associated with hypertension, including RAS-mediated blood pressure increases in rats (139). Bilateral NF-κB inhibition within the PVN attenuates Ang II-induced hypertensive response by reducing pro-inflammatory cytokines and ROS (140). Central administration of the ROS scavenger tempol attenuates Ang II-induced hypertension by decreasing the expression of pro-inflammatory cytokines in PVN (141). These findings highlight how pro-inflammatory signal transduction involving ROS drives central RAS-mediated hypertension.
Several pathways have been implicated in this response. Inhibiting the P44/42 mitogen-activated protein kinase (MAPK) signaling pathway in the PVN lessens Ang II-induced hypertension by reducing SNS activity (142). However, the expression of pro-inflammatory cytokines in this study failed to decrease after disruption of PVN P44/42 MAPK signaling, suggesting an alternative source for these cytokines. Possible cytokine-secreting cell types that contribute to the development of hypertension include glia and neurons (143–145). Supporting this, overexpression of anti-inflammatory IL-10 reduces both activated microglia and blood pressure in rats (146). Interestingly, Ang II can directly pass through the BBB to affect neuronal circuits (147, 148), or alternatively, increase BBB permeability, further contributing to baroreceptor reflex dysfunction and hypertension (149). IL-1β and TNF-α can also increase BBB permeability via disruption of tight junctions (150, 151). This finding is particularly intriguing considering that prorenin can increase the expression of TNF-α and IL-1β in the NTS via the NF-κB complex (152). TNF-α stimulation of the NF-κB pathway in POMC neurons leads to increased blood pressure by increasing SNS outflow (131, 132). Taken together, Ang-II and prorenin increase the expression of pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) and decrease the expression of anti-inflammatory cytokines in the hypothalamus. Subsequent activation of NF-κB signaling augments the pro-inflammatory response and increases permeability of BBB in the Ang-II-induced hypertension. This results in further inflammation and SNS activity further increasing blood pressure.
Hypothalamic endoplasmic reticulum (ER) stress in hypertension
The ER is a cellular organelle that regulates protein synthesis and secretion. The unfolded protein response (UPR) is an intracellular stress response to a buildup of newly synthesized, unfolded proteins in the ER. Several inflammatory signaling systems, including JAK-AP1 and NF-κB pathways, interact with the three prototypical branches of the UPR that regulate metabolism and SNS activity (153–158). Overnutrition-related ER stress in the hypothalamus activates NF-κB and is sufficient to cause insulin and leptin resistance, which increases SNS outflow and hypertension (131, 132). Similarly, reduced ER capacity in the hypothalamus of mice on a high-fat diet results in severe leptin resistance and leads to increased obesity (159). Intracerebroventricular injection of the ER stress inducer thapsigargin induces systemic insulin resistance and hypertension (132), while blocking ER stress induces leptin sensitization (159) and reduces obesity-related hypertension (132). In line with these findings, acute induction of hypertension by hypothalamus ER stress can also be abrogated by NF-κB inhibition (132). In summary, brain ER stress is likely involved in some form of hypothalamic inflammation and certain aspects of neurogenic hypertension involving increased SNS activity.
Hypothalamic oxidative stress in hypertension
As mentioned above, ROS help to drive hypertension both locally and systemically. Mitochondrial oxidative stress is frequent in overnutrition conditions and high levels of ROS in the PVN can modulate SNS activity as well as hypertension (160). Chronic Ang II infusion into the PVN leads to membrane mobilization of p47phox, a cytoplasmic NADPH oxidase subunit required to initiate ROS production (161). ROS reduce nitric oxide signal transduction in the PVN and increase glutamatergic signaling, which can contribute to neural dysfunction. However, enhanced nitric oxide signaling reduces blood pressure, decreases SNS activity, and shows anti-hypertensive effect via adrenomedullin receptors (162). Anti-oxidative treatments, such as overexpression of superoxide dismutase 1 (SOD1), or bilateral infusion of the radical scavenger tempol into the PVN inhibit ROS-driven SNS activation and hypertension (163). Regarding the mechanisms involved, inflammation is likely involved in the mitochondrial dysfunction (164). Mitochondrial dysfunction itself can also directly lead to the overexpression of pro-inflammatory cytokines, resulting in a feedforward loop characterized by increasing neuronal dysfunction. Notably, NF-κB inhibition in the PVN also abrogates the ROS production, which reduces inflammation in the hypothalamus and attenuates Ang II-dependent hypertension (140).
Hypothalamic pro-inflammatory IKKβ/NF-κB signaling in hypertension
Hypothalamic inflammation is frequently observed in overnutrition or obesity and is associated with IKKβ/NF-κB signaling pathway activation in the brain (131). Besides various cytokines and various intracellular stress responses that lead to activation of hypothalamic NF-κB, it can also be activated by excess leptin (165). Thus, while leptin’s anorexic effects are blunted in obese mice (166), the resultant chronic elevation of leptin levels may contribute to activating the NF-κB pathway in the hypothalamus. Activation of the NF-κB complex is a critical modulator for the expression of the suppressor of cytokine signaling 3 (SOCS3), which plays an important role in the development of leptin and insulin resistance in feeding dysregulation (167), as indeed SOCS3 deficiency in the hypothalamus causes elevated leptin sensitivity and resistance to diet-induced obesity (168–170). Activation of IKKβ/NF-κB pathway is also responsible for the upregulation of protein tyrosine phosphatase 1B (PTP1B), which further inhibits leptin and insulin signaling in a manner similar to SOCS3 (170). Therefore, there appears to be a vicious cycle consisting of inflammation, leptin resistance and pathological increase in leptin release. Of interest, while leptin resistance caused by hypothalamic IKKβ/NF-κB activation leads to impaired function in controlling appetite, the action of leptin in elevating blood pressure is abnormally augmented under this inflammatory condition of obesity. The underling divergence remains puzzling, but possibly involves different downstream molecular events and neural circuitries in the hypothalamus and brain.
Control of hypertension via targeting inflammatory-sympathetic mechanism
As hypothalamic and neuroinflammation related hypertension continues to attract more attention by researchers, treatments targeting central mechanisms of hypertension may be promising in the near future. The animal studies discussed above support the potential for novel pharmacological therapies and lifestyle modifications. Given that high sodium-mediated activation of RAS leads to expression of pro-inflammatory cytokines (55, 56), one promising option includes local inhibition of epithelial sodium channels in the CNS to prevent hypertension (171). Inhibition of RAS by renin inhibitors, ACEIs, angiotensin receptor blockers or MR blockers already shows benefit in clinical practice and treatment with such drugs can prevent future cardiovascular complications (171). Animal studies indicate that systematic administration of an angiotensin receptor blocker has anti-hypertensive effects that also prevent the SNS hyperactivity (172). Additionally, a leptin antagonist was recently shown to reduce blood pressure independent of body weight changes (97). ROS scavengers and immunosuppressive agents can also reduce blood pressure and have shown promise in both experimental models and humans (173, 174). Finally, epigallocatechin-3-O-gallate is a polyphenol present in green tea that is currently being tested for its antioxidant and anti-inflammatory properties. It has been shown to prevent hypertension and sympathetic outflow (175).
Conclusion
Over the last several decades, the understanding of the brain’s role in the development of hypertension has dramatically increased. Current understanding postulates that neurogenic hypertension involves dysregulation of different neural cell types and signaling pathways. As outlined in this review, hypothalamic inflammation is one such signaling pathway that can result in cellular dysfunction that is detrimental to blood pressure homeostasis. Future studies should be aimed at delineating hypothalamic inflammatory pathways and their cross talk as it pertains to neurogenic hypertension. Further recognition of the underlying mechanisms of hypertension will help generate more therapeutic targets for further treatment of human hypertension.
Acknowledgments
The authors sincerely thank the members of the Cai laboratory for their contributions to projects that were related to this review. These projects were supported by NIH R01 DK078750, R01 AG031774, R01 HL113180, and R01 DK099136 (all awarded to Dr. Cai).
Acronyms
- 11βHSD2
11beta-hydroxy steroid dehydrogenase 2
- α-MSH
Alpha-melanocyte-stimulating hormone
- ACE
Angiotensin converting enzyme
- ACEI
Angiotensin-converting enzyme inhibitor
- AGRP
Agouti-related peptide
- Ang II
Angiotensin II
- AP-1
Activator protein-1
- ARC
Arcuate nucleus
- AT1
Angiotensin II type 1
- AVP
Arginine vasopressin
- BBB
Blood-brain barrier
- CNS
Central nervous system
- CVO
Circumventricular organ
- ER
Endoplasmic reticulum
- GABA
Gamma-amino butyric acid
- GR
Glucocorticoid receptor
- HPA
Hypothalamic-pituitary-adrenal
- IKKβ
Inhibitor of nuclear factor-kappa B kinase subunit beta
- IL-1β
Interleukin-1β
- IL-10
Interleukin-10
- JAK
Janus kinase
- MC4
Melanocortin receptor 4
- MR
Mineralocorticoid receptor
- NF-κB
Nuclear factor-kappa B
- NTS
Nucleus tractus solitarius (solitary nucleus)
- PVN
Paraventricular nucleus
- PTSD
Posttraumatic stress disorder
- POMC
Proopiomelanocortin
- PTP1B
Protein tyrosine phosphatase 1B
- RAS
Renin-angiotensin-system
- ROS
Reactive oxygen species
- SCN
Suprachiasmatic nucleus
- SOCS3
Suppressor of cytokine signaling 3
- SON
Supraoptic nucleus
- SNS
Sympathetic nervous system
- TNF-α
Tumor necrosis factor-alpha
- UPR
Unfolded protein response
References
- 1.Lackland DT, Weber MA. Global burden of cardiovascular disease and stroke: hypertension at the core. The Canadian journal of cardiology. 2015;31(5):569–71. doi: 10.1016/j.cjca.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Yoon SS, Gu Q, Nwankwo T, Wright JD, Hong Y, Burt V. Trends in blood pressure among adults with hypertension: United States, 2003 to 2012. Hypertension. 2015;65(1):54–61. doi: 10.1161/HYPERTENSIONAHA.114.04012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA. 2010;303(20):2043–50. doi: 10.1001/jama.2010.650. [DOI] [PubMed] [Google Scholar]
- 4.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. The lancet. 2013;380(9859):2224–60. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet (London, England) 2005;365(9455):217–23. doi: 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 6.Chobanian AV. Shattuck Lecture. The hypertension paradox–more uncontrolled disease despite improved therapy. N Engl J Med. 2009;361(9):878–87. doi: 10.1056/NEJMsa0903829. [DOI] [PubMed] [Google Scholar]
- 7.DiBona GF. Sympathetic nervous system and hypertension. Hypertension. 2013;61(3):556–60. doi: 10.1161/HYPERTENSIONAHA.111.00633. [DOI] [PubMed] [Google Scholar]
- 8.de Wardener HE. The hypothalamus and hypertension. Physiological reviews. 2001;81(4):1599–658. doi: 10.1152/physrev.2001.81.4.1599. [DOI] [PubMed] [Google Scholar]
- 9.Hirooka Y, Kishi T, Ito K, Sunagawa K. Potential clinical application of recently discovered brain mechanisms involved in hypertension. Hypertension. 2013;62(6):995–1002. doi: 10.1161/HYPERTENSIONAHA.113.00801. [DOI] [PubMed] [Google Scholar]
- 10.Parati G, Esler M. The human sympathetic nervous system: its relevance in hypertension and heart failure. European heart journal. 2012;33(9):1058–66. doi: 10.1093/eurheartj/ehs041. [DOI] [PubMed] [Google Scholar]
- 11.Poulter NR, Prabhakaran D, Caulfield M. Hypertension. Lancet (London, England) 2015;386(9995):801–12. doi: 10.1016/S0140-6736(14)61468-9. [DOI] [PubMed] [Google Scholar]
- 12.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104(4):545–56. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 13.Lifton RP. Molecular genetics of human blood pressure variation. Science (New York, NY) 1996;272(5262):676–80. doi: 10.1126/science.272.5262.676. [DOI] [PubMed] [Google Scholar]
- 14.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, Pylypenko O, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nature genetics. 2012;44(4):456–60. s1–3. doi: 10.1038/ng.2218. [DOI] [PubMed] [Google Scholar]
- 16.Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (New York, NY) 2011;331(6018):768–72. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nature genetics. 2013;45(4):440–4. 4e1–2. doi: 10.1038/ng.2550. [DOI] [PubMed] [Google Scholar]
- 18.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478(7367):103–9. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wain LV, Verwoert GC, O’Reilly PF, Shi G, Johnson T, Johnson AD, et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nature genetics. 2011;43(10):1005–11. doi: 10.1038/ng.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munroe PB, Barnes MR, Caulfield MJ. Advances in blood pressure genomics. Circ Res. 2013;112(10):1365–79. doi: 10.1161/CIRCRESAHA.112.300387. [DOI] [PubMed] [Google Scholar]
- 21.Tragante V, Barnes MR, Ganesh SK, Lanktree MB, Guo W, Franceschini N, et al. Gene-centric meta-analysis in 87,736 individuals of European ancestry identifies multiple blood-pressure-related loci. American journal of human genetics. 2014;94(3):349–60. doi: 10.1016/j.ajhg.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nature genetics. 2015;47(11):1282–93. doi: 10.1038/ng.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiological reviews. 2010;90(2):513–57. doi: 10.1152/physrev.00007.2009. [DOI] [PubMed] [Google Scholar]
- 24.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7(5):335–46. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- 25.Doba N, Reis DJ. Acute fulminating neurogenic hypertension produced by brainstem lesions in the rat. Circulation Research. 1973;32(5):584–93. doi: 10.1161/01.res.32.5.584. [DOI] [PubMed] [Google Scholar]
- 26.Thrasher TN. Effects of chronic baroreceptor unloading on blood pressure in the dog. Am J Physiol Regul Integr Comp Physiol. 2005;288(4):R863–71. doi: 10.1152/ajpregu.00489.2004. [DOI] [PubMed] [Google Scholar]
- 27.Ferrario CM, McCubbin JW, Page IH. Hemodynamic characteristics of chronic experimental neurogenic hypertension in unanesthetized dogs. Circulation research. 1969;24(6):911–22. doi: 10.1161/01.res.24.6.911. [DOI] [PubMed] [Google Scholar]
- 28.Nathan MA, Reis DJ. Chronic labile hypertension produced by lesions of the nucleus tractus solitarii in the cat. Circulation research. 1977;40(1):72–81. doi: 10.1161/01.res.40.1.72. [DOI] [PubMed] [Google Scholar]
- 29.Snyder DW, Doba N, Reis DJ. Regional distribution of blood flow during arterial hypertension produced by lesions of the nucleus tractus solitarii in rats. Circulation research. 1978;42(1):87–91. doi: 10.1161/01.res.42.1.87. [DOI] [PubMed] [Google Scholar]
- 30.Doba N, Reis DJ. Role of central and peripheral adrenergic mechanisms in neurogenic hypertension produced by brainstem lesions in rat. Circulation research. 1974;34(3):293–301. doi: 10.1161/01.res.34.3.293. [DOI] [PubMed] [Google Scholar]
- 31.Lohmeier TE, Iliescu R. Chronic lowering of blood pressure by carotid baroreflex activation: mechanisms and potential for hypertension therapy. Hypertension. 2011;57(5):880–6. doi: 10.1161/HYPERTENSIONAHA.108.119859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zandberg P, De Jong W, De Wied D. Effect of catecholamine-receptor stimulating agents on blood pressure after local application in the nucleus tractus solitarii of the medulla oblongata. European journal of pharmacology. 1979;55(1):43–56. doi: 10.1016/0014-2999(79)90146-8. [DOI] [PubMed] [Google Scholar]
- 33.Lippoldt A, Paul M, Fuxe K, Ganten D. The brain renin-angiotensin system: molecular mechanisms of cell to cell interactions. Clinical and Experimental Hypertension. 1995;17(1–2):251–66. doi: 10.3109/10641969509087069. [DOI] [PubMed] [Google Scholar]
- 34.Phillips M. Angiotensin in the brain. Neuroendocrinology. 1978;25(6):354–77. doi: 10.1159/000122756. [DOI] [PubMed] [Google Scholar]
- 35.Andersson B, ERIDSSON S, Rundgren M. Angiotensin and the brain. Acta physiologica scandinavica. 1995;155(2):117–25. doi: 10.1111/j.1748-1716.1995.tb09956.x. [DOI] [PubMed] [Google Scholar]
- 36.McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, et al. The brain renin-angiotensin system: location and physiological roles. Int J Biochem Cell Biol. 2003;35(6):901–18. doi: 10.1016/s1357-2725(02)00306-0. [DOI] [PubMed] [Google Scholar]
- 37.Ganten D, Fuxe K, Phillips MI, Mann JF, Ganten U. The brain isorenin-angiotensin system: biochemistry, localization, and possible role in drinking and blood pressure regulation. Frontiers in neuroendocrinology. 1978;5:61–99. [Google Scholar]
- 38.Fink GD, Haywood JR, Bryan WJ, Packwood W, Brody MJ. Central site for pressor action of blood-borne angiotensin in rat. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 1980;239(3):R358–R61. doi: 10.1152/ajpregu.1980.239.3.R358. [DOI] [PubMed] [Google Scholar]
- 39.Montezano AC, Touyz RM. Molecular mechanisms of hypertension–reactive oxygen species and antioxidants: a basic science update for the clinician. The Canadian journal of cardiology. 2012;28(3):288–95. doi: 10.1016/j.cjca.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 40.Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95(2):210–6. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- 41.Peterson JR, Sharma RV, Davisson RL. Reactive oxygen species in the neuropathogenesis of hypertension. Curr Hypertens Rep. 2006;8(3):232–41. doi: 10.1007/s11906-006-0056-1. [DOI] [PubMed] [Google Scholar]
- 42.Ito S, Komatsu K, Tsukamoto K, Kanmatsuse K, Sved AF. Ventrolateral medulla AT1 receptors support blood pressure in hypertensive rats. Hypertension. 2002;40(4):552–9. doi: 10.1161/01.hyp.0000033812.99089.92. [DOI] [PubMed] [Google Scholar]
- 43.Osborn JW, Hendel MD, Collister JP, Ariza-Guzman PA, Fink GD. The role of the subfornical organ in angiotensin II-salt hypertension in the rat. Exp Physiol. 2012;97(1):80–8. doi: 10.1113/expphysiol.2011.060491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishihara M, Hirooka Y, Matsukawa R, Kishi T, Sunagawa K. Oxidative stress in the rostral ventrolateral medulla modulates excitatory and inhibitory inputs in spontaneously hypertensive rats. J Hypertens. 2012;30(1):97–106. doi: 10.1097/HJH.0b013e32834e1df4. [DOI] [PubMed] [Google Scholar]
- 45.Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109(19):2357–62. doi: 10.1161/01.CIR.0000128695.49900.12. [DOI] [PubMed] [Google Scholar]
- 46.Nishihara M, Hirooka Y, Kishi T, Sunagawa K. Different role of oxidative stress in paraventricular nucleus and rostral ventrolateral medulla in cardiovascular regulation in awake spontaneously hypertensive rats. J Hypertens. 2012;30(9):1758–65. doi: 10.1097/HJH.0b013e32835613d7. [DOI] [PubMed] [Google Scholar]
- 47.Morimoto S, Cassell MD, Beltz TG, Johnson AK, Davisson RL, Sigmund CD. Elevated blood pressure in transgenic mice with brain-specific expression of human angiotensinogen driven by the glial fibrillary acidic protein promoter. Circulation research. 2001;89(4):365–72. doi: 10.1161/hh1601.094988. [DOI] [PubMed] [Google Scholar]
- 48.Adrogue HJ, Madias NE. Sodium and potassium in the pathogenesis of hypertension. N Engl J Med. 2007;356(19):1966–78. doi: 10.1056/NEJMra064486. [DOI] [PubMed] [Google Scholar]
- 49.Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, et al. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol. 2012;302(5):H1031–49. doi: 10.1152/ajpheart.00899.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi H, Yoshika M, Komiyama Y, Nishimura M. The central mechanism underlying hypertension: a review of the roles of sodium ions, epithelial sodium channels, the renin-angiotensin-aldosterone system, oxidative stress and endogenous digitalis in the brain. Hypertens Res. 2011;34(11):1147–60. doi: 10.1038/hr.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woods JW, Pittman AW, Pulliam CC, Werk EE, Jr, Waider W, Allen CA. Renin profiling in hypertension and its use in treatment with propranolol and chlorthalidone. N Engl J Med. 1976;294(21):1137–43. doi: 10.1056/NEJM197605202942101. [DOI] [PubMed] [Google Scholar]
- 52.Minami J, Ishimitsu T, Matsuoka H. Is there overlap in blood-pressure response to the blockers of the renin-angiotensin system between lower and higher renin subjects? Am J Hypertens. 2008;21(2):130–1. doi: 10.1038/ajh.2007.40. author reply 2. [DOI] [PubMed] [Google Scholar]
- 53.Sumners C, Phillips MI. Central injection of angiotensin II alters catecholamine activity in rat brain. Am J Physiol. 1983;244(2):R257–63. doi: 10.1152/ajpregu.1983.244.2.R257. [DOI] [PubMed] [Google Scholar]
- 54.Nishimura M, Nanbu A, Ohtsuka K, Takahashi H, Iwai N, Kinoshita M, et al. Sodium intake regulates renin gene expression differently in the hypothalamus and kidney of rats. J Hypertens. 1997;15(5):509–16. doi: 10.1097/00004872-199715050-00006. [DOI] [PubMed] [Google Scholar]
- 55.Nishimura M, Ohtsuka K, Takahashi H, Yoshimura M. Role of FMRFamide-activated brain sodium channel in salt-sensitive hypertension. Hypertension. 2000;35(1 Pt 2):443–50. doi: 10.1161/01.hyp.35.1.443. [DOI] [PubMed] [Google Scholar]
- 56.Nishimura M, Ohtsuka K, Nanbu A, Takahashi H, Yoshimura M. Benzamil blockade of brain Na+ channels averts Na(+)-induced hypertension in rats. Am J Physiol. 1998;274(3 Pt 2):R635–44. doi: 10.1152/ajpregu.1998.274.3.R635. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Huang BS, Leenen FH. Brain sodium channels and ouabainlike compounds mediate central aldosterone-induced hypertension. Am J Physiol Heart Circ Physiol. 2003;285(6):H2516–23. doi: 10.1152/ajpheart.00299.2003. [DOI] [PubMed] [Google Scholar]
- 58.Pascual-Le Tallec L, Lombes M. The mineralocorticoid receptor: a journey exploring its diversity and specificity of action. Molecular endocrinology (Baltimore, Md) 2005;19(9):2211–21. doi: 10.1210/me.2005-0089. [DOI] [PubMed] [Google Scholar]
- 59.Grossmann C, Ruhs S, Langenbruch L, Mildenberger S, Stratz N, Schumann K, et al. Nuclear shuttling precedes dimerization in mineralocorticoid receptor signaling. Chem Biol. 2012;19(6):742–51. doi: 10.1016/j.chembiol.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 60.Huang BS, Wang H, Leenen FH. Enhanced sympathoexcitatory and pressor responses to central Na+ in Dahl salt-sensitive vs. -resistant rats. Am J Physiol Heart Circ Physiol. 2001;281(5):H1881–9. doi: 10.1152/ajpheart.2001.281.5.H1881. [DOI] [PubMed] [Google Scholar]
- 61.Chen J, Gomez-Sanchez CE, Penman A, May PJ, Gomez-Sanchez E. Expression of mineralocorticoid and glucocorticoid receptors in preautonomic neurons of the rat paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol. 2014;306(5):R328–40. doi: 10.1152/ajpregu.00506.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gomez-Sanchez EP, Gomez-Sanchez CE. Central regulation of blood pressure by the mineralocorticoid receptor. Mol Cell Endocrinol. 2012;350(2):289–98. doi: 10.1016/j.mce.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shekhtman E, Geerling JC, Loewy AD. Aldosterone-sensitive neurons of the nucleus of the solitary tract: multisynaptic pathway to the nucleus accumbens. J Comp Neurol. 2007;501(2):274–89. doi: 10.1002/cne.21245. [DOI] [PubMed] [Google Scholar]
- 64.Geerling JC, Engeland WC, Kawata M, Loewy AD. Aldosterone target neurons in the nucleus tractus solitarius drive sodium appetite. J Neurosci. 2006;26(2):411–7. doi: 10.1523/JNEUROSCI.3115-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Funder JW. Mineralocorticoid receptor activation and oxidative stress. Hypertension. 2007;50(5):840–1. doi: 10.1161/HYPERTENSIONAHA.107.098012. [DOI] [PubMed] [Google Scholar]
- 66.Xue B, Beltz TG, Yu Y, Guo F, Gomez-Sanchez CE, Hay M, et al. Central interactions of aldosterone and angiotensin II in aldosterone- and angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol. 2011;300(2):H555–64. doi: 10.1152/ajpheart.00847.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang HW, Huang BS, White RA, Chen A, Ahmad M, Leenen FH. Mineralocorticoid and angiotensin II type 1 receptors in the subfornical organ mediate angiotensin II – induced hypothalamic reactive oxygen species and hypertension. Neuroscience. 2016;329:112–21. doi: 10.1016/j.neuroscience.2016.04.050. [DOI] [PubMed] [Google Scholar]
- 68.Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, et al. Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ Res. 2004;95(9):937–44. doi: 10.1161/01.RES.0000146676.04359.64. [DOI] [PubMed] [Google Scholar]
- 69.Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, et al. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res. 2002;91(11):1038–45. doi: 10.1161/01.res.0000043501.47934.fa. [DOI] [PubMed] [Google Scholar]
- 70.Campese VM, Shaohua Y, Huiquin Z. Oxidative stress mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension. 2005;46(3):533–9. doi: 10.1161/01.HYP.0000179088.57586.26. [DOI] [PubMed] [Google Scholar]
- 71.Zhang ZH, Yu Y, Kang YM, Wei SG, Felder RB. Aldosterone acts centrally to increase brain renin-angiotensin system activity and oxidative stress in normal rats. Am J Physiol Heart Circ Physiol. 2008;294(2):H1067–74. doi: 10.1152/ajpheart.01131.2007. [DOI] [PubMed] [Google Scholar]
- 72.De Kloet ER. Hormones and the stressed brain. Ann N Y Acad Sci. 2004;1018:1–15. doi: 10.1196/annals.1296.001. [DOI] [PubMed] [Google Scholar]
- 73.Joels M, Sarabdjitsingh RA, Karst H. Unraveling the time domains of corticosteroid hormone influences on brain activity: rapid, slow, and chronic modes. Pharmacol Rev. 2012;64(4):901–38. doi: 10.1124/pr.112.005892. [DOI] [PubMed] [Google Scholar]
- 74.Gomez-Sanchez EP. Brain mineralocorticoid receptors in cognition and cardiovascular homeostasis. Steroids. 2014;91:20–31. doi: 10.1016/j.steroids.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kellendonk C, Gass P, Kretz O, Schutz G, Tronche F. Corticosteroid receptors in the brain: gene targeting studies. Brain Res Bull. 2002;57(1):73–83. doi: 10.1016/s0361-9230(01)00638-4. [DOI] [PubMed] [Google Scholar]
- 76.Harris AP, Holmes MC, de Kloet ER, Chapman KE, Seckl JR. Mineralocorticoid and glucocorticoid receptor balance in control of HPA axis and behaviour. Psychoneuroendocrinology. 2013;38(5):648–58. doi: 10.1016/j.psyneuen.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 77.Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23(5):477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 78.De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- 79.Yi SS, Kim HJ, Do SG, Lee YB, Ahn HJ, Hwang IK, et al. Arginine vasopressin (AVP) expressional changes in the hypothalamic paraventricular and supraoptic nuclei of stroke-prone spontaneously hypertensive rats. Anat Cell Biol. 2012;45(2):114–20. doi: 10.5115/acb.2012.45.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han SY, Bouwer GT, Seymour AJ, Korpal AK, Schwenke DO, Brown CH. Induction of hypertension blunts baroreflex inhibition of vasopressin neurons in the rat. European Journal of Neuroscience. 2015;42(9):2690–8. doi: 10.1111/ejn.13062. [DOI] [PubMed] [Google Scholar]
- 81.Kim YB, Kim YS, Kim WB, Shen FY, Lee SW, Chung HJ, et al. GABAergic excitation of vasopressin neurons: possible mechanism underlying sodium-dependent hypertension. Circ Res. 2013;113(12):1296–307. doi: 10.1161/CIRCRESAHA.113.301814. [DOI] [PubMed] [Google Scholar]
- 82.Choe KY, Han SY, Gaub P, Shell B, Voisin DL, Knapp BA, et al. High salt intake increases blood pressure via BDNF-mediated downregulation of KCC2 and impaired baroreflex inhibition of vasopressin neurons. Neuron. 2015;85(3):549–60. doi: 10.1016/j.neuron.2014.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marosi K, Mattson MP. Hold the Salt: Vasopressor Role for BDNF. Cell metabolism. 2015;21(4):509–10. doi: 10.1016/j.cmet.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davisson RL, Yang G, Beltz TG, Cassell MD, Johnson AK, Sigmund CD. The brain renin-angiotensin system contributes to the hypertension in mice containing both the human renin and human angiotensinogen transgenes. Circulation Research. 1998;83(10):1047–58. doi: 10.1161/01.res.83.10.1047. [DOI] [PubMed] [Google Scholar]
- 85.Morimoto S, Cassell MD, Sigmund CD. The brain renin-angiotensin system in transgenic mice carrying a highly regulated human renin transgene. Circulation research. 2002;90(1):80–6. doi: 10.1161/hh0102.102272. [DOI] [PubMed] [Google Scholar]
- 86.Crofton JT, Share L, Shade RE, Lee-Kwon W, Manning M, Sawyer WH. The importance of vasopressin in the development and maintenance of DOC-salt hypertension in the rat. Hypertension. 1979;1(1):31–8. doi: 10.1161/01.hyp.1.1.31. [DOI] [PubMed] [Google Scholar]
- 87.Ribeiro N, do Nascimento Panizza H, dos Santos KM, Ferreira-Neto HC, Antunes VR. Salt-induced sympathoexcitation involves vasopressin V1a receptor activation in the paraventricular nucleus of the hypothalamus. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2015;309(11):R1369–R79. doi: 10.1152/ajpregu.00312.2015. [DOI] [PubMed] [Google Scholar]
- 88.Littlejohn NK, Siel RB, Ketsawatsomkron P, Pelham CJ, Pearson NA, Hilzendeger AM, et al. Hypertension in mice with transgenic activation of the brain renin-angiotensin system is vasopressin dependent. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2013;304(10):R818–R28. doi: 10.1152/ajpregu.00082.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gomez-Sanchez EP, Gomez-Sanchez CM, Plonczynski M, Gomez-Sanchez CE. Aldosterone synthesis in the brain contributes to Dahl salt-sensitive rat hypertension. Exp Physiol. 2010;95(1):120–30. doi: 10.1113/expphysiol.2009.048900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takahashi H, Takeda K, Ashizawa H, Inoue A, Yoneda S, Yoshimura M, et al. Centrally induced cardiovascular and sympathetic responses to hydrocortisone in rats. Am J Physiol. 1983;245(6):H1013–8. doi: 10.1152/ajpheart.1983.245.6.H1013. [DOI] [PubMed] [Google Scholar]
- 91.Geerling JC, Loewy AD. 11beta-hydroxysteroid dehydrogenase 2 vs. transgene: discrepant loci of expression in the adult brain. Am J Physiol Renal Physiol. 2007;293(1):F440–1. doi: 10.1152/ajprenal.00517.2006. author reply F2–3. [DOI] [PubMed] [Google Scholar]
- 92.Geerling JC, Kawata M, Loewy AD. Aldosterone-sensitive neurons in the rat central nervous system. J Comp Neurol. 2006;494(3):515–27. doi: 10.1002/cne.20808. [DOI] [PubMed] [Google Scholar]
- 93.Robson AC, Leckie CM, Seckl JR, Holmes MC. 11 Beta-hydroxysteroid dehydrogenase type 2 in the postnatal and adult rat brain. Brain Res Mol Brain Res. 1998;61(1–2):1–10. doi: 10.1016/s0169-328x(98)00161-2. [DOI] [PubMed] [Google Scholar]
- 94.Leenen FH. Actions of circulating angiotensin II and aldosterone in the brain contributing to hypertension. Am J Hypertens. 2014;27(8):1024–32. doi: 10.1093/ajh/hpu066. [DOI] [PubMed] [Google Scholar]
- 95.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1(11):1155–61. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 96.Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31(1 Pt 2):409–14. doi: 10.1161/01.hyp.31.1.409. [DOI] [PubMed] [Google Scholar]
- 97.Simonds SE, Pryor JT, Ravussin E, Greenway FL, Dileone R, Allen AM, et al. Leptin mediates the increase in blood pressure associated with obesity. Cell. 2014;159(6):1404–16. doi: 10.1016/j.cell.2014.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mark AL. Selective leptin resistance revisited. Am J Physiol Regul Integr Comp Physiol. 2013;305(6):R566–81. doi: 10.1152/ajpregu.00180.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harlan SM, Morgan DA, Agassandian K, Guo DF, Cassell MD, Sigmund CD, et al. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ Res. 2011;108(7):808–12. doi: 10.1161/CIRCRESAHA.111.240226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.do Carmo JM, da Silva AA, Cai Z, Lin S, Dubinion JH, Hall JE. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension. 2011;57(5):918–26. doi: 10.1161/HYPERTENSIONAHA.110.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tanida M, Yamamoto N, Shibamoto T, Rahmouni K. Involvement of hypothalamic AMP-activated protein kinase in leptin-induced sympathetic nerve activation. PloS one. 2013;8(2):e56660. doi: 10.1371/journal.pone.0056660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harlan SM, Guo D-F, Morgan DA, Fernandes-Santos C, Rahmouni K. Hypothalamic mTORC1 signaling controls sympathetic nerve activity and arterial pressure and mediates leptin effects. Cell metabolism. 2013;17(4):599–606. doi: 10.1016/j.cmet.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31(4):506–43. doi: 10.1210/er.2009-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li P, Cui BP, Zhang LL, Sun HJ, Liu TY, Zhu GQ. Melanocortin 3/4 receptors in paraventricular nucleus modulate sympathetic outflow and blood pressure. Exp Physiol. 2013;98(2):435–43. doi: 10.1113/expphysiol.2012.067256. [DOI] [PubMed] [Google Scholar]
- 105.Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension. 2011;57(3):435–41. doi: 10.1161/HYPERTENSIONAHA.110.160671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.da Silva AA, do Carmo JM, Kanyicska B, Dubinion J, Brandon E, Hall JE. Endogenous melanocortin system activity contributes to the elevated arterial pressure in spontaneously hypertensive rats. Hypertension. 2008;51(4):884–90. doi: 10.1161/HYPERTENSIONAHA.107.100636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rahmouni K, Haynes WG, Morgan DA, Mark AL. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J Neurosci. 2003;23(14):5998–6004. doi: 10.1523/JNEUROSCI.23-14-05998.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. 1989;79(4):733–43. doi: 10.1161/01.cir.79.4.733. [DOI] [PubMed] [Google Scholar]
- 109.Takeda N, Maemura K. Circadian clock and vascular disease. Hypertens Res. 2010;33(7):645–51. doi: 10.1038/hr.2010.68. [DOI] [PubMed] [Google Scholar]
- 110.Sheppard JP, Hodgkinson J, Riley R, Martin U, Bayliss S, McManus RJ. Prognostic significance of the morning blood pressure surge in clinical practice: a systematic review. Am J Hypertens. 2015;28(1):30–41. doi: 10.1093/ajh/hpu104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, et al. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med. 2010;16(1):67–74. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- 112.Gubin DG, Gubin GD, Gapon LI, Weinert D. Daily Melatonin Administration Attenuates Age-Dependent Disturbances of Cardiovascular Rhythms. Curr Aging Sci. 2016;9(1):5–13. doi: 10.2174/1874609809666151130220011. [DOI] [PubMed] [Google Scholar]
- 113.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404(6778):661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 114.Blouet C, Schwartz GJ. Hypothalamic nutrient sensing in the control of energy homeostasis. Behav Brain Res. 2010;209(1):1–12. doi: 10.1016/j.bbr.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 115.Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J. 1998;17(17):5085–94. doi: 10.1093/emboj/17.17.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: perception and integration. Nat Rev Genet. 2002;3(8):589–600. doi: 10.1038/nrg862. [DOI] [PubMed] [Google Scholar]
- 117.Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36(2):199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 118.Schwartz MW, Woods SC, Seeley RJ, Barsh GS, Baskin DG, Leibel RL. Is the energy homeostasis system inherently biased toward weight gain? Diabetes. 2003;52(2):232–8. doi: 10.2337/diabetes.52.2.232. [DOI] [PubMed] [Google Scholar]
- 119.Zigman JM, Elmquist JK. Minireview: From anorexia to obesity–the yin and yang of body weight control. Endocrinology. 2003;144(9):3749–56. doi: 10.1210/en.2003-0241. [DOI] [PubMed] [Google Scholar]
- 120.Tang Y, Purkayastha S, Cai D. Hypothalamic microinflammation: a common basis of metabolic syndrome and aging. Trends Neurosci. 2015;38(1):36–44. doi: 10.1016/j.tins.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Purkayastha S, Cai D. Neuroinflammatory basis of metabolic syndrome. Mol Metab. 2013;2(4):356–63. doi: 10.1016/j.molmet.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mathieu P, Poirier P, Pibarot P, Lemieux I, Després J-P. Visceral obesity the link among inflammation, hypertension, and cardiovascular disease. Hypertension. 2009;53(4):577–84. doi: 10.1161/HYPERTENSIONAHA.108.110320. [DOI] [PubMed] [Google Scholar]
- 123.Cai D. NFkappaB-mediated metabolic inflammation in peripheral tissues versus central nervous system. Cell Cycle. 2009;8(16):2542–8. doi: 10.4161/cc.8.16.9386. [DOI] [PubMed] [Google Scholar]
- 124.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 125.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121(6):2111–7. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118(9):2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shoelson SE, Goldfine AB. Getting away from glucose: fanning the flames of obesity-induced inflammation. Nat Med. 2009;15(4):373–4. doi: 10.1038/nm0409-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem. 2011;286(37):32324–32. doi: 10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296(5):E1003–12. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat Med. 2011;17(7):883–7. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2011;108(7):2939–44. doi: 10.1073/pnas.1006875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, et al. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology. 2011;152(4):1314–26. doi: 10.1210/en.2010-0659. [DOI] [PubMed] [Google Scholar]
- 134.Horvath TL, Sarman B, Garcia-Caceres C, Enriori PJ, Sotonyi P, Shanabrough M, et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A. 2010;107(33):14875–80. doi: 10.1073/pnas.1004282107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122(1):153–62. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Felder RB. Mineralocorticoid receptors, inflammation and sympathetic drive in a rat model of systolic heart failure. Exp Physiol. 2010;95(1):19–25. doi: 10.1113/expphysiol.2008.045948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Felder RB, Yu Y, Zhang ZH, Wei SG. Pharmacological treatment for heart failure: a view from the brain. Clin Pharmacol Ther. 2009;86(2):216–20. doi: 10.1038/clpt.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schiltz JC, Sawchenko PE. Signaling the brain in systemic inflammation: the role of perivascular cells. Front Biosci. 2003;8:s1321–9. doi: 10.2741/1211. [DOI] [PubMed] [Google Scholar]
- 139.Qi J, Zhang DM, Suo YP, Song XA, Yu XJ, Elks C, et al. Renin-angiotensin system modulates neurotransmitters in the paraventricular nucleus and contributes to angiotensin II-induced hypertensive response. Cardiovasc Toxicol. 2013;13(1):48–54. doi: 10.1007/s12012-012-9184-9. [DOI] [PubMed] [Google Scholar]
- 140.Cardinale JP, Sriramula S, Mariappan N, Agarwal D, Francis J. Angiotensin II-induced hypertension is modulated by nuclear factor-kappaBin the paraventricular nucleus. Hypertension. 2012;59(1):113–21. doi: 10.1161/HYPERTENSIONAHA.111.182154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Su Q, Qin DN, Wang FX, Ren J, Li HB, Zhang M, et al. Inhibition of reactive oxygen species in hypothalamic paraventricular nucleus attenuates the renin-angiotensin system and proinflammatory cytokines in hypertension. Toxicol Appl Pharmacol. 2014;276(2):115–20. doi: 10.1016/j.taap.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 142.Yu Y, Xue BJ, Zhang ZH, Wei SG, Beltz TG, Guo F, et al. Early interference with p44/42 mitogen-activated protein kinase signaling in hypothalamic paraventricular nucleus attenuates angiotensin II-induced hypertension. Hypertension. 2013;61(4):842–9. doi: 10.1161/HYPERTENSIONAHA.111.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Saavedra JM. Angiotensin II AT(1) receptor blockers as treatments for inflammatory brain disorders. Clin Sci (Lond) 2012;123(10):567–90. doi: 10.1042/CS20120078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wu KL, Chan SH, Chan JY. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation. 2012;9:212. doi: 10.1186/1742-2094-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.de Kloet AD, Krause EG, Shi PD, Zubcevic J, Raizada MK, Sumners C. Neuroimmune communication in hypertension and obesity: a new therapeutic angle? Pharmacol Ther. 2013;138(3):428–40. doi: 10.1016/j.pharmthera.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56(2):297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience. 2010;171(3):852–8. doi: 10.1016/j.neuroscience.2010.09.029. [DOI] [PubMed] [Google Scholar]
- 148.Paton JF, Wang S, Polson JW, Kasparov S. Signalling across the blood brain barrier by angiotensin II: novel implications for neurogenic hypertension. J Mol Med (Berl) 2008;86(6):705–10. doi: 10.1007/s00109-008-0324-4. [DOI] [PubMed] [Google Scholar]
- 149.Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood-brain barrier. Hypertension. 2014;63(3):572–9. doi: 10.1161/HYPERTENSIONAHA.113.01743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Labus J, Hackel S, Lucka L, Danker K. Interleukin-1beta induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J Neurosci Methods. 2014;228:35–45. doi: 10.1016/j.jneumeth.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 151.Rochfort KD, Collins LE, Murphy RP, Cummins PM. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS One. 2014;9(7):e101815. doi: 10.1371/journal.pone.0101815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zubcevic J, Jun JY, Lamont G, Murca TM, Shi P, Yuan W, et al. Nucleus of the solitary tract (pro)renin receptor-mediated antihypertensive effect involves nuclear factor-kappaB-cytokine signaling in the spontaneously hypertensive rat. Hypertension. 2013;61(3):622–7. doi: 10.1161/HYPERTENSIONAHA.111.199836. [DOI] [PubMed] [Google Scholar]
- 153.Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10(4):249–59. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sabio G, Cavanagh-Kyros J, Barrett T, Jung DY, Ko HJ, Ong H, et al. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev. 2010;24(3):256–64. doi: 10.1101/gad.1878510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–6. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 156.Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science (New York, NY) 2008;322(5907):1539–43. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, et al. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009;10(6):491–8. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6(5):386–97. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 159.Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9(1):35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 160.Carmichael CY, Wainford RD. Hypothalamic signaling mechanisms in hypertension. Curr Hypertens Rep. 2015;17(5):39. doi: 10.1007/s11906-015-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Coleman CG, Wang G, Faraco G, Marques Lopes J, Waters EM, Milner TA, et al. Membrane trafficking of NADPH oxidase p47(phox) in paraventricular hypothalamic neurons parallels local free radical production in angiotensin II slow-pressor hypertension. J Neurosci. 2013;33(10):4308–16. doi: 10.1523/JNEUROSCI.3061-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhou YB, Sun HJ, Chen D, Liu TY, Han Y, Wang JJ, et al. Intermedin in paraventricular nucleus attenuates sympathetic activity and blood pressure via nitric oxide in hypertensive rats. Hypertension. 2014;63(2):330–7. doi: 10.1161/HYPERTENSIONAHA.113.01681. [DOI] [PubMed] [Google Scholar]
- 163.Yuan N, Zhang F, Zhang LL, Gao J, Zhou YB, Han Y, et al. SOD1 gene transfer into paraventricular nucleus attenuates hypertension and sympathetic activity in spontaneously hypertensive rats. Pflugers Arch. 2013;465(2):261–70. doi: 10.1007/s00424-012-1173-0. [DOI] [PubMed] [Google Scholar]
- 164.Vitale G, Salvioli S, Franceschi C. Oxidative stress and the ageing endocrine system. Nat Rev Endocrinol. 2013;9(4):228–40. doi: 10.1038/nrendo.2013.29. [DOI] [PubMed] [Google Scholar]
- 165.Jang PG, Namkoong C, Kang GM, Hur MW, Kim SW, Kim GH, et al. NF-kappaB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. J Biol Chem. 2010;285(13):9706–15. doi: 10.1074/jbc.M109.070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Morgan DA, Thedens DR, Weiss R, Rahmouni K. Mechanisms mediating renal sympathetic activation to leptin in obesity. Am J Physiol Regul Integr Comp Physiol. 2008;295(6):R1730–6. doi: 10.1152/ajpregu.90324.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. 2006;17(9):365–71. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 168.Reed AS, Unger EK, Olofsson LE, Piper ML, Myers MG, Jr, Xu AW. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes. 2010;59(4):894–906. doi: 10.2337/db09-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, et al. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006;4(2):123–32. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 170.Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10(7):739–43. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 171.Romero CA, Orias M, Weir MR. Novel RAAS agonists and antagonists: clinical applications and controversies. Nat Rev Endocrinol. 2015;11(4):242–52. doi: 10.1038/nrendo.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Horiuchi M, Mogi M, Iwai M. Signaling crosstalk angiotensin II receptor subtypes and insulin. Endocr J. 2006;53(1):1–5. doi: 10.1507/endocrj.53.1. [DOI] [PubMed] [Google Scholar]
- 173.Bencini A, Failli P, Valtancoli B, Bani D. Low molecular weight compounds with transition metals as free radical scavengers and novel therapeutic agents. Cardiovasc Hematol Agents Med Chem. 2010;8(3):128–46. doi: 10.2174/187152510791698389. [DOI] [PubMed] [Google Scholar]
- 174.Berzigotti A, Bosch J. Pharmacologic management of portal hypertension. Clin Liver Dis. 2014;18(2):303–17. doi: 10.1016/j.cld.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 175.Yi QY, Qi J, Yu XJ, Li HB, Zhang Y, Su Q, et al. Paraventricular Nucleus Infusion of Epigallocatechin-3-O-Gallate Improves Renovascular Hypertension. Cardiovasc Toxicol. 2015 doi: 10.1007/s12012-015-9335-x. [DOI] [PubMed] [Google Scholar]