

Abstract
We are developing Chlamydomonas strains that can be used for safe and sustainable control of mosquitoes, because they produce proteins from Bacillus thuringiensis subsp. israelensis (Bti) in the chloroplast. Chlamydomonas has a number of advantages for this approach, including genetic controls that are not generally available with industrial algae. The Bti toxin has been used for mosquito control for > 30 years and does not engender resistance; it contains three Cry proteins, Cry4Aa (135 kDa), Cry4Ba (128 kDa) and Cry11Aa (72 kDa), and Cyt1Aa (25 kDa). To express the Cry proteins in the chloroplast, the three genes were resynthesized and cry4Aa was truncated to the first 700 amino acids (cry4Aa700); also, since they can be toxic to host cells, the inducible Cyc6:Nac2-psbD expression system was used. Western blots of total protein from the chloroplast transformants showed accumulation of the intact polypeptides, and the relative expression level was Cry11Aa > Cry4Aa700 > Cry4Ba. Quantitative western blots with purified Cry11Aa as a standard showed that Cry11Aa accumulated to 0.35% of total cell protein. Live cell bioassays in dH20 demonstrated toxicity of the cry4Aa700 and cry11Aa transformants to larvae of Aedes aegypti and Culex quinquefasciatus. These results demonstrate that the Cry proteins that are most toxic to Aedes and Culex mosquitoes, Cry4Aa and Cry11Aa, can be successfully expressed in the chloroplast of Chlamydomonas.
Keywords: green algae, Bacillus thuringiensis subsp. israelensis, Chlamydomonas, Cry toxins, mosquito control, West Nile, Zika
Introduction
The re-emergence of dengue and the spread of West Nile, chikungunya and now Zika arboviruses signals an increasing global danger of vector-borne infectious diseases, driven by factors such as globalization and climate change (Morens and Fauci 2013). In the absence of treatments for the aforementioned viral diseases, vector control is the principal means of limiting these diseases. Mosquito control is a proven approach that is uniquely powerful, because it can block the transmission of multiple diseases simultaneously. The preferred strategy is integrated pest management (IPM), which uses a combination of practices based on data, and targets all stages of the insect life cycle. An important part of IPM is the use of pesticides to bring down damaging pest populations. In most places, the pesticides used most often are chemicals that kill the adult insects (e.g., DDT, Malathion and Naled), although methoprene is used to block larval development. The problems with these compounds are their toxicity to non-target organisms, such as honey bees, and the increasing resistance to them among mosquito populations (e.g., Ibrahim et al. 2013; Kioulos et al. 2014; Koou et al. 2014); indeed, all the chemical pesticides eventually fail due to the development of resistance. In addition, there are growing concerns about effects that long-term low-dose exposure to chemical pesticides may have on human beings (Kamel 2013). Thus, many leading public health organizations have called for new approaches and products for mosquito control.
Unbeknownst to most outside of the vector control arena, there are safe biolarvacides made from the bacterium Bacillus thuringiensis subsp. israelensis (Bti) that are also unique in that they have not produced strong resistance in > 30 years of use against mosquitoes (Ben-Dov 2014). Moreover, controlling mosquitoes at the larval stage, before they become disease-transmitting adults, has obvious advantages. However, Bti has drawbacks that have limited its use: mainly it does not persist very long in many environments, and in polluted or organic-rich waters its efficacy is diminished requiring much higher amounts (Ohana et al. 1987; de Barjac and Sutherland 1990; Liu et al. 1993; Glare and O'Callaghan 2000; Bravo et al. 2011; Tetreau et al. 2012). These drawbacks arise because the Bti toxin is a protein complex that is produced during sporulation but is outside of the spore, in what is known as a parasporal body. The Bti spores themselves do not reproduce well in most aquatic habitats.
The fact that algae and mosquito larvae co-occur in the same habitats (Laird 1988), and that most algae are excellent food sources for larvae (Marten 1986; Kaufman et al. 2006) raises the possibility of using them as a biocontrol mechanism. In an early effort, algae that are indigestible for mosquito larvae were identified, with the idea of using them to starve the larvae (Marten 1986); this study also identified many algae that could sustain larval development, including Chlamydomonas reinhardtii. Recently, Chlamydomonas strains expressing double-stranded RNA (RNAi) for a segment of a gene from Anopheles gambiae were shown to be detrimental to Anopheles stephensi when they were fed to the emerging larvae (Kumar et al. 2013). Whether or not the RNAi will effect non-target species, or if the mosquitoes will develop resistance to RNAi are not known. A marine chlorophyte, Chlorella dessicata, was stably engineered to produce a regulatory peptide (trypsin modulating oostatic factor) that represses trypsin in the larval gut (Borovsky et al. 2016). The transgenic Chlorella strains were lethal to A. aegypti larvae; however, the assays had extraordinarily high algal cell numbers (3 × 108 cells mL-1).
The most effort has been based on modifying cyanobacteria to synthesize the larvacidal proteins of Bti (Angsuthanasombat and Panyim 1989; Stevens et al. 1994; Soltes-Rak et al. 1995; Zaritsky et al. 2010). In the most successful series to date, Zaritsky and colleagues developed a larvicidal Anabaena strain with chromosomally-expressed (ie, not plasmid-born) genes for cry4Aa and cry11Aa that was quite toxic to Aedes and Culex larvae (Wu et al. 1997). However, the Anabaena strain has not, to our knowledge, been employed for mosquito control, due partly to concerns over the bacterial antibiotic-resistance gene(s) that it contains (Zaritsky et al. 2010). It also lacked the cyt1Aa gene needed to prevent the development of resistance, although cyt1Aa gene could be present on a plasmid with these genes in other strains (Khasdan et al. 2003).
Our approach to this problem is to develop a biological platform for mosquito control based on the eukaryotic green alga Chlamydomonas reinhardtii, and, at least initially, the Bti larvacide. Chlamydomonas is an edible alga that can swim and multiply in larval habitats, but also depends on sexual reproduction to survive in the environment, as the zygote is the tough stage of the life cycle (Harris 1989). The sexual phase of its life cycle has a genetic feature that we can use to prevent the formation of genetically modified progeny should it mate with a local indigenous strain, namely that the chloroplast genome is inherited from only one parent (Harris 1989). Also, chloroplast engineering in Chlamydomonas can be accomplished without leaving behind a bacterial antibiotic-resistance gene (Boynton and Gillham 1993; Fischer et al. 1996; Odom et al. 2001; Chen and Melis 2013; reviewed in Purton et al. 2013). Besides their greater ability to recycle, algal strains producing the Bti proteins would be more effective than Bti in organic-rich and/or polluted waters, because the toxin will be intracellular. Moreover, they should be cheaper to produce, since they would not require processing, and could be grown locally anywhere.
The Bti toxin is a crystal-like inclusion composed of 4 main proteins - three Cry proteins (Cry4Aa, Cry4Ba, Cry11Aa) and Cyt1Aa - that act synergistically to destroy the gut membrane following ingestion by the larvae. Each of the individual proteins have activity, but the Cry proteins are considerably more toxic than Cyt1Aa; however, Cyt1Aa synergizes the Cry proteins and blocks the development of strong resistance. The Cry proteins differ considerably in their toxicity toward different mosquito genera; e.g., Cry4Ba is highly toxic to Anopheles, whereas Cry11Aa and Cry4Aa are very active against Aedes and Culex. These differences largely reflect the specific binding properties of the Cry proteins to receptors in the larval gut membrane (reviewed in Ben-Dov 2014).
The Bti toxin genes can be quite challenging to express in other organisms, as the Cry proteins are all large (ranging from 75 to 130 kDa) and can be toxic to host cells at high levels, even though they are considered to be protoxins in the bacterium. Hence, as a first step, we have used an inducible expression system (Surzycki et al. 2007) to produce the three Cry proteins in the chloroplast of Chlamydomonas. The inducible system relies on a metal (Cu2+) repressible protein that goes into the chloroplast and activates the introduced cry gene via its 5′ UTR, which is from the psbD gene. With this system, we could express all 3 Cry proteins from Bti in the chloroplast, and we demonstrated larvacidal activity for the Cry11Aa and Cry4Aa-producing strains.
Materials and methods
Chlamydomonas strains, growth, and chlorophyll determination
The Ind41_18 strain of C. reinhardtii was obtained from Jean-David Rochaix (University of Geneva, Switzerland). The cultures were maintained on agar plates of Tris-acetate-phosphate medium (TAP) (Gorman and Levine 1965) in light at 23°C. Since TAP contains copper it was used as the +Cu2+ repression medium (TAP + Cu2+), while the inducing medium (TAP − Cu2+) was made by removing copper sulfate from the Hutner's trace solution. The flasks and graduate cylinders (glass) used to make the − Cu2+ medium were washed sequentially with 6 N HCl, dH20 (7×), and MilliQ-water (3×) before use (Quinn and Merchant 1998).
Liquid cultures were in flasks that were ∼40% full and mixed continuously on an orbital shaker at 125 rpm. Cell counts were made with a hemocytometer after killing the cells with iodine, or by estimating total chlorophyll (below) and using the reference value of 4 mg chlorophyll per 1 × 109 cells (Harris 1989). Culture wet weight were measured in some cases after centrifuging cell cultures in a 50 mL tube, and weighing of the cell pellets.
Growth rates of the Cry transformants and the parental Ind41_18 strain under uninduced and induced conditions were determined in liquid medium. First, agar cultures were used to inoculate flasks of TAP medium, and the cells were grown to near-stationary phase (4 × 106 cells mL-1). These cultures were used to inoculate, in parallel, 50-ml cultures of TAP and TAP − Cu2+ at 5 × 104 cells mL-1, and grown as described above. Aliquots were removed every 12 h for determination of total chlorophyll.
Total chlorophyll was measured as described by Wintermans and De Mots (1965). Culture aliquots (1 mL) were centrifuged in a microfuge for 5 min, and the resulting cell pellet was extracted with 1 mL of 95% EtOH and mixing. After recentrifugation (2 min), the supernatant was removed and its absorbance at 665 and 649 nm was measured and used to calculate total chlorophyll.
Design and preparation of synthetic cry genes
We elected to express the cry4Ba and cry11Aa genes as full-length and to truncate cry4Aa after amino acid 700 (cry4A700), since studies showed that the toxicity resided in the first 700 amino acids (Yoshida et al. 1989). The FLAG peptide DYKDDDDK was added to the C-terminus of each protein. The sequences of the native Bti genes (gene IDs: cry4Aa - 5759905, cry4Ba - 5759934, cry11Aa - 5759849) were optimized with the program Optimizer (Puigbò et al., 2007) using a custom codon-usage table. After analyzing the predicted RNA structures at the 5′ end using Mfold, the third codon in cry4Ba was changed from AAC to CAA, which changed the amino acid from asparagine (N) to glutamine (Q); this was done to prevent the start codon from being in a predicted paired region. The genes were synthesized commercially (Integrated DNA Technologies) and we obtained them as cloned plasmids.
Construction of Cry plasmids
The plasmids were carried in E. coli host strain DH5α (Invitrogen), and were assembled in the low-copy pET-16b vector. The Cry genes were excised by digesting with Xba I (on the 3′ side), blunting with Klenow DNA polymerase, and then digesting with Nde I (on the 5′ side). To accept cry4Aa700 and cry11Aa, pET-16b was cut with XhoI, blunted with Klenow, and then cut with Nde I. However, the Nde I digestion was incomplete, so the clones turned out to have 9 extra nucleotides (3 amino acids, MLD) at the beginning of the coding sequence that included an intact Nde I site. For cry4Ba, pET-16B was digested with BamH I before blunting and digestion with Nde I, so the cry4Ba clone did not have extra nucleotides at the 5′ end. The new plasmids were pET-4A700, pET-4B, and pET-11A.
The 5′ and 3′ expression signals (from psbD and psbA, respectively) were added sequentially to the cry genes as PCR products made with the high-fidelity Phusion DNA polymerase (New England Biolabs) according to the manufacturer's instructions. The thermocycling program was as follows: 94°C (3 min); 33 cycles of 52°C (1 min), 72°C (3.5 min), and 94°C (30 sec); 52°C (1 min); and then 72°C (5 min). And the PCR products were analyzed on 1% agarose gels before restriction digestion and cloning.
The 5′ region from psbD, including the promoter and 5′ UTR, was amplified from plasmid p108-14 (Surzycki et al. 2007), obtained from Jean-David Rochaix (University of Geneva). The forward primer (847 in Table 1) contained overlapping Nco I and BamH I sites; the NcoI site was used to attach it to the coding regions (as a Nco I-Nde I fragment) and the BamH I site was used later to excise the whole gene. The reverse primer (850 in Table 1) contained the Nde I site, but also altered a possible Shine-Dalgano sequence at nucleotides -13 to -9 from GGAG to AAAG (Nickelsen et al. 1999); this mutation was introduced to block translation in E. coli. The altered 5′ region was named psbDm and the resulting PCR product was double-digested with Nco I and Nde I and cloned into the Nco I+Nde I-digested pET-Cry plasmids (above); the new plasmids were named pET-5D4A700, pET-5D4B, and pET-5D11A. The psbA 3′ region was amplified from plasmid P-322 (Newman et al. 1992) with primers 860 and 861; both primers contained a BlpI site and the reverse primer (861) contained a BamHI site for subcloning the whole construct into a chloroplast transformation plasmid (below). The PCR product was cut with BlpI and cloned into Bpu1102I-cut pET-5D4A700, pET-5D4B, and pET-5D11A, where it attaches in only one direction. This added ∼50 nucleotides of vector sequence between the coding region and the 3′ UTR-proper from psbA. The psbDm-Cry-psbA constructs were confirmed by sequencing of the new plasmids (pET-5D4A7003A, pET-5D4B3A, and pET-5D11A3A). To create the chloroplast transformation plasmids, the Cry gene constructs were excised with BamHI and cloned into the BamHI site of p322.1, creating pCry4A700, pCry4B, and pCry11A.
Table 1. Oligonucleotide sequences.
| ID no. | Namea | Sequenceb |
|---|---|---|
| 795 | cry4A F | GTCAACAAAACCAACAATACG |
| 796 | cry4A R | TTAGTGTAGTCAGTACCTGAG |
| 797 | cry4B F | AACGACTTACAAGGTTCAATG |
| 798 | cry4B R | TGTCTGGGAATACGTCTACAG |
| 799 | cry11A F | GGAAGACTCATCATTAGACAC |
| 800 | cry11A R | AGTAGCAGTGTTGAAACCAGT |
| 847 | psbD 5′ F | gctcccatggatccTCATAATAATAAAACCTTTATTCAT Nco I BamH I |
| 850 | psbD 5′ R | ccggcatatgGTGTATCTTTAAAATAAAAAAACAACTCATCGTTACG Nde I |
| 860 | psbA 3′ F | cggggctgAGCTCAAACAACTAATTTTTTTTTAAAC Blp I |
| 861 | psbA 3′ R | cagtgctcagcggaTCCTGCCAACTGCCTATGGTAGC Blp I BamH I |
| 864 | Integration site F | TGGAATTGGATATGGACTAG |
| 865 | Integration site R | GGTACTTGCATTTCATAAGT |
| 1001 | cry4A RT F | TGGTGGTGACTTAATTGACTTC |
| 1002 | cry11A RT F | AGACTTCTACTCAAAAAAATCACACTAC |
| 1003 | cry4A RT R | GGGAATAATACTGGAATTAAAGTACCGA |
| 1004 | cry11A RT R | GAGAAAGCAGCTTTAGCAGCCCATTT |
F, forward; R, reverse
Upper case nucleotides, cry or chloroplast gene nucleotides; lower case, additional nucleotides; underlined nucleotides, restriction sites; bold and gray-shaded TT nts in psbD 5′R are substitutions of the normal CC nts, in order to eliminate the Shine-Dalgarno-like sequence.
Chloroplast transformation
The Ind41_18 strain was grown in TAP under a light flux of ∼40 μmol phitons m-2 s-1 until the culture reached 2 - 4 × 106 cells mL-1. The cells were harvested by centrifugation and then biolistically transformed with the Cry plasmids as described previously (Odom et al. 2001) using co-transformation with plasmid pB4CC110; pB4CC110 contains the spectinomycin-resistance marker, spr-u-1-6-2, in the 16S rrn gene (Newman et al. 1990). Typically, 5 μg of pB4CC110 and an equal amount of one of the Cry plasmids were co-precipitated onto 3 mg of tungsten particles (M17, Bio-Rad), and particles containing ∼600 ng of DNA was shot at each plate of cells embedded in 0.25% soft agar (Boynton and Gillham, 1993). The bombarded plates were incubated overnight in dim light (∼2 μmol photons m-2 s-1), then the cell layer was scraped off and split onto two TAP-agar plates containing 100 μg mL-1 spectinomycin. These selection plates were incubated under bright light (∼40 μmol photons m-2 s-1) at 23°C, and spectinomycin-resistant colonies appeared in 2-4 weeks. The colonies were re-streaked and grown several times on TAP-agar containing 300 μg mL-1 spectinomycin until they reached homoplasmicity as judged by PCR.
Transformants for PCR analysis were grown on TAP-agar plates with 300 μg mL-1 spectinomycin, and total DNA was extracted as described by Kwon et al. (2014). To check for homoplasmicity, primers 864 and 865 (Table 1), which amplify the site of integration site in chloroplast DNA (CpDNA) were used; homoplasmicity was indicated by the absence of untransformed copies of the genome. PCR with gene-specific primers was also performed using the following oligos: 795 and 796 for cry4Aa700, 797 and 798 for cry4Ba, and 799 and 800 cry11Aa (Table 1). Standard PCR procedures with Taq DNA polymerase (NEB) and the manufacturer's buffer were used. The thermocycle program was: 94°C (3 min); 33 cycles of 52°C (1 min), 72°C (3.5 min), and 94°C (30 sec); 52°C (1 min); and 72°C (5 min).
Protein extraction, measurement, and western blotting
For the extraction of total cellular protein, 50 mL of culture was pelleted by centrifugation at 900 × g for 10 min at RT, and resuspended in 1 mL of 100 mM Tris-HCl pH 8.5, 100 mM DTT, 7 mM benzamidine, and 5 mM EDTA pH 8.0. Then, 0.6 mL was added to 0.4 mL of LDS solution (5% lithium dodecylsulfate, 30% sucrose, 0.025% bromophenol blue), and the lysates were stored at -70°C. Protein was determined with the Bradford reagent (Bio-Rad) on the remaining 0.4 mL of cells, after removing the chlorophyll. The cells were pelleted by centrifugation at 14000 × g for 5 min (RT), and then resuspended in 90% acetone. After extraction for 2-3 min, the tubes were recentrifuged, and the protein pellet was resuspended in 10 mM Tris-HCl pH 8.0, 1 mM EDTA plus 1% SDS, by heating at 60 °C for 3 min. Protein was measured with the Bradford assay (Bio-Rad) using IgG for the standard curve.
For SDS-PAGE, the solubilized cells and purified Cry11A (from E. coli) in 17 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 0.01% bromphenol blue, 0.1 M DTT were heated to 100°C for 3 min. A prestained protein ladder (PageRuler, Fermentas) was used as size markers, and the samples were separated at RT in 10% acrylamide SDS-PAGE gels (Laemmli 1970). After electrophoresis, the gels were soaked in cold transfer buffer (25 mM Tris base, 192 mM glycine, 5% methanol) for 15 min and then electrotransferred to a PVDF membrane (Hybond-P, GE HealthCare) for 1.5 h at 4°C (Memon et al. 1993). The blots were stained with ponceau S to check for even protein transfer, blocked for 1 h with 5% nonfat dried milk in TBS-T (20 mM Tris-HCl pH 7.6,150 mM NaCl plus 0.05% Tween 20), and washed 2× with TBS-T for 5 min each (with shaking). The blots were incubated for 1 h at RT with an anti-FLAG monoclonal antibody (M2) coupled to alkaline phosphatase (Sigma A9469) that was diluted 1:4,000 in TBS-T. The blots were washed 6× (for 5 min each) by agitation with TBS-T. The bound antibodies were detected using a chemiluminescent reaction with Lumi-Phos WB (Thermo Scientific) or CDP-Star (Invitrogen) substrates as described by the manufacturer. The chemiluminescence was detected by exposing the blots to X-Ray film and/or detection with a Fluorchem Q imager (Protein Simple), and images were quantified with Image J (NIH).
Production and purification of a Cry11Aa standard for quantitative western blots
The codon-adapted cry11Aa gene was also expressed in E. coli and purified by virtue of an N-terminal His tag. Plasmid pET-11Aca (above) was transformed into E. coli Rosetta (DE3) carrying pLysS/RARE (Novagen), and the transformants selected on carbenicillin (200 μg mL-1) and chloramphenicol (34 μg mL-1). For expression, a transformant was grown in LB plus the same antibiotics at 37°C and induced with IPTG (1 mM) for 3 h. The cells were pelleted, washed with cold 10 mM Tris–HCl pH 7.4, 150 mM NaCl and frozen at -70°C. After thawing, the cell pellet was resuspended in 50 mM NaH2PO4 pH 8, 0.3 M NaCl plus Triton X-100 (0.1%), and sonicated in 1-mL aliquots (on ice) using twenty 30-s pulses. The lysate was centrifuged at 10,000 × g for 30 min (4°C), the supernatant was removed and the pellet was saved. Cry11Aa was purified from the pellet (after solubilization with 8 M urea) on a Ni2+ affinity column (PrepEase, USB) under denaturing conditions as described by the manufacturer. Analysis by SDS-PAGE showed the protein to be at least 95% pure, and the concentration was estimated by SDS-PAGE and the Bradford assay (Bio-Rad) using BSA as standard.
RT-PCR
Total nucleic acids (TNA) were extracted as described previously (Kwon et al. 2014) from cultures (50 mL) grown in +Cu2+ and −Cu2+ medium (in the light), and 10 μg of each was treated with Turbo DNase (Ambion) in total volume of 55 μL. Subsequently, 4 μL aliquots were used for reverse transcription with Superscript III (Invitrogen) and internal reverse primers (796 for cry4Aa700 and 800 for cry11Aa) in a total volume of 20 μL at 65°C (for 5 min). Then, 1 μL aliquots of the cDNA products were used in PCR reactions (25 μL total volume) with Taq DNA polymerase and primer sets specific for cry4Aa700 (795/796, 1001/796, 795/1003) and cry11Aa (799/800, 1002/800, 799/1004) (Table 1). The thermocycle program was similar to that described above for analyzing transformants, except the number of cycles was varied so as to evaluate product appearance with time.
Larval bioassays
Bioassays with mosquito larvae followed guidelines of the World Health Organization (2005) with some modifications. The cry4Aa700 and cry11Aa transformants were grown under inducing and noninducing conditions, and Ind41_18 was grown under inducing conditions (described above) until they reached stationary phase. The algal cells were harvested by centrifugation (1000 × g), washed with dH2O, recentrifuged, and resuspended in dH2O. The bioassays were performed in triplicate at 27°C under 12 h:12 h light/dark cycles with 10 live mosquito larvae in 5 mL of dH2O plus/minus Chlamydomonas cells; dH2O was used so the algae would not grow during the assay. The larvae, 3rd instar Culex quinquefasciatus (C. quinquefasciatus) and 4th instar Aedes aegypti (A. aegypti), were transferred to dH2O the day before the assay and starved overnight; larval deaths were counted visually after 24, 48, and 72 h with the live algae. For the determination of the median lethal concentration (LC50) of the cry11Aa transformant against 4th-instar A. aegypti larvae, 10 different concentrations of induced-cry11Aa cells were used (in triplicate) in the bioassay, and the LC50 was calculated with Microsoft Excel using Probit analysis (Finney 1971).
Results
The Cry genes
Early on, we attempted to express the native cry11Aa gene in the chloroplast and no protein was detected with an anti-Cry11Aa antibody (unpublished). Also, Juntadech et al. (2012) expressed a cry4Ba gene in the Chlamydomonas chloroplast with a similar result. Since codon bias is one of the limiting factors for expressing foreign genes in the Chlamydomonas chloroplast (Franklin et al. 2002; Mayfield et al. 2003), codon-optimized genes for the 3 Cry proteins of Bti were synthesized. Also, since there is a negative correlation between protein size and expression level (Raghava and Han 2005), cry4Aa (which is 1180 amino acids) was C-terminally truncated, containing only amino acids 1-700; the 3 essential domains (I-III) found in many Cry proteins are contained in this region (Yoshida et al. 1989). Although the Cry4Ba protein is nearly as gigantic as Cry4Aa, we did not truncate it, in part to find out if the chloroplast could produce a foreign protein of that size. The 8-amino acid FLAG tag (Einhauer and Jungbauer 2001) was added to the C-terminus of all three proteins; it was expected to have little effect on toxicity as both ends of Cry protoxins are cleaved in the gut. Before attempting chloroplast expression, we expressed all 3 genes in E. coli using the inducible pET system (Studier et al. 1990), and the expected protein sizes were obtained in all 3 cases (data not shown).
Using the inducible expression system
Initially, we tried to express the Cry genes using a high-expression system developed by Steve Mayfield and colleagues (Rasala et al. 2010), where the psbA gene's coding region is replaced with the transgene coding region. However, the transformants remained heteroplasmic, indicative of host cell toxicity. Although the knock-out of photosynthesis that accompanies this expression system could have been a major part of the problem, we decided to use an inducible system (Surzycki et al. 2007) to express these genes in the chloroplast.
To use the copper-repressible system (Surzycki et al. 2007), a modified psbD 5′ region was added to the cry genes, to enable control by the nuclear Cyc6:NAC2 gene, and the 3′ region from psbA was added to stabilize the mRNA. The psbDm:Cry:psbA gene constructs were cloned into an intergenic site in p322.1 (Fig 2), which when co-transformed into Ind41_18 with pB4C110 (which confers spectinomycin resistance) places the Cry gene in the inverted repeat region of CpDNA. Spectinomycin-resistant colonies were restreaked on spectinomycin plates several times prior to analysis of their CpDNA by PCR (Fig. 3). Primer pair 864/865 amplifies the integration site in CpDNA and was used to judge the homoplasmicity of transformants: a 100-bp product with these primers is indicative of genome copies that have not been transformed, whereas the large product is from copies that have been transformed; thus, the absence of the small product indicates homoplasmicity. Also, internal primer pairs were used to verify the presence of the specific cry gene and in every case they gave the correct-size product (data not shown). The results indicate that: one of the two cry4Aa700 transformants (Fig. 3a) was homoplasmic (4A-2), while the other (4A-5) still had some untransformed copies; the cry4Ba transformant (Fig. 3b) was homoplasmic; and one of the two Cry11Aa transformants (Fig. 3c) was homoplasmic (11A-8), while the other (11A-6) still had some untransformed CpDNA copies. Thus, we obtained at least one homoplasmic transformant for each cry gene. It should also be noted that the wild-type (WT) CpDNA that was used as the control DNA in Fig. 3, is the same as Ind41_18 in that region of the genome.
Fig. 2.
Diagrams of the cry gene constructs and the site of integration in the chloroplast genome of Ind41_18. Expression of the Cry genes is controlled by a modified psbD promoter/5′-UTR (psbDm) and 3′ region from psbA. The locations of primers used for PCR screening of the transformants (see Fig 3) are indicated; note that primers 864 and 865, which give a product of 102 bp, are located upstream and downstream, respectively, of the integration site in CpDNA. Some parts of the diagram are not drawn to scale
Fig. 3.
PCR analysis of chloroplast transformants. The locations of the primer pair 864/865 are shown in Fig. 2, and the fluorescence gel images were inverted. All of the lanes in the panels are PCR reactions except the size markers (M); these are given in bp. Wild-type (WT) DNA was used to visualize the size of the PCR product (with 864/865) from untransformed CpDNA as it is identical to Ind41_18 in this region. The reactions labeled (-) had no DNA added. a Analysis of two cry4Aa700 transformants. pCry4A is the plasmid that was shot into the chloroplast, and serves as a product size marker. b Analysis of a cry4Ba transformant. c Analysis of two cry11Aa transformants. pCry11A is the plasmid that was shot into the cells, and provides a product size marker
Analysis of Cry protein accumulation
Accumulation of the Cry proteins in the transformants grown with (U, Uninduced) and without (I, Induced) Cu2+ was assessed using western blotting of total cell protein with an anti-FLAG antibody (Fig. 4). Since both cry4Aa700 transformants (Fig. 3a) gave similar results, as did both cry11Aa transformants (Fig. 3b), only results with the homoplasmic transformants are shown in Fig. 4. A protein of the predicted size (74 kDa for Cry4Aa700, 130 kDa for Cry4Ba, and 73 kDa for Cry11Aa) was induced, or increased under inducing (I) conditions (− Cu2+), for each of the Cry gene transformants. In Fig. 4a,b, the same western blot that was exposed to X-Ray film for the image in Fig. 4a, was then imaged with a CCD-based imager in Fig. 4b; the film was more sensitive and facilitated the visualization of Cry4Ba, whereas the imager had a greater dynamic range and gave a more accurate Cry11Aa signal. Cry11Aa accumulation was significantly greater than Cry4Aa700 and much greater (at least 5-fold) than Cry4Ba. It should be noted that because very large proteins like Cry4Ba do not transfer efficiently during blotting, its accumulation could be underestimated as much as 2-fold. The figure also shows that there was significant accumulation of Cry11Aa even without induction, although the protein level did increase ∼2.5 fold with induction.
Fig. 4.
Western blot analysis of Cry proteins in the transformants. The blots were probed with an anti-FLAG monoclonal antibody coupled to alkaline phosphatase. a and b, Analysis of Cry proteins in total cell protein, either 10 or 20 μg as indicated, from transformants for cry4Aa700 (4A), cry4Ba (4B), and cry11Aa (11A) grown under uninducing (U) and inducing (I) conditions. a The blot was exposed to X-Ray film. b The same blot was imaged with a Fluorochem imager. c Quantitation of Cry11Aa in the cry11Aa transformant grown under inducing conditions. Lanes 1-4 contained the indicated amounts (in nanograms) of purified Cry11Aa expressed in E. coli, and lanes 5-9 contained the indicated amounts (in μg) of total cell protein from the cry11Aa transformant grown under inducing conditions (11A-Induced)
To obtain an absolute measure of Cry11Aa accumulation, we used the FLAG-tagged Cry11Aa protein purified from an E. coli culture as a standard, and quantitative western blotting (Fig. 4c). From 3 of these blots we estimated that Cry11Aa was accumulating to 0.33% ± 0.04% of total protein, or 90 μg g-1 wet weight of cells.
RT-PCR analysis of Cry4Aa700 and Cry11Aa gene expression
Although Cry4Aa700 and Cry11Aa are similar size proteins and the genes had the same expression signals and chromosomal location, there were significant differences in protein accumulation and induction (Fig. 4). So, we decided to examine the mRNA expression by RT-PCR, by using primers that amplified either most of the mRNA (Fig. 5a), as a measure of mRNA intactness, or primers that gave short products (Fig. 5b), which can be more quantitative. Both analyses indicated that the levels of cry11Aa mRNA (11A in Fig. 5a,b) were higher than cry4Aa700 mRNA, which is consistent with the protein findings. The data also shows that both mRNAs were present in the uninduced cells, as were the proteins (Fig. 3), suggesting that the accumulation and translation of these mRNAs is not totally dependent on the NAC2 protein (which is repressed by Cu2+). The results also indicate an incongruous correlation between cry11Aa mRNA and Cry11Aa protein, with the mRNA induction being stronger than the protein induction. Taken together with the significant level of Cry11Aa protein in the uninduced cells with low mRNA, the data suggests that cry11Aa mRNA is translated more efficiently at low mRNA levels, and less efficiently at higher mRNA levels after induction.
Fig. 5.
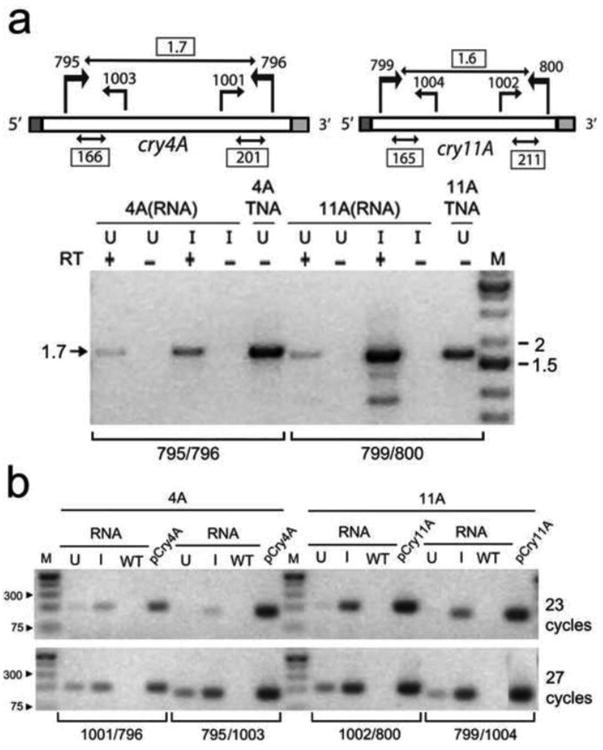
RT-PCR analysis of the cry4Aa700 (4A) and cry11Aa (11A) transformants. An equal amount of RNA from cultures grown for 72 hrs under uninduced (U) and induced (I) conditions was used for reverse transcription with gene-specific primers; 796 for cry4Aa700 and 799 for cry11Aa. The resulting cDNAs were amplified using the indicated primer sets; the locations of the primers are shown in the diagrams at top. a RT-PCR with primers near the ends of the mRNAs. RT-PCR reactions without reverse transcriptase (RT) served as negative controls, and reactions with total nucleic acids (TNA), in place of RNA, served as a positive control for the PCR step. Lane M contained size markers, and the gel image was inverted. b RT-PCR with primers that give short products. The primer sets are indicated below the gel, whose image was inverted. The same RNAs from uninduced (U) and induced (I) cultures used in (a) were employed and the PCR products were visualized on the gel after 23 and 27 cycles, when the yield was still increasing. RNA from wild-type cells (WT) was used as a negative control, and a PCR reaction with the plasmid that was shot into the cells provided a size marker for the expected RT-PCR products
Effect of Cry gene expression on Chlamydomonas growth rates
As a test of possible toxicity of the Cry proteins to the host Chlamydomonas cells, the growth rates of the transformants (and parental strain) under induced (−Cu2+) and uninduced (+Cu2+) conditions were examined. Fig. 6 shows that the growth curves obtained under both conditions were quite similar for the cry4Aa700, cry4Ba, and cry11Aa transformants, suggesting that the proteins are not very toxic to the Ind41_18 cells when expressed under these conditions.
Fig. 6.
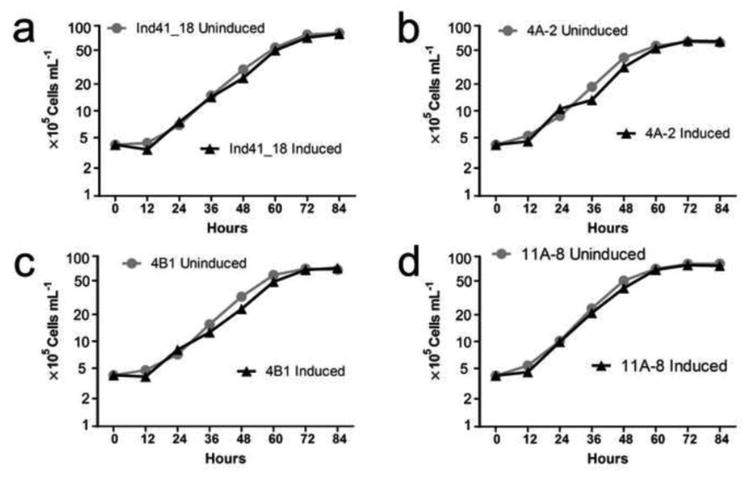
Effect of inducing the Cry4A700, Cry4B, and Cry11Aa proteins on the growth rate of the algal cells. The cells were grown under Uninduced (TAP + Cu2+) and Induced (TAP − Cu2+) conditions. a Ind41_18 parental strain. b cry4Aa700 transformant 4A-2. c cry4Ba transformant 4B-1. d cry11Aa transformant 11A-8
Larvacidal activity of the Cry transformants
To determine whether the algae were larvacidal, live cell bioassays with larvae of Culex quinquefasciatus (3rd instar stage) and Aedes aegypti (4th instar stage) were performed. The bioassay was performed in dH2O to prevent the algae from growing during the assay period. Preliminary tests showed that larvae raised on the parental strain (Ind41_18) were very active and developed into pupae and adults, confirming that Chlamydomonas can be used as sole food source (Marten 1986; Kaufman et al. 2006). Cry4Ba is known to have low toxicity against Culex spp. (Angsuthanasombat et al. 1992; Delécluse et al. 1993), and since initial tests of this transformant also showed low toxicity against A. aegypti, it was not pursued further. However, as Fig. 7 shows, both the cry4Aa700 (4A) and cry11Aa (11A) transformants were lethal to C. quinquefasciatus (Fig. 7a) and A. aegypti (Fig. 7b) larvae, with cry11Aa exhibiting ∼3.5-fold greater toxicity against C. quinquefasciatus and ∼6-fold greater toxicity than cry4Aa700 against A. aegypti at 1 × 106 cells mL-1 (or 1×). The effect of induced cry4Aa700 cells on C. quinquefasciatus also showed clear evidence of toxicity inhibition at high algal cell numbers (see 4A-I series in Fig. 7a), an effect that was not seen with A. aegypti larvae over the same range (compare to the 4A-I series in Fig. 7b). Although not clearly understood, the decreased toxicity of cry4Aa700 against C. quinquefasciatus at high cell numbers may be analogous to the reduced toxicity of Bti when there is excess food or non-food particles (Ben-Dov et al. 2003). The relatively low lethality of the uninduced cry11Aa cells (11A-U) at the 10× cell number against A. aegypti (Fig. 7b) may result from the same effect, but it seems to be overcome if there is sufficient Cry11Aa protoxin in the cells, because the 10× number of induced cells (11A-I) was highly lethal. Finally, the high lethality of C. quinquefasciatus in the dH2O-alone treatment, compared to A. aegypti (Water in Fig. 7a, b), is consistent with the former's known susceptibility to starvation.
Fig. 7.
Lethality of the cry4Aa700 and cry11Aa transformants to Culex quinquefasciatus and Aedes aegypti larvae. The cry4Aa700 (4A-2) and cry11Aa (11A-8) transformants were grown under uninduced (U) and induced (I) conditions, whereas the parental strain (Ind41_18) was grown only under inducing conditions, 1× = 1 × 106 algal cells mL-1. The assays were performed in dH2O to prevent the algae from growing during the assay, and a dH2O-only control (Water) was included. Larval mortality was checked every 24 hrs; the data are from 48 hrs. The assays were performed in triplicate with 10 larvae per test. a Bioassay with Culex quinquefasciatus. b Bioassay with Aedes aegypti
To estimate LC50 for the induced cry11Aa transformant, a more extended series of cell concentrations were used in the bioassay with 4th instar A. aegypti larvae. After Probit analysis of the data, the LC50 was found to be 3.3 × 105 cells mL-1. Given that the Cry11Aa content of the cells is ∼65 ng per 106 cells we can infer an LC50 for in vivo Cry11Aa as ∼22 ng mL-1.
Discussion
We have demonstrated that it is possible to produce, individually, the three Cry proteins from Bti (Cry4Aa, Cry4Ba and Cry11Aa) in the chloroplast of Chlamydomonas, and for Cry11Aa and Cry4Aa700, at high enough levels to kill mosquito larvae. Cry11Aa was produced at the highest level among the three, which is fortuituous because it is also the most toxic to Aedes and Culex larvae (Frankenhuyzen, 2009). Using purified Cry11Aa and quantitative westerns, the Cry11Aa level in the algal cells was determined to be 0.35% of total cell protein, or 90 μg g-1 wet weight of cells. That level compares favorably to the Cry protein levels in insect-resistant transgenic plants (e.g., Wang et al. 2014). Moreover, the toxicity of this (cry11Aa) transformant to A. aegypti larvae (LC50 was 3.3 × 10-5 cells mL-1) was nearly as high (in cells mL-1) as transgenic Anabaena expressing cry11Aa and cry4Aa (Wu et al. 1999), further supporting the conclusion of high-level expression of cry11Aa in Chlamydomonas. Although these are not the strains that one would actually use for mosquito control, they did seem to be stable genetically (based on DNA and protein analysis) for at least 1 year.
It should be noted that this is the first report of successful expression of Cry proteins from Bti in a eukaryotic alga or organelle (chloroplast). A previously published attempt to express a Cry4Ba variant in Chlamydomonas that was not codon-adapted resulted in mRNA but no detectable protein (Juntadech et al. 2012). Even with the codon-adapted gene, however, we obtained low levels of Cry4Ba protein and low toxicity against A. aegypti; we did not evaluate mRNA levels for the Cry4Ba transformant. Low levels of Cry4Ba protein were also observed in transgenic cyanobacteria, even with strong promoters (Angsuthanasombat and Panyim, 1989; Soltes-Rak et al., 1995; Khasdan et al., 2003). Hence, further work will be necessary to achieve high-level expression of cry4Ba, which would be useful in controlling Anopheles spp mosquitoes.
Cry4Aa, like Cry4Ba, is a very large protein (135 kDa), and given the negative correlation between size and protein expression levels (Raghava and Han 2005), we truncated the cry4Aa gene to create cry4Aa700. Although cry4Aa700 expression was lower than cry11Aa, it was at least 0.1% of total protein, and the cells were were toxic to A. aegypti. Since we did not try expressing full-length cry4Aa, we cannot be certain that truncating the protein to 74 kDa increased protein levels, but it seems likely that it did. Truncating Cry1 proteins, which are also quite large, increased their expression and toxicity in higher plants (reviewed in Deist et al. 2014). Hence, truncating cry4Ba may improve its expression, and an attempt to do that is underway in the laboratory.
The RT-PCR analysis of the cry4Aa700 and cry11Aa strains suggests that mRNA levels play a role in the corresponding protein levels, but the relationship is certainly not simple; this was evident in the case of cry11Aa, whose mRNA seemed to be translated more efficiently at lower levels prior to full induction. Nonetheless, the results suggest that it may be possible to increase the expression of these proteins further by using a stronger promoter than psbD, such as that from 16S rRNA. The problem that one can run into, however, is toxicity to E. coli if the expression level is too high in the bacterium, which it can be with some chloroplast gene expression signals. Thus, it will be important to minimize translation in E. coli in order to make constructs with the 16S rRNA promoter.
The growth rates of the cry transformants under induced and uninduced conditions were very similar, indicating that the Cry proteins, including Cry11Aa, were not toxic to the Ind41_18 host strain. This result was somewhat surprising, since our initial attempt to engineer very high level expression by replacing the psbA gene (Rasala et al. 2010) was indicative of toxicity problems. Perhaps there is a threshold for toxic effects of cry genes in the chloroplast that we did not reach with these expression levels. Alternatively, there are differences among Chlamydomonas host strains in their sensitivity to these proteins. Further work will be needed to sort out that particular aspect of Cry gene expression in the chloroplast of Chlamydomonas.
Finally, we would like to make a cautionary statement about quantitating tagged proteins using an antibody against the tag. In a number of papers describing transgenic tagged-protein expression, researchers have used a heterologous tagged protein as a quantitation standard for western blots (e.g., Surzycki et al. 2009; Zedler et al. 2014; Braun-Galleani et al. 2015). We tried that approach also, at first, by using a FLAG-tagged BAP (bacterial alkaline phosphatase) from a commercial source as the standard and obtained values for Cry11Aa that were 10-fold higher, ∼3% of total protein (unpublished results). However, that would be an extraordinary level of Cry protoxin for a growing organism. Moreover, it probably should have produced a visible change in the Coomassie stained gel pattern, but that was not observed. Thus, we believe that our quantitation using the homologous tagged protein prepared by us, is the more accurate estimate. Although it may be fine in some, or even most cases it is possible that the binding of the antibody could be affected by the protein that is fused to the tag. Thus, we suggest that caution is warranted, especially when the values obtained using the heterologous standard are very high.
Fig. 1.
Synthetic codon-optimized cry genes, cry4Aa700, cry4Ba, and cry11Aa were designed using the native amino acid sequences, the program Optimizer, and a codon-usage table based on highly-expressed Chlamydomonas chloroplast genes; with optimization, the codon adaptive index (CAI) for each gene increased from ∼0.5 to 1. Cry4Aa700 is a truncated version of the native gene, containing only the first 700 amino acids of the 1180 total residues in the Cry4Aa protein, whereas Cry4Ba and Cry11Aa are full-length proteins. The 8-residue FLAG epitope was added to the C-terminus of all three genes for antibody detection
Acknowledgments
We are grateful to Dr. Jean-David Rochaix for providing the Ind41_18 strain, and BEI Resources (NIAID) for Culex quinquefasciatus. This work was supported by the Bill and Melinda Gates Foundation (grant OPP1024635 to DLH) and the National Institutes of Health (grant R41-AI115854A to DLH).
Footnotes
Conflict of interest statement: The senior author (DLH) is the founder and owner of Pond Life Technologies LLC. Also, a patent application based in part on this work has been filed with the US Patent Office (15/142,865).
References
- Angsuthanasombat C, Panyim S. Biosynthesis of 130-kilodalton mosquito larvicide in the cyanobacterium Agmenellum quadruplicatum PR-6. Appl Environ Microbiol. 1989;55:2428–2430. doi: 10.1128/aem.55.9.2428-2430.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angsuthanasombat C, Crickmore N, Ellar DJ. Comparison of Bacillus thuringiensis subsp. israelensis CryIVA and CryIVB cloned toxins reveals synergism in vivo. FEMS Microbiol Lett. 1992;94:63–68. doi: 10.1016/0378-1097(92)90584-b. [DOI] [PubMed] [Google Scholar]
- de Barjac H, Sutherland DJ. Bacterial control of mosquitoes and black flies: Biochemistry, genetics, & applications of Bacillus thuringiensis israelensis and Bacillus sphaericus. Rutgers University Press; New Brunswick, NJ: 1990. [Google Scholar]
- Ben-Dov E, Saxena D, Wang Q, Manasherob R, Boussiba S, Zaritsky A. Ingested particles reduce susceptibility of insect larvae to Bacillus thuringiensis. J Appl Entomol. 2003;127:146–152. [Google Scholar]
- Ben-Dov E. Bacillus thuringiensis subsp. israelensis and its Dipteran-specific toxins. Toxins. 2014;6:1222–1243. doi: 10.3390/toxins6041222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovsky D, Sterner A, Powell CA. Cloning and expressing trypsin modulating oostatic factor in Chlorella desiccata to control mosquito larvae. Arch Insect Biochem Physiol. 2016;91:17–36. doi: 10.1002/arch.21306. [DOI] [PubMed] [Google Scholar]
- Boynton JE, Gillham NW. Chloroplast transformation in Chlamydomonas. In: Ray W, editor. Meth Enzymol. Vol. 217. Academic Press; New York: 1993. pp. 510–536. [DOI] [PubMed] [Google Scholar]
- Bravo A, Likitvivatanavong S, Gill SS, Soberon M. Bacillus thuringiensis:A story of a successful bioinsecticide. Insect Biochem Mol Biol. 2011;41:423–431. doi: 10.1016/j.ibmb.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun-Galleani S, Baganz F, Purton S. Improving recombinant protein production in the Chlamydomonas reinhardtii chloroplast using vivid Verde Fluorescent Protein as a reporter. Biotechnol J. 2015;10:1289–1297. doi: 10.1002/biot.201400566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Melis A. Marker-free genetic engineering of the chloroplast in the green microalga Chlamydomonas reinhardtii. Plant Biotechnol J. 2013;11:818–828. doi: 10.1111/pbi.12073. [DOI] [PubMed] [Google Scholar]
- Deist B, Rausch M, Fernandez-Luna M, Adang M, Bonning B. Bt toxin modification for enhanced efficacy. Toxins. 2014;6:3005. doi: 10.3390/toxins6103005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delécluse A, Poncet S, Klier A, Rapoport G. Expression of cryIVA and cryIVB genes, independently or in combination, in a crystal-negative strain of Bacillus thuringiensis subsp. israelensis. Appl Environ Microbiol. 1993;59:3922–3927. doi: 10.1128/aem.59.11.3922-3927.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einhauer A, Jungbauer A. The FLAG™ peptide, a versatile fusion tag for the purification of recombinant proteins. J Biochem Biophys Meth. 2001;49:455–465. doi: 10.1016/s0165-022x(01)00213-5. [DOI] [PubMed] [Google Scholar]
- Finney DJ. Probit analysis. Cambridge University Press; Cambridge, U.K: 1977. [Google Scholar]
- Fischer N, Stampacchia O, Redding K, Rochaix JD. Selectable marker recycling in the chloroplast. Mol Gen Genet. 1996;251:373–380. doi: 10.1007/BF02172529. [DOI] [PubMed] [Google Scholar]
- Frankenhuyzen KV. Insecticidal activity of Bacillus thuringiensis crystal proteins. J Invert Pathol. 2009;101:1–16. doi: 10.1016/j.jip.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Franklin S, Ngo B, Efuet E, Mayfield SP. Development of a GFP reporter gene for the Chlamydomonas reinhardtii chloroplast. Plant J. 2002;30:733–744. doi: 10.1046/j.1365-313x.2002.01319.x. [DOI] [PubMed] [Google Scholar]
- Glare TR, O'Callaghan M. Bacillus thuringiensis: Biology, Ecology and Safety. Wiley; West Sussex: 2000. [Google Scholar]
- Gorman DS, Levine RP. Cytochrome f and plastocyanin: their sequence in the photosynthetic electron transport chain of Chlamydomonas reinhardi. Proc Natl Acad Sci USA. 1965;54(6):1665–1669. doi: 10.1073/pnas.54.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EH. The Chlamydomonas sourcebook: A comprehensive guide to biology and laboratory use. Academic Press; San Diego, CA: 1989. [DOI] [PubMed] [Google Scholar]
- Ibrahim MA, Griko NB, Bulla LA. The Cry4B toxin of Bacillus thuringiensis subsp. Israelensis kills permethrin-resistant Anopheles gambiae, the principal vector of malaria. Exp Biol Med. 2013;238:350–359. doi: 10.1177/1535370213477973. [DOI] [PubMed] [Google Scholar]
- Juntadech T, Yokthon K, Tangphatsornruang S, Yap YK, Katzenmeier G, Angsuthanasombat C. Efficient transcription of the larvicidal cry4Ba gene from Bacillus thuringiensis in transgenic chloroplasts of the green algal Chlamydomonas reinhardtii. Adv Biosci Biotech. 2012;3:362–369. [Google Scholar]
- Kamel F. Paths from pesticides to Parkinson's. Science. 2013;341:722–723. doi: 10.1126/science.1243619. [DOI] [PubMed] [Google Scholar]
- Kaufman MG, Wanja E, Maknojia S, Bayoh MN, Vulule JM, Walker ED. Importance of algal biomass to growth and development of Anopheles gambiae larvae. J Med Entomol. 2006;43:669–676. doi: 10.1603/0022-2585(2006)43[669:ioabtg]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Khasdan V, Ben-Dov E, Manasherob R, Boussiba S, Zaritsky A. Mosquito larvicidal activity of transgenic Anabaena strain PCC 7120 expressing toxin genes from Bacillus thuringiensis subsp. israelensis. FEMS Microbiol Lett. 2003;227:189–195. doi: 10.1016/S0378-1097(03)00679-7. [DOI] [PubMed] [Google Scholar]
- Kioulos I, Kampouraki A, Morou E, Skavdis G, Vontas J. Insecticide resistance status in the major West Nile virus vector Culex pipiens from Greece. Pest Manag Sci. 2014;70:623–627. doi: 10.1002/ps.3595. [DOI] [PubMed] [Google Scholar]
- Koou SY, Chong CS, Vythilingam I, Ng LC, Lee CY. Pyrethroid resistance in Aedes aegypti larvae (Diptera: Culicidae) from Singapore. J Med Entomol. 2014;51:170–181. doi: 10.1603/me13113. [DOI] [PubMed] [Google Scholar]
- Kwon T, Odom OW, Qiu W, Herrin DL. PCR Analysis of chloroplast double-strand break (DSB) repair products induced by I-CreII in Chlamydomonas and Arabidopsis. In: Edgell DR, editor. Homing Endonucleases. Vol. 1123. Humana Press; 2014. pp. 77–86. Meth Mol Biol. [DOI] [PubMed] [Google Scholar]
- Kumar A, Wang S, Ou R, Samrakandi M, Beerntsen BT, Sayre RT. Development of an RNAi based microalgal larvicide to control mosquitoes. Malaria World J. 2013;4(6):1–6. doi: 10.5281/zenodo.10894766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Laird M. The natural history of larval mosquito habitats. Academic Press; London: 1988. [Google Scholar]
- Liu YT, Sui MJ, Dar-Der JI, Wu IH, Chou CC, Chen CC. Protection from ultraviolet irradiation by melanin of mosquitocidal activity of Bacillus thuringiensis var. israelensis. J Invert Pathol. 1993;62:131–136. doi: 10.1006/jipa.1993.1088. [DOI] [PubMed] [Google Scholar]
- Marten GG. Mosquito control by plankton management: the potential of indigestible green algae. J Trop Med Hyg. 1986;89:213–222. [PubMed] [Google Scholar]
- Mayfield S, Franklin S, Lerner R. Expression and assembly of a fully active antibody in algae. Proc Natl Acad Sci USA. 2003;100:438–442. doi: 10.1073/pnas.0237108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memon AR, Herrin DL, Thompson GA., Jr Intracellular translocation of a 28 kDa GTP-binding protein during osmotic shock-induced cell volume regulation in Dunaliella salina. Biochim Biophys Acta. 1993;1179:11–22. doi: 10.1016/0167-4889(93)90070-6. [DOI] [PubMed] [Google Scholar]
- Morens DM, Fauci AS. Emerging infectious diseases: Threats to human health and global stability. PLoS Pathog. 2013;9:e1003467. doi: 10.1371/journal.ppat.1003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman SM, Boynton JE, Gillham NW, Randolph-Anderson BL, Johnson AM, Harris EH. Transformation of chloroplast ribosomal RNA genes in Chlamydomonas: molecular and genetic characterization of integration events. Genetics. 1990;126:875–888. doi: 10.1093/genetics/126.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman SM, Harris EH, Johnson AM, Boynton JE, Gillham NW. Nonrandom distribution of chloroplast recombination events in Chlamydomonas reinhardtii: evidence for a hotspot and an adjacent cold region. Genetics. 1992;132:413–429. doi: 10.1093/genetics/132.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickelsen J, Fleischmann M, Boudreau E, Rahire M, Rochaix JD. identification of cis-acting RNA leader elements required for chloroplast psbD gene expression in Chlamydomonas. Plant Cell. 1999;11:957–970. doi: 10.1105/tpc.11.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana B, Margalit J, Barak Z. Fate of Bacillus thuringiensis subsp. israelensis under simulated field conditions. Appl Environ Microbiol. 1987;53:828–831. doi: 10.1128/aem.53.4.828-831.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom OW, Holloway SP, Deshpande NN, Lee J, Herrin DL. Mobile self-splicing group I introns from the psbA gene of Chlamydomonas reinhardtii: Highly efficient homing of an exogenous intron containing its own promoter. Mol Cell Biol. 2001;21:3472–3481. doi: 10.1128/MCB.21.10.3472-3481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigbò P, Guzmán E, Romeu A, Garcia-Vallvé S. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucl Acids Res. 2007;35:W126–W131. doi: 10.1093/nar/gkm219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton S, Szaub JB, Wannathong T, Young R, Economou CK. Genetic engineering of algal chloroplasts: Progress and prospects. Russ J Plant Physiol. 2013;60:491–499. [Google Scholar]
- Quinn JM, Merchant S. Copper-responsive gene expression during adaptation to copper deficiency. In: Lee M, editor. Meth Enzymol. Vol. 297. Academic Press; 1998. pp. 263–279. [DOI] [PubMed] [Google Scholar]
- Raghava GPS, Han JH. Correlation and prediction of gene expression level from amino acid and dipeptide composition of its protein. BMC Bioinformatics. 2005;6:59. doi: 10.1186/1471-2105-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasala BA, Muto M, Lee PA, Jager M, Cardoso RMF, Behnke CA, Kirk P, Hokanson CA, Crea R, Mendez M, Mayfield SP. Production of therapeutic proteins in algae, analysis of expression of seven human proteins in the chloroplast of Chlamydomonas reinhardtii. Plant Biotechnol J. 2010;8:719–733. doi: 10.1111/j.1467-7652.2010.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltes-Rak E, Kushner D, Williams DD, Coleman J. Factors regulating cryIVB expression in the cyanobacterium Synechococcus PCC 7942. Mol Gen Genet. 1995;246:301–308. doi: 10.1007/BF00288602. [DOI] [PubMed] [Google Scholar]
- Stevens SE, Murphy RC, Lamoreaux WJ, Coons LB. A genetically engineered mosquitocidal cyanobacterium. J Appl Phycol. 1994;6:187–197. [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Meth Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Surzycki R, Cournac L, Peltier G, Rochaix JD. Potential for hydrogen production with inducible chloroplast gene expression in Chlamydomonas. Proc Natl Acad Sci USA. 2007;104:17548–17553. doi: 10.1073/pnas.0704205104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surzycki R, Greenham K, Kitayama K, Dibal F, Wagner R, Rochaix JD, Ajam T, Surzycki S. Factors effecting expression of vaccines in microalgae. Biologicals. 2009;37:133–138. doi: 10.1016/j.biologicals.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Tetreau G, Stalinski R, Kersusan D, Veyrenc S, David JP, Reynaud S, Després L. Decreased toxicity of Bacillus thuringiensis subsp. israelensis to mosquito larvae after contact with leaf litter. Appl Environ Microbiol. 2012;78:5189–5195. doi: 10.1128/AEM.00903-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang L, Li Y, Liu Y, Han L, Zhu Z, Wang F, Peng Y. Expression of Cry1ab protein in a marker-free transgenic Bt rice line and its efficacy in controlling a target pest, Chilo suppressalis (Lepidoptera: Crambidae) Environ Entomol. 2014;43:528–536. doi: 10.1603/EN13254. [DOI] [PubMed] [Google Scholar]
- WHO. Guidelines for laboratory and field testing of mosquito larvicides. World Health Organization; Geneva, Switzerland: 2005. [Google Scholar]
- Wintermans JFGM, De Mots A. Spectrophotometric characteristics of chlorophylls a and b and their phenophytins in ethanol. Biochim Biophys Acta. 1965;109:448–453. doi: 10.1016/0926-6585(65)90170-6. [DOI] [PubMed] [Google Scholar]
- Wu X, Vennison SJ, Huirong L, Ben-Dov E, Zaritsky A, Boussiba S. Mosquito larvicidal activity of transgenic Anabaena strain PCC 7120 expressing combinations of genes from Bacillus thuringiensis subsp. israelensis. Appl Environ Microbiol. 1997;63:4971–4974. doi: 10.1128/aem.63.12.4971-4974.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Matsushima YK, Sen HS, Komano T. Insecticidal activity of a peptide containing the 30th to 695th amino acid residues of the 130-kDa protein of Bacillus thuringiensis var. israelensis. Agricul Biol Chem. 1989;53:2121–2127. [Google Scholar]
- Zaritsky A, Ben-Dov E, Borovsky D, Boussiba S, Einav M, Gindin G, Horowitz AR, Kolot M, Melnikov O, Mendel Z, Yagil E. Transgenic organisms expressing genes from Bacillus thuringiensis to combat insect pests. Bioeng Bugs. 2010;1:341–344. doi: 10.4161/bbug.1.5.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zedler JA, Gangl D, Hamberger B, Purton S, Robinson C. Stable expression of a bifunctional diterpene synthase in the chloroplast of Chlamydomonas reinhardtii. J Appl Phycol. 2015;27:2271–2277. [Google Scholar]