

Abstract
The long non-coding RNA X-inactive specific transcript (XIST) mediates the transcriptional silencing of genes on the X chromosome. Here we show that in human cells XIST is highly methylated with at least 78 N6-methyladenosine (m6A) residues—a reversible base modification of unknown function in long non-coding RNAs. We show that m6A formation in XIST, as well as in cellular mRNAs, is mediated by RNA binding motif protein 15 (RBM15) and its paralogue RBM15B, which bind the m6A-methylation complex and recruit it to specific sites in RNA. This results in the methylation of adenosine nucleotides in adjacent m6A consensus motifs. Furthermore, we show that knockdown of RBM15 and RBM15B, or knockdown of methyltransferase like 3 (METTL3), an m6A methyltransferase, impairs XIST-mediated gene silencing. A systematic comparison of m6A-binding proteins shows that YTH domain containing 1 (YTHDC1) preferentially recognizes m6A residues on XIST and is required for XIST function. Additionally, artificial tethering of YTHDC1 to XIST rescues XIST-mediated silencing upon loss of m6A. These data reveal a pathway of m6A formation and recognition required for XIST-mediated transcriptional repression.
Introduction
XIST is a long non-coding RNA (lncRNA) that mediates the silencing of gene transcription on the X chromosome during female mammalian development1 via the recruitment of specific protein complexes. These complexes have been identified in studies of the genetic domains involved in XIST silencing2 as well as by recent unbiased proteomic screens that identified direct XIST-binding proteins using zero-distance ultraviolet irradiation-based crosslinking methods3,4. Proteins that are bound directly or indirectly to XIST via protein intermediates have also been identified using crosslinking reagents such as formaldehyde5. These include HNRNPU (also known as SAF-A), which anchors XIST to the X chromosome6, SHARP (SPEN), which recruits HDAC3 (ref. 3), as well as PRC2, which introduces repressive chromatin marks7.
Here we show that XIST-mediated gene silencing requires adenosine methylation, a reversible RNA-modification pathway that forms m6A. Although the m6A modification is well-studied in mRNAs, m6A mapping studies8 have shown that m6A is also present in lncRNAs. Our data show that XIST is highly methylated and that m6A modifications are required for XIST-mediated gene silencing. Formation of m6A in XIST and mRNAs is mediated by two previously unknown components of the m6A methylation complex, RBM15 and RBM15B. These proteins bind and recruit the m6A-methylation complex to specific sites within XIST, leading to m6A formation at adjacent sites. Furthermore, we show that m6A in XIST recruits the m6A reader, YTHDC1 (hereafter DC1), and that the binding of DC1 to XIST promotes XIST-mediated gene repression. These studies reveal a role for m6A and DC1 as mediators of transcriptional repression via the lncRNA XIST.
RBM15 and RBM15B are required for gene silencing
Recent studies have shown that RBM15 binds to XIST3,4. Previously, we found that the knockdown of RBM15 did not block XIST-mediated gene silencing3; however, another study found the opposite to be true9. We therefore considered the possibility that another protein compensated for the function of RBM15 in our RBM15 knockdown experiments. RBM15 possesses notable similarity to another protein, RBM15B, in sequence and domain organization, making it a suitable candidate for compensation of RBM15 function (Extended Data Fig. 1a).
To test the functional redundancy of these proteins, we first investigated whether RBM15 and RBM15B show similar binding patterns in XIST by mapping their binding sites using individual-nucleotide resolution UV crosslinking and immunoprecipitation (iCLIP)10 in human embryonic kidney 293T (HEK293T) cells. For all iCLIP experiments, we examined only the endogenous protein and identified antibodies that selectively precipitated each protein. We also confirmed that there was consistency between the transcriptome-wide iCLIP data set replicates (Extended Data Fig. 1b–g and Supplementary Tables 1, 2).
RBM15 and RBM15B showed a similar distribution of iCLIP tags (that is, processed reads; see Methods for further details) along the length of XIST (Fig. 1a and Extended Data Fig. 1h), including at the A-repeat region, an evolutionarily conserved region in the 5′ region that is essential for the initiation of silencing11. Additionally, RBM15 and RBM15B showed similar distributions of iCLIP tag clusters, which represent regions of enriched binding, and crosslinking-induced truncation sites (CITS), which represent direct contacts with XIST (Supplementary Tables 3, 4).
Figure 1. RBM15 and RBM15B are necessary for XIST-mediated gene silencing.

a, RBM15 and RBM15B show similar binding patterns in XIST. Shown is the distribution of normalized RBM15 and RBM15B iCLIP tags (in unique tags per million, uTPM) and statistically significant CITS. Light blue vertical lines, RBM15; dark blue vertical lines, RBM15B; P < 0.0001. b, c, Knockdown of both Rbm15 and Rbm15b (siRBM15/15B) impair XIST-mediated gene silencing. XIST expression was induced by doxycycline, and the X-linked genes Gpc4 (green) and Atrx (red) were quantified by RNA-FISH (b). Representative FISH images are shown with DAPI nuclear counterstain (blue) (c). The number of detected RNA spots for both genes are indicated on each image. Scale bars, 5 μm. Data are mean ± s.e.m. for 50 cells from one experiment. ***P < 0.001, *****P < 0.0001, relative to siControl by unpaired two-sample t-test. NS, not significant.
To assess whether RBM15 and RBM15B are required for XIST-mediated gene silencing, we used male mouse embryonic stem (ES) cells that express XIST on the X chromosome in a doxycycline-dependent manner 3. XIST-mediated gene silencing is induced by 16 h of doxycycline (Dox)-induced XIST expression and is measured by quantifying the expression of two X-linked genes, Gpc4 and Atrx, using single-molecule RNA fluorescence in situ hybridization (FISH)3. In these assays, we knocked down mRNAs using short interfering RNAs (siRNAs) and confirmed that each examined cell showed successful depletion of both the siRNA-targeted mRNA as well as Dox-induced XIST expression.
In wild-type siRNA-transfected cells, we observed the expected silencing of the X-linked genes. Gpc4 transcript levels decreased from 21 copies (−Dox) to 1 copy (+Dox) per cell and Atrx transcript levels decreased from 17 to 1 copy per cell (Fig. 1b, c and Extended Data Fig. 2a, b). Knockdown of both Rbm15 and Rbm15b, but not knockdown of either gene individually, prevented XIST-mediated gene silencing in these cells (Fig. 1b, c). This was also seen in a female mouse ES cell line that similarly exhibits Dox-inducible XIST expression on one X chromosome (Extended Data Fig. 2c). RBM15 and RBM15B therefore have redundant function in mediating XIST-mediated transcriptional silencing.
RBM15/RBM15B link the methylation complex to XIST
RBM15 and RBM15B were recently identified as high-confidence interactors with Wilms tumour-associated protein (WTAP) in a proteomic analysis12. WTAP binds METTL3 (refs 13–15), the methyltransferase that mediates methylation of m6A in mRNA16, and is recruited to RNAs via an unknown adaptor protein to trigger m6A formation14.
We therefore investigated whether RBM15 and/or RBM15B is a component of the WTAP–METTL3 complex, targeting it to RNA. Immunoprecipitation of RBM15 or RBM15B from HEK293T nuclear lysates co-precipitated METTL3 (Fig. 2a). Knockdown of WTAP reduced the interaction between METTL3 and both RBM15 and RBM15B (Fig. 2a), indicating that this interaction is mediated by WTAP. A reciprocal immunoprecipitation similarly indicated that METTL3 binds RBM15 and RBM15B in a WTAP-dependent manner (Extended Data Fig. 3a–c).
Figure 2. RBM15 and RBM15B recruit METTL3 to XIST.

a, RBM15 and RBM15B interact with METTL3 in a WTAP-dependent manner. RBM15 (left) and RBM15B (right) were immunoprecipitated from HEK293T nuclear extracts. Co-immunoprecipitation of METTL3 was reduced in siWTAP-transfected cells. The IgG heavy chain (H chain) prevents visualization of WTAP; however, knockdown is seen in the input sample. NE, nuclear extracts. b, Quantification of METTL3-bound XIST upon knockdown of methylation machinery components. XIST was quantified by qRT–PCR using regions selected based on the presence or absence of RBM15- and RBM15B-binding sites (indicated with light blue and blue lines, respectively). Data are mean ± s.e.m. from three independent experiments. **P < 0.001, ***P < 0.0001, relative to siControl by unpaired two-sample t-test.
To determine whether both RBM15 and RBM15B (RBM15/15B) recruit WTAP–METTL3 to XIST, we treated HEK293T cells with formaldehyde to crosslink XIST to any bound proteins. We then immunoprecipitated METTL3 from the cell lysates and measured the amount of bound XIST by quantitative reverse transcription PCR (qRT–PCR) at regions with and without RBM15/15B-binding sites. METTL3 immunoprecipitates contained significantly higher levels of XIST than control immunoprecipitates at these binding sites (Fig. 2b and Extended Data Fig. 3d, e). This interaction was impaired after knockdown of WTAP, RBM15, and/or RBM15B, with the greatest loss following knockdown of both RBM15 and RBM15B (Fig. 2b). This led us to believe that RBM15/15B is the component of the methylation complex that accounts for its recruitment to XIST.
RNA-anchored methylation complexes
Our initial m6A mapping studies, using methylated RNA immunoprecipitation followed by sequencing (MeRIP–seq), showed XIST contained m6A modifications8, although this approach was at low resolution. More recently, we mapped m6A at single-nucleotide resolution using m6A iCLIP (miCLIP)17. Analysis of the miCLIP data set shows 78 putative m6A residues in XIST, some of which are localized at-or-near the A-repeat region (Fig. 3a and Extended Data Fig. 4a). To investigate whether RBM15 and RBM15B mediate m6A formation in XIST, we measured m6A levels in XIST in wild-type control and RBM15/15B-deficient cells. Methylated XIST was precipitated with an m6A-specific antibody and XIST levels were quantified from three m6A-containing regions (Fig. 3a). Knockdown of METTL3, RBM15, RBM15B, and both RBM15 and RBM15B resulted in significantly reduced levels of methylated XIST, with the largest reduction in m6A levels following RBM15/RBM15B double knockdown (Fig. 3b and Extended Data Fig. 3d–f). This indicates that RBM15 and RBM15B promote XIST methylation by recruiting WTAP–METTL3.
Figure 3. N6-adenosine methylation is necessary for XIST-mediated gene silencing.

a, m6A residues (red lines) identified via miCLIP are broadly distributed along XIST. Normalized miCLIP17 tags are shown in purple. b, Methylation of XIST requires RBM15 and RBM15B. m6A levels in XIST were quantified by m6A-RNA immunoprecipitation followed by qRT–PCR of three m6A regions of XIST. Data are mean ± s.e.m. from six samples coming from three technical replicates of two biological replicates. ***P < 0.0001, **P < 0.001 relative to siControl by unpaired two-sample t-test. c, d, m6A promotes XIST-mediated gene silencing. XIST expression was induced by Dox, and X-linked genes Gpc4 (green) and Atrx (red) were quantified by RNA-FISH (c). Representative FISH images are shown (d). The number of detected RNA spots is indicated on each image. Scale bars, 5 μm. Data are mean ± s.e.m. for 50 cells from one experiment. ****P < 0.005 relative to siControl by an unpaired two-sample t-test.
We observed that m6A residues are typically located in the vicinity of RBM15 and RBM15B iCLIP clusters on XIST (Extended Data Fig. 4b). Indeed, the median distance between each RBM15 or RBM15B CITS in XIST and the closest m6A was 45 or 28.5 nucleotides, respectively (Extended Data Fig. 4c). By contrast, the distance between m6A and randomly picked sites along XIST was approximately 70–90 nucleotides (P = 0.0026, RBM15; P = 0.0001, RBM15B). Thus, m6A residues are positioned significantly closer to RBM15 and RBM15B sites than would be expected by chance. This proximity suggests that RBM15/15B recruits the WTAP–METTL3 complex to methylate adenosine bases that lie in proximal m6A consensus sites.
We next asked whether RBM15/15B binds next to m6A bases in mRNA. Using our single-nucleotide-resolution m6A data set in mRNA17, we calculated the spatial relationship of RBM15/15B-binding sites relative to m6A residues. As a control, we measured the binding of RBM15 and RBM15B relative to non-methylated adenosines that fall within the m6A consensus DRACH sequence (where D denotes A/G/U, R denotes A/G and H denotes A/C/U)17. These sites lack miCLIP reads and thus are non-methylated. Transcriptome-wide analysis shows that RBM15/15B-binding sites are significantly enriched on either side of m6A residues, while minimal enrichment is seen at the nearest non-methylated DRACH site (Extended Data Fig. 5a). RBM15/15B-binding sites are characterized by U-rich motifs (Extended Data Fig. 5c–e) that are readily detected adjacent to m6A residues on individual transcripts (Extended Data Fig. 5b).
Notably, knockdown of both RBM15 and RBM15B resulted in a substantial drop in m6A levels in poly(A) RNA (Extended Data Fig. 5f, g), indicating that RBM15 and RBM15B direct methylation of adenosine residues at sites in both mRNA and XIST.
XIST m6A is required for gene silencing
XIST has more mapped m6A residues than any other RNA (Supplementary Tables 5, 6), raising the possibility that m6A may mediate important aspects of XIST function. The role of m6A in XIST-mediated gene silencing cannot be tested in Mettl3−/− mouse ES cells because these cells do not express XIST owing to the persistent expression of XIST-suppressing pluripotency genes18. We thus used the Dox-inducible XIST-expression system to assess the role of METTL3 in XIST-mediated transcriptional silencing. METTL3 knockdown reduces m6A levels across the transcriptome, including in XIST19. In control siRNA-transfected cells, we observed the expected silencing of X-linked genes upon XIST induction (Fig. 3c, d and Extended Data Fig. 2c–e). However, in siMettl3-treated cells, XIST was induced but failed to silence Gpc4 and Atrx expression (Fig. 3c, d and Extended Data Fig. 2d, e). A similar silencing defect was seen in a female mouse ES cell line with Dox-inducible XIST expression (Extended Data Fig. 2c). Therefore, m6A is required for XIST-mediated transcriptional silencing.
DC1 binds XIST to mediate gene silencing
We next investigated the mechanism by which m6A in XIST is recognized in order to mediate transcriptional silencing. m6A residues are recognized by the YTH proteins20 which comprise three members of the YTHDF family (DF1, DF2, and DF3), YTHDC1 (DC1) and YTHDC2 (DC2) (Extended Data Fig. 6a). DF1, DF2, DF3 and DC2 are primarily cytoplasmic21–24, whereas DC1 is located primarily in the nucleus24.
Using iCLIP, we assessed the transcriptome-wide binding properties of the endogenous YTH proteins and determined whether any interacted preferentially with m6A in XIST (Extended Data Figs 6, 7 and Supplementary Table 1, 2). In this analysis, we quantified the binding of YTH proteins at each of the 78 mapped m6A residues in XIST as well as the other 11,452 mapped m6A residues in the transcriptome. Each m6A residue was assigned an intensity value that was defined as the normalized number of miCLIP tags for each m6A residue17. This value is influenced by both the transcript abundance and the m6A stoichiometry. Next, the binding of each YTH protein to each m6A residue was determined using the normalized number of mapped iCLIP tags at the m6A site. For most m6A residues, the miCLIP intensity value increased with the amount of bound YTH protein (Fig. 4a); however, only DC1 showed clear preferential binding for XIST m6A residues (Fig. 4a, b and Extended Data Fig. 8a–c).
Figure 4. DC1 binds XIST m6A residues and promotes XIST-mediated gene silencing.
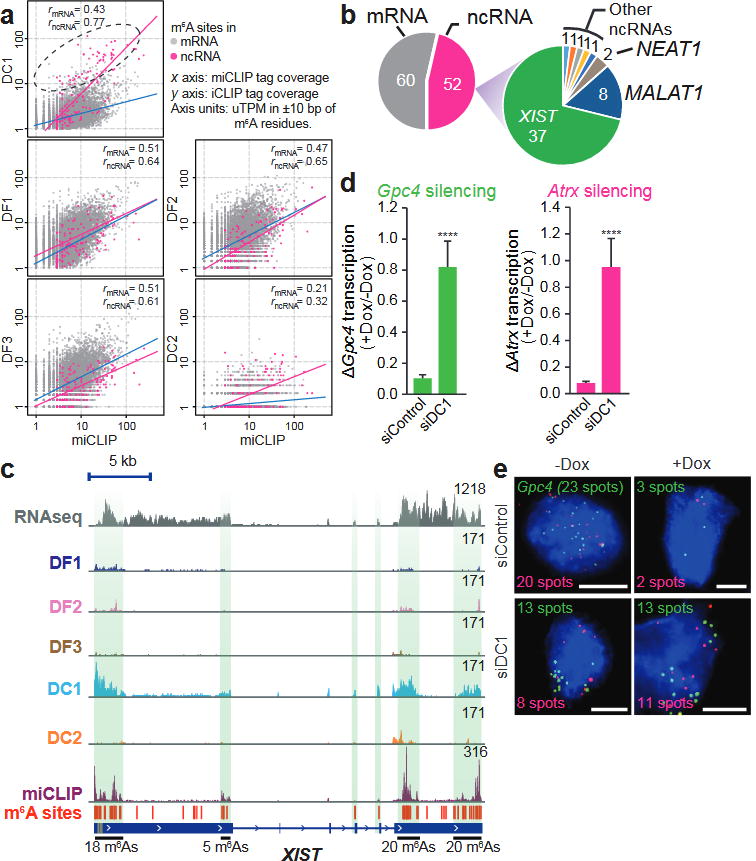
a, YTH iCLIP tag coverage at 11,530 annotated m6A residues. Correlation coefficients for mRNA m6A (grey) and non-coding RNA (ncRNA) m6A (magenta) are indicated. DF1, DF2 and DF3 show similar correlations between m6A abundance and YTH binding for mRNAs (blue line) and ncRNAs (magenta line). DC1 shows preference for ncRNA m6A, with the top 1% of DC1-bound m6A indicated (dotted ellipse). b, mRNA/ncRNA distribution of the top 1% of DC1-bound m6A sites. Most detected ncRNA m6A are present on XIST (indicated in green). c, Normalized tag distributions for each YTH protein on XIST shows predominantly DC1 binding. High-density m6A regions are indicated by green shading. d, e, Ythdc1 knockdown (siDC1) impairs XIST-mediated gene silencing. XIST was induced by Dox, and X-linked genes Gpc4 (green) and Atrx (red) were quantified by RNA-FISH. Representative FISH images are shown (e). The number of detected RNA spots is indicated on each image. Scale bars, 5 μm. Data are mean ± s.e.m. ****P < 0.005 relative to siControl by an unpaired two-sample t-test.
A direct comparison of iCLIP tags on XIST also showed that DC1 is the only YTH protein to exhibit prominent XIST binding (Fig. 4c and Extended Data Fig. 8a–d). Notably, the DC1 iCLIP tag clusters overlap with the XIST m6A miCLIP tag clusters, consistent with the binding of DC1 to m6A residues in XIST (Fig. 4c and Extended Data Fig. 8d).
The binding of DC1 to XIST could also be confirmed through the co-immunoprecipitation of DC1 and XIST using antibodies against DC1, with XIST detected by qRT–PCR using primers that detect either of the two regions with a high DC1 iCLIP signal (Extended Data Fig. 9a). XIST pulldown was reduced following the knockdown of methylation machinery components (METTL3, WTAP, RBM15, RBM15B, and RBM15 and RBM15B double knockdown). Furthermore, DC1 was enriched in the XIST nuclear subcompartment in comparison to autosomal domains as measured by 3D structured illumination super-resolution microscopy (3D-SIM) (Extended Data Fig. 9b–d). This localization was reduced following knockdown of METTL3 or both RBM15 and RBM15B (Extended Data Fig. 9e). Together, these data show that DC1 binds to XIST in an m6A-dependent manner.
We then assessed whether DC1 is required for XIST-mediated transcriptional silencing. Knockdown of DC1 but not of DF1, DF2, DF3 or DC2 prevented XIST-mediated gene silencing in cells with Dox-induced XIST expression (Fig. 4d, e and Extended Data Fig. 2f–j) and in differentiating female mouse ES cells (Extended Data Fig. 2i). To determine whether DC1 binding to XIST promotes XIST-mediated gene silencing, we tethered DC1 to XIST using an XIST transcript with three BoxB hairpins appended to the 3′ end (XIST–(BoxB)3) (Fig. 5a). These hairpins bind the λN peptide fused to the C terminus of DC1, allowing the BoxB hairpins to bind the λN peptide. Dox-induced expression of XIST–(BoxB)3 caused transcriptional repression of Gpc4 and this silencing was lost following knockdown of Mettl3 or both Rbm15 and Rbm15b (Fig. 5b, c). However, XIST-mediated gene silencing was rescued when DC1–λN was expressed (Fig. 5b, c). Thus, recruitment of DC1 to XIST is sufficient to induce its repressive function in the absence of the methylation machinery. Taken together, these data suggest that m6A methylation of XIST triggers binding to DC1, which promotes XIST-mediated transcriptional silencing.
Figure 5. m6A-independent tethering of DC1 to XIST is sufficient to exert XIST-mediated gene silencing.

a, Schematic of tethering approach. The 3′ end of XIST was genomically modified with three BoxB sequences (XIST–(BoxB)3). m6A-dependent recruitment of DC1 is blocked in methylation-deficient cells; however, artificial tethering can be achieved with DC1–λN, which binds to the BoxB elements in XIST–(BoxB)3. b, c, Dox-induced expression of XIST–(BoxB)3 results in gene silencing in siControl-transfected cells, but not in siMETTL3 or siRBM15 and siRBM15B co-transfected cells. DC1–λN rescued silencing in these cells, suggesting that the primary function of m6A in XIST-mediated gene silencing is to recruit DC1 to XIST. Quantification of Gpc4 expression is shown in b. Representative FISH images showing DAPI-stained nuclei (blue), Gpc4 RNA (green), and XIST (pink) are shown in c. Scale bars, 5 μm. Data are mean ± s.e.m. in b for 50 cells from one experiment. *****P < 0.0001 by unpaired two-sample t-test.
Discussion
Although the m6A modification has been well characterized in mRNA, no function for m6A in lncRNAs has previously been demonstrated. Here we show that m6A functions to enable the transcriptional repression effects of XIST. XIST is highly enriched in m6A throughout its length, enabling the recruitment of the nuclear m6A binding protein DC1. The importance of m6A in XIST function is highlighted by the fact that diverse components of the m6A methylation complex bind XIST and are required for XIST-mediated gene silencing. Together, these discoveries reveal a role for RNA modification in lncRNA function and describe the assembly of XIST into a transcriptionally repressive ribonucleoprotein complex (Extended Data Fig. 10a).
Recent proteomic studies have revealed large numbers of XIST-binding proteins3–5,9, several of which we now recognize as contributing to m6A formation or recognition. For example, WTAP was identified in a proteomic analysis of XIST-associated proteins5 and was shown to be required for XIST-mediated gene silencing in a functional screen9. Although WTAP has numerous functions, our data support the idea that its m6A methylation-promoting effects are required for XIST-mediated gene silencing. DC1 was also observed in a proteomic analysis of formaldehyde-crosslinked proteins bound to XIST5.
Similarly, RBM15 was shown to be required for XIST-mediated gene silencing9 and was also identified as an XIST-binding protein3,5,9. Our data suggest that RBM15/15B is a component of the m6A methylation complex that binds XIST, and that it is this methylation role that is essential in bringing about the silencing defect observed when both are knocked down. RBM15 and RBM15B appear to have redundant functions as both need to be knocked down in order to deplete m6A to sufficient levels to impair XIST function. The large number of m6A residues in XIST ensures that at least a few will bind to DC1 to activate gene-silencing mechanisms.
The identification of the WTAP–METTL3 complex13 and its role in m6A formation14 raised several important questions. First, why are some RNAs methylated, while others lack m6A? Second, why are only a subset of DRACH-adenosine residues selected for methylation, despite the high prevalence of DRACH consensus sites in RNA25. Our data sheds light on these questions. RBM15 and RBM15B, proteins that associate with WTAP–METTL3 and contain RNA-binding domains, enable the binding of WTAP–METTL3 to specific mRNAs, as well as, XIST. The localized binding at specific sites in the RNA sequence allows for the selective methylation of adjacent DRACH sites while leaving distant DRACH sites unmethylated. The three-dimensional RNA structure of XIST could promote further adenosine methylation by bringing distant DRACH consensus sites into the proximity of the RBM15/15B-anchored methylation complex.
Our single-nucleotide-resolution map of m6A (ref. 17) showed that RBM15/15B is found adjacent to methylated but not non-methylated DRACH sequences in the mRNA transcriptome. The double knockdown of RBM15 and RBM15B markedly reduce m6A levels in mRNA, supporting the idea that RBM15/15B-binding determines which DRACH sites are methylated in the transcriptome.
How DC1 binding to XIST leads to gene silencing remains unclear. However, a recent proteomics study exploring DC1 binding partners27 may provide initial mechanistic insights. These partners include SHARP, LBR, HNRNPU and HNRNPK which each have distinct roles in the initiation of transcriptional silencing (Extended Data Fig. 10b–e). Analysis of the DC1 interaction network, based on an independent protein–protein interaction database28, also identifies additional interactions with components of the PRC1 and PRC2 complexes (Extended Data Fig. 10b–e and Supplementary Table 7). Various XIST-interacting gene-silencing proteins may bind to DC1 and utilize the ability of DC1 to bind m6A residues on XIST to achieve additional specificity in the binding of precise locations on XIST. Further experiments are required both to determine whether DC1 directly affects binding of these silencing proteins and to explore the mechanisms used by DC1 to enable m6A-dependent transcriptional silencing.
METHODS
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Cell culture
HEK293T/17 (ATCC CRL-11268) cells were maintained in 1× DMEM (11995-065, Life Technologies) with 10% FBS, 100 U ml−1 penicillin and 100 μg ml−1 of streptomycin under standard tissue culture conditions. Cells were split using TrypLE Express (Life Technologies) according to manufacturer’s instructions. Mouse ES cells expressing Xist RNA from the endogenous locus under a Tet-driven promoter (pSM33 ES cell line) were maintained as previously described29. Cell lines were not tested for mycoplasma contamination.
Generation of female pSM33 cell line
The Tet-regulated promoter was inserted at the promoter region of the endogenous Xist locus of mouse female ES cell line (F1 2-1 line, derived from a 129 × castaneous F1 mouse cross) using CRISPR-mediated homologous recombination. Clonal cell lines derived from single cells were screened for the presence of Tet-inducible promoter by PCR. Promoter integration was confirmed by Sanger sequencing with primers flanking the insertion site. Recombinant Xist alleles were further identified by SNP analysis. A clonal line with promoter insertion in the 129 allele was used for studying Xist-mediated gene silencing.
Insertion of BoxB sequence elements in Xist
Three BoxB sequence elements were inserted at the 3′ end of the endogenous Xist loci in the male pSM33 cell line using CRISPR-mediated homologous recombination. In brief, cells were first co-transfected with a plasmid expressing Cas9 under a CAG promoter, a short guide RNA (Target sequence: 5′-CCTCATCCTCATGTCTTCTC-3′), and a ssDNA ultramer (IDT) containing three BoxB elements (5′-GGGCCCTGAAGAAGGGCCCATGGGCCCTGAAGAAGGGCCCATAGGGCCCTGAAGAAGGGCCC-3′; underlined bases mark the BoxB sequence) flanked by 70-nucleotide-long DNA sequence identical to the upstream and downstream genomic DNA sequence at the point of BoxB insertion. Cells were sorted and single colonies were screened for the insertion of BoxB elements by PCR. Insertion was further confirmed by Sanger sequencing. Recombinant clones were tested for X-chromosome silencing by induction of Xist expression and Gpc4 and Atrx RNA-FISH. A clone showing silencing identical to the non-recombinant cell line was used for DC1–λN–XIST tethering functional assay.
Construction of λN–3×Flag epitope-tagged DC1 expression construct
A human YTHDC1-encoding open reading frame (ORF) was PCR-amplified from oligo-(dT)18-primed HEK293T cDNA using hYTHDC1–EcoRI-F and hYTHDC1–XhoI-R primers (Supplementary Table 8). The PCR fragment was initially cloned in pcDNA3-Flag-HA (1436 pcDNA3-Flag-HA was a gift from W. Sellers; Addgene plasmid 10792) plasmid at EcoRI and XhoI sites. Full-length YTHDC1 was then PCR amplified and subcloned into pCAG-GW-λN-3×Flag-BSD construct using the Gateway entry cloning system (Invitrogen). This plasmid (pCAG-GW-hYTHDC1-λN-3×Flag-BSD) expresses human YTHDC1 protein with a C-terminal λN–3×Flag tag under CAG promoter. We verified that λN–3×Flag-tagged DC1 protein was still functional by ensuring that it could rescue knockdown of the endogenous protein.
Generation of Ythdc1+/− female ES cells
Ythdc1+/− female ES cell line was generated using the CRISPR–Cas9 system. In brief, female ES cells were co-transfected with a Cas9-expressing pCAG plasmid and a pool of short guide RNAs targeting the region around the first codon of the Ythdc1 ORF at the endogenous loci to generate frameshift mutations causing disruption in the reading frame. Target DNA sequences were 5′A-AAGCCGGAGGGCAGCCATGG-3′,5′-GCGGTGGCGGCGGCGGAAGC-3′ and 5′-CGGCGGAAGCCGGAGGGCAG-3′. We screened 24 colonies derived from single cells for the presence of frame-shift mutations at the desired location in Ythdc1 gene using PCR and Sanger sequencing, with primers flanking the target site. No clone showed a homozygous frame-shift mutation, suggesting that homozygous Ythdc1 deletion is lethal. Only clones with heterozygous frame shift mutations were detected. Confirmation of the presence of a heterozygous knockout of Ythdc1 (Ythdc1+/−) was performed by RNA-FISH and immunofluorescence. A clonal cell line showing a 50% reduction in the expression level of Ythdc1 mRNA and protein were used for assaying X-chromosome silencing.
Antibodies
Details of the antibodies used in this study are given in Supplementary Table 1.
siRNA and shRNA transfection
Target sequences of siRNA and short hairpin RNA (shRNA) used in this study are listed in Supplementary Table 9. For validation of antibodies for iCLIP, 20 nM siRNA was transfected using Pepmute transfection reagent (Signagen) and pSuperior–EGFP shRNA plasmid (OligoEngine) was transfected using Fugene HD transfection reagent (Promega) according to the manufacturer’s instructions. Forty-eight hours after the first transfection, a second transfection was performed. Cells were maintained at 70–80% confluency and collected 96 h after the first transfection. Knockdown was confirmed by western blot analysis (list of antibodies and dilutions used are given in Supplementary Table 1).
For studying the effect of Rbm15, Rbm15b, Mettl3, Ythdf1, Ythdf2, Ythdf3, Ythdc1 and Ythdc2 knockdown on XIST-mediated gene silencing, 20 nM of siRNA targeting each gene were transfected into 100,000 pSM33 ES cells using the Neon transfection system (settings: 1,200 V, 40 ms width, 1 pulse; Invitrogen). At the time of XIST induction, the observed knockdown efficiency for all the target genes was greater than 70%. For Mettl3, the efficiency was 95%.
Construction of iCLIP libraries
All iCLIP studies were performed on the endogenous proteins. Previous CLIP-based analyses of YTH proteins used overexpressed proteins. Since this can affect the localization and assembly of proteins into multiprotein complexes, we identified antibodies that bound the endogenous proteins for these studies (described later). iCLIP libraries were constructed as described elsewhere with minor modifications30. To improve the efficiency of cell lysis and dissolution of RNA–protein conjugates, cells were lysed in 1% SDS as described previously31. In brief, 9 × 106 HEK293T cells were seeded per 10 cm dish 12 h before UV irradiation. Media was discarded and 6 ml of ice-cold PBS was gently added to the cells. Cells were maintained on ice and immediately irradiated once with UV at 254 nm (150 mJ cm−2) in a UV crosslinker (Stratagene 2400). Cells were scraped in PBS using a cell scraper and collected by centrifugation at 200g for 10 min at 4 °C. Supernatant was discarded, and cells were gently suspended in 100 μl of 1% SDS with 10 mM DTT and 10× protease inhibitors (EDTA-free cOmplete mini, Roche) and incubated at 25 °C for 10 min to denature the protein complexes. SDS was neutralized with 900 μl of iCLIP lysis buffer (CLB) without SDS (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate). Lysates were sonicated using a Branson Digital Sonifier Model 450 fitted with 3.125 mm tapered microtip probe on ice at 20% amplitude for 30 s with 2 s ON and 10 s OFF cycle. DNase I and RNase I digestion was performed with 2 μl of Turbo DNase I (AM2238, Life Technologies) and 10 μl of different dilutions of RNase I per ml of lysate for 3 min at 37 °C. For validation of antibodies for iCLIP and the construction of iCLIP libraries, 1:5 dilution of RNase I (AM2295, Life Technologies) was used as high (H) and 1:150 dilution was used as low (L) concentration RNase. Antibodies were first bound to CLB-washed Protein A/G beads (88803, Thermo Fisher) in CLB (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) followed by incubation at 25 °C for 30 min with mixing. Beads were washed twice with CLB.
For validation of antibodies for iCLIP, 500 ng of antibody was used per immunoprecipitation and for the construction of iCLIP libraries, 2–10 μg of antibody was used. Clarified RNase- and DNase-digested lysates were incubated with antibody bound to Protein A/G-beads at 4 °C for 12 h. Further steps of iCLIP library preparation were carried out as described previously30. To avoid cross-contamination of RNA and library PCR products, electrophoresis equipment was treated with 10% commercial bleach for 20 min at 25 °C and thoroughly washed with nuclease-free water before use. Replicates were tagged with unique barcodes using the 5′ Rtclip primer in reverse transcription. Low-, medium- and high-molecular-mass cDNA libraries were mixed at 1:5:5 molar ratio and sequenced on Illumina HiSeq 2500 from a single end for 50 bases.
Analysis of iCLIP sequence data
Low-quality bases, reads with more than two ambiguous base calls, and adaptor sequences were all removed using FLEXBAR tool (--max-uncalled 2--min-read-length 15--pre-trim-phred 20, 3′ adaptor: AGATCGGAAGAGCGGTTCAG). Reads were demultiplexed based on 5′ barcodes for individual replicates using an in-house Linux shell script. Reads were processed in pooled or separate replicate modes using the CITS analysis pipeline32. In brief, reads were converted to fasta format using fastq_to_fasta tool from FASTX-toolkit and then collapsed to remove PCR amplified duplicates based on sequence using CIMS/fasta2collapse.pl script. The barcode was stripped and added to the name of the read. Reads were aligned to the human genome (hg19) using Novoalign (v3.02.12, NovoCraft Technologies) (Options: -t 85 -l 16 -s 1 -r None). Further analysis until the identification of CITS (P < 0.0001) was performed as described previously32. Unique sequence reads that are free of PCR duplicates represent unique RNA-protein binding events. These processed reads are referred to as iCLIP/miCLIP tags (or just tags), and the mapped cluster of processed reads are referred to as tag clusters throughout this study.
Motif enrichment analysis
Analysis of motif enrichment was performed on the sense DNA sequence 20 nucleotides up- and down-stream of the called truncation sites using the MEME suite33. For this analysis the top 20% of the sites identified as statistically significant (those with P < 0.0001), with the highest number of crosslinking induced truncations, were used. Since fewer sites were detected for DC2, all of the sites were used for MEME analysis of DC2-binding sites.
Metagene analysis
Metagenes were constructed for the called CITS/miCLIP-identified m6A residues using an in-house Perl annotation pipeline and an R script. In brief, the single-nucleotide sites were mapped to different RNA features (5′ UTR, CDS and 3′ UTR) of the human genome (hg19). The position of the sites was normalized to the median feature length of the transcripts to which the sites mapped. A frequency distribution plot was generated by counting the number of sites in contiguous bins on a virtual mRNA transcript, sites whose feature lengths represent the median feature lengths of transcripts under analysis. A Gaussian estimate of kernel density was then plotted as a metagene. For YTH, RBM15 and RBM15B proteins, all statistically significant CITS (P < 0.0001) were used and for miCLIP m6A, residues identified from poly(A) RNA from ref. 17 were used.
Comparison of iCLIP and miCLIP tag coverage
For comparing iCLIP tags, we calculated normalized tag counts using a previously described approach with minor modifications34. Instead of using read counts per million mapped reads (RPM) normalization to reduce PCR amplification bias, we used unique tag counts obtained from CITS analysis. Each iCLIP tag represents a unique RNA–protein or antibody–m6A binding event. The number of unique events from a million such events is proportional within replicates and also comparable across different CLIP libraries. For this, the number of iCLIP tags per million uniquely mapped tags (unique tags per million, uTPM) was calculated at every coordinate on the human genome using the following formula: where t = number of unique CLIP tags at a base, T = total number of uniquely mapped unique CLIP tags in the whole CLIP library.
For comparing replicates, the normalized mean tag counts (in uTPM) between replicates at randomly selected ten-thousand 100-bp bins on the human genome were compared. For comparing various iCLIP/HITS-CLIP/miCLIP data sets, iCLIP data analysed in pooled mode was used. Here, the normalized total tag count in the 10-bp flanking region of 11,530 miCLIP-identified m6A residues mapping to mRNA and ncRNA (includes snoRNAs, lncRNA and other ncRNAs) were calculated. Only m6A residues in non-BCANN consensus sequence were considered for this analysis. These represent unique sites obtained from merging (mergeBed -s -d 2) of CIMS- and CITS-based m6A site calls from ref. 17. All rRNA, tRNA, and mitochondrial genomic miCLIP sites were removed. Tag counting was performed using the bedtools suite. Tag counts (uTPM + 1) were compared using scatter plots and Pearson correlation coefficients (r) were determined in R.
For identification of DC1-preferred m6A residues, residuals of simple linear regression model were calculated and sorted in R. The top 1% of sites with highest residuals were selected and annotated. HNRNPA2B1 HITS-CLIP data was obtained from a previously published study35 (GEO accession numbers: GSE70061, SRR2071655 and SRR2071656).
For representation of miCLIP tracks in Figs 3a, 4c and Extended Data Figs 4a, b, 5b, 8d, tag counts from miCLIP data sets using poly(A) RNA and miCLIP data sets using total RNA were added at every genomic position (GEO accession number: GSE63753).
Annotation of CITS
Normalized iCLIP tag-abundance was determined in the 20-bp flanking regions of the RefSeq RNA mapping CITS. Sites were then sorted based on tag abundance, and the top 1,000 sites with the highest normalized tag abundance were annotated using the annotatePeaks.pl script from the Homer package36.
Statistical significance of overlap of RNA-binding sites
To determine the statistical significance of overlap of RBM15 and RBM15B CITS (RBM15, n = 37; RBM15B, n = 56; P < 0.0001 for both) on XIST, random sites were generated on the RNA and an overlap with the RBM15 CITS was calculated (± 20 nucleotides) using the bedtools window tool. This was repeated 10,000 times to generate a null distribution for overlap counts. The P-value for the observed overlap between RBM15 and RBM15B was estimated from the null distribution (two-sided). For clusters, random clusters of equal size (median length = 91 nucleotides) were generated on XIST and a similar null distribution to CITS was generated. For both comparisons, the same number of random sites or clusters were generated as in RBM15B data set (n = 30). Clusters showing a minimum overlap of half-cluster length with the RBM15B clusters were counted. All RBM15 clusters (n = 30) overlapped with RBM15B clusters (n = 30) on XIST (P < 0.0001). The RBM15 cluster overlaps with randomly permuted RBM15B clusters, while maintaining the mean cluster size of 91 nucleotides, did not show a similar or greater percentage overlap.
RBM15/15B binding at m6A residues
Unique iCLIP tags were aligned to the hg19 genome using STAR aligner (STAR–outSAMtype BAM SortedByCoordinate --outSAMattributes All --outFilterMultimapNmax 1 --outFilterMismatchNmax 2). For determination of the average RBM15/15B-binding at m6A and non-m6A sites (both of which are DRACH-consensus sequences), sequence alignment (BAM) files were further processed using deepTools37. Methylated DRACH sites (n = 14,209) were obtained by merging miCLIP sites from HEK293 poly(A) and total RNA from ref. 17. A non-methylated DRACH site was identified near each methylated DRACH site within a distance of 20–200 nucleotides in the same transcript in the refseq transcriptome) using an in-house python script. For this purpose, DRACH sites on transcripts with no miCLIP tags were considered to be non-methylated. Heat maps were generated using the plotHeatmap script from the deepTools suite.
Immunoprecipitation of RBM15, RBM15B and METTL3
HEK293T cells were transfected with 10 nM siRNA (Supplementary Table 9) using the Pepmute transfection reagent and then grown to 80% confluency in a 150 mm dish. After 72 h, cells were washed twice with cold PBS, scraped, and collected by centrifugation. The cell pellet was then resuspended in three packed cell volumes of hypotonic buffer (10 mM HEPES pH 7.6, 10 mM KCl, 1 mM EDTA, 0.1 mM EGTA, protease and phosphatase inhibitor cocktail (Pierce)), and incubated on ice for 10 min. Triton X-100 was added to a final concentration of 0.3%, the lysate was briefly vortexed and centrifuged at 15,000g for 1 min at 4 °C. Supernatant (cytoplasm) was discarded, and the nuclear pellet was washed with 3 packed cell volumes of hypotonic buffer and centrifuged as before. The pellet was resuspended in 1 ml NP-40 lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, protease and phosphatase inhibitor cocktail) and passed through a 21-gauge syringe several times, followed by treatment with 100 U benzonase for 30 min at 37 °C. Nuclear lysates were centrifuged at 21,000g for 15 min at 4 °C. Immunoprecipitations were carried out with 500 μg of nuclear extracts with 5 μg of antibody at 4 °C overnight, followed by a 2 h incubation with 25 μl of Pierce Protein A/G magnetic beads at 4 °C. For the co-immunoprecipitation of METTL3–RBM15B, 250 μg of nuclear lysate was used per 5 μg of the METTL3 antibody. Beads were washed five times with NP-40 lysis buffer and proteins were eluted with 1× Novex Loading buffer with 50 mM dithiothreitol (DTT). The eluent was heat-denatured, electrophoresed, and transferred to a PVDF membrane and probed for different proteins. A list of antibodies and dilutions used for immunoprecipitation and western blot analysis are given in Supplementary Table 1. Quantification of band intensities was performed by the relative quantitation approach using Image Lab software (Bio-Rad, v5.2.1).
RNP immunoprecipitation and quantification of XIST
METTL3/DC1/RBM15/RBM15B-bound XIST RNA was quantified in the immunoprecipitates obtained from formaldehyde-crosslinked cells using a method previously described38 with some modifications. In brief, siRNA-transfected cells were washed with ice-cold PBS and fixed with 1% formaldehyde in PBS for 10 min at 25 °C with gentle rocking. Formaldehyde was quenched by adding glycine to a final concentration of 0.25 M and then incubating at 25 °C for 5 min. Fixed cells were washed three times with ice-cold PBS and resuspended in 0.5 ml of RIPA buffer (50 mM Tris–HCl pH 7.4, 100 mM NaCl, 1% Igepal CA-630, 0.1% SDS, 0.5% sodium deoxycholate) with protease inhibitors (Roche) and 1 mM DTT per 3 million cells. DNA was sheared by sonication on ice twice at 15% amplitude for 2 s ON, 10 s OFF for a total of 30 s. Lysates were incubated on ice for 10 min, and subjected to DNase I- and partial RNase I-digestion for 3 min at 37°C with mixing (2 μl Turbo DNase I and 5 μl of 1 to 25 times diluted RNase I in PBS per 0.5 ml of lysate). Tubes were immediately transferred to ice and incubated for 5 min. Lysates were then clarified by centrifugation at 21,000g at 4 °C for 10 min. Protein (200 μg) was supplemented with SUPERase In RNase inhibitor (100 U ml−1, Thermo Fisher) and then subjected to immunoprecipitation in RIPA buffer. Antibodies targeting METTL3, DC1, RBM15 or RBM15B (2 μg per 10 μl beads; Supplementary Table 1) were first bound to RIPA-buffer-washed Protein A/G magnetic beads (Thermo Fisher). Antibody-bound beads were then washed with RIPA buffer, added to the lysate for immunoprecipitation and incubated at 4 °C for 12 h. Rabbit IgG antibody was used as a control. Beads were washed five times with 500 μl RIPA buffer containing 1 M NaCl and 1 M Urea at 25 °C and resuspended in 100 μl eGFP-RNA (100 pg)-containing RNA elution buffer (50 mM Tris-HCl pH 7.4, 5 mM EDTA, 10 mM DTT, 1% SDS). Formaldehyde-induced crosslinks were reversed by incubation at 70 °C for 30 min with mixing. Supernatant was mixed with Trizol LS (Thermo Fisher) and co-immunoprecipitated RNA was purified according to the manufacturer’s instructions. Glycoblue (Thermo Fisher) was used to visualize the RNA pellet. Purified RNA was then reverse-transcribed with random hexamers using SuperScript III reverse transcriptase. XIST RNA levels were detected by qRT–PCR and normalized to the spike-in eGFP RNA levels. Relative XIST RNA enrichment was calculated as the ratio of normalized XIST RNA levels in protein immunoprecipitation to levels in IgG immunoprecipitates. A very low level of XIST RNA was detected in the immunoprecipitate of non-crosslinked cells compared to the crosslinked cells (<1%). Quantification of XIST was performed using primer pairs directed against three regions in XIST, selected based on the presence of RBM15- and RBM15B-binding sites (see Figs 2b, 3a). These regions were: region 1 (chrX:73,072,444–73,072,560), region 2 (chrX:73,046,651–73,046,776), and region 3 (chrX:73,067,594–73,067,714). Region 1 and 2 contain RBM15/15B-binding sites whereas region 3 lacks RBM15/15B-binding sites. Primers used for quantification are given in Supplementary Table 8. Primer PCR amplification efficiency was between 90 and 100%.
MeRIP qRT–PCR of XIST RNA
Total RNA was isolated from HEK293T cells by Trizol extraction according to the manufacturer’s instructions and poly(A) RNA was isolated using oligo-d(T)25 magnetic beads (NEB). In total, 5 μg of anti-m6A antibody (ab190886, Abcam) was pre-bound to Protein A/G magnetic beads in immunoprecipitation buffer (20 mM Tris-HCl pH 7.5, 140 mM NaCl, 0.05% Triton X-100) for 2 h. A total of 2.5 μg of poly(A) RNA was mixed with 100 pg of non-m6A (eGFP, 0.7 kb) and m6A-containing spike-in RNAs in 400 μl of immunoprecipitation buffer. Protein A/G beads were then added and incubated at 4 °C for 2 h. Samples were washed five times with immunoprecipitation buffer, and RNA was eluted from the beads by incubating with 400 μl of 0.5 mg ml−1 m6ATP for 1 h at 4 °C. Following ethanol precipitation, the input RNA and eluted poly(A) RNA were reverse transcribed with random hexamers and enrichment was determined by qRT–PCR. The spike-in control RNAs were synthesized by in vitro transcription. Non-m6A RNA (eGFP) was transcribed using an eGFP-ORF-containing plasmid in the presence of ATP (no m6ATP). The m6A-containing RNA was transcribed from an artificially synthesized dsDNA template that encoded a 1.6-kb RNA with only one adenosine residue in the presence of m6ATP and no ATP.
X-chromosome silencing assay
For this assay, a previously described method3 was used. In brief, siRNA-transfected male or female pSM33 cells were plated on poly-L-lysine or poly-D-lysine (Sigma-Aldrich) and 0.2% gelatin (Sigma-Aldrich)-coated coverslips in wells of a 24-well plate in 2i media. After 48 h, Xist RNA expression was induced with doxycycline (2 μg ml−1) (Sigma-Aldrich) in fresh media for 16 h. Control cells received only media. Immediately following incubation, cells were fixed for FISH staining.
For inducing differentiation and induction of Xist expression in the female ES cells, 2i media was replaced with MEF media (DMEM, 10% BenchMark FBS; Gemini Bio-products, 1× L-glutamine, 1× NEAA, 1× penicillin and streptomycin; Life Technologies) 12 h after transfection. After another 12 h, cells were treated with 1 μM retinoic acid (Sigma-Aldrich) for 24 h. Untreated cells were maintained in 2i media until fixing.
Cells were then fixed in Histochoice (Sigma-Aldrich) for 10 min, washed with PBS, and subjected to FISH staining and imaging. Atrx, Gpc4, Mettl3, Rbm15, Rbm15b, Xist, Ythdc1, Ythdc2, Ythdf1, Ythdf2, and Ythdf3 RNAs were stained by single-molecule RNA-FISH. They were then imaged and quantified as described in ref. 3. Probe sets and conjugated fluorophores (excitation wavelengths) for FISH probes were TYPE 1-XIST (550 nm), TYPE 4-Gpc4 (488 nm), TYPE 10-Atrx, Rbm15b (740 nm), and TYPE 6-Mettl3, Rbm15, Ythdc1, Ythdc2, Ythdf1, Ythdf2, and Ythdf3 (650 nm). Imaging was performed using Nikon Ti Eclipse microscope with the Nikon CFI Plan Apochromat λ DM 60×/1.40 oil objective. Images were processed in Fiji (ImageJ v1.51d)39. To enhance the FISH spot size, Maximum Filter plugin with a radius of 2.0 pixels was applied to the Gpc4 and/or Atrx channels.
DC1–λN–XIST–(BoxB)3 RNA tether function assay
For this assay, male mouse pSM33 cells expressing Xist–(BoxB)3 RNA under doxycycline control were used. Cells (1.5 × 105) were co-transfected with 20 nM siMETTL3 or siRBM15/15B and 0.75 μg of pCAG-GW-hYTHDC1-λN-3×Flag-BSD plasmid using Neon transfection system (10 μl tip, settings: 1,200 V, 40 ms width, 1 pulse) and seeded on coverslips as described for the X-chromosome silencing assay. For the identification of DC1–λN–3×Flag expressing cells, fixed cells were first subjected to immunofluorescence using mouse anti-Flag antibody (Sigma-Aldrich). Briefly, fixed cells were permeabilized with 0.1% Triton X-100 in PBS at room temperature for 10 min, and blocked with 5% normal goat serum in 0.1% Triton X-100 in PBS at room temperature for 30 min. Cells were then incubated with anti-Flag M2 antibody (Sigma-Aldrich; F3165; dilution 1 to 50) for 1 h at room temperature, followed by washes with 0.1% Triton X-100 in PBS and incubation with secondary antibody (goat anti-mouse IgG antibody-Alexa Fluor 750 conjugate, Thermo Fisher, dilution 1:200) at room temperature for 1 h. The samples were then processed using the RNA-FISH protocol, as described above.
Protein–protein interaction (PPI) network analysis
PINA2 (ref. 28) was used to mine the PPI networks of DC1, its immediate neighbours, the proteins regulating XIST-mediated gene silencing (SHARP, HDAC3, HNRNPK, HNRNPU, NCOR2/SMRT, LBR), and components of PRC (polycomb repressor complexes). Protein sub-networks showing interaction with DC1 and an enrichment of transcription repressor gene ontogology terms (false discovery rate < 0.05, P < 0.05) were curated and filtered for visualization. Networks were imported, visualized, and edited in Cytoscape (v3.3.0)40 for image production. To identify potentially novel interactions between DC1 and the proteins contributing to XIST-mediated gene silencing, publically available mass spectrometry data of DC1-associated proteins (PeptideAtlas accession number PASS00835) from ref. 27 was mined. Peptides were first identified by comparing the mass spectrometry spectra with references from the human proteome database (SwissProt) according to ref. 41 (15 p.p.m. peptide mass tolerance and 20 m.m.u. fragment mass tolerance). Identified peptides with natural log(e) scores below −1 and more than two unique peptides were further mined for peptides from proteins known to regulate XIST-mediated gene silencing. Identified proteins were manually added to the PPI network.
Determination of relative m6A levels by thin layer chromatography
Levels of internal m6A in mRNA were determined by thin layer chromatography (TLC) as previously described42. In brief, poly(A) RNA (100 ng) was digested with 2 U RNase T1 (Thermo Fisher) for 2 h at 37 °C in the presence of RNasin RNase Inhibitor (Promega). Five prime ends were subsequently labelled with 10 U T4 PNK (NEB) and 0.4 mBq [γ-32P]ATP at 37 °C for 30 min followed by removal of the γ-phosphate of ATP by incubation with 10 U apyrase (NEB) at 30 °C for 30 min. After phenol-chloroform extraction and ethanol precipitation, RNA samples were resuspended in 10 μl of water and digested to mononucleotides with 2 U of P1 nuclease (Sigma-Aldrich) for 3 h at 37 °C. Following this, 2 μl of the released 5′ monophosphates from this digest were then analysed by 2D-TLC on glass-backed PEI-cellulose plates (Merck-Millipore). The nucleotides were first separated in the first dimension in isobutyric acid with 0.5 M NH4OH (5:3, v/v), followed by isopropanol, HCl and water at a ratio of 70:15:15 (v/v/v) in the second dimension. Signal acquisition was carried out using a storage phosphor screen (GE Healthcare Life Sciences) at 200 μm pixel size on a Typhoon scanner (GE Healthcare Life Sciences). For quantification, m6A was calculated as a percentage of the total of the A, C and U spots, as described previously42.
Structured Illumination Microscopy (3D-SIM) and image analysis
HEK293T cells were fixed and subjected to immunofluorescence and single-molecule RNA-FISH staining using a protocol from ref. 43 with some modifications. In brief, siRNA-transfected and non-transfected HEK293T cells were seeded on poly-L-lysine-coated no. 1.5 H (170 μm ± 5 μm) coverslips (poly-L-lysine: 3438-100-01, Trevigen; coverslips: 474030-9000-000, Carl Zeiss) in 6-well plates. After 12–24 h of incubation, cells were washed twice with PBS at 25 °C and fixed with 2% methanol-free formaldehyde (28906, Thermo Fisher) in PBS for 10 min at room temperature. Cells were then washed three times with PBS and permeabilized with permeabilization buffer (1% acetylated BSA (Sigma-Aldrich), 0.3% Triton X-100, 2 mM vanadyl ribonucleoside complexes (NEB) in 1× PBS) at 25 °C for 60 min. Following permeabilization, cells were incubated with rabbit anti-YTHDC1 (ab122340, Abcam, dilution 1:1,000) in permeabilization buffer for 2 h at 25 °C in a humidified chamber. Cells were then washed with immunofluorescence-wash buffer (0.5% Tween-20 in PBS) three times at room temperature. Each wash was maintained for 5 min on cells with gentle shaking. Cells were further incubated with donkey anti-Rabbit IgG antibody-Alexa Fluor 488 conjugate (A-21206, Thermo Fisher, dilution 1:1,000) for 30 min at 25 °C in a humidified, dark chamber. Following the incubation, cells were washed as before, fixed with 4% formaldehyde in PBS for 10 min at room temperature, and washed with PBS three times. The second formaldehyde fixation immobilizes the primary and secondary antibodies at the target antigen. This step avoids loss of antibodies during the probe-hybridization step of RNA-FISH. Probe hybridization in RNA-FISH uses organic solvent such as formamide that may alter antibody structure thereby affecting its ability to bind the target antigen.
After PBS wash, cells were equilibrated in FISH-wash buffer (10% formamide in 2× SSC buffer diluted from a 20x stock (S6639, Sigma-Aldrich)) for 10 min at room temperature, and then incubated with fluorescently labelled DNA probes against XIST (Stellaris FISH probes hXIST w/ Q570, SMF-2038-1, Biosearch Technologies) in Hybridization buffer (10% formamide, 10% dextran sulfate in 2× SSC buffer) at a concentration of 100 nM in a humidified chamber at 37 °C for overnight. Following the incubation, cells were washed twice with FISH-wash buffer at 37 °C for 30 min without shaking. Cells were further washed three times with PBS, and then incubated with DAPI (2 μg ml−1 in PBS) for 15 min at room temperature with gentle shaking. Cells were further washed and maintained in PBS until mounting. Coverslips with fixed and stained cells were mounted in mounting media (Prolong Diamond, P36961, Life Technologies) and quickly sealed with a nail polish. After drying of nail polish, the slides were temporarily stored at 4 °C until imaging.
Cells were imaged by super-resolution 3D-SIM on OMX Blaze 3D-SIM super-resolution microscope (Applied Precision) equipped with a 100×/1.40 numerical aperture UPLSAPO oil objective (Olympus), EMCCD cameras (Photometrics), and 405, 488, 568 nm lasers. Fifteen raw images per plane (5 phases at 3 angles) were captured with a Z-spacing of 0.125 μm using an oil with a refractive index of 1.515. To reduce spherical aberrations, an oil of optimal refractive index was first identified. Image reconstruction and registration was performed using SoftWoRx (GE, v6.5) employing channel-specific optical transfer functions (OTFs) and Wiener filter (settings: 0.0020 for red and green channel, 0.0050 for blue channel). Further processing of 32-bit images was performed using Fiji (ImageJ v1.51d) with in-house JavaScript scripts. Images were converted to 16-bit images. A mask for the XIST signal (red) was created on all the slices in Fiji using the thresholding menu option. DC1 (green signal) in the mask was extracted using Fiji’s math menu options. 3D Object Counter plugin was then used to count the green objects (DC1 signal) in the XIST of the nucleus (n = 5, 2 XIST and 2 autosomal domains per nucleus). For autosomal domains, areas showing dense DAPI staining were manually selected at the region of interest, DC1 signal (green) was obtained, and 3D objects were counted. Objects here refer to 3D objects identified based on distribution and centre of mass of red or green signal across contiguous image slices. To calculate the percentage fraction of DC1 signal that is localized in the XIST territory in various knockdowns, total red (XIST) and green (DC1) objects were also counted in each nucleus separately. Percentage DC1 per XIST object was calculated using the following formula:
where ngx = number of green objects (DC1) in XIST domain, Trx = total number of red (XIST) objects, and Tg = total number of green (DC1) objects in the nucleus. Two-tailed Mann–Whitney test was used to calculate statistical significance.
Validation of anti-YTHDC1 antibody for immunofluorescence imaging
HEK293T cells were transfected with pSuperior-EGFP constructs expressing shLacZ or shDC1 shRNA and incubated for 48 h. These cells (20,000 per well) were then seeded on a poly-L-lysine-coated coverslips (coverslips: 1.5H, 12 mm, round, NC9455457, Fisher Scientific) in 24-well plates. Following a 12-h incubation, cells were processed for immunostaining using the immunofluorescence staining protocol of the 3D-SIM method given above. After the second formaldehyde fixation step, cells were washed three times with 1× PBS and stained with DAPI, washed, and mounted on slides in mounting media following a method similar to the 3D-SIM method. Slides were stored at 4 °C until imaging. DC1 was stained with rabbit anti-YTHDC1 antibody (ab122340, Abcam, 1:1,000) and eGFP (expressed from shRNA expressing plasmid) was stained with chicken anti-GFP antibody (ab13970, Abcam, 1:1,000). Donkey anti-rabbit IgG antibody-Alexa Fluor 568 conjugate (A10042, Thermo Fisher, 1:1,000) and goat anti-chicken IgY antibody-Alexa Fluor 488 conjugate (A-11039, Thermo Fisher, 1:1,000) were used to probe the primary antibodies. Images were captured on a wide-field fluorescence microscope (Nikon Eclipse Ti) using a 60× oil immersion objective. Images were processed on Fiji (ImageJ v1.51d).
Bacterial expression of His6-DF proteins
Full-length DF family cDNA ORFs were PCR amplified from HEK293T oligo-d(T)25-primed cDNA and cloned at NheI and XhoI for DF1 and DF3, NdeI and XhoI for DF2 in pET-28c(+) (Novagen) plasmid. These plasmids were transformed into Rosetta 2(DE3) Singles (Novagen) Escherichia coli cells. Bacteria were grown until they reached an OD600 nm of 0.5 and treated with 0.1 mM IPTG at 18 °C for 1–4 h to allow a comparable level of protein expression. Time points showing a similar level of protein expression for all the DF proteins were only analysed by western blot. DNA oligonucleotides used for amplification of the cDNA ORFs are given in Supplementary Table 8.
Enrichment of DC1-binding RNA motifs in different RNA and genomic features
For this analysis, all the 35,823 CIT sites were used. CITS were first mapped to the different genomic and RNA features in the hg19 genome using the annotation script, annotatePeaks.pl, from the Homer package. Sites mapping to rRNA, tRNA and the mitochondrial genome were discarded. For every site, strand-specific DNA sequence (± 20 nucleotides) was obtained from the hg19 genome. An enrichment of DC1-binding RNA motifs (DRACH, MTTAH, and KTCAHC) in different RNA/genomic features was determined using Centrimo tool in the MEME suite.
Confirmation of X-chromosome silencing by RT-qPCR
Total RNA was extracted and purified from 1 × 106 siRNA-transfected pSM33 cells using RNeasy Mini Kit (Qiagen) and DNA was removed by digestion with RNase-free DNase Set (Qiagen). DNA-free RNA (500 ng) was used to make cDNA with random hexamer using SuperScript III reverse transcriptase (Invitrogen) following the manufacturer’s instructions. Relative expression of genes Gpc4 and Atrx relative to Gapdh was quantified by qPCR using the LightCycler 480 SYBR Green I Master Mix (Roche). Primer information is given in Supplementary Table 8.
Extended Data
Extended Data Figure 1. Validation of RBM15 and RBM15B antibodies for iCLIP, construction and comparison of iCLIP library replicates.

a, RBM15 and RBM15B exhibit high sequence homology. RBM15 and RBM15B comprise three RRM domains (RRM1, 2 and 3, all in purple) and a C-terminal SPOC domain (green). These domains show high sequence identity between RBM15 and RBM15B (indicated on the shaded areas that connect the compared regions). RRM, RNA recognition motif; SPOC, Spen paralogue and orthologue C-terminal. b, c, Validation of specificity of RBM15 and RBM15B antibodies for iCLIP, performed using immunoprecipitation. In each experiment, we used high (H) and low (L) RNase, as per the iCLIP validation protocol30 (see Methods). The bottom western blots are loading control (GAPDH). To confirm knockdown, RBM15 and RBM15B protein levels are shown. Additionally, we show the amount of protein in the anti-RBM15 or anti-RBM15B pulldowns. These experiments confirm that the RBM15 and RBM15B are knocked down after siRNA transfection. d, e, Autoradiograms of the samples used for the RBM15 and RBM15B iCLIP experiments. Shown are the representative autoradiograms from the nitrocellulose blots of samples used for preparing the RBM15 and RBM15B iCLIP library. The excised portion of the membrane is shown (red square). The red arrow indicates the position of RBM15 and RBM15B protein after high RNase treatment that matches with the size seen in b and c respectively. Both RBM15 and RBM15B show specific RNA–protein conjugates of expected size with a minimal contamination of RNA–protein conjugates of other sizes. f, g, RBM15 and RBM15B iCLIP replicates show reproducible iCLIP tag coverage on the human genome. Three iCLIP library replicates were prepared for RBM15 and RBM15B. We compared the normalized tag counts of replicates in 100 nucleotide bins in the human genome on scatter plots, and estimated the Pearson correlation coefficient (r). Shown are the representative scatter plots (left), and heat maps (right) showing the obtained r value in multiple pairwise replicate comparisons. rep1–rep3, replicate 1–replicate 3 for each protein; RBM15 in f and RBM15B in g. The x and y axes of the scatter plots represent normalized tag counts in uTPM in 100 nucleotide bins on the human genome in rep1 and rep3, respectively. Correlation values are indicated on each tile. From this analysis, RBM15 and RBM15B iCLIP replicates show a similar, highly reproducible iCLIP tag coverage on the human genome. The diagonal dashed line in scatter plots represents reference trend line for a perfect correlation (r = 1, x = y). h, RBM15 and RBM15B show similar binding preferences on XIST. Each of the 30 clusters in the RBM15 data set overlapped with the clusters in the RBM15B data set. We also examined the CITS induced by RBM15 and RBM15B. CITS are single-nucleotide sites that represent direct contacts of these proteins with XIST (Supplementary Tables 3, 4). Most RBM15 CITS (23 out of 37) overlapped with RBM15B CITS (top). This overlap was statistically significant (P < 0.0001) based on a permutation analysis in which we measured the overlap of randomly selected sites on XIST for RBM15 and RBM15B (see Methods). Lastly, a pairwise analysis of iCLIP tag density at each CITS showed that RBM15 and RBM15B binding was highly correlated (bottom).
Extended Data Figure 2. Quantification of X-linked gene silencing upon knockdown of m6A readers and writers.

a, b, Quantification of Gpc4 spots upon Rbm15 and Rbm15b knockdown (Fig. 1b, c). The number of Gpc4 spots before and after XIST induction (−Dox and +Dox, respectively) (a). Representative RNA-FISH images with DAPI-stained nuclei with Gpc4 spots (green) and XIST staining (pink, last column) are shown (b). The number of Gpc4 spots is indicated on each FISH image. Scale bar, 5 μm. Data in a are mean ± s.e.m. NS, not significant; *****P < 0.0001 relative to Dox-deficient control by unpaired two-sample t-test. c, m6A modification is necessary for XIST-mediated gene silencing in female pSM33 cells. Quantification of Gpc4 RNA spots with and without induction of XIST expression (left). Representative RNA-FISH images showing Gpc4 RNA spots (green) with DAPI-stained nuclei (right). Wild-type (WT) cells show a normal XIST-induced silencing whereas Gpc4 spots are partially reduced (24 to 17 spots). Similar to male ES pSM33 cells, female ES cells fail to show XIST-mediated gene silencing upon knockdown of Rbm15/15b or Mettl3. Error bars mean ± s.e.m. for 50 cells per sample. NS, not significant; ****P < 0.0001, relative to no-doxycycline control by unpaired two-sample t-test. d, e, Similar to Fig 3c, d, shown is an siRNA pool that targets a (different) region on Mettl3. The data from Fig. 3c, d for the siRNA pool 1 is also shown here for comparison. In both the siControl and siMETTL3-transfected cells, XIST shows aggregation consistent with its interaction with the X chromosome. Thus, early steps of XIST interaction with the X chromosome may not require m6A. Gpc4 counts (d, top) and the change in transcription, as measured by the ratio of Gpc4 +Dox/−Dox. Notably, there is a reduction in Gpc4 and Atrx spots (see Fig. 3d) in siMETTL3-transfected cells, even in the absence of XIST expression. Representative FISH images with DAPI nuclear stain in blue, Gpc4 in green and XIST in pink (e). Following Dox treatment, the number of Gpc4 spots is markedly reduced in the control-transfected cells. However, after knockdown of Mettl3, the number of Gpc4 mRNA spots remain unchanged. Scale bars, 5 μm. Data in c are mean ± s.e.m.across 50 cells. NS, not significant; *****P < 0.0001 relative to no-doxycycline control (top graph) and siControl (bottom graph) by unpaired two-sample t-test. f, g, Similar to d and e, we show a defect in XIST-mediated silencing upon silencing of Ythdc1 as shown in Fig. 4d, e using multiple siRNA pools from different vendors. Targeting a different region of DC1 using a siRNA pool (siDC1-Q) prevents XIST-mediated gene silencing. The data from Fig. 4d, e for the Dharmacon siRNA pool is shown alongside. Data in f are mean ± s.e.m across 50 cells. NS, not significant; ****P < 0.005 relative to no-doxycycline control (top graph) and siControl (bottom graph) by unpaired two-sample t-test. h, DF1, DF2, DF3 and DC2 do not mediate XIST-mediate gene silencing. Quantification of Gpc4 (top left) and Atrx (bottom left) RNA-FISH spots is shown. Representative FISH images with DAPI-stained nuclei (blue) with Gpc4 (green) and Atrx (red) spots are shown (right). The number of detected RNA spots for both the genes are indicated on each FISH image. Scale bars, 5 μm. Data are mean ±s.e.m. across 50 cells from one experiment. ****P < 0.0001 relative to control (−Dox) by unpaired two-sample t-test. i, RBM15/15B and DC1 mediate XIST-mediate gene silencing in differentiating wild-type female ES cells. Quantification of Gpc4 RNA expression was performed in female mouse ES cells in response to retinoic acid-induced (+RA) differentiation by RNA-FISH (left). Representative FISH images showing DAPI-stained nuclei (blue), Gpc4 RNA (green), and XIST (pink) are shown (right). Wild-type cells exhibit normal Gpc4 silencing in response to retinoic acid treatment. Single knockdown of either Rbm15 or Rbm15b also exhibited normal silencing of Gpc4. Double knockdown resulted in no XIST expression (C.-K.C. and M.G., data not shown), reminiscent of the lack of XIST expression in METTL3-deficient ES cells45. CRISPR-mediated homozygous knockout of DC1 (Ythdc1−/ −) cells could not be recovered, suggesting that deletion of this gene is lethal. However, heterozygous knockout of DC1 (Ythdc1−/+) impaired Gpc4 silencing in response to retinoic acid in these cells. These data support the idea that DC1 is required for silencing of X-linked genes during ES cell differentiation. ****P < 0.0001 relative to control by unpaired two-sample t-test. j, qRT–PCR-based validation of effects of RBM15/15B and DC1 on XIST-mediated gene silencing. Gene expression level after XIST induction (+Dox) was normalized to Gapdh before XIST induction (−Dox) in both the siControl and siRbm15/siRbm15b double-knockdown sample. Quantification of the change in gene transcript levels upon expression of XIST is shown for Gpc4 and Atrx. Dox-induced XIST expression led to reduced transcription of both the genes in Control knockdown cells. However, Rbm15 and Rbm15b double knockdown and DC1 knockdown failed to show XIST-induced silencing. **P < 0.01 relative to siControl-transfected cells by unpaired two-sample t-test.
Extended Data Figure 3. Reciprocal co-immunoprecipitation of METTL3–RBM15/15B complex, validation of WTAP, RBM15 and RBM15B knockdown and their lack of effect on XIST levels.

a, b, Confirmation of WTAP-dependent METTL3–RBM15/15B interaction by reciprocal co-immunoprecipitation. METTL3 was immunoprecipitated using an antibody against the endogenous protein from nuclear extracts of the siControl- and siWTAP-transfected HEK293T cells under native conditions. Both RBM15 and RBM15B were detected in the METTL3 immunoprecipitates by western blot. The binding of both these proteins was significantly reduced in siWTAP-transfected cells, indicating that METTL3 interacts with RBM15/15B in a WTAP-dependent manner to form a RBM15/15B–WTAP–METTL3 complex. IgG heavy chain signal prevents visualization of WTAP; however, knockdown is seen in the input sample. c, Relative protein band intensities for RBM15/15B–METTL3 co-immunoprecipitation experiments. Shown here are the relative protein band intensities obtained in western blots of RBM15/15B–METTL3 and reciprocal co-immunoprecipitation experiments shown in Fig. 2a and Extended Data Fig. 3a, b, respectively. For METTL3 in RBM15 IP, n = 3; METTL3 in RBM15B IP, n = 3; RBM15 in METTL3 IP, n = 7; and RBM15B in METTL3, n = 3. d, Confirmation of WTAP, RBM15, and RBM15B knockdown. siRNA-transfected HEK293T cell lysates used for assays in Figs 2b, 3b were probed for protein levels using western blot analysis. Knockdown resulted in a significant reduction in the corresponding proteins. None of the siRNA affect METTL3 levels. The antibody for RBM15B recognizes a doublet, but only the lower band is lost after the knockdown. The specificity of this antibody for iCLIP is demonstrated in Extended Data Fig. 1b–e. e, Knockdown of WTAP, RBM15 and RBM15B, as well as double knockdown of RBM15 and RBM15B do not affect XIST RNA levels. Quantification of XIST levels by qRT–PCR from RNA purified from siRNA-transfected cells shows no significant change in XIST RNA levels. f, Validation of the anti-m6A antibody approach for pulldown of methylated XIST RNA. To validate XIST quantification used in Fig. 3b, we used a control spike-in RNAs with a single m6A, and a eGFP control RNA with no m6A residues. Unlike the m6A RNA (left), the non-methylated RNA (right) is de-enriched in the immunoprecipitation sample. NS, not significant. g, RBM15/15B bind XIST in m6A-independent manner. RBM15/15B binding of XIST in cells deficient in components of the m6A methylation machinery (METTL3 and WTAP) is shown. RBM15 and RBM15B were immunoprecipitated and XIST levels were determined by qRT–PCR at three regions (regions 1–3 refer to Fig. 2b, 3a and Extended Data Fig. 4a). XIST binding to RBM15 and RBM15B remains unchanged upon METTL3 and WTAP knockdown at region 1 and 2 where RBM15/15B both show binding. Thus, RBM15 and RBM15B are not binding to XIST in an m6A-dependent manner and are not m6A readers. At region 3, where both proteins do not show any binding, a basal level of amplification was seen similar to the level detected in IgG control. NS, not significant relative to siControl transfected cells by unpaired two-sample t-test.
Extended Data Figure 4. Zoomed-in views of miCLIP, RBM15 and RBM15B iCLIP tracks on XIST.

a, m6A residues are broadly distributed along XIST. Shown are m6A residues mapped in XIST using miCLIP17;these sites are indicated with red lines. Total RNA at every genomic position are shown in purple. RNA-seq read distribution is shown in grey. Many of the m6A sites are clustered in a 2 kb domain surrounding the A-repeat (yellow) region. The zoomed-in region shows m6A sites (red lines) and miCLIP tag distribution in a 1-kb region closest to the A-repeat region. Region 1, which contains RBM15/15B-binding sites (see Fig. 2b) is also indicated. b, c, RBM15 and RBM15B bind XIST near m6A sites. To determine whether RBM15/15B-binding sites are in proximity to known m6A sites, we compared the iCLIP tag clusters with m6A sites on XIST. Shown in b are the RBM15 and RBM15B iCLIP, and miCLIP tag distributions on XIST. m6A sites are marked with red bars above the XIST gene model. Vertical green shaded boxes mark the regions of miCLIP and RBM15/15B iCLIP tag cluster alignments. A zoomed-in view of a region with high-tag abundance (bottom left) and another with low-tag abundance (bottom right), show examples of m6A sites that are in proximity to RBM15B and RBM15B tag clusters. Normalized tags are shown in uTPM. In c, the median distance of RBM15 (left) and RBM15B (right) CITS to the nearest m6A site on XIST was determined and compared with a randomly permuted data set of RBM15- and RBM15B-binding sites. RBM15/15B-binding sites show a marked proximity to m6A compared to randomly positioned RBM15/15B sites (RBM15, **P = 0.0026, number of permutations, 10,000; RBM15B, ***P = 0.0001, number of permutations, 10,000). This proximity is not due to RBM15 or RBM15B itself binding m6A as its binding to XIST was unaffected by METTL3 or WTAP knockdown (Extended Data Fig. 3g). The red dashed line indicates the location of m6A sites.
Extended Data Figure 5. RBM15 and RBM15B bind near m6A sites on mRNA.

a, RBM15/15B binds at-or-near-to m6A sites throughout the transcriptome, including at m6A sites in XIST and ACTB mRNA. Shown are plots with an average binding-per-base around m6A (red curve) or non-m6A DRACH (green curve) sites for RBM15 (top left) and RBM15B (top right). The bottom two panels present the tag count per base around m6A or non-m6A DRACH sites as heat maps. Each row in the heat map is an m6A or non-m6A site. RBM15 and RBM15B show increased binding at or near m6A sites than at non-methylated DRACH sites (~3–4-fold higher). b, RBM15 and RBM15B bind near m6A sites on mRNA. Shown is the RNA-seq read (grey), and iCLIP (light blue, RBM15; dark blue, RBM15B) and miCLIP (purple) tag distribution on ACTB mRNA. iCLIP CITS sites are indicated below their respective tracks. miCLIP-identified m6A sites are indicated with red bars. Both proteins (light versus dark blue tracks) show a similar binding profile on ACTB mRNA, with considerable overlap of miCLIP tags at various regions along the sequence (vertical green shading). A zoomed-in view of the tag distribution is shown in the bottom panel. The sense DNA sequence of the zoomed-in region is shown above the gene model. A vertical dotted black line running through the middle of the tracks connects the RBM15/15B-binding sites with the DNA sequence that indicates the sequence at the binding site (highlighted yellow). At single-nucleotide resolution RBM15/15B binds a U-rich sequence near m6A sites on mRNA also. The binding sites show a clear separation (5 nucleotides) from the putative m6A-containing GAC sequence (red bars). RNA-seq reads are shown in absolute read counts, iCLIP and miCLIP tags are shown in uTPM. c, d, Genomic and transcriptomic distribution of RBM15- and RBM15B-RNA binding sites. To determine the types of RNA sequence that contain bound RBM15 and RBM15B, the top 1,000 iCLIP CITS (P < 0.0001) with the highest iCLIP tag coverage (in uTPM) were mapped to different features of the human genome and the overall distribution was determined. Sites mapped to mRNA (blue) represent roughly an equal fraction of all the binding sites of the proteins (~35%). To determine the overall distribution of the RNA-binding sites in mRNA, we further plotted the distribution of all the RBM15- and RBM15B-binding sites on a virtual transcript (shown in d). Metagenes for both RBM15- and RBM15B-binding sites show a similar distribution of the binding sites on the different features of mRNA. Although this metagene shows coverage all along mRNA, as is seen with m6A, this distribution does not match the m6A metagene. CDS, coding sequence; UTR, untranslated region. e, RBM15 and RBM15B bind U-rich RNA consensus motif. Shown are motifs enriched in both RBM15- and RBM15B-binding sites and the percentage distribution of the sites containing the identified motif is indicated below each motif. U-rich RNA binding motifs (shown as T in this genome-based alignment) were significantly enriched in the sequence at or around the iCLIP-identified RBM15- and RBM15B-binding sites (P < 0.0001). The absence of an m6A-like DRACH motif for both the proteins indicates that RBM15/15B does not directly bind m6A or DRACH sequences. Notably, the U-rich motif seen with RBM15/15B resembles the uracil-rich HNRNPC-binding motif, which may account for the previously observed proximity between m6A and HNRNPC-binding sites46. f, g, Knockdown of RBM15B and RBM15B reduced m6A levels in cellular mRNA. Schematic diagram of a 2D-TLC (left, f) showing the migration pattern of monophosphate nucleotides after TLC separation. Shown are relative positions of m6A (orange dotted circle) and those of adenosine (A), cytosine (C), and uracil (U) (black dotted circles). Arrows indicate the direction of solvent migration in the two dimensions. Middle and right panels show radiochromatograms obtained from 2D-TLC of poly(A) RNA from control and RBM15/RBM15B double-knockdown HEK293T cells. Double knockdown of RBM15 and RBM15B leads to a considerable decrease in m6A levels in mRNA (spots marked with black arrow in the middle and right panel). Quantification of m6A levels calculated using m6A:A + C + U ratio from mononucleotide intensity in two independent biological replicates (g).
Extended Data Figure 6. Validation experiments for iCLIP of YTH proteins: anti-YTH antibodies, and library construction.

a, Schematic representation of domain structures of human YTH proteins: DF1, DF2, DF3, DC1 and DC2. The YTH domain (blue) is located internally in DC1, while it is at the C-terminal region in the other proteins. DC1 has a different domain organization to DC2 and the similar DF proteins. The low-complexity and Glu-rich regions are indicated, as are the R3H, DEXDc, ankyrin repeats (ANK), HELICc, HA2 and OB-fold domains. The length of the protein is indicated next to each protein name. b, Validation of DF1, DF2 and DF3 antibody specificity via western blot. Full-length DF1, DF2, and DF3 were expressed as His6-fusion proteins in E. coli. IPTG was used as an inducer of protein expression (-, non-IPTG-treated; +, IPTG-treated). For anti-DF1, His6-DF1 was the major band detected but trace levels of His6-DF2 and His6-DF3 could be detected at longer exposure times. Thus, anti-DF2 and anti-DF3 antibodies are highly specific, while anti-DF1 shows a strong preference towards DF1 over the other DF proteins. c–g, Confirmation of iCLIP antibody pulldown specificity. Autoradiograms of the 32P-labelled RNA-crosslinked protein conjugates on nitrocellulose membrane (top) for DF1 (c), DF2 (d), DF3 (e), DC1 (f) and DC2 (g) are shown. High (H) and low (L) RNase are used in accordance with the iCLIP validation protocol30 (see Methods). The red arrow indicates the expected size of the YTH protein. In each case, knockdown of the YTH protein mRNA (lanes 3 and 4) abolished RNA pulldown. GAPDH was used as a loading control. To confirm knockdown, YTH protein levels in the input samples and in the anti-YTH pulldown is shown. Antibodies and their antigenic peptide regions on the target proteins are provided in Supplementary Table 1. siRNA and shRNA target sequences in mRNA are listed in Supplementary Table 9. h–l, Autoradiograms from the nitrocellulose blots of samples used for each iCLIP library replicate. For each YTH protein, four biological replicates (rep1–4) were prepared. The red arrow confirms the position of the YTH protein after high RNase treatment and matches the size seen in c–g. Typically, UV crosslinking causes an increase in the intensity of the 32P signal at the expected size of the YTH proteins (red arrow), indicating the formation of RNA-protein conjugates (lane 1 versus 3 in all panels). In the case of DC1, there is some 32P signal even in the absence of UV crosslinking (lane 1 versus 3 in k). This type of background signal is due to autophosphorylation activity of the protein or of a co-immunoprecipitating protein kinase that phosphorylates DC1. RNase-sensitive smears were obtained for all of the YTH proteins (compare lanes 4–7 to lane 3 in h–l). Experiments using protein A/G beads that did not include the antibody (lane 2) did not show any signal in the region of interest. Overall, all the replicates of each YTH protein show highly specific RNA–protein conjugates of expected size with a minimal contamination of RNA–protein conjugates of other sizes. The eluted RNA material was used for constructing iCLIP libraries. Shown below the autoradiograms are western-blot loading controls (GAPDH and each YTH protein) as well as the controls that confirm the presence of the YTH protein in the immunoprecipitates.
Extended Data Figure 7. Comparison of transcriptome-wide RNA-binding sites of endogenous YTH proteins by iCLIP.

a–e, YTH iCLIP library replicate reproducibility. For each YTH protein (DF1, DF2, DF3, DC1 and DC2), four independent biological replicate iCLIP libraries were constructed. Reproducibility of iCLIP tag coverage was assessed as in Extended Data Fig. 1f, g. Normalized iCLIP tag counts in uTPM from different replicates were compared on a scatter plot and the Pearson correlation coefficient (r) was determined. Scatter plots comparing mean tag counts of rep1 and rep2 (x axis), and rep3 and rep4 (y axis) are shown (left). A similar analysis was carried out for pairwise comparison of all the iCLIP replicates. The obtained correlation coefficients are shown on the heat map (right). The colour of the tiles in the heat map indicates the r value. YTH iCLIP replicates show similar and highly reproducible iCLIP tag coverage. The diagonal dashed line represents reference trend line for a perfect correlation (r = 1, x = y). f, Enriched motifs for each YTH protein based on transcriptome-wide iCLIP binding data. Motif analysis of the binding sites recognized by DF1, DF2, DF3 and DC1 proteins in the iCLIP data showed a DRACH sequence as the most prominent motif, which matches the known consensus motif for m6A in the transcriptome47. DC2 also showed the DRACH motif, as well as other motifs, which probably reflects its numerous RNA-binding domains (Extended Data Fig. 6a). DC1 predominantly bound DRACH in various transcriptomic and genomic features (Supplementary Table 10). These data suggest YTH proteins bind m6A in cells. RNA-binding motifs were identified using MEME analysis (see Methods). The percentage of analysed sites containing the identified motifs is shown in the top right. P values were obtained using the MEME CentriMo tool by a one-tailed binomial test. g, Global comparison of the distribution of YTH-binding sites and m6A sites on mRNA. We compared the metagene for each YTH protein binding site to the previously reported metagene of single-nucleotide resolution miCLIP-identified m6A sites on mRNA17 (YTH protein, green; miCLIP, orange). Each curve represents a kernel density (y axis) plot of CITS distribution on a virtual transcript (x axis). Transcription start site, 5′ UTR, start codon (AUG), CDS, stop codon, and 3′ UTR are indicated on the virtual transcript. Vertical dashed lines mark UTR-CDS boundaries. Owing to a small number of DC2 sites that map to mRNA, a metagene for DC2 is not shown. h, Pairwise comparison of YTH iCLIP tag coverage at m6A sites shows distinct binding preferences for DC1. To determine whether YTH proteins recognize similar m6A sites on the basis of iCLIP tag coverage, we estimated the correlation coefficient of iCLIP tag coverage at each of 11,530 m6A sites in the transcriptome from a pairwise comparison of two YTH iCLIP libraries at miCLIP-identified m6A sites. The Pearson correlation coefficients are shown as a heat map. To identify YTH proteins that show similar binding preferences, libraries were hierarchically clustered based on the obtained correlation coefficients (see dendrogram below the heat map). This indicates that the DF proteins cluster together and show a similar binding pattern, and these proteins target similar m6A sites. Both DC1 and DC2 have atypical m6A site preferences; DC2 has a weak correlation with known m6A sites. Scatter plots used for this comparison are shown in Extended Data Fig. 8a. i, Genomic distribution of RNA-binding sites. To determine the genomic distribution of preferred YTH protein-RNA binding sites, the statistically significant top 1,000 iCLIP CITS (P < 0.0001) with the highest iCLIP tag coverage (in uTPM) were mapped to different features of the human genome. DC1 exhibits prominent binding to ncRNAs (19% of top thousand CITS), while less than 2% of the DF1, DF2 or DF3 CITS lie within annotated ncRNAs, including lncRNAs. Most DF1-, DF2- and DF3-binding sites are located in mRNAs and introns. DC2 had negligible coverage in mRNAs, and predominantly bound to introns and intergenic sequences.
Extended Data Figure 8. DC1 preferentially binds to a subset of m6A sites that are primarily localized to XIST and other ncRNAs.

a, Pairwise comparison of YTH iCLIP libraries, and identification of DC1 preferred m6A sites. Shown are data used to generate the heat map in Extended Data Fig. 7h. In each pairwise analysis, two YTH proteins were compared for their binding to each m6A residue using normalized tag counts (see Methods), providing an estimate of the preferred binding partner for each m6A site for each YTH protein comparison. Tag counts in a window surrounding each m6A genomic coordinate (10 bp upstream and downstream) were determined for each YTH protein. Scatter plots are shown for each pair of indicated YTH proteins. m6A sites are plotted as points in which x and y coordinates represent the tag counts in the compared libraries. The DF family of proteins show highly similar binding preferences as indicated by their high Pearson correlation coefficients (r, top right corner of each plot). Hierarchical clustering as shown in Extended Data Fig. 7h supports the overall relatedness of the binding preferences of DF proteins. However, DC1 and DC2 show a pattern different from the DF proteins. DC2 shows low tag coverage on most m6A sites, and thus yields low r values. Notably, DC1 shows a global de-enrichment of binding at DF1, DF2 and DF3-preferred sites as seen by the flattened trend line (green). Additionally, DC1 shows enrichment at a unique set of m6A sites (the 1% of sites furthest from the trend line is highlighted with a red dashed ellipse in the comparison between DC1 and DF1, DF2 and DF3). b, A Venn diagram showing the number of sites preferred for DC1 over DF1, DF2 and DF3. The vast majority (105, white shaded area) are the same between each comparison, meaning these sites are preferred by DC1 over any DF protein. The rightward projection shows that most of these m6A sites are in ncRNA, constituted primarily of XIST m6A sites. c, Sequence logo analysis shows that the DC1-preferred m6A sites conform to the DRACH-like m6A consensus motif seen throughout the transcriptome, not in a novel DC1-specific motif. d, Zoomed-in views of iCLIP tag distribution on XIST for the five YTH proteins on XIST. The miCLIP tag distribution also identifies regions enriched in m6A. Only DC1 (blue) exhibits prominent iCLIP tags on XIST, the other YTH proteins do not. Vertical green shading marks the regions of XIST that contain the highest density of m6A sites. RNA-seq reads are shown in read counts, iCLIP and miCLIP tags are shown in uTPM. Regions 1 and 2 contain RBM15/15B-binding sites, region 3 does not. These sites are indicated by coloured boxes. DC1 shows a higher number of iCLIP tags at regions 1 and 2, areas containing several m6A sites. Although region 3 (grey) shows a putative m6A site, DC1 shows poor binding, possibly owing to the structural organization of XIST. e, HNRNPA2B1 does not bind m6A sites on XIST. HNRNPA2B1 was previously shown to bind m6A sites on primary micro RNA (pri-miRNA) transcripts35. We compared HNRNPA2B1 HITS-CLIP and miCLIP17 tag coverage (±10 bp in uTPM) at 11,530 annotated m6A sites, and determined correlation coefficients for m6A sites in mRNA (red) and in ncRNA (blue). HNRNPA2B1 does not show any significant binding to m6A sites on mRNA and ncRNA. Notably, the miCLIP-identified m6A sites17 used in this analysis lacks m6A sites from pri-miRNAs.
Extended Data Figure 9. DC1 binds XIST m6A in an METTL3-, RBM15-, and RBM15B-dependent manner.

a, DC1 interacts with XIST in an RBM15/15B-dependent manner. Quantification of XIST in DC1 immunoprecipitates at regions 1 and 2 (left) by RNA immunoprecipitation followed by qPCR. Western blot analysis of protein from the siRNA-transfected cells (right). Knockdown of METTL3, WTAP, RBM15 and RBM15B leads to a significant decrease in XIST enrichment from DC1 immunoprecipitates with RBM15/RBM15B double knockdown exhibiting the greatest decrease. These data indicate that DC1 binds XIST RNA in a METTL3/RBM15/15B-dependent manner. Region 3 showed no reproducible and detectable amplification, possibly owing to the poor binding of DC1. In Extended Data Fig. 8d, region 3 shows a very low DC1 iCLIP tag coverage. Data are mean ± s.e.m. for three independent experiments. ***P < 0.0001 relative to XIST levels in siControl-transfected cells by unpaired two-sample t-test. b, Validation of DC1 antibody for immunofluorescence. Images of shLacZ- and shDC1-transfected HEK293T cells probed with DC1 antibody. DC1 exhibits a nuclear localization (red). In eGFP-expressing shDC1-transfected cells (arrow), the DC1 antibody signal is substantially lower than in a non-transfected cell in the same field (compare red signal, bottom row). Control knockdown with shLacZ-expressing plasmid shows DC1 staining similar to the non-transfected cells in the same view (red channel, arrow). Nuclei were stained with DAPI. Scale bars, 10 μm. c, d, DC1 preferentially localizes to the XIST subnuclear compartment. 3D-SIM was used to examine the levels of DC1 in the XIST subnuclear compartment compared to an autosomal domain in HEK293T cells following DC1 immunofluoresence labelling and XIST RNA-FISH. HEK293T cells are triploid, and thus exhibit two inactive X chromosomes48,49. Left, a representative image showing DC1 (green), XIST (red) and DAPI (nucleus, grey-white) staining. Right, 2× magnification of highlighted regions (squares). DC1 is enriched in the XIST domains over similar dense autosomal compartments (right, top two versus bottom two rows). A distribution analysis of 3D-object counts performed on the DC1 signal in the XIST and autosomal domains also shows a significant enrichment (d, number of nuclei = 5, total XIST domains = 10, total autosomal domains = 10). Regions A and B (yellow squares) highlight two DAPI-stained inactivated X-chromosome territories marked by the presence of XIST (red). Areas C and D (blue squares) mark DAPI-stained autosomal domains. Scale bar, 5 μm. In d, **P = 0.0023 using two-tailed Mann–Whitney test. e, Localization of DC1 in XIST territory is METTL3- and RBM15/15B-dependent. To determine whether DC1 localizes to the XIST subnuclear compartment in an m6A-dependent manner, the number of DC1 spots in the XIST domain after METTL3 and RBM15/RBM15B knockdown was assessed by 3D-SIM, followed by image analysis. Knockdown of METTL3 and RBM15/RBM15B led to a significant decrease in the XIST-localized DC1. Box plot shows distribution of percentage of DC1 molecules (green objects) in XIST domain from the different knockdown cells. 10 nuclei per knockdown; ***P = 0.0011, ****P = 0.0147 relative to control knockdown in a two-tailed Mann–Whitney test.
Extended Data Figure 10. Model for the role of m6A in XIST-mediated transcriptional silencing and DC1 protein–protein interaction network analysis.

a, A model for m6A-dependent XIST-mediated gene silencing. RBM15/RBM15B is the portion of the m6A methylation complex (that is, RBM15/RBM15B–WTAP–METTL3) that binds XIST. This binding enables methylation of adjacent adenosine residues in DRACH consensus sites. The m6A residues act as recruitment sites for DC1, which may facilitate and stabilize the assembly of silencing proteins on XIST. b, Protein–protein interaction (PPI) network analysis identifies a multi-component pathway that might mediate efficient XIST-mediated gene silencing. DC1 has no known protein domain that could directly mediate repression of gene transcription. We mined the PINA2 database28 for the PPI network of DC1, as well as for proteins that interact with DC1-binding proteins and proteins that regulate XIST-mediated gene silencing (SHARP, HDAC3, HNRNPK, HNRNPU, NCOR2 (also known as SMRT), LBR, PRC1 and PRC2). A network of proteins that interact with DC1 is shown, as are the interactions of these proteins (subnetworks). Proteins that are linked to XIST-mediated silencing are indicated in pink (the PRC components) or orange. c–e, Subnetworks showing the presence of proteins involved in transcription repression. Gene Ontology terms were filtered from the main network in b. In c, the DC1-BMI subnetwork is shown. This interaction is based on co-immunoprecipitation of DC1 with BMI1, a component of the PRC complex required to maintain gene repression50. BMI1 may recruit SHARP, which directly binds XIST and mediates the recruitment of HDAC3 on the X chromosome. The EMD (emerin) subnetwork shown in d, is significantly enriched in proteins involved in transcription repression (false discovery rate < 0.05, P < 0.05) (Supplementary Table 7). DC1 interacts with EMD51, which is linked to proteins that are known to be necessary for XIST-mediated gene silencing (interactions indicated with bold red lines). A separate analysis of DC1 co-immunoprecipitated proteins identified by tandem immunoprecipitation followed by mass spectrometry analysis also shows the presence of SHARP and LBR proteins (interactions indicated with red dotted lines). Protein-binding partners of another DC1-interacting protein, KHDRBS1 (ref. 52) is shown in e. KHDRBS1 (also known as SAM68) is a well-known transcriptional repressor. Here KHDRBS1 is shown to interact with PRC component proteins SUZ12, EZH2, and RNF2. SUZ12 and EZH2 are components of the PRC2/EED–EZH2 complex that mediates histone methylation at K9 and K27 residues, leading to transcriptional repression. KHDRBS1 also interacts with XIST5. RNF2 is a component of PRC1 complex. RNF2 has E3 ubiquitin-protein ligase activity that mediates monoubiquitination of Lys119 of histone H2A (H2AK119Ub). Components of PRC1/2 and are found to be enriched on the inactivated X chromosome4.
Supplementary Material
Acknowledgments
We thank members of the Jaffrey and Guttman laboratories for comments and suggestions, A. Olarerin-George for discussions on computational analysis, J. -Y. Kim for discussions on immunostaining and imaging, the members of the Weill Cornell Epigenomics Core for their assistance in high-throughput sequencing, A. North and other staff members of the Bio-Imaging Resource Center at the Rockefeller University for their assistance with 3D-SIM imaging, and S. Mukherjee for input on image analysis. This work was supported by NIH grants R01CA186702 (to S.R.J.); T32CA062948 (B.F.P.); T32GM07616 (C.-K.C.) and an NIH Director’s Early Independence Award (DP5OD012190), the Rose Hills Foundation, Edward Mallinckrodt Foundation, Sontag Foundation, Searle Scholars Program, the Pew-Stewart Scholars program, and funds from the California Institute of Technology (M.G.).
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions All authors designed the experiments and analysed data. D.P.P., B.F.P. and S.R.J. primarily oversaw iCLIP, biochemistry, and SIM experiments; C.-K.C. and M.G. oversaw experiments on XIST-mediated silencing in mouse ES cells. D.P.P. performed the iCLIP experiments, collected data, performed 3D-SIM imaging, analysed data, and prepared figures. C.-K.C. and C.J. performed XIST mouse ES experiments, collected data, and prepared figures. A.C. made and characterized the female pSM33 ES cell line. D.P.P. and S.R.J. wrote the manuscript and all authors provided input and comments.
Author Information: iCLIP datasets generated in this study have been deposited in the Gene Expression Omnibus database under accession number GSE78030.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
Reviewer Information Nature thanks S. Schwartz and the other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X-chromosome inactivation. Nature. 1996;379:131–137. doi: 10.1038/379131a0. [DOI] [PubMed] [Google Scholar]
- 2.Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet. 2011;12:542–553. doi: 10.1038/nrg3035. [DOI] [PubMed] [Google Scholar]
- 3.McHugh CA, et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521:232–236. doi: 10.1038/nature14443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minajigi A, et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science. 2015;349:aab2276. doi: 10.1126/science.aab2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu C, et al. Systematic discovery of Xist RNA binding proteins. Cell. 2015;161:404–416. doi: 10.1016/j.cell.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasegawa Y, et al. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev Cell. 2010;19:469–476. doi: 10.1016/j.devcel.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moindrot B, et al. A pooled shRNA screen identifies Rbm15, Spen, and Wtap as factors required for Xist RNA-mediated silencing. Cell Reports. 2015;12:562–572. doi: 10.1016/j.celrep.2015.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.König J, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wutz A, Rasmussen TP, Jaenisch R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet. 2002;30:167–174. doi: 10.1038/ng820. [DOI] [PubMed] [Google Scholar]
- 12.Horiuchi K, et al. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. 2013;288:33292–33302. doi: 10.1074/jbc.M113.500397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong S, et al. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20:1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwala SD, Blitzblau HG, Hochwagen A, Fink GR. RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLoS Genet. 2012;8:e1002732. doi: 10.1371/journal.pgen.1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ping XL, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem. 1994;269:17697–17704. [PubMed] [Google Scholar]
- 17.Linder B, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plath K, Lowry WE. Progress in understanding reprogramming to the induced pluripotent state. Nat Rev Genet. 2011;12:253–265. doi: 10.1038/nrg2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz S, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Reports. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morohashi K, et al. Cyclosporin A associated helicase-like protein facilitates the association of hepatitis C virus RNA polymerase with its cellular cyclophilin B. PLoS One. 2011;6:e18285. doi: 10.1371/journal.pone.0018285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berglund L, et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–2027. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon SC, et al. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013;20:1122–1130. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- 27.Xiao W, et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Cowley MJ, et al. PINA v2.0: mining interactome modules. Nucleic Acids Res. 2012;40:D862–D865. doi: 10.1093/nar/gkr967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engreitz JM, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huppertz I, et al. iCLIP: protein-RNA interactions at nucleotide resolution. Methods. 2014;65:274–287. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer KD, et al. 5′UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weyn-Vanhentenryck SM, et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Reports. 2014;6:1139–1152. doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma W, Noble WS, Bailey TL. Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat Protocols. 2014;9:1428–1450. doi: 10.1038/nprot.2014.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh G, et al. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151:750–764. doi: 10.1016/j.cell.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alarcón CR, et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA Processing events. Cell. 2015;162:1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–91. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods. 2002;26:182–190. doi: 10.1016/S1046-2023(02)00021-X. [DOI] [PubMed] [Google Scholar]
- 39.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Link AJ, LaBaer J. Proteomics: A Cold Spring Harbor Laboratory Course Manual. Ch. 8. Cold Spring Harbor; 2009. pp. 117–142. [Google Scholar]
- 42.Jia G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kochan J, Wawro M, Kasza A. Simultaneous detection of mRNA and protein in single cells using immunofluorescence-combined single-molecule RNA-FISH. Biotechniques. 2015;59:209–212. 214, 216. doi: 10.2144/000114340. passim. [DOI] [PubMed] [Google Scholar]
- 44.Uranishi H, et al. The RNA-binding motif protein 15B (RBM15B/OTT3) acts as cofactor of the nuclear export receptor NXF1. J Biol Chem. 2009;284:26106–26116. doi: 10.1074/jbc.M109.040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geula S, et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 46.Liu N, et al. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei CM, Gershowitz A, Moss B. 5′-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry. 1976;15:397–401. doi: 10.1021/bi00647a024. [DOI] [PubMed] [Google Scholar]
- 48.Bylund L, Kytölä S, Lui WO, Larsson C, Weber G. Analysis of the cytogenetic stability of the human embryonal kidney cell line 293 by cytogenetic and STR profiling approaches. Cytogenet Genome Res. 2004;106:28–32. doi: 10.1159/000078556. [DOI] [PubMed] [Google Scholar]
- 49.Lin YC, et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun. 2014;5:4767. doi: 10.1038/ncomms5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao Q, et al. The central role of EED in the orchestration of polycomb group complexes. Nat Commun. 2014;5:3127. doi: 10.1038/ncomms4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilkinson FL, et al. Emerin interacts in vitro with the splicing-associated factor, YT521-B. Eur J Biochem. 2003;270:2459–2466. doi: 10.1046/j.1432-1033.2003.03617.x. [DOI] [PubMed] [Google Scholar]
- 52.Hartmann AM, Nayler O, Schwaiger FW, Obermeier A, Stamm S. The interaction and colocalization of Sam68 with the splicing-associated factor YT521-B in nuclear dots is regulated by the Src family kinase p59(fyn) Mol Biol Cell. 1999;10:3909–3926. doi: 10.1091/mbc.10.11.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.