

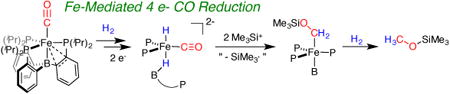
Abstract
One of the major challenges associated with developing molecular Fischer-Tropsch catalysts is the design of systems that promote the formation of C-H bonds from H2 and CO while also facilitating the release of the resulting CO-derived organic products. To this end, we describe the synthesis of reduced iron-hydride/carbonyl complexes that enable an electrophile-promoted hydride migration process, resulting in the reduction of coordinated CO to a siloxymethyl (LnFe-CH2OSiMe3) group. Intramolecular hydride-to-CO migrations are extremely rare, and to our knowledge the system described herein is the first example where such a process can be accessed from a thermally stable M(CO)(H) complex. Further addition of H2 to LnFe-CH2OSiMe3 releases CH3OSiMe3, demonstrating net four-electron reduction of CO to CH3OSiMe3 at a single Fe site.
Graphical abstract
In the industrial Fischer-Tropsch (FT) process, pressurized mixtures of CO and H2 are heated over a heterogeneous transition metal catalyst, most commonly Fe or Co, to yield a mixture of liquid organic products.1 Interestingly, it was recently demonstrated that nitrogenase enzymes also facilitate some degree of FT reactivity, with H-atom equivalents provided as protons and electrons.2 Well-defined model complexes can be used to explore viable pathways for mechanistically relevant CO reductions under controlled reaction conditions. In particular, Fe-mediated model systems are of interest owing to the role of iron in both industrial and (presumably) biological FT systems.
One of the major challenges associated with molecular CO reduction reactivity is early stage C-H bond formation from H2.3 Migratory insertion of CO into M-H bonds is generally presumed to be thermodynamically unfavorable.4,5 For early transition metal systems, C-H bond formation is concomitant with strong M-O bond formation.6 Mid-to-late transition metal systems are thought to lack this driving force, and C-H bond forming steps have instead been established using strong hydride donors that are not generated from H2.7,8 Recently, it has been demonstrated that weaker, H2-derived hydride sources can be used to facilitate C-H bond formation from H2 in Lewis acid-activated Re-CO complexes.9,10 C-H bond formation in this latter case is driven by the formation of strong B-O bonds.
Our group has previously studied reduced Fe-CO complexes that react with silyl electrophiles at oxygen to generate carbyne products (e.g., Fe-CO- + SiMe3+ → Fe≡C-OSiMe3).11 We wondered whether related O-functionalization at reduced Fe(CO)(H)n species might promote concomitant hydride migration to carbon. Herein, we describe the synthesis of a series of reduced Fe(CO)(H)n complexes. One of these complexes serves as a key intermediate from which functionalization of the coordinated CO promotes the migration of two hydride equivalents to carbon.
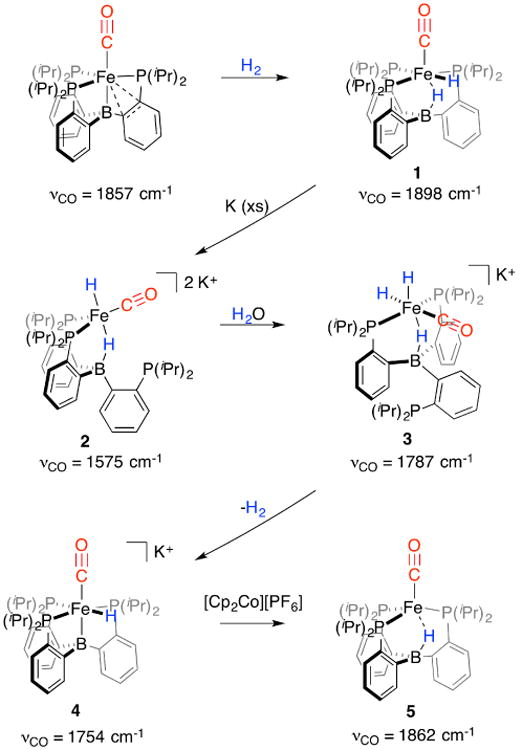
In this study we employ the P3B Fe-system12 (P3B = B(o-iPr2PC6H4)3)13, for which its monocarbonyl complex, P3B Fe-CO, was previously shown to activate H2 to generate P3B (μ-H)Fe(H)(CO), 1.14 Two-electron reduction of 1 can be readily accomplished with excess potassium metal in THF, leading to a distinct color change from yellow to dark red-brown. NMR spectroscopy reveals that a new diamagnetic species, 2, is cleanly generated, with hydridic resonances observed in the 1H NMR spectrum at -19.12 ppm (B-H-Fe, br, 1H) and -20.76 ppm (Fe-H, t, 2JHP = 65 Hz, 1H). The 31P NMR spectrum has two resonances at 102.3 and -1.5 ppm (2:1 integration), indicating that one of the phosphine arms has dechelated from the Fe center, while the thin film IR spectrum of 2 indicates a highly activated CO ligand with a stretch at 1575 cm-1 . Structural characterization of dianionic 2 confirms that the product has two coordinated phosphine ligands and the CO ligand in an approximate trigonal geometry (average Σ(trigonal plane) = 359.4),15 with the axial sites presumably occupied by the hydride and borohydride ligands that are not crystallographically observed (Figure 1). Among Fe-CO complexes, 2 has an unusually short Fe-C distance (1.70 Å; average) with corresponding elongation of the C-O bond (1.24 Å; average), consistent with significant contribution of carbyne character in this species. Note that 2 is ion-paired with the K+ cations (see SI for details), contributing to elongation of the C-O bond. For relevant comparison, P3B Fe-CO (v(CO) = 1857 cm-1 ) has an Fe-C distance of 1.752(3) Å and a C-O distance of 1.167(3) Å,12 while the carbyne complex P3SiFe≡C-OSiMe3 has an Fe-C distance of 1.671(2) Å and a C-O distance of 1.278(3) Å.11a
Figure 1.
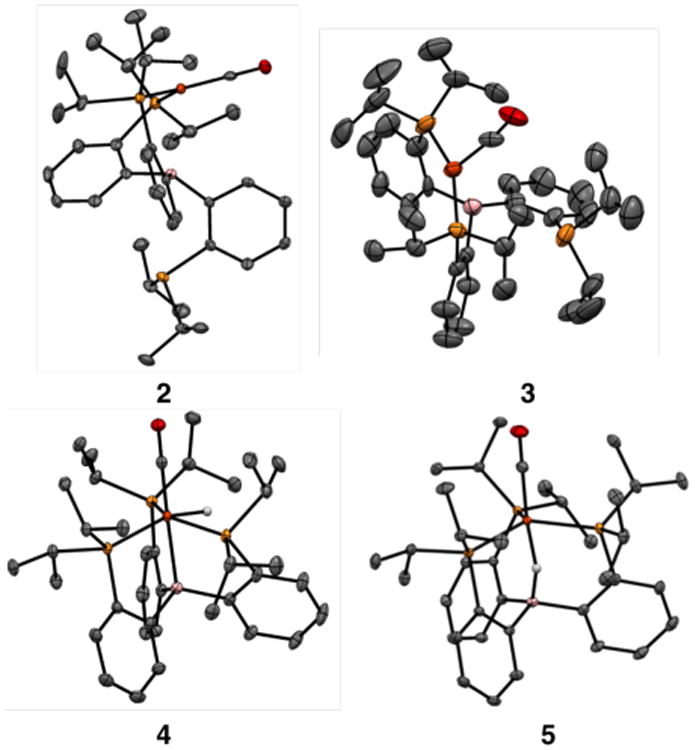
Crystal structures of Fe(CO)(H)n complexes 2-5. Displacement ellipsoids shown at 50% probability; hydrogen atoms (except for hydride ligands found in the difference map), solvent molecules, counter cations, and disorder of 3 (see SI) omitted for clarity.
A number of structurally unusual reduced hydride species are readily available from 2. For example, complex 2 can be protonated selectively by water via addition of wet N2 (produced by bubbling N2 through water) yielding the anionic trihydride complex [P3B (μ-H)Fe(H)2(CO)][K(THF)n], 3. NMR spectroscopy reveals inequivalent 31P NMR resonances at 106.7 and -9.6 ppm (2:1 integration) and three hydridic resonances in the 1H NMR spectrum at -8.40 ppm (Fe-H, td, 2JHP = 66 Hz, 2JHH = 16 Hz, 1H), -14.69 ppm (B-H-Fe, br, 1H), and -20.53 ppm (Fe-H, t, 2JHP = 48 Hz, 1H). An IR stretch is observed for Fe-ligated CO at 1787 cm-1 . The coordinated phosphine ligands are located trans to one another in the solid state, with one of the terminal hydride ligands trans to the terminal CO (Figure 1).16
The trihydride complex 3 is unstable to the loss of H2 in solution, with clean, irreversible conversion to the anionic and diamagnetic monohydride complex [P3BFe(H)(CO)][K(THF)n], 4, observed over 2 d (v(CO) = 1754 cm-1; see SI for NMR details). In the solid state, 4 adopts an approximately octahedral geometry, with the hydride ligand located in the difference map as a terminal Fe-H (Figure 1). Oxidation of 4 using [Cp2Co][PF6] generates the doublet product (P3B-H)Fe(CO), 5, featuring a broad, axial EPR signal at 77 K. The IR spectrum of 5 shows an intense CO stretch at 1862 cm-1 and a broad hydride stretch at 2588 cm-1, consistent with a boron-coordinated hydride, as revealed in its solid-state structure (Figure 1).
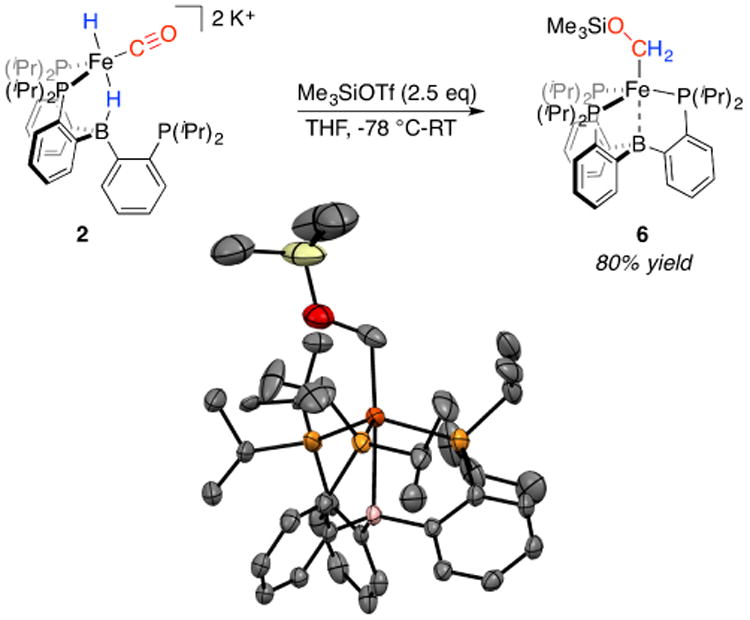
With these complexes in hand, we next canvassed their reactivity with electrophiles. Accordingly, clean in situ generation of a THF solution of dianion 2 followed by treatment with at least two equivalents of Me3SiOTf at low temperature generates a new paramagnetic species, P3BFe-CH2OSiMe3, 6, as the major product (Scheme 2).17 57Fe Mössbauer spectroscopy indicates that 6 is generated in ∼80% chemical yield and its parameters (δ = 0.49 mm/s; ΔEQ = 2.05 mm/s) are consistent with an S = 3/2 P3BFe species.18 For representative comparison, the previously reported complex P3BFe-Me has the following parameters: δ = 0.50 mm/s, ΔEQ = 1.84 mm/s.19 Structure determination of 6 by XRD analysis confirms its assignment and clearly establishes that the carbonyl ligand has been O-functionalized by the silyl electrophile, with both hydride equivalents having migrated to the carbonyl carbon; one equivalent of electrophile (Me3Si+) presumably serves as a one-electron oxidant. Despite repeated attempts, we have been unable to isolate complex 6 in analytically pure form owing to its high hydrocarbon solubility. Persistent S = 1/2 Fe-containing impurities that are similarly soluble, including 5 as an oxidation side-product, are consistently present in preparative scale, worked-up reactions.20
Scheme 2. Reaction of 2 with Me3SiOTf.
We next surveyed conditions for the release of the CO-derived organic fragment.21 For related P3BFe-Me, treatment with acid results in methane release.19 For complex 6, treatment with acid ([H(OEt2)2][BArF], [LutH][Cl] and H2O, for example) invariably results in unproductive decomposition to regenerate P3B(μ-H)Fe(H)(CO), 1, as the major product, presumably via acidic cleavage of the Si-O bond.22 We therefore canvassed the reactivity of 6 towards hydrogen and silanes (Scheme 3).
Scheme 3. E-H promoted product release.
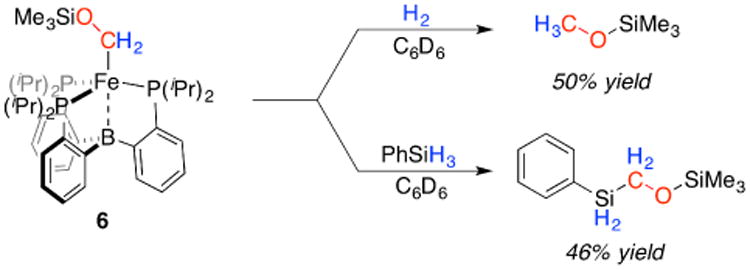
Treatment of 6 with an atmosphere of H2 at RT results in the release of CH3OSiMe3 over 24 h in moderate yields (50%, average of three runs; assumes 6 present at 80% purity initially based on Mössbauer analysis; vide supra). Product release can also be affected by the addition of excess PhSiH3 (5 equiv), with C-Si bond formation in this case and similar yields of the CO-derived product, PhSiH2CH2OSiMe3 (46%, average of three runs; again assumes 6 present at 80% purity initially). The Fe-containing products of these reactions have not yet been identified; spectroscopic analysis indicates complex mixtures of P3BFe-containing products.23
To track the origin of the H equivalents delivered to the terminal organic product, CH3OSiMe3, upon H2 addition we undertook a series of labeling studies. These experiments are particularly valuable given our inability to isolate 6 in analytically pure form.
Use of the dianionic complex 2 as a precursor to the alkyl complex 6 resulted in full H incorporation into the free organic product, CH3OSiMe3 as determined by 1H NMR spectroscopy (Scheme 4A). Likewise, using the 2H-labeled analog 2-D2 to generate 6-D2, followed by treatment with D2, resulted in (nearly) complete incorporation of deuterium in the released product CD3OSiMe3 (Scheme 4B; ∼5% CD2HOSiMe3 was also detected). These results suggest that the three H equivalents delivered to the CO C-atom are derived from the hydride ligands and/or the added H2 gas; scrambling into the alkyl phosphine substituents or incorporation of H-equivalents from solvent, is therefore not kinetically relevant. Interestingly, when 6-D2 was treated with an atmosphere of H2, the partially-deuterated organic products CDH2OSiMe3 and CD2HOSiMe3 were obtained in a 5:1 ratio (Scheme 4C). Similarly, when 6 was treated with D2 a mixture of products was observed, with CD2HOSiMe3 and CDH2OSiMe3 obtained in a 3:1 ratio, and ∼5% CH3OSiMe3 also detected (Scheme 4D). These results are suggestive of a facile exchange process between the alkyl C-H bonds and the added H2 or D2 prior to product release. One scenario by which such an exchange could occur involves reversible alpha elimination from 6-D2 to generate a carbene-deuteride intermediate (i.e., P3BFe-CD2OSiMe3 ⇌ P3B(μ-D)Fe=C(D)(OSiMe3)) that then reacts with H2.24
Scheme 4. Summary of Labeling Experiments.
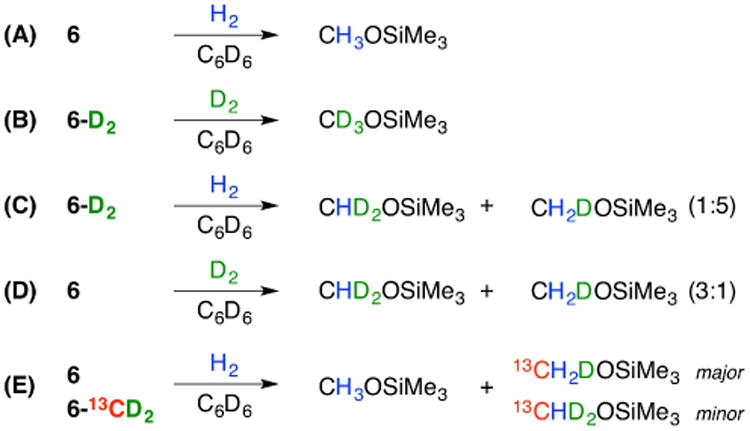
To probe the possibility of intermolecular C-H bond forming steps we undertook a crossover experiment wherein the dianionic complex 2 was synthesized as a 50:50 mixture of its [P3B(μ-D)Fe(D)(13CO)]2- and [P3B(μ-H)Fe(H)(12CO)]2- variants. Standard functionalization of this labeled mixture to generate the corresponding alkyl complexes 6, and product release via the addition of H2, showed deuterium incorporation exclusively in the 13C-labeled complex (Scheme 4E). This result is fully consistent with an intramolecular pathway for the initial C-H bond-forming steps to generate the alkyl complex 6, and also the subsequent C-H bond formation from H2 to promote the release of the organic product.
To conclude, using a mononuclear iron system we have explored a silyl electrophile promoted hydride-to-CO migration process that allows for the net 4-electron reduction of CO, releasing CH3OSiMe3 upon hydrogenolysis. Use of the bifunctional tris(phosphine)borane ligand, P3B, is key to the hydride migration step as it helps to stabilize the unusual bis(phosphine)-dihydride-carbonyl precursor, 2, via a bridging interaction of one of the hydride ligands with the borane. Isotopic labeling studies establish that the C-H bond forming steps are unimolecular. Intramolecular hydride-to-CO migrations are extremely rare; to our knowledge the iron system described herein is the first thermally stable M(CO)(H) complex to exhibit such reactivity. Future studies will be aimed at expanding the scope of this reactivity, for example by replacing the silyl electrophile Me3Si+ with H+ as a route to CH3OH generation.
Supplementary Material
Synthetic and spectroscopic details: Crystallographic information:
Scheme 1. Synthesis Of P3BFe(Co)(H)N Complexes.
Acknowledgments
This work was supported by the NIH (GM070757) and the Gordon and Betty Moore Foundation. We thank Larry Henling and Mike Takase for crystallographic assistance.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
References
- 1.(a) Rofer-DePoorter CK. Chem Rev. 1981;81:447. [Google Scholar]; (b) Maitlis PM, Zanotti V. Chem Commun. 2009:1619. doi: 10.1039/b822320n. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lee CC, Hu Y, Ribbe MW. Science. 2010;329:642. doi: 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu Y, Lee CC, Ribbe MW. Science. 2011;333:753. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For a recent review of molecular FT model chemistry see: West NM, Miller AJM, Labinger JA, Bercaw JE. Coord Chem Rev. 2011;255:881.
- 4.(a) Berke H, Hoffmann R. J Am Chem Soc. 1978;100:7224. [Google Scholar]; (b) Ziegler T, Versluis L, Tschinke V. J Am Chem Soc. 1986;108:612. [Google Scholar]
- 5.There is a single reported example of reversible CO migratory insertion into a metal-hydride (Th-H) bond. Th-O bond formation helps to drive the insertion. See: Fagan PJ, Moloy KG, Marks TJ. J Am Chem Soc. 1981;103:6959.
- 6.Select examples: Wolczanski PT, Bercaw JE. Acc Chem Res. 1980;13:121.Toreki R, LaPointe RE, Wolczanski PT. J Am Chem Soc. 1987;109:7558.
- 7.For a review of CO reduction reactivity supported by the Fp fragment (Fp = (η5-C5H5)Fe(CO)2) see: Cutler AR, Hanna PK, Vites JC. Chem Rev. 1988;88:1363. For additional examples see Ref. 3.
- 8.For exceptions see: Wayland BB, Woods BA. J Chem Soc, Chem Commun. 1981:700.Wayland BB, Woods BA, Pierce R. J Am Chem Soc. 1982;104:302.Paonessa RS, Thomas NC, Halpern J. J Am Chem Soc. 1985;107:4333.Grimmett DL, Labinger JA, Bonfiglio JN, Masuo ST, Shearin E, Miller JS. J Am Chem Soc. 1982;104:6858.Lambic NS, Lilly CP, Sommer RD, Ison EA. Organometallics. 2016;35:3060.
- 9.Lewis acids are known to promote migratory insertion reactions to generate metal-acyl complexes and can also stabilize metal-acyl and metal-formyl complexes. Select examples: Butts SB, Holt EM, Strauss SH, Alcock NW, Stimson RE, Shriver DF. J Am Chem Soc. 1979;101:5864.Anderson GDW, Boys OJ, Cowley AR, Green JC, Green MLH, Llewellyn SA, von Beckh CM, Pascu SI, Vei IC. J Organomet Chem. 2004;689:4407.Elowe PR, West NM, Labinger JA, Bercaw JE. Organometallics. 2009;28:6218.
- 10.(a) Miller AJM, Labinger JA, Bercaw JE. J Am Chem Soc. 2008;130:11874. doi: 10.1021/ja805108z. [DOI] [PubMed] [Google Scholar]; (b) Miller AJM, Labinger JA, Bercaw JE. J Am Chem Soc. 2010;132:3301. doi: 10.1021/ja100574n. [DOI] [PubMed] [Google Scholar]
- 11.(a) Lee Y, Peters JC. J Am Chem Soc. 2011;133:4438. doi: 10.1021/ja109678y. [DOI] [PubMed] [Google Scholar]; (b) Suess DLM, Peters JC. J Am Chem Soc. 2013;135:12580. doi: 10.1021/ja406874k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moret ME, Peters JC. Angew Chem Int Ed. 2011;50:2063. doi: 10.1002/anie.201006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bontemps S, Bouhadir G, Dyer PW, Miqueu K, Bourissou D. Inorg Chem. 2007;46:5149. doi: 10.1021/ic7006556. [DOI] [PubMed] [Google Scholar]
- 14.Fong H, Moret ME, Lee Y, Peters JC. Organometallics. 2013;32:3053. doi: 10.1021/om400281v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The dianionic dihydride complex 2 crystallizes as a cluster with four crystallographically distinct iron centers and eight associated potassium countercations (See SI).
- 16.Analysis of the detailed bond metrics of this complex is complicated by the presence of a constitutional disorder, with the trihydride complex 3 cocrystallizing with a ∼10% impurity of the monoanionic hydride complex 4.
- 17.When one equivalent of silyl electrophile is used a new diamagnetic product is observed as the major species. This new complex does not appear to be an intermediate in the formation of 6 (see SI).
- 18.For a discussion of the trend observed relating spin-state and isomer shift in P3B-supported Fe complexes see: Del Castillo TJ, Thompson NB, Peters JC. J Am Chem Soc. 2016;138:5341. doi: 10.1021/jacs.6b01706.
- 19.Anderson JS, Moret ME, Peters JC. J Am Chem Soc. 2013;135:534. doi: 10.1021/ja307714m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The product mixture generated in the conversion of 2 to 6 is highly soluble in nonpolar solvents (e.g. alkanes, hexamethyldisiloxane, tetramethylsilane), even at low temperature. While the mixtures show some insolubility in polar solvents (e.g. MeCN), reaction impurities and 6 exhibit similar solubility properties, precluding further purification of 6.
- 21.Note: Owing to our inability to obtain analytically pure 6, reactions were by necessity carried-out in the presence of remaining iron impurities.
- 22.Treatment of 6 with MeOTf or H-atom sources (e.g. TEMPO-H, Bu3Sn-H) similarly leads to unproductive decomposition of 6 Me3SiOTf did not react with 6 under conditions we have explored.
- 23.The addition of PMe3 to these reactions leads simpler product mixtures, but the major species has not yet been identified (see SI).
- 24.A normal KIE is observed for the reaction of 6 with H2/D2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthetic and spectroscopic details: Crystallographic information: