

Summary
Regulatory T cells (Tregs) are a barrier to anti-tumor immunity. Neuropilin-1 (Nrp1) is required to maintain intratumoral Treg stability and function but is dispensable for peripheral immune tolerance. Treg-restricted Nrp1 deletion results in profound tumor resistance due to Treg functional fragility. Thus, identifying the basis for Nrp1 dependency and the key drivers of Treg fragility could help to improve immunotherapy for human cancer. We show that a high percentage of intratumoral NRP1+ Tregs correlates with poor prognosis in melanoma and head and neck squamous cell carcinoma. Using a mouse model of melanoma where Nrp1-deficient (Nrp1−/−) and wild-type (Nrp1+/+) Tregs can be assessed in a competitive environment, we find that a high proportion of intratumoral Nrp1−/− Tregs produce interferon-γ (IFNγ), which drives the fragility of surrounding WT Tregs, boosts anti-tumor immunity, and facilitates tumor clearance. We also show that IFNγ-induced Treg fragility is required for response to anti-PD1, suggesting that cancer therapies promoting Treg fragility may be efficacious.
Graphical abstract
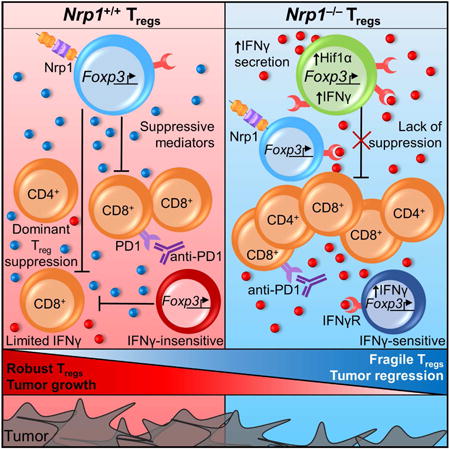
Introduction
Regulatory T cells (Tregs), characterized by their expression of the forkhead box transcription factor, Foxp3, are required to maintain immune homeostasis and prevent excessive tissue damage (Fontenot et al., 2005; Kim et al., 2007; Miyara and Sakaguchi, 2007; Vignali et al., 2008). Humans that lack a functional Treg population develop a lethal autoimmune disorder, termed Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked (IPEX) syndrome, which can be recapitulated in mice through Foxp3 deletion. While Tregs are required to limit autoimmunity and maintain immune regulation, they can be deleterious in cancer through suppression of anti-tumor immunity (Chaudhry and Rudensky, 2013; Facciabene et al., 2012; Liu et al., 2016). Indeed, high numbers of Tregs and a low CD8+ T cell:Treg ratio are considered poor prognostic factors for many tumor types, including melanoma, head and neck squamous cell carcinoma (HNSCC), ovarian cancer and colorectal carcinoma (Curiel et al., 2004; Drennan et al., 2013; Jacobs et al., 2012; Nishikawa and Sakaguchi, 2010; Saito et al., 2016). Although targeting intratumoral Tregs could be an effective therapeutic approach for multiple tumor types, perturbation of peripheral Treg number or function could lead to life-threatening autoimmune or inflammatory complications. Therefore, identifying pathways that could be targeted to selectively undermine intratumoral Tregs is essential.
We have previously shown that Neuropilin-1 (Nrp1) is expressed by ∼90% of tumor infiltrating Tregs in mouse models of cancer and is critical for their function in the tumor microenvironment (Delgoffe et al., 2013). Indeed, mice with a Treg-restricted deletion of Nrp1 are highly resistant to B16 melanoma, which is normally refractory to immune-mediated clearance, yet remarkably do not exhibit any autoimmune or inflammatory disease. Although we have previously described Nrp1-deficient Tregs as ‘unstable’, due to their loss of function (Delgoffe et al., 2013), previous studies and data included here clearly show that they retain Foxp3 expression. Thus, we now refer to this phenotype as Treg ‘fragility’ consistent with their retention of Foxp3 expression yet loss of function ex vivo (as exhibited by loss of suppressive activity in vitro) and tumor tolerance in vivo (as exhibited by tumor growth reduction/clearance). While our previous data demonstrated the importance of Nrp1 in maintaining intratumoral Treg function, many questions remain including the fate of these fragile Tregs and their contribution to anti-tumor immunity, the drivers of Treg fragility, the expression, contribution, and impact of NRP1 on human intratumoral Tregs, and the broader implications for Treg function and cancer immunotherapy.
Results
Increased NRP1 expression on Tregs in human cancer
While Nrp1 has been shown to prevent Treg fragility in mice, its presence and role in human Tregs remains unclear. Previous studies have been controversial, with some suggesting peripheral human Tregs do not express NRP1 while other suggest that NRP1+ Tregs are potent suppressors (Battaglia et al., 2009; Battaglia et al., 2008; Chaudhary and Elkord, 2015; Chaudhary et al., 2014; Gao et al., 2016; Milpied et al., 2009; Piechnik et al., 2013; Tatura et al., 2015). Indeed, very few human Tregs in peripheral blood lymphocytes (PBL) from healthy donors express NRP1 (Fig. 1A and B). Remarkably, most patients with metastatic melanoma and head and neck squamous cell carcinoma (HNSCC) possessed a reasonably high percentage of intratumoral NRP1+ Tregs (Fig. 1A and B). This varied considerably from 3-90% in melanoma and 35-90% in HNSCC. The percentage of NRP1+ Tregs in PBL was also substantially enhanced. Interestingly, NRP1 expression in intratumoral Tregs appeared to correlated with poor prognosis in both melanoma and HNSCC (Fig. 1C).
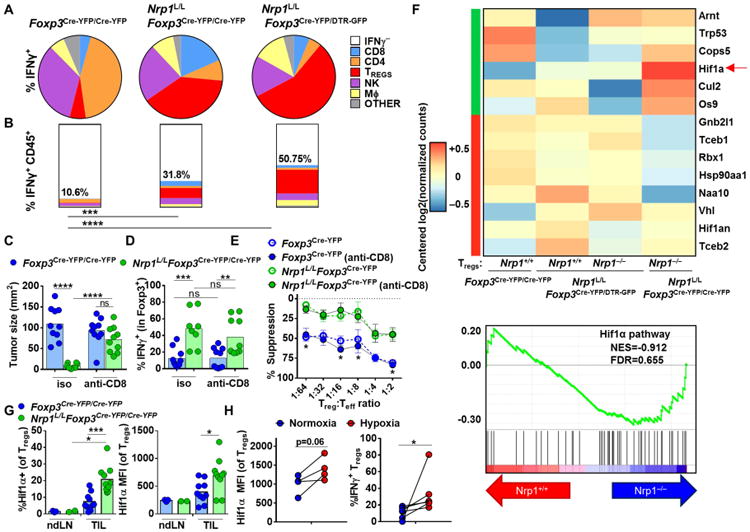
Figure 1. Decreased Nrp1 expression leads to tumor regression and enhanced survival.
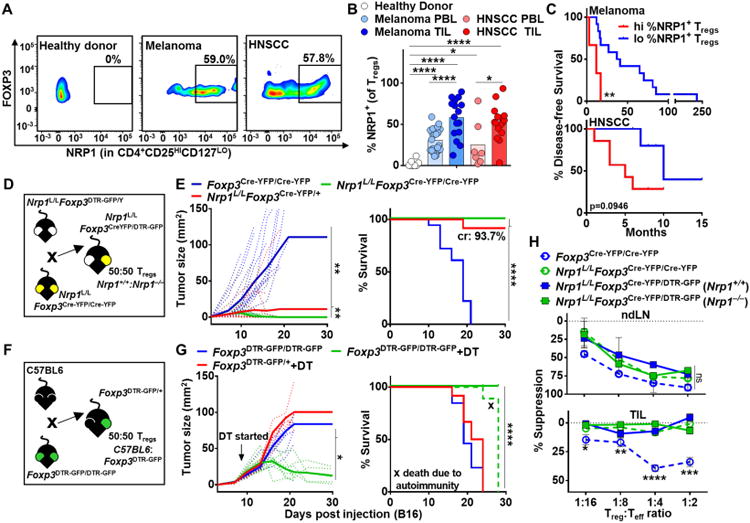
(A-C) Lymphocytes were harvested from PBL of healthy donors (n=8) or from PBL and TIL of Head and Neck Squamous Cell Carcinoma (HNSCC) and metastatic melanoma (3-5 experiments, n=16-23) and frozen or stained fresh. Frozen TIL and PBL were thawed and stained directly without stimulation. (D-G) Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/DTR-GFP, Foxp3DTR-GFP/DTR-GFP, and Foxp3DTR-GFP/+ mice were injected with B16.F10 melanoma tumor cells ID on day 0. Tumor growth was measured with digital calipers every three days. Mice were removed from study when tumor growth reached a diameter of 2cm in any direction or when necrosis was observed, and survival plots were generated (4 experiments, n=9-18). (F-G) Foxp3DTR-GFP/DTR-GFP, and Foxp3DTR-GFP/+ mice were treated with 100μg Diptheria Toxin IP every three days starting on day 7. (H) Tregs were isolated on day 12 post B16 injection from ndLN and TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice and cultured with effector T cells and APCs for 72 hours in a classical microsuppression assay. Tregs were pooled from 3 mice with 5-6 mice per group per experiment. Proliferation was measured and percent suppression was calculated as described in methods. Data represent 3-5 (A-C), 4 (D-G), or 3 (H) independent experiments. Error bars represent the mean ± SEM. Student unpaired t test (Fig. 1B, H), 2 way ANOVA (Fig. 1E, G), and Kaplan-Meier tests (Fig. 1E, G) were used (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Nrp1−/− Tregs block wild type Treg function and promote anti-tumor immunity
We have previously shown that anti-Nrp1 substantially limits the growth of a B16 mouse model of human melanoma (Delgoffe et al., 2013). Given the heterogeneous nature of NRP1 expression on human tumor infiltrating Tregs, where only a proportion express NRP1, we questioned what impact Nrp1 loss on only a proportion of mouse Tregs might have on the function of the remaining wild-type (WT) counterparts, and by extension anti-tumor immunity and tumor growth. Also, as Nrp1-deficient Tregs show a reduction in suppressive function but also an increase in effector phenotype (Delgoffe et al., 2013), we questioned whether these cells had an active role in re-shaping the tumor microenvironment, or whether reduced tumor growth was instead due to reduction of a major suppressive cell population.
Foxp3 is on the X chromosome and is thus subject to X inactivation (Briggs and Reijo Pera, 2014; Galupa and Heard, 2015; Lee and Bartolomei, 2013), rendering only one allele active in each Treg. Consequently in heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP female mice, 50% of Tregs have a Cre-mediated deletion of Nrp1, are marked with YFP [herein referred to as Nrp1−/− Tregs] and exhibit functional fragility, and the other 50% express DTR-GFP and are WT Tregs, as they carry the Nrp1L/L allele but not Foxp3Cre-YFP [herein referred to as Nrp1+/+ Tregs] (Fig. 1D). We first assessed tumor growth in heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP female mice, with Nrp1L/LFoxp3Cre-YFP/Cre-YFP [all Tregs are Nrp1−/−] and Foxp3Cre-YFP/Cre-YFP [all Tregs are WT/Nrp1+/+] female mice as controls. Strikingly, Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice exhibited dramatically reduced tumor growth, enhanced survival, and increased intratumoral lymphocyte and CD8+ T cell number, phenocopying Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice (Fig. 1E and fig. S1A) (Delgoffe et al., 2013). This occurred despite the presence of Nrp1+/+ Tregs in similar numbers to Nrp1−/− Tregs in the tumor (fig. S1A). A previous study had suggested that the absence of Nrp1 leads to reduced influx of Tregs into certain tumor types (Hansen et al., 2012). However, there did not seem to be a significant difference in the number of intratumoral Nrp1−/− versus Nrp1+/+ Tregs, even in Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice (fig. S1A). One might also argue that the Nrp1L/LFoxp3Cre-YFP mutation causes a basal inflammatory state that impacts the establishment of a tumor mass. To rule this out and any potential impact Nrp1 loss may have on Treg development, migration and function, the impact of Nrp1 temporal deletion in Tregs following the establishment of B16 tumor growth was determined. Nrp1L/LFoxp3CreERT2, but not Foxp3CreERT2, mice exhibited substantially reduced tumor growth following Nrp1 deletion induced by daily tamoxifen treatment on Days 7-11 (fig. S1B-C).
To rule out the possibility that the inability of Nrp1+/+ Tregs to block anti-tumor immunity and tumor clearance was due to their reduced number in heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice, we also assessed tumor growth in heterozygous Foxp3DTR-GFP/+ female mice in which diphtheria toxin (DT) treatment reduced peripheral and intratumoral Treg number by approximately half (Fig. 1F and fig. S1D). In stark contrast to tumor growth in heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice, DT-treated heterozygous Foxp3DTR-GFP/+ mice exhibited tumor growth that was indistinguishable from the untreated control mice (Fig. 1G). While Foxp3DTR-GFP/DTR-GFP mice treated with DT largely cleared their tumors, they ultimately succumbed to autoimmunity due to an absence of Tregs in contrast to heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFP female mice that never exhibited an autoimmune or inflammatory phenotype (Fig. 1D and 1E, and data not shown) (Delgoffe et al., 2013). Taken together, our data suggest that if half the Tregs are depleted tumors grow unrestrained, as the reduced number of WT Tregs are still capable of blocking anti-tumor immunity. In contrast, if half the Tregs lose Nrp1, tumors are controlled, suggesting that Nrp1−/− Tregs are playing an active role in re-shaping the tumor microenvironment.
It is possible that Nrp1−/− Tregs impact the tumor microenvironment by losing Foxp3 and becoming so-called ex-Tregs with an altered functional phenotype (Overacre and Vignali, 2016; Zhou et al., 2009b). Indeed, we have previously shown that Nrp1 contributes to Treg stability in the tumor microenvironment by undermining the pAKT:FOXO pathway and preventing the expression of T helper lineage-defining transcription factors, raising the possibility that this may also result in the loss of Foxp3 expression. We analyzed Foxp3 fate mapping mice in which Tregs either possessed (Foxp3Cre-YFPRosaL-Tom-L-GFP) or lacked Nrp1 expression (Nrp1L/LFoxp3Cre-YFPRosaL-Tom-L-GFP) (fig. S2A). In this mouse model, non-Tregs are Tomato+GFP−YFP−, Foxp3+ Tregs are Tomato−GFP+YFP+ and Foxp3− ex-Tregs are Tomato−GFP+YFP−. Interestingly, there were very few (<5%) ex-Tregs present in the periphery or in the tumor, regardless of Nrp1 expression (fig. S2B and C). These data suggest that the absence of Nrp1 does not affect Foxp3 expression and does not result in the generation of ex-Tregs.
Previous reports have shown that Tregs can display alternative functions in vivo while maintaining Foxp3 expression (Hori, 2014; Sharma et al., 2013). We hypothesized that Nrp1−/− Tregs not only lost their suppressive activity but may also negatively impact the function of surrounding intratumoral Nrp1+/+ Tregs. In order to assess this possibility, we determined the suppressive capacity of Nrp1+/+ (GFP+) and Nrp1−/− (YFP+) Tregs from Nrp1L/LFoxp3Cre-YFP/DTR-GFP heterozygous and control mice using a microsuppression assay (Turnis et al., 2016). While all Treg populations isolated from non-draining lymph nodes (ndLN) were equally capable of suppressing effector T cells (Fig. 1H), Nrp1−/− Tregs isolated from homozygous Nrp1L/LFoxp3Cre-YFP/Cre-YFP and heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP tumors lacked suppressive activity (Fig. 1H). Interestingly, intratumoral Nrp1+/+ Tregs isolated from heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice were also unable to suppress. Two hallmarks of Treg fragility are elevated pAkt and reduced ICOS expression (Delgoffe et al., 2013). Elevated pAkt and reduced ICOS were observed in Nrp1−/− Tregs in Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice and Nrp1L/LFoxp3Cre-YFP/DTR-GFP heterozygous mice relative to Nrp1+/+ Tregs in Foxp3Cre-YFP/Cre-YFP mice as expected (fig. S2D and E). However, elevated pAkt and, to a lesser extent, reduced ICOS was also observed in Nrp1+/+ Tregs in Nrp1L/LFoxp3Cre-YFP/DTR-GFP heterozygous mice. Taken together, these data suggest that Nrp1−/− Tregs have a negative impact on the suppressive function of intratumoral Nrp1+/+ Tregs.
Fragile and wild type Tregs have a reciprocal impact on their transcriptome
We next used transcriptomic analysis of Nrp1+/+ and Nrp1−/− Tregs from Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice to evaluate the cell intrinsic and extrinsic impact of Nrp1 loss (fig. S3). Significant alterations in the Treg transcriptome were observed between Nrp1+/+ and Nrp1−/− Tregs. Principal Component Analysis (PCA) based on differentially expressed genes clearly separated Teff and Tregs based on both location and genotype. Interestingly, intratumoral Nrp1+/+ and Nrp1−/− Tregs from heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice were similar to each other and yet distinct from their genotypically identical counterparts from control Foxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice (Fig. 2A, fig. S4A-B). A similar impact was observed when Treg signature genes were assessed wherein it became evident that all four populations were district and yet bore a transcriptional relationship (fig. S4C). These data suggest that Nrp1+/+ and Nrp1−/− Tregs in heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice impact each other's transcriptome in a reciprocal manner.
Figure 2. Nrp1 alters the Treg transcriptome.
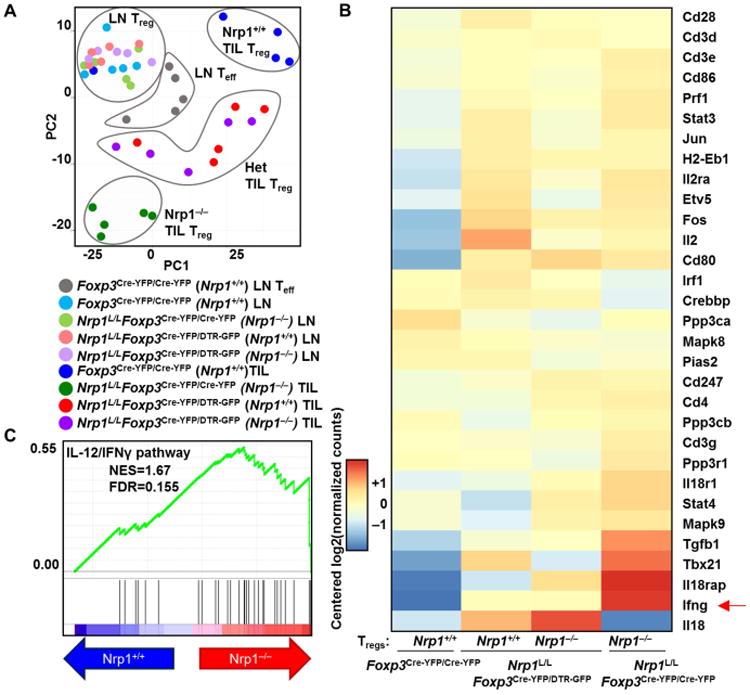
(A-C) Tregs were purified based on CD4+, and GFP or YFP expression from Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice on D12, cDNA and libraries were generated using the Clontech SmartER Ultra-Low and Illumina Nextera XT Library Prep kits. Samples were normalized to 2nM and sequenced on a NextSeq500. (A) Differentially expressed genes are determined by the genes that have q-value of 0.2 between any two of the four Treg groups in the TIL. PCA was computed using the “prcomp()” R functions using the normalized voom data restricted to the same differentially expressed genes as shown in figure. (B-C) Significant genes were cross-referenced with those that were annotated to “plasma membrane” or “extracellular part” in the Cellular Component Gene Ontology. The Gene Ontology annotations were obtained from mSigDB. A number of genes associated with the Ifng/Il12/Il18 pathways were upregulated in the Nrp1−/− samples. Data represent 5 independent experiments with 3-5 mice pooled per experiment.
Pathway analysis highlighted a potential role for IFNγ/IL-12-related transcriptional programs. Of particular interest was an increase in Ifng and its targets in Nrp1−/− Tregs in Nrp1L/LFoxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/DTR-GFP as well as in Nrp1+/+ Tregs in Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice (Fig. 2B-C, fig. S4D), implicating a role for the IFNγ pathway in modulating Treg function and function in the tumor microenvironment.
While previous reports have suggested that a small subset of Tregs produce IFNγ during inflammation (Duhen et al., 2012; Koenecke et al., 2012; Pandiyan and Zhu, 2015), the expression of IFNγ by Tregs in tumors and its impact on their suppressive function remains unclear. Using flow cytometry, we found that there was increased expression of IFNγ by Nrp1−/− Tregs in both Nrp1L/LFoxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice (Fig. 3A-B). Interestingly, an increased percentage of Nrp1+/+ Tregs from Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice also expressed IFNγ. Interferon-γ receptor 1 (IFNγR) expression showed an elevated trend in Nrp1−/− Tregs in Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice, and both Nrp1−/− and Nrp1+/+ Tregs in Nrp1L/LFoxp3Cre-YFP/DTR-GFP heterozygous mice (Fig. 3B). In addition, several type 1 helper T cell markers were upregulated in Nrp1−/− Tregs, such as Tbet, Cxcr3, and Eomes (fig. S5A–F) as well as downstream pSTAT1 in Nrp1−/− Tregs from Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice and pSTAT4 in all Nrp1−/− Tregs (fig. S5G, H).
Figure 3. Nrp1−/− Tregs display increased IFNγ in the tumor microenvironment.
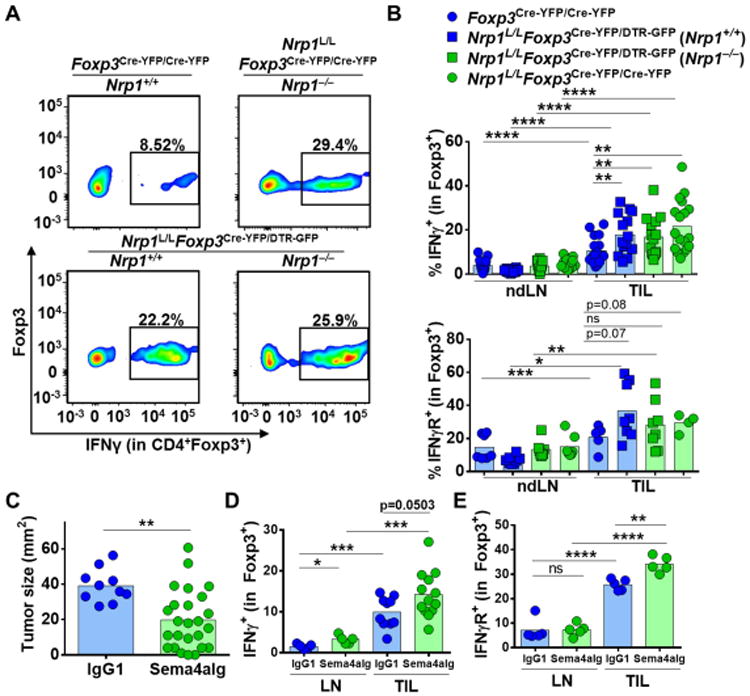
(A-B) Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice were injected with B16.F10 melanoma tumor cells ID on day 0 and sacrificed on day 12. Lymphocytes were isolated from ndLN and TIL of mice noted, stimulated and stained for IFNγ and IFNγR. (n=8-18). (C-E) C57BL/6 mice were injected with B16.F10 melanoma tumor cells ID on day 0. Mice were treated with either Sema4aIg or IgG1 every 3 days until sacrifice starting on day 5 (400ug, 200ug, 200ug, 200ug). (C) Tumors were measured on day 12 for prior to sacrifice (n=10-25). (D) Lymphocytes were isolated from ndLN and TIL, stimulated and stained for IFNγ (n=5-13). (E) Lymphocytes were isolated from ndLN and TIL, and stained for IFNγR (n=5). Data represent 3-4 independent experiments. Student unpaired t test was used. (*p<0.05, **p < 0.01, ***p <0.001, ****p < 0.0001).
In order to determine whether modulation of IFNγ and IFNγR expression also occurred following blockade of the Nrp1:Sema4a axis, we treated B16 tumor-bearing mice with Sema4aIg (Delgoffe et al., 2013). Indeed, Sema4aIg treatment decreased tumor size, and led to an unstable Treg phenotype as assessed by increased IFNγ production and higher IFNγR expression (Fig. 3C-E). Surprisingly, we found that the majority of the IFNγ+ cells in the TIL of Nrp1L/LFoxp3Cre-YFP and Nrp1L/LFoxp3Cre-YFP/DTR-GFP were Tregs (Fig. 4A) and that the total percentage of IFNγ+ cells in TIL was small in the absence of Treg-restricted Nrp1 deletion (Fig. 4B), raising the possibility that IFNγ production may be a dominant feature of Treg fragility and thus could be affecting surrounding cells in the tumor microenvironment including Nrp1+/+ Tregs.
Figure 4. Hypoxia sensitizes intratumoral Tregs to IFNγ-mediated fragility.
(A-B) Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice were injected with B16.F10 melanoma tumor cells ID on day 0 and sacrificed on day 12. Lymphocytes were isolated from TIL of mice noted, stimulated and stained for IFNγ (n=5). (C-D) Mice were treated with anti-CD8 or isotype (200ug) every 3 days starting on day 5. Tumor size was measured on day of sacrifice (D12), lymphocytes were isolated from TIL of mice noted, stimulated, and stained for IFNγ (n=7-10). (E) Lymphocytes were isolated from mice noted and used in a microsuppression assay. (F) Tregs were purified, processed, and analyzed as in Fig. 2 (n=5). Heatmap includes genes previously shown to be positive or negative regulators in the Hif1α pathway. Pathway analysis includes all genes in pathway. (G) Foxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice were injected with B16.F10 melanoma tumor cells ID on day 0 and sacrificed on day 12. Lymphocytes were isolated from ndLN and TIL and stained for Hif1α (n=10). (H) Tregs were isolated from LN of Foxp3Cre-YFP/Cre-YFP mice, stimulated for 3 days in hypoxia or normoxia, and stained (n=4-6). Data represent 2-5 independent experiments. Student unpaired t test was used. (*p<0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
While our data show that Tregs were the predominant source of intratumoral IFNγ in Nrp1L/LFoxp3Cre-YFP and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice, we could not rule out the possibility that IFNγ production by fragile Tregs was initially triggered, or potentiated by, the altered tumor microenvironment. Indeed, tumor size is greatly reduced and lymphocyte infiltration increased in Nrp1L/LFoxp3Cre-YFP and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice (Fig. 1E and fig. S1A). In order to address this possibility, we blocked the anti-tumor immune response and prevented tumor shrinkage by depleting CD8+ T cells (using anti-CD8) in B16 tumor bearing mice, and assessed Treg phenotype and function. We found that CD8+ T cell depletion had no effect on the suppressive capacity in Nrp1L/LFoxp3Cre-YFP and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice. Indeed, Nrp1−/− Tregs exhibited increased IFNγ and Tbet protein expression along with reduced in suppressive capacity in an in vitro suppression assay, suggesting that Treg fragility due to Nrp1 loss is primarily due to a cell intrinsic mechanism rather than an extrinsic environmental effect due to increased CD8+ T cell infiltration, the ensuing anti-tumor response, and tumor size (Fig. 4C-E and fig. S5I). However, a role for CD8+ T cell-derived IFNγ in promoting Treg fragility in this system cannot be ruled out.
Although cell-intrinsic processes downstream of Nrp1 loss appeared to drive Treg fragility, it is still possible that cell-extrinsic, environmental factors facilitated intratumoral Treg fragility. Further analysis of our RNAseq data highlighted enhanced expression of Hif1a (hypoxia-inducible factor 1 alpha) in Nrp1−/− compared to Nrp1+/+ intratumoral Tregs (Fig. 4F). Indeed, the percentage of Hif1α+ Nrp1−/− intratumoral Tregs and Hif1α protein expression within those Tregs was higher than their Nrp1+/+ counterparts (Fig. 4G). Interestingly, Hif1α has been shown to be upregulated by Akt signaling which in turn led to increased IFN-γ production by Tregs (Dang et al., 2011; Lee et al., 2015). As Hif1α is upregulated in hypoxic conditions, we wondered whether hypoxia was capable of inducing Treg fragility, analogous to the environment in which intratumoral Tregs reside. Remarkably, LN-derived Tregs from a naïve mouse showed increased IFN-γ and Tbet expression, and an elevated trend in Hif1α expression while retaining Foxp3 expression after being cultured for 3 days in hypoxic versus normoxic conditions (Fig. 4H and fig. S5J and K). These data suggest that the hypoxia:Hif1α axis may prime Tregs to become functionally fragile in the tumor microenvironment.
IFNγ is required and sufficient to drive intratumoral Treg fragility
In order to test whether Nrp1−/− Tregs could directly impact the function of Nrp1+/+ Tregs, and if this was mediated by IFNγ, we co-cultured ndLN- or tumor-derived Nrp1+/+ Tregs from either Thy1.1 Foxp3Cre-YFP or Ifngr1−/−Foxp3Cre-YFP mice (Tregs which lack the IFNγ receptor) with Nrp1−/− Tregs from Thy1.2 Nrp1L/LFoxp3Cre-YFP mice prior to assessing their regulatory activity in a microsuppression assay (Fig. 5A). All cell populations were also cultured alone under the same conditions as controls, and as expected, Treg populations cultured alone exhibited the expected suppressive capacity (Fig. 1H compared with Fig. 5B, left side columns). Note that APCs were not included in the 72h pre-culture prior to the Treg microsuppression assay. Interestingly, TIL-derived Nrp1+/+ Tregs that were co-cultured with tumor-derived Nrp1−/− Tregs lost their ability to suppress effector T cells, in contrast to ndLN-derived Tregs (Fig. 5B, right side columns). Loss of suppressive activity did not require cell-cell contact, but was dependent on IFNγR expression (Fig. 5B-D). In order to confirm that IFNγ was the sole cytokine responsible for Nrp1+/+ Treg fragility following co-culture with Nrp1−/− Tregs, we co-cultured Nrp1+/+ and Nrp1−/− Tregs in the presence of different concentrations of anti-IFNγ for 72 hours, and then assessed the suppressive capacity of the purified Nrp1+/+ Tregs in the absence of anti-IFNγ. IFNγ neutralization prevented the loss of tumor-derived Nrp1+/+ Treg suppression in a dose-dependent manner (Fig. 5E and fig. S6A). The increased sensitivity of tumor- versus ndLN-derived Nrp1+/+ Tregs to Nrp1−/− Tregs appeared to correlate with IFNγR expression (fig. S6B).
Figure 5. IFNγ reduces Treg suppression.
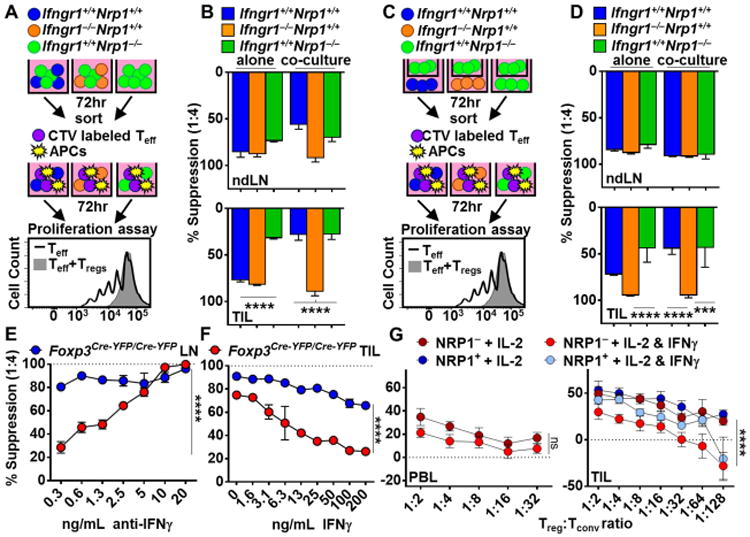
(A, B) Tregs were isolated from ndLN and TIL of mice noted, stimulated with PMA and Ionomycin, cultured with IL-2 alone or with IL-2 and Nrp1−/− Tregs for 72 hours and used in a microsuppression assay in absence of cytokine (n=6-7). (C, D) Tregs were isolated and stimulated as in (A), cultured in the bottom of a transwell plate with IL-2 alone or with IL-2 and Nrp1−/− Tregs in the top well for 72 hours and used in a microsuppression assay in the absence of cytokine (n=6-7). (E) Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP/Cre-YFP mice, co-cultured with Nrp1−/− Tregs and IL-2 in the presence or absence of anti-IFNγ, re-sorted and used in a microsuppression assay in the absence of cytokine (n=6). (F) Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP/Cre-YFP mice, treated with IL-2 and IFNγ for 72 hours, re-sorted and used in a microsuppression assay in the absence of cytokine (n=6). (G) Tregs were isolated from HNSCC PBL and TIL, cultured with IL-2 +/- IFNγ for 3 days, then used in a microsuppression assay in the absence of cytokine (n=2-14). Data represent 3-5 experiments. Error bars represent the mean ± SEM. Student unpaired t test (A-D) and 2 Way Anova (E-G) were used. (*p<0.05, **p <0.01, ****p <0.0001).
While previous studies have suggested that IL-12 can impact Treg suppression and induce IFNγ expression (Koch et al., 2012; Zhao et al., 2012), whether IFNγ has a direct effect on Tregs and if so what impact that might have in their function remains obscure. In order to determine whether IFNγ was sufficient to limit suppressive capacity, we treated Nrp1+/+ Tregs from ndLN and TIL with IFNγ for 72 hours plus IL-2 in stimulating conditions prior to assessing their functional capacity in a microsuppression assay in the absence of cytokine. IFNγ substantially limited the suppressive capacity of TIL-derived, and to a lesser extent ndLN-derived, Nrp1+/+ Tregs in a dose-dependent manner (Fig. 5F). This effect was lost if Ifngr1−/− Tregs were used (fig. S6C). Pre-treatment with IFNγ also induced IFNγ expression by WT Tregs but not Ifngr1−/− Tregs (fig. S6D, and data not shown). Given that IFNγ limits the function of murine Tregs, we asked whether IFNγ could also impact human Tregs and whether this was enhanced by the human tumor microenvironment. Indeed, intratumoral human Tregs showed an increased sensitivity to IFNγ in comparison to PBL Tregs when cultured with the cytokine 72 hours prior to assessment of their suppressive capacity in the absence of cytokine (Fig. 5G). Furthermore, intratumoral NRP1+ Tregs appeared to be less sensitive to the effects of IFNγ than NRP1− Tregs. Taken together, these data suggest that IFNγ can undermine the function of murine and human Tregs in vitro.
We next sought to determine if Nrp1−/− Treg-derived IFNγ from could drive Nrp1+/+ Treg fragility in vivo. To address this, we used Foxp3−/− mice that lack Tregs and succumb to a scurfy-like phenotype if Tregs are not adoptively transferred within 48 hours of birth (Workman et al., 2011). Tregs of a single genotype or a 50:50 mixture of two different Treg genotypes were adoptively transferred into two-day-old Foxp3−/− mice. At 4 weeks of age, mice were injected with B16 melanoma and tumor growth assessed over time (Fig. 6A). Mice were monitored after Treg injection and removed from study prior to B16 injection if any signs of autoimmunity were observed (Workman et al., 2011). Tumor growth in Foxp3−/− mice that received either Nrp1+/+ Tregs, Nrp1−/− Tregs or a 50:50 mixture of Nrp1+/+ and Nrp1−/− Tregs phenocopied tumor growth in Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice, respectively (Fig. 1E and 6B-D and fig. S6E). We then assessed the impact of Treg-derived IFNγ on tumor growth by transferring either [a] Nrp1+/+ Tregs that cannot respond to IFNγ with Nrp1−/− Tregs (50% Nrp1+/+Ifngr1−/− + 50% Nrp1−/−Ifngr1+/+), or [b] Nrp1−/− Tregs that cannot produce IFNγ with Nrp1+/+ Tregs (50% Nrp1+/+Ifng+/+ + 50% Nrp1−/−Ifng−/−). Strikingly, tumor growth was completely restored with either combination (Fig. 6E-H), revealing a critical role for IFNγ produced by fragile Nrp1−/− Tregs in mediating Nrp1+/+ Treg dysfunction, and thereby facilitating anti-tumor immunity and limiting tumor growth.
Figure 6. IFNγ uptake by Tregs is required for Treg fragility and tumor clearance.
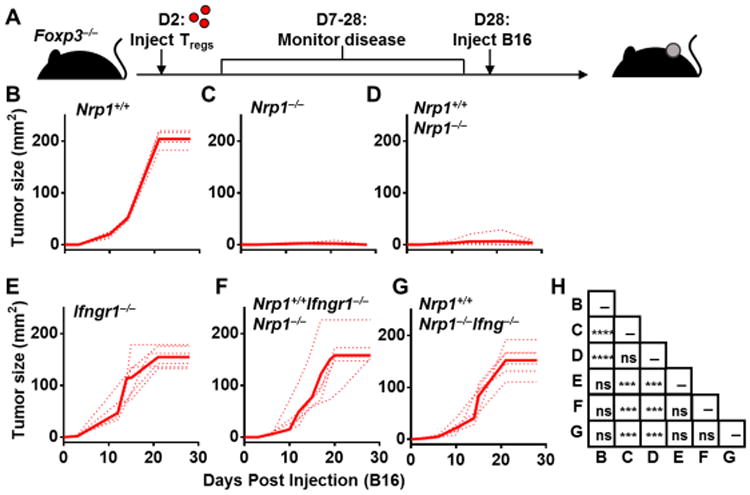
(A-G) Foxp3−/− mice were injected with 106 Tregs on day 2 post-birth, and monitored for 28 days for the onset of any autoimmune symptoms [4 of 34 mice were removed from study], no more than 1 per experimental group. B16.F10 was injected ID on day 28 and tumor size was measured every 3 days. (H) Statistics of Foxp3−/− mice tumor growth. Data represent 5-7 independent experiments with 5-7 mice per experimental group. Error bars represent the mean ± SEM. 2 way ANOVA was used. (ns, not significant, ***p < 0.001, ****p < 0.0001).
IFN-γ-induced Treg fragility is required for effective PD1-targeted immunotherapy
While our previous data suggested a prominent role for IFN-γ in driving Treg fragility, the importance for this observation in the broader context of an immunotherapeutic response is unknown. We sought to address this question using mice that lack IFN-γR on Tregs (Ifngr1L/LFoxp3Cre-YFP) (fig. S7A and B). Ifngr1L/LFoxp3Cre-YFP or Foxp3Cre-YFP mice were injected with 5×105 MC38 (an anti-PD1 sensitive tumor cell line) subcutaneously (SC) and then treated with either anti-PD1 or Armenian Hamster IgG control (200ug) on Days 6, 9 and 12 post-tumor injection. Strikingly, Ifngr1L/LFoxp3Cre-YFP mice were completely resistant to PD1 blockade in comparison to Foxp3Cre-YFP mice, as exhibited by tumor growth and survival (Fig. 7A and fig. S7C). Consistent with the loss of IFN-γR expression preventing the development of Treg fragility, no increase in percentage of IFNγ+ Tregs was observed in Ifngr1L/LFoxp3Cre-YFP mice in contrast to Foxp3Cre-YFP mice following anti-PD1 treatment (Fig. 7B). Taken together, these data suggest that IFN-γ-induced Treg fragility is required for an effective response to PD1-targeted immunotherapy.
Figure 7. IFNγ-mediated Treg fragility is required for antiPD1 response.
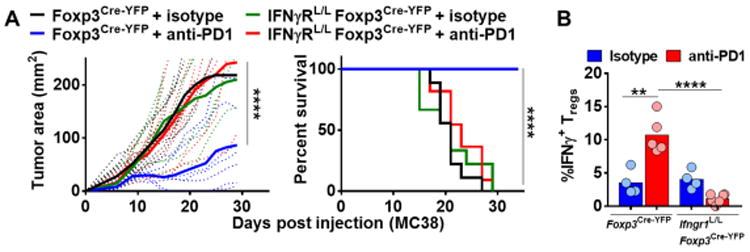
(A-B) Foxp3Cre-YFP and Ifngr1L/LFoxp3Cre-YFP mice were injected with MC38 SC on day 0 and treated with either anti-PD1 or isotype on days 6, 9, and 12 (200ug, 200ug, 200ug). (A) Tumor growth was measured with digital calipers every three days. Mice were removed from study when tumor growth reached a diameter of 2cm in any direction or when necrosis was observed, and survival plots were generated. (B) Lymphocytes were isolated from TIL on day 12 from Foxp3Cre-YFP and Ifngr1L/LFoxp3Cre-YFP mice and were stimulated and stained for IFNγ. Data represent 2 independent experiments with 4-11 mice per experimental group. 2 way ANOVA (Fig. 7A), Kaplan-Meier test (Fig. 7A), and Student unpaired t test (Fig. 7B) were used (**p < 0.01, ***p<0.001, ****p < 0.0001).
Discussion
In summary, our data highlight seven key observations. [i] A high proportion of human Tregs expressed NRP1 in two tumor types: melanoma and HNSCC. It is also noteworthy that PBL Tregs from these cancer patients also possessed a clear population of NRP1+ Tregs in contrast to healthy donor PBL Tregs, which exhibited little to no NRP1 expression. Interestingly, the percentage of intratumoral NRP1+ Tregs appeared to correlate with poor disease prognosis. [ii] B16 tumors were rapidly cleared in mice harboring a 50:50 mixture of Nrp1-deficient and WT Tregs due to increased functional fragility and loss of suppressive activity of both Treg populations without loss of Foxp3 expression. This was the result of Nrp1-deficient Treg fragility rather than the generation of Foxp3− ex-Tregs. [iii] Treg fragility had a reciprocal impact on the transcriptomes of Nrp1-deficient and WT Tregs, highlighting the previously unappreciated fact that Tregs can impact other Treg populations directly as well as many other cell types. [iv] The induction of IFNγ pathway genes was a dominant feature of Treg fragility in tumors. Intratumoral Tregs were more susceptible to this functionally fragile phenotype due to the hypoxic tumor microenvironment, which led to increased Hif1α expression and IFNγ production (Lee et al., 2015). While it is possible that IFNγ derived from other sources could lead to Treg fragility, we have shown that hypoxia promoted IFNγ production and Treg fragility. [v] IFNγ, exogenously-provided (human or mouse) or intratumoral Nrp1−/− Treg-derived (mouse), was capable of driving the fragility of tumor-derived WT Tregs and loss of mouse and human Treg suppressive activity in vitro. This was a direct effect of IFNγ or Nrp1−/− Treg-derived IFNγ as no other cells types were included in the in vitro experiments. Previous studies have suggested that IL12 can impact Treg suppression and induce IFNγ expression (Dominguez-Villar et al., 2011; Koch et al., 2012; Zhao et al., 2012), while others show that IFNγ can limit Treg expansion (Deligne et al., 2015; Olalekan et al., 2015; St Rose et al., 2013; Visperas et al., 2014). However, the direct effect of IFNγ on Treg function in vivo had surprisingly not been appreciated. While some studies have shown that IFNγ+ Tregs can maintain suppressive function, this seems to be largely disease specific and has not been carefully assessed in the context of the tumor microenvironment (Oldenhove et al., 2009; Zhao et al., 2011). It is possible that the tumor microenvironment plays a critical role in driving IFNγ-mediated Treg fragility, as suggested by the role of Hif1α in this process. In addition, while we anticipate that IL-12 may be playing a role in this process given that we see increased pSTAT4 expression, we have shown that IFNγ is capable of driving Treg fragility both in vitro and in vivo in tumor-derived Tregs exposed to a hypoxic environment. (Figs. 5E-F and 6), suggesting that IL-12 may not be essential. [vi] Intratumoral Treg fragility was mediated by IFNγ derived from Nrp1−/− Tregs that acted on WT intratumoral Tregs, thereby leading to their fragility and loss of suppressive activity. This was supported by the fact that inclusion of WT Tregs that could not respond to IFNγ or Nrp1−/− Tregs that could not produce IFNγ restored Treg function, block anti-tumor immunity, and promote tumor growth. While these data suggest that Nrp1−/− Treg-derived IFNγ is required, we do not yet know if it is sufficient. These observations were consistent with a model in which fragile Nrp1−/− Tregs produce large amounts of IFNγ in the tumor microenvironment that directly promotes the fragility of intratumoral WT Tregs without loss of Foxp3 expression. Importantly, this occurred without any detectable peripheral Treg fragility and without impacting their maintenance of peripheral tolerance, suggesting that this was a proximally- and locally-driven event, likely induced by inflammation. While we only observed this in the context of the tumor microenvironment, it is possible that Treg fragility could indeed occur in other inflammatory settings where exposure to IFNγ is increased. We would argue that the mechanism of IFNγ-induced Treg fragility is mediated directly between Nrp1−/− and WT Tregs as either loss of IFNγ or IFNγR expression, respectively, impacts fragility. While it is possible that IFNγ derived from other cell populations, such as CD8+ T cells or NK cells, or the ensuing anti-tumor response and altered tumor microenvironment contributed to Treg fragility, it is noteworthy that the dominant IFNγ-producing cell type was Nrp1−/− Tregs and CD8+ T cell depletion did not impact the enhanced IFNγ production and loss of suppressive activity observed. [vii] IFNγR expression on intratumoral Tregs was required for an effective response to PD1 blockade. Strikingly, Ifngr1L/LFoxp3Cre-YFP mice were completely resistant to anti-PD1 immunotherapy. Whereas WT Tregs showed a significant increase in IFNγ expression after PD1 blockade, Ifngr1−/− Tregs showed no increase in IFNγ, suggesting that IFNγ-driven Treg fragility may need to be induced for an effective immunotherapeutic response.
A previous study had suggested that the absence of Nrp1 leads to reduced influx of Tregs into certain tumor types (Vegfa+/+ or Vegfa−/− fibrosarcomas and Ret melanoma models) (Hansen et al., 2012). However, we did not observe any defect in the migration of Nrp1-deficient Tregs even in the competitive environment of B16 tumors in heterozygous Nrp1L/LFoxp3Cre-YFP/DTR-GFP female mice. These discrepancies could be due to the different tumor models analyzed. Alternatively, their study primarily utilized Nrp1L/LCD4Cre mice in which Nrp1 would be removed in all T cells, which could have many direct and indirect effects on intratumoral Tregs (Hansen et al., 2012). Indeed, we and others have shown that Nrp1 is expressed on a number of cell types, especially in the tumor (Jackson et al., 2014) (data not shown).
Whether Treg fragility is a feature of certain diseases and the extent to which this can be prevented or utilized therapeutically remains largely unknown and highly controversial (Rubtsov et al., 2010; Sakaguchi et al., 2013; Zhou et al., 2009a; Zhou et al., 2009b). We speculate that the IFNγ pathway may drive Treg fragility in certain inflammatory environments. The loss of Nrp1 not only results in Treg fragility but also results in substantial IFNγ expression, which in turn induces fragility in other Tregs regardless of their Nrp1 expression in a feed-forward manner; a process we refer to as “infectious fragility”. As IFNγ production is a hallmark of a productive T cell-mediated immune response, our observations also raise the possibility that IFNγ-induced Treg fragility may be a physiologically important regulatory mechanism to locally limit Treg function and promote a productive immune response. Given the profound consequences of Treg-derived IFNγ production, our data emphasize the importance of the Nrp1 pathway in limiting Treg fragility in the tumor microenvironment but also highlights that this pathway can be overcome when sufficient IFNγ is induced. Indeed, our data show that the IFNγ response induced by PD1 blockade appears to be sufficient to drive intratumoral Treg fragility despite expression of Nrp1. However, loss of IFNγR expression on Tregs, renders mice completely resistant to anti-PD1 immunotherapy. This raises the provocative possibility that an essential component of effective immunotherapy is to induce sufficient IFNγ in the tumor microenvironment to drive Treg fragility. However, the impact of Treg fragility and IFNγ expression by intratumoral Tregs on tumor growth and responsiveness to immunotherapy in murine and human tumors needs to be investigated further.
As a high frequency of intratumoral Tregs in cancer patients is largely considered a negative prognostic factor (Facciabene et al., 2012; Ichihara et al., 2003; Knol et al., 2011), identifying approaches to selectively target intratumoral Tregs while maintaining peripheral tolerance is critical. Although expression of NRP1 in peripheral, tissue-resident Tregs remains unclear, our findings highlight the surprisingly extensive and variable expression of NRP1 on human intratumoral Tregs (Battaglia et al., 2009; Battaglia et al., 2008; Chaudhary et al., 2014; Milpied et al., 2009). Importantly, given that our mouse model experiments suggest that NRP1 may not need to be targeted in all human intratumoral Tregs to derive a therapeutic effect, it is possible that targeting NRP1+ intratumoral Tregs with an NRP1 mAb may be therapeutic. As we show that the impact of the IFNγ pathway on human Tregs is conserved, it is possible that by blocking NRP1 on Tregs, one could induce functional fragility in surrounding Tregs to further enhance the therapeutic effect and overall outcome. Our identification of IFNγ as the critical mediator of Treg fragility, whether driven by manipulation of the Nrp1 pathway in Tregs or PD1 blockade highlights the potential importance of this mechanism in promoting anti-tumor immunity and provides a pathway to develop immunotherapeutic approaches that could lead to tumor reduction while maintaining peripheral tolerance.
Star Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Dario A.A. Vignali (dvignali@pitt.edu).
Experimental Model and Subject Details
Mice
Nrp1L/L mice were obtained from D. Cheresh (UC San Diego). Foxp3Cre-YFP/Cre-YFP, Foxp3DTR-GFP/DTR-GFP, Foxp3−/− mice were obtained from A.Y. Rudensky (Memorial Sloan Kettering). (Fontenot et al., 2003; Kim et al., 2007; Rubtsov et al., 2008). Ifngr1−/−, Ifng−/−, Ifngr1L/L and RosaL-Tomato-L-GFP mice were obtained from Jackson Laboratories (Dalton et al., 1993; Huang et al., 1993; Lee et al., 2013). All animal experiments were performed in the American Association for the Accreditation of Laboratory Animal Care-accredited, specific-pathogen-free facilities in Animal Resource Center, St. Jude Children's Research Hospital (SJCRH), and Division of Laboratory Animal Resources, University of Pittsburgh School of Medicine (UPSOM). Female and male mice were used. Animal protocols were approved by the Institutional Animal Care and Use Committees of SJCRH and UP.
Human T-cell populations
All HNSCC and melanoma tissues were acquired under a University of Pittsburgh Cancer Institute Institutional Review Board (IRB)-approved protocol with written informed consent obtained from each patient in conjunction with the University of Pittsburgh Cancer Institute HNSCC and Melanoma SPOREs. There were no restrictions on cancer subtype, smoking status, age, race, or prior adjuvant therapy. Control donor peripheral blood (PBL) was collected through an approved MTA protocol with the Western Pennsylvania Bloodbank. Human HNSCC PBL and TIL samples (unmatched) as well as healthy donor PBL samples were provided by R. Ferris from patients with high-risk, advanced (stage III or IV) resectable HNSCC treated with surgery. Most tumors were from oral cavity or laryngeal sites, and all were HPV-negative. Tumor specimens were obtained at the time of surgical resection, prior to adjuvant therapy. TIL were isolated, frozen, and thawed prior to staining for NRP1. Freshly processed samples were used in functional assays. Human melanoma TIL and PBL samples were provided by J. Kirkwood from an accrual trail (96-099) of patients with metastatic melanoma.
Method Details
Antibodies and flow cytometry
Single cell suspensions were stained with antibodies against CD4 (clone# GK1.5, Biolegend), CD8a (clone# YTS156.7.7, Biolegend; clone# H35-17.2, eBioscience), TCRβ (clone# H57-597, Biolegend), Thy1.1 (clone# OX-7, Biolegend), Thy1.2 (clone# 30-H12, Biolegend), Foxp3 (clone# FJK-16s, eBioscience; clone# 150D, Biolegend), IFNγ (clone# XMG1.2, Biolegend), ICOS (clone# C398.4A, Biolegend), phosphor-Stat1 (Clone# 4a, BD Biosciences) and phosphor-Stat4 (Clone# 38, BD Biosciences). Surface staining was performed on ice for 15min. For cytokine expression analysis, cells were activated with 0.1ng/ml PMA (Sigma) and 0.5ng/ml Ionomycin (Sigma) in RPMI containing 10% FBS and Monensin (eBioscience) for 8hr. For intracellular staining of cytokines and transcription factors, cells were stained with surface markers, fixed in Fix/Perm buffer (eBioscience) for 15 minutes, washed in permeabilization buffer (eBioscience) twice and stained intracellular factors in permeabilization buffer for 30min on ice. For phosphoprotein staining, cells were fixed with 1.5% PFA (Alfa Aesar) at 37°C for 15min, permeabilized with ice cold Methanol for 1hr, and stained on ice for 1hr. Cells were sorted on Aria II (BD Biosciences) or analyzed on Fortessa (BD Biosciences), and data analysis was performed on FlowJo (Tree Star).
Tumor models
Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, or Foxp3DTR-GFP/DTR-GFP mice were injected with B16.F10 melanoma (1.25 × 105 cells intradermally). Tumors were measured every 3 days with digital calipers and tumor size was calculated; this was performed in blinded manner but not randomized. 100ug Diptheria Toxin was injected every 3 days starting on day 7 in Foxp3DTR-GFP/DTR-GFP and Foxp3DTR-GFP/+ mice. Sema4aIg was injected every 3 days starting on day 5 (400ug, 200ug, 200ug), and anti-CD8 (YTS) was injected every 3 days starting on day 5 (200ug). Foxp3Cre-YFP and Ifngr1L/LFoxp3Cre-YFP mice were injected with MC38 (5×105 cells subcutaneously). Tumors were measured as above and mice were treated with either anti-PD1 (clone G4) or isotype (Armenian Hamster IgG). Tumors and non-tumor draining lymph nodes were collected for analysis on day 12. TILs were prepared with enzymatic (collagenase IV and dispase, 1mg/mL) and mechanical disruption. To achieve reasonable power, at least 5 mice were used in each group, at least 3 mice per group per experiment. Group means were compared with Student's t tests. Tumor growth over time was analyzed using two-way ANOVA with multiple comparisons. Event-free survival (moribund) estimates were calculated with the Kaplan-Meier method. Groups of mice were compared by log-rank test. All p values are two-sided, and statistical significance was assessed at the 0.05 level. Analyses were conducted using GraphPad Prism software.
Foxp3−/− model
CD45.1+ Foxp3+/− female mice were bred with CD45.1+Foxp3+/+ male mice in timed matings. Male progeny were genotyped at birth for Foxp3−/− status. Tregs from Thy1.1+ Foxp3Cre-YFP/Cre-YFP, Thy1.2+ Nrp1L/LFoxp3Cre-YFP/Cre-YFP, Thy1.2+ Ifng−/−Nrp1L/LFoxp3Cre-YFP/Cre-YFP, Thy1.1+ Ifngr1−/− Foxp3Cre-YFP/Cre-YFP mice were purified by FACS and 106 cells injected intraperitoneally into Foxp3−/− male pups within 2 days of birth (Workman et al., 2011). When a 50:50 mixture of Tregs was injected the total was maintained at 106 cells. Mice were monitored for the autoimmune phenotype ‘scurfy’ (scaly skin, eye inflammation, runted phenotype, and lack of mobility) (Workman et al., 2011). Any mice exhibiting any autoimmune or inflammatory symptoms prior to B16 injection, even if mild, were removed from further study. Mice were injected with 1.25×105 B16.F10 cells at 4 weeks of age and tumor growth was monitored every 3 days.
Gene expression profiling by RNAseq and bioinformatics analyses
Tregs (5×103) were either single (n=3) or double sorted (n=2) and cDNAs were prepared using the SMARTer® Ultra™ Low Input RNA Kit for Sequencing - v3 user manual (Clontech Laboratories). We reasoned that double sorting results in higher purity but has lower yield and may alter the expression profile. Though melanoma genes were found at lower levels in double sorted samples no other substantial differences, such as activation of stress response genes, were observed. Tregs were sorted on the following markers: Foxp3Cre-YFP/Cre-YFP (Nrp1+/+) on YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP (Nrp1−/−) on YFP, Nrp1L/LFoxp3Cre-YFP/DTR-GFP (Nrp1−/−) on YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP (Nrp1+/+) on GFP. Sequencing libraries were prepared using Nextera XT DNA Library Preparation kit (Illumina), normalized at 2nM using Tris-HCl (10mM, pH 8.5) with 0.1% Tween20, diluted and denatured to a final concentration of 1.8nM using the Illumina Denaturing and Diluting libraries for the NextSeq 500 protocol Revision D (Illumina). Cluster generation and 75bp paired-end dual-indexed sequencing was performed on Illumina NextSeq 500 system.
RNAseq data was aligned to the mm10 genome using the STAR aligner (Dobin et al., 2013) and quantified using featureCounts (Liao et al., 2014). The raw counts data were processed using the “voom” function (Law et al., 2014) in the limma R package (Ritchie et al., 2015; Wu and Smyth, 2012), which normalizes the data and assigns a weight for each measurement for subsequent linear model fitting. Unsupervised analysis of the date revealed a small cluster of melanoma specific genes that reasoned were caused by contaminations. Following a previous approach (Battle et al., 2014), we removed five melanoma specific genes from all downstream analysis (Mlana, Syt4, Tyr, Tyrp1, Dct). To filter for low expression genes we defined a cutoff of 90 reads per gene based on visual inspection of the bimodal count distribution. Only genes that meat this threshold in at least 5 samples (∼11,000 out of ∼23,000) were kept for further analysis. Differential expression was assessed using the limma moderated T statistic. The differences between the intratumoral Treg populations were subtle and in order to increase the power of our study we included technical factors as covariates in our differential expression analysis. Following the approach outlined in a recent human RNAseq study (Battle et al., 2014) we included three Picard RNAseq metrics (“PCT_INTERGENIC_BASES” “MEDIAN_3PRIME_BIAS”, “MEDIAN_CV_COVERAGE”) (Picard) as well GC correlation (computed as the sample specific Pearson correlation between each gene's GC content and its expression value). Normalization for replicate number and technical parameters was also applied directly to the voom result to obtain “normalized counts”, which were used for data visualization. Geneset enrichment was performed using the “RankSumWithCorrelation” function in the limma R package, which automatically corrects enrichment statistic inflation due to correlation among genes (with the immune genesets restricted to those relevant to T cells). p-values were combined from the “Nrp1L/LFoxp3Cre-YFP/Cre-YFP (Nrp1−/−) vs Foxp3Cre-YFP/Cre-YFP (Nrp1+/+)” and “Nrp1L/LFoxp3Cre-YFP/DTR-GFP (Nrp1+/+) vs Nrp1L/LFoxp3Cre-YFP/DTR-GFP (Nrp1−/−) using the Fisher's “log sum” method [Fisher] to define significant genes. Genesets with a q-value FDR of <0.2 were considered significant. In order to assess the alterations in Treg specific expression profile we relied on the Treg signature genes defined in a previous study (Hill et al., 2007). For pathway analysis bar charts, results of geneset enrichment analysis were depicted with colors representing the effect size and height representing the corresponding p-values. The effect size is defined as AUC (area under receiver operating curve) – 0.5 which provides a normalized ranksum statistic that is comparable across genesets of different sizes. The plot is restricted to the top 10 pathways (based on their Nrp1+/+ vs Nrp1−/− significance) from the “canonical” mSigDB geneset. We additionally restricted this analysis to genesets and pathways that were deemed relevant to intercellular signaling (defined at least half of the genes in the geneset having an extracellular or membrane annotation).
In vitro assays
Splenic CD4+YFP− (CD4+Foxp3− T cells) cells from Foxp3Cre-YFP mice were sorted as responder cells and labeled with 5μM CellTrace Violet (Life Technology). CD4−CD8− splenocytes from Foxp3Cre-FP mice were treated with 20μg/mL mytomycin C (Sigma) at 37°C for 30min, washed five times with PBS, and then used as antigen presenting cells (APCs). Responder cells (4×103), APCs (8×103), and different concentrations of Tregs (1:2-1:16 Treg:Teff ratio, 500-2000 Tregs) were activated with 2μg/ml anti-CD3 (Biolegend) in a 96-well round bottom plate with 100ul RPMI for 3 days (Turnis et al., 2016). Suppression was calculated as previously described (McMurchy and Levings, 2012). Briefly, cells were acquired by BD Fortessa, and the division index of responder cells was analyzed using FlowJo based on the division of CellTrace Violet. Suppression was then calculated with the formula % Suppression = (1-DITreg/DICtrl) × 100% (DITreg stands for the division index of responder cells with Tregs, and DICtrl stands for the division index of responder cells activated without Tregs). Human microsuppression assays were performed similarly to mouse assays with the following changes: 0.5μg/ml anti-CD3 is used for activation, and cells are cultured in assay conditions (200uL) for 4 days.
For co-culture assays, sorted Treg populations were cultured together in a 96 well round-bottom plate or in a 96 well transwell plate (Millipore) for 72 hours prior to being resorted and used in a suppression assay. Cells were treated with 100ng/mL PMA (Sigma), 500ng/mL lonomycin (Sigma) and 10001U hlL-2 (Prometheus) for co-culture. For some experiments, sorted Tregs were cultured in the presence of 0.3-20ng/mL anti-IFNγ (BioXcell) or 0-200ng/mL IFNγ (Biolegend) to the microsuppression assay.
For the hypoxia assays, Tregs were stimulated with PMA/Ionomycin and IL-2 for 3 days in normoxic (5% CO2 and 20% O2) or hypoxic (5.5% CO2 and 1.5% O2) conditions at 37°C. Cells were then stained in the same condition in the absence of cytokine.
Quantification and Statistical Analysis
Statistics were performed using Prism v6.07. Student t tests were used in Figures 1B,H; 3B-E; 4B-D, F-G; 5B,D; 7B-C and supplemental figures 1A-B; 2E-F, 5, 6B-D, 7C. ANOVA was used in Figures 1C, E, G; 5E-G; 6; 7A. Kaplan Meier was used in Figures 1C, E, F; 7A. “n” represents the number of mice used in the experiment, with the number of individual experiments listed in the legend. Graphs show individual samples. Samples are shown with the mean with or without error bars showing the SEM. Significance was defined as p=0.05.
Data and Software Availability
The RNASeq datasets have been deposited in the Gene Expression Omnibus (GEO) under code GSE97939.
Supplementary Material
Supplemental Figure 1. Temporal Nrp1 deletion leads to reduced tumor growth, Related to Figure 1. (A) Lymphocytes were isolated from TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice, stained and quantified for cell numbers (n=2-6). (B) Foxp3CreERT2-GFP RosaLSL-YFP or Nrp1L/LFoxp3CreERT2-GFP RosaLSL-YFP mice were injected with B16.F10 ID on day 0 and treated with Tamoxifen on days 7-11. Tumors were measured every 3 days (n=5-11). (C) Foxp3CreERT2-GFPRosaLSL-YFP or Nrp1L/LFoxp3CreERT2-GFPRosaLSL-YFP mice were bled on days indicated and stained. Mice were treated with Tamoxifen on days 1-5 (n=3). (D) Lymphocytes were isolated from ndLN and TIL of Foxp3DTR-GFP/DTR-GFP, Foxp3DTR-GFP/DTR-GFP + DT, and Foxp3DTR-GFP/+ + DT mice (n=3-6). Data represent 2-4 independent experiments. Student unpaired t test (S1A), 2-way ANOVA and Kaplan-Meier tests (S1B) were used. (*p < 0.05, ****p<0.0001).
Supplemental Figure 2. Nrp1−/− Tregs maintain Foxp3 expression, Related to Figure 1. (A-C) Lymphocytes were isolated from ndLN, dLN, and TIL of Foxp3Cre-YFP/Cre-YFPRosaL-Tom-L GFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFPRosaL-Tom-L GFP mice and stained (n=3-5). (D-E) Lymphocytes were isolated from TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/+ mice and stained. Tregs from Nrp1L/LFoxp3Cre-YFP/+ mice were stained with Foxp3 and Nrp1+ and Nrp1− Tregs were distinguished as YFP+Foxp3+ (Nrp1−) and YFP−Foxp3+ (Nrp1+). This was also verified by Nrp1 staining (n=5). Data represent 2-3 independent experiments. Student unpaired t test was used. (ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Supplemental Figure 3. RNASeq sorting scheme, Related to Figure 2. Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice and checked for purity. Cells were either single or double sorted. No significant difference was noted between single or double sorted samples. Data represent 5 independent experiments.
Supplemental Figure 4. Nrp1−/− display an altered Treg signature, Related to Figure 2. Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice and checked for purity. (A) Volcano plots. (B) Global heatmap. All genes that passed a threshold of q-value FDR <0.2 are shown. (C) Treg signature gene heatmap. Green/red coloring represents genes that are upregulated/downregulated in Tregs. (D) Bar chart of top differentially expressed pathways. Data represent 5 experiments.
Supplemental Figure 5. Nrp1 loss leads to an increase in TH1 markers, Related to Figures 3-4. (A-H) Lymphocytes were isolated from ndLN, dLN, or TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/+ mice and stained. Tregs from Nrp1L/LFoxp3Cre-YFP/+ mice were stained with Foxp3 and Nrp1+ and Nrp1− Tregs were distinguished as YFP+Foxp3+ (Nrp1−) and YFP−Foxp3+ (Nrp1+) (n=5-15). (I) Foxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice were injected with B16 on day 0 and treated with isotype or anti-CD8 (n=4-7). Lymphocytes were isolated from TIL and stained. (J-K) Tregs were isolated from Foxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice and stimulated in hypoxia or normoxia for 3 days and stained (n=6). Data represent 3-6 independent experiments. Student unpaired t test was used. (ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Supplemental Figure 6. Treg suppression is crippled by IFNγ, Related to Figures 5-6. (A) Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP mice, co-cultured with Nrp1−/− Tregs and IL-2 in the presence or absence of anti-IFNγ, resorted and used in a microsuppression assay (n=6). (B) Tregs were isolated from the ndLN and TIL of Foxp3Cre-YFP/Cre-YFP mice, treated with IL-12 and IL-2 for 72 hours, sorted based on Ifngr1 expression, co-cultured with Nrp1−/− Tregs for 72 hours with or without anti-IFNγ, and re-isolated for a microsuppression assay (n=3). (C) Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP and Ifngr1−/−Foxp3Cre-YFP mice, treated with IL-2 and IFNγ for 72 hours, resorted and used in a microsuppression assay (n=3-4). (D) Tregs were isolated from TIL of Foxp3Cre-YFP mice, treated with IFNγ and IL-2 for 72 hours, washed and stained (n=3). (E) Foxp3−/− mice were injected with 106 of a 50:50 mix of Nrp1+/+Thy1.1 and Nrp1−/−Thy1.2 Tregs on day 2 post-birth. B16.F10 was injected ID on day 28. Lymphocytes were isolated and stained (n=3). Data represent 2-3 independent experiments. Error bars represent the mean ± SEM. Student unpaired t test was used. (ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001).
Supplemental Figure 7. Ifngr1L/LFoxp3Cre-YFP mice are non-responsive to PD1 blockade, Related to Figure 7. (A) Lymphocytes were isolated from ndLN of Foxp3Cre-YFP or Ifngr1L/LFoxp3Cre-YFP mice and stained for IFNγR (n=2). (B) Tconv, Tregs, CD8s, or B cells were isolated from LN and spleen of Foxp3Cre-YFP or Ifngr1L/LFoxp3Cre-YFP mice, DNA was extracted for endpoint PCR testing for the presence or absence of the loxP sites or excision of loxP-targeted sequence (n=2). (C) Foxp3Cre-YFP or Ifngr1L/LFoxp3Cre-YFP mice were injected with MC38 on day 0, sacrificed on day 13 (n=3-6). Data represent 2 experiments. Student unpaired t test was used. (ns, not significant, **p < 0.01).
Acknowledgments
The authors would like to thank A. Rudensky and D. Cheresh for mice, A. Yates, D. Falkner, H. Shen from the Immunology Flow Core for cell sorting, the staff of the Division of Laboratory Animal Services for the animal husbandry, and the Immunology Department at the University of Pittsburgh for helpful discussions. This work was supported by the National Institutes of Health (R01 AI091977 to D.A.A.V.; F31 CA189441 to A.E.O.), NCI Comprehensive Cancer Center Support CORE grant (CA047904 and CA21765, to D.A.A.V.), and the American Lebanese Syrian Associated Charities (ALSAC, to D.A.A.V.). Part of this project used the UPCI Flow Core that is supported in part by P30 CA047904.
Footnotes
Author Contributions: Conceptualization, A.E.O.D., and D.A.A.V.; Formal Analysis, M.C., D.P.N., Y.S.; Investigation, A.E.O.D., H.Y., R.E.D., E.A.B., G.S., W.H., G.M.D., T.C.B.; Resources, J.M.M., C.S., J.M.K., R.L.F.; Writing – Original Draft, A.E.O.D. and D.A.A.V.; Writing – Review & Editing, A.E.O.D., M.C., R.E.D., H.Y,. E.A.B., G.S., W.H., J.M.M., J.K.K., J.M.K., R.L.F., C.S., D.P.N., Y.S., G.M.D., T.C.B., C.J.W., D.A.A.V.; Supervision, J.K.K., T.C.B., C.J.W., D.A.A.V.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Battaglia A, Buzzonetti A, Baranello C, Ferrandina G, Martinelli E, Fanfani F, Scambia G, Fattorossi A. Metastatic tumour cells favour the generation of a tolerogenic milieu in tumour draining lymph node in patients with early cervical cancer. Cancer immunology, immunotherapy : CII. 2009;58:1363–1373. doi: 10.1007/s00262-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia A, Buzzonetti A, Monego G, Peri L, Ferrandina G, Fanfani F, Scambia G, Fattorossi A. Neuropilin-1 expression identifies a subset of regulatory T cells in human lymph nodes that is modulated by preoperative chemoradiation therapy in cervical cancer. Immunology. 2008;123:129–138. doi: 10.1111/j.1365-2567.2007.02737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Mostafavi S, Zhu X, Potash JB, Weissman MM, McCormick C, Haudenschild CD, Beckman KB, Shi J, Mei R, et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome research. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs SF, Reijo Pera RA. X chromosome inactivation: recent advances and a look forward. Current opinion in genetics & development. 2014;28:78–82. doi: 10.1016/j.gde.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary B, Elkord E. Novel expression of Neuropilin 1 on human tumor-infiltrating lymphocytes in colorectal cancer liver metastases. Expert opinion on therapeutic targets. 2015;19:147–161. doi: 10.1517/14728222.2014.977784. [DOI] [PubMed] [Google Scholar]
- Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer immunology, immunotherapy : CII. 2014;63:81–99. doi: 10.1007/s00262-013-1500-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Rudensky AY. Control of inflammation by integration of environmental cues by regulatory T cells. J Clin Invest. 2013;123:939–944. doi: 10.1172/JCI57175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013 doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deligne C, Metidji A, Fridman WH, Teillaud JL. Anti-CD20 therapy induces a memory Th1 response through the IFN-gamma/IL-12 axis and prevents protumor regulatory T-cell expansion in mice. Leukemia. 2015;29:947–957. doi: 10.1038/leu.2014.275. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med. 2011;17:673–675. doi: 10.1038/nm.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drennan S, Stafford ND, Greenman J, Green VL. Increased frequency and suppressive activity of CD127(low/-) regulatory T cells in the peripheral circulation of patients with head and neck squamous cell carcinoma are associated with advanced stage and nodal involvement. Immunology. 2013;140:335–343. doi: 10.1111/imm.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood. 2012;119:4430–4440. doi: 10.1182/blood-2011-11-392324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72:2162–2171. doi: 10.1158/0008-5472.CAN-11-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Galupa R, Heard E. X-chromosome inactivation: new insights into cis and trans regulation. Current opinion in genetics & development. 2015;31:57–66. doi: 10.1016/j.gde.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Gao YL, Chai YF, Qi AL, Yao Y, Liu YC, Dong N, Wang LJ, Yao YM. Neuropilin-1highCD4(+)CD25(+) Regulatory T Cells Exhibit Primary Negative Immunoregulation in Sepsis. Mediators of inflammation. 2016;2016:7132158. doi: 10.1155/2016/7132158. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, Albert J, Sparwasser T, Sakaguchi S, Westendorf AM, et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. The Journal of experimental medicine. 2012;209:2001–2016. doi: 10.1084/jem.20111497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Hori S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev. 2014;259:159–172. doi: 10.1111/imr.12175. [DOI] [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res. 2003;9:4404–4408. [PubMed] [Google Scholar]
- Jackson SR, Berrien-Elliott M, Yuan J, Hsueh EC, Teague RM. Neuropilin-1 expression is induced on tolerant self-reactive CD8+ T cells but is dispensable for the tolerant phenotype. PLoS One. 2014;9:e110707. doi: 10.1371/journal.pone.0110707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JF, Nierkens S, Figdor CG, de Vries IJ, Adema GJ. Regulatory T cells in melanoma: the final hurdle towards effective immunotherapy? The Lancet Oncology. 2012;13:e32–42. doi: 10.1016/S1470-2045(11)70155-3. [DOI] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Knol AC, Nguyen JM, Quereux G, Brocard A, Khammari A, Dreno B. Prognostic value of tumor-infiltrating Foxp3+ T-cell subpopulations in metastatic melanoma. Experimental dermatology. 2011;20:430–434. doi: 10.1111/j.1600-0625.2011.01260.x. [DOI] [PubMed] [Google Scholar]
- Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity. 2012;37:501–510. doi: 10.1016/j.immuni.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenecke C, Lee CW, Thamm K, Fohse L, Schafferus M, Mittrucker HW, Floess S, Huehn J, Ganser A, Forster R, et al. IFN-gamma production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol. 2012;189:2890–2896. doi: 10.4049/jimmunol.1200413. [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome biology. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Elly C, Park Y, Liu YC. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1alpha to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity. 2015;42:1062–1074. doi: 10.1016/j.immuni.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–1323. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- Lee SH, Carrero JA, Uppaluri R, White JM, Archambault JM, Lai KS, Chan SR, Sheehan KC, Unanue ER, Schreiber RD. Identifying the initiating events of anti-Listeria responses using mice with conditional loss of IFN-gamma receptor subunit 1 (IFNGR1) J Immunol. 2013;191:4223–4234. doi: 10.4049/jimmunol.1300910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (Oxford, England) 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu C, Workman CJ, Vignali DA. Targeting regulatory T cells in tumors. The FEBS journal. 2016;283:2731–2748. doi: 10.1111/febs.13656. [DOI] [PubMed] [Google Scholar]
- McMurchy AN, Levings MK. Suppression assays with human T regulatory cells: a technical guide. Eur J Immunol. 2012;42:27–34. doi: 10.1002/eji.201141651. [DOI] [PubMed] [Google Scholar]
- Milpied P, Renand A, Bruneau J, Mendes-da-Cruz DA, Jacquelin S, Asnafi V, Rubio MT, MacIntyre E, Lepelletier Y, Hermine O. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol. 2009;39:1466–1471. doi: 10.1002/eji.200839040. [DOI] [PubMed] [Google Scholar]
- Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends in molecular medicine. 2007;13:108–116. doi: 10.1016/j.molmed.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. International journal of cancer Journal international du cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- Olalekan SA, Cao Y, Hamel KM, Finnegan A. B cells expressing IFN-gamma suppress Treg-cell differentiation and promote autoimmune experimental arthritis. Eur J Immunol. 2015;45:988–998. doi: 10.1002/eji.201445036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O'Brien S, Blank R, Lamb E, Natarajan S, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overacre AE, Vignali DA. Treg stability: to be or not to be. Curr Opin Immunol. 2016;39:39–43. doi: 10.1016/j.coi.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiyan P, Zhu J. Origin and functions of pro-inflammatory cytokine producing Foxp3+ regulatory T cells. Cytokine. 2015;76:13–24. doi: 10.1016/j.cyto.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard. picard. In Picard (Broad Institute).
- Piechnik A, Dmoszynska A, Omiotek M, Mlak R, Kowal M, Stilgenbauer S, Bullinger L, Giannopoulos K. The VEGF receptor, neuropilin-1, represents a promising novel target for chronic lymphocytic leukemia patients. International journal of cancer Journal international du cancer. 2013;133:1489–1496. doi: 10.1002/ijc.28135. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667–1671. doi: 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22:679–684. doi: 10.1038/nm.4086. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol. 2013;13:461–467. doi: 10.1038/nri3464. [DOI] [PubMed] [Google Scholar]
- Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, Pan F, Blazar BR, Pardoll DM, Mellor AL, et al. An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos. Immunity. 2013;38:998–1012. doi: 10.1016/j.immuni.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Rose MC, Taylor RA, Bandyopadhyay S, Qui HZ, Hagymasi AT, Vella AT, Adler AJ. CD134/CD137 dual costimulation-elicited IFN-gamma maximizes effector T-cell function but limits Treg expansion. Immunology and cell biology. 2013;91:173–183. doi: 10.1038/icb.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatura R, Zeschnigk M, Hansen W, Steinmann J, Vidigal PG, Hutzler M, Pastille E, Westendorf AM, Buer J, Kehrmann J. Relevance of Foxp3(+) regulatory T cells for early and late phases of murine sepsis. Immunology. 2015;146:144–156. doi: 10.1111/imm.12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, Beres AJ, Vogel P, Workman CJ, Vignali DA. Interleukin-35 Limits Anti-Tumor Immunity. Immunity. 2016;44:316–329. doi: 10.1016/j.immuni.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visperas A, Shen B, Min B. gammadelta T cells restrain extrathymic development of Foxp3+-inducible regulatory T cells via IFN-gamma. Eur J Immunol. 2014;44:2448–2456. doi: 10.1002/eji.201344331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman CJ, Collison LW, Bettini M, Pillai MR, Rehg JE, Vignali DA. In vivo Treg suppression assays. Methods Mol Biol. 2011;707:119–156. doi: 10.1007/978-1-61737-979-6_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Smyth GK. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic acids research. 2012;40:e133. doi: 10.1093/nar/gks461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhao J, Fett C, Trandem K, Fleming E, Perlman S. IFN-gamma- and IL-10-expressing virus epitope-specific Foxp3(+) T reg cells in the central nervous system during encephalomyelitis. The Journal of experimental medicine. 2011;208:1571–1577. doi: 10.1084/jem.20110236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhao J, Perlman S. Differential effects of IL-12 on Tregs and non-Treg T cells: roles of IFN-gamma, IL-2 and IL-2R. PLoS One. 2012;7:e46241. doi: 10.1371/journal.pone.0046241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol. 2009a;21:281–285. doi: 10.1016/j.coi.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009b;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Temporal Nrp1 deletion leads to reduced tumor growth, Related to Figure 1. (A) Lymphocytes were isolated from TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice, stained and quantified for cell numbers (n=2-6). (B) Foxp3CreERT2-GFP RosaLSL-YFP or Nrp1L/LFoxp3CreERT2-GFP RosaLSL-YFP mice were injected with B16.F10 ID on day 0 and treated with Tamoxifen on days 7-11. Tumors were measured every 3 days (n=5-11). (C) Foxp3CreERT2-GFPRosaLSL-YFP or Nrp1L/LFoxp3CreERT2-GFPRosaLSL-YFP mice were bled on days indicated and stained. Mice were treated with Tamoxifen on days 1-5 (n=3). (D) Lymphocytes were isolated from ndLN and TIL of Foxp3DTR-GFP/DTR-GFP, Foxp3DTR-GFP/DTR-GFP + DT, and Foxp3DTR-GFP/+ + DT mice (n=3-6). Data represent 2-4 independent experiments. Student unpaired t test (S1A), 2-way ANOVA and Kaplan-Meier tests (S1B) were used. (*p < 0.05, ****p<0.0001).
Supplemental Figure 2. Nrp1−/− Tregs maintain Foxp3 expression, Related to Figure 1. (A-C) Lymphocytes were isolated from ndLN, dLN, and TIL of Foxp3Cre-YFP/Cre-YFPRosaL-Tom-L GFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFPRosaL-Tom-L GFP mice and stained (n=3-5). (D-E) Lymphocytes were isolated from TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/+ mice and stained. Tregs from Nrp1L/LFoxp3Cre-YFP/+ mice were stained with Foxp3 and Nrp1+ and Nrp1− Tregs were distinguished as YFP+Foxp3+ (Nrp1−) and YFP−Foxp3+ (Nrp1+). This was also verified by Nrp1 staining (n=5). Data represent 2-3 independent experiments. Student unpaired t test was used. (ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Supplemental Figure 3. RNASeq sorting scheme, Related to Figure 2. Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice and checked for purity. Cells were either single or double sorted. No significant difference was noted between single or double sorted samples. Data represent 5 independent experiments.
Supplemental Figure 4. Nrp1−/− display an altered Treg signature, Related to Figure 2. Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, and Nrp1L/LFoxp3Cre-YFP/DTR-GFP mice and checked for purity. (A) Volcano plots. (B) Global heatmap. All genes that passed a threshold of q-value FDR <0.2 are shown. (C) Treg signature gene heatmap. Green/red coloring represents genes that are upregulated/downregulated in Tregs. (D) Bar chart of top differentially expressed pathways. Data represent 5 experiments.
Supplemental Figure 5. Nrp1 loss leads to an increase in TH1 markers, Related to Figures 3-4. (A-H) Lymphocytes were isolated from ndLN, dLN, or TIL of Foxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/Cre-YFP, Nrp1L/LFoxp3Cre-YFP/+ mice and stained. Tregs from Nrp1L/LFoxp3Cre-YFP/+ mice were stained with Foxp3 and Nrp1+ and Nrp1− Tregs were distinguished as YFP+Foxp3+ (Nrp1−) and YFP−Foxp3+ (Nrp1+) (n=5-15). (I) Foxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice were injected with B16 on day 0 and treated with isotype or anti-CD8 (n=4-7). Lymphocytes were isolated from TIL and stained. (J-K) Tregs were isolated from Foxp3Cre-YFP/Cre-YFP and Nrp1L/LFoxp3Cre-YFP/Cre-YFP mice and stimulated in hypoxia or normoxia for 3 days and stained (n=6). Data represent 3-6 independent experiments. Student unpaired t test was used. (ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Supplemental Figure 6. Treg suppression is crippled by IFNγ, Related to Figures 5-6. (A) Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP mice, co-cultured with Nrp1−/− Tregs and IL-2 in the presence or absence of anti-IFNγ, resorted and used in a microsuppression assay (n=6). (B) Tregs were isolated from the ndLN and TIL of Foxp3Cre-YFP/Cre-YFP mice, treated with IL-12 and IL-2 for 72 hours, sorted based on Ifngr1 expression, co-cultured with Nrp1−/− Tregs for 72 hours with or without anti-IFNγ, and re-isolated for a microsuppression assay (n=3). (C) Tregs were isolated from ndLN and TIL of Foxp3Cre-YFP and Ifngr1−/−Foxp3Cre-YFP mice, treated with IL-2 and IFNγ for 72 hours, resorted and used in a microsuppression assay (n=3-4). (D) Tregs were isolated from TIL of Foxp3Cre-YFP mice, treated with IFNγ and IL-2 for 72 hours, washed and stained (n=3). (E) Foxp3−/− mice were injected with 106 of a 50:50 mix of Nrp1+/+Thy1.1 and Nrp1−/−Thy1.2 Tregs on day 2 post-birth. B16.F10 was injected ID on day 28. Lymphocytes were isolated and stained (n=3). Data represent 2-3 independent experiments. Error bars represent the mean ± SEM. Student unpaired t test was used. (ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001).
Supplemental Figure 7. Ifngr1L/LFoxp3Cre-YFP mice are non-responsive to PD1 blockade, Related to Figure 7. (A) Lymphocytes were isolated from ndLN of Foxp3Cre-YFP or Ifngr1L/LFoxp3Cre-YFP mice and stained for IFNγR (n=2). (B) Tconv, Tregs, CD8s, or B cells were isolated from LN and spleen of Foxp3Cre-YFP or Ifngr1L/LFoxp3Cre-YFP mice, DNA was extracted for endpoint PCR testing for the presence or absence of the loxP sites or excision of loxP-targeted sequence (n=2). (C) Foxp3Cre-YFP or Ifngr1L/LFoxp3Cre-YFP mice were injected with MC38 on day 0, sacrificed on day 13 (n=3-6). Data represent 2 experiments. Student unpaired t test was used. (ns, not significant, **p < 0.01).