

Abstract
Collagen deposition is a hallmark of atherosclerosis. Although compromised collagen I degradation has been implied in the pathogenesis of atherosclerosis, the molecular mechanisms are still unclear. Thus, we determined the role of CD38, an enzyme involved in cellular calcium modulation and autophagic flux, in the regulation of collagen I degradation in coronary arterial myocytes (CAMs). In primary cultured CAMs from CD38−/− mice, collagen I protein accumulation but not mRNA abundance was significantly increased compared with cells from CD38+/+ mice either under control or upon TGF-β stimulation. Pharmacological inhibition of the formation of autophagosomes with 3-methyladenine or of autophagolysosomes with a lysosomal functional blocker, bafilomycin A1, induced a similar increase in collagen protein levels, while inhibition of the proteasome by MG132 had no effects on collagen I accumulation. In addition, CD38-deficiency did not change the protein expression of matrix metalloprotein-9 (MMP-9) or tissue inhibitor of metalloproteinase-1 (TIMP-1) in CAMs. Confocal microscopy showed that collagen I deposition was mainly lied within lysosomes or autophagosomes in CD38−/− or TGF-β treated CAMs. Collagen I deposition increased when CAMs lack CD38 expression or if autophagy was blocked, which is associated with impaired autophagic degradation of collagen I. This CD38 regulation of autophagic flux may represent a novel mechanism for extracellular matrix (ECM) plasticity of coronary arteries upon atherogenic stimulation.
Keywords: CD38, Collagen, Autophagy, Coronary Artery, Mouse
2. INTRODUCTION
Vascular fibrosis, characterized by arterial wall thickening and excessive deposition of extracellular matrix (ECM), is one of the hallmarks of cardiovascular diseases such as atherosclerosis, restenosis, and heart failure (1). Collagens are the main component of ECM which contains several subtypes as I, III, IV, V and VI, with type I being most abundant in the vascular systems (2). Excess formation or decreased degradation or both of collagen I may result in increased collagen I deposition, an event observed in the early stage of atherosclerosis (3). Although many studies have been conducted to pursue the molecular mechanisms of collagen I degradation, the molecular mechanisms remain poorly understood.
Cluster of differentiation 38 (CD38) expresses broadly in immune cells, endothelial cells, vascular smooth muscle cells and endothelial progenitor cells which exerts multiple functions mainly by the production and metabolism of cyclic ADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP) (4, 5). The CD38/cADPR pathway contributes to the M1 receptor agonist-induced Ca2+ release from the sarcoplasmic reticulum (SR) in coronary arterial myocytes (CAMs) (6). NAADP has been reported to be the most potent intracellular Ca2+-mobilizing signaling. It has been proved that in arterial SMCs, NAADP mobilizes intracellular Ca2+ through a two-pool mechanism by which NAADP releases Ca2+ from a lysosome-dependent Ca2+ store that in turn serves as a localized signal to trigger Ca2+-induced Ca2+ release leading to global Ca2+ increase in the cytosol (7). It has been reported that Angiotensin II, endothelin-1, and norepinephrine cause vasoconstriction in renal, coronary or pulmonary arteries, depending on CD38 mediated Ca2+ release (8). CD38-mediated Ca2+ mobilization in immunocyte has been reported to contribute to chemokine production, immunocyte infiltration, neutrophil adhesion and chemotaxis associated with cerebral injury after cardiac surgery (9, 10) and temporary ischemia and reperfusion of brain (11). CD38-activated NAD(P)ase depletes endothelial NADPH profoundly inhibiting NO generation and endothelium-dependent relaxation of coronary artery, which contributes to contractile dysfunction and increases infarction of postischemic heart (12). In addition, the CD38 immunopositive hepatic stellate cells (HSCs) were found to be attributed to liver fibrosis in patients with chronic hepatitis (13). Recently, a growing body of evidence have shown that NAADP-sensitive lysosome-derived Ca2+ and consequent increases in cytosolic Ca2+ promoted autophagosome trafficking and fusion with lysosomes, a process named autophagic flux in CAMs, under both normal condition and atherogenic stimulation (14), indicating that autophagic flux derangement may be a contributing factor to atherogenesis.
It has been known that autophagy is a critical cellular homeostatic process through which unnecessary or dysfunctional components of cells are degraded and/or recycled (15). Autophagic process begins with the induction and formation of autophagosome that the cytoplasmic components are sequestrated in double-membrane vesicles. Then, the autophagic flux consisting of autophagosomes trafficking and fusion with lysosomes to form autophagolysosomes and breakdown of autophagic contents in autophagolysosomes would happen (14). With respect to the pathogenic role of autophagic derangement, it has been reported that the kidneys from mice with deficient autophagic protein, beclin 1 exhibited profibrotic phenotype as shown by increased collagen deposition (16, 17). The degradation of collagen by autophagy has also been suggested in alveolar epithelial cells and fibrotic lung tissues, hepeatic stellate cell as well as cardiac fibroblasts during fibrosis (18). Rats treated with iloprost had a striking reduction of the right ventricular collagen deposition, which resulted from increased expression of autophagic genes accounting for collagen degradation (19). TGF-β1 has been shown as an inducer of both collagen I synthesis and autophagy-mediated collagen I degradation, and in TGF-β1 treated cells inhibition of autophagy enhanced the collagen I deposition significantly (16). In fibroblasts, a defective autophagy induced by alterations in the metabolic pathways or pharmacological handling could contribute to the excessive production of extracellular matrix by altering the turnover of collagen (20). Moreover, enhanced autophagy was shown to exert a potent anti-aging influence on the arteries by increasing NO bioavailability, reducing oxidative stress and modifying structural factors (21). Based on the central role of CD38 in regulating lysosomal transport and lysosomal fusion with autophagosome and the role of autophagy defects in fibrosis of various tissues, we hypothesized that the regulation of the autophagic flux by CD38 promotes collagen I degradation in CAMs resulting in cellular dysfunction that finally elicits fibrogenesis during the development of atherosclerosis.
In the present study, several in vivo and in vitro approaches were carried out to test this hypothesis. Our data demonstrate that CD38-mediated signaling pathway mediates an important autophagic regulatory mechanism by which collagen metabolism is fine controlled and that collagen deposition and arterial wall thickening in CD38−/− mice received a high fat Western diet (WD) are associated with a deficient autophagic degradation of collagen in CAMs.
3. MATERIALS AND METHODS
3.1. Animal procedure
Both CD38−/− and wild-type (CD38+/+) mice on C57BL/6 background were purchased from Jackson Laboratory. CD38+/+ and CD38−/− mice (8 weeks of age; male) were fed for 10 weeks with normal diet or Western diet containing (g%): protein 20, carbohydrate 50, and fat 21 (Dytes, Bethlehem, PA, USA), as described previously (22). Mice were sacrificed by cervical dislocation under ether anaesthesia. The hearts with the coronary artery were obtained, dissected or frozen in liquid nitrogen for preparation of frozen section slides. All experimental protocols were reviewed and approved by the Animal Care Committee of Virginia Commonwealth University. All animals were provided standard rodent chow and water ad libitum in a temperature-and humidity controlled room.
3.2. Western blot analysis
Western blot analysis was performed as described previously (14). Primary antibodies used are rabbit anti-collagen 1 (1:1000, Cell Signaling Technology), rabbit anti-LC3B (1:1000, Abcam), rabbit anti- matrix metalloprotein-9 (MMP-9) (1:1000, Abcam), 20S Proteasome α1 Antibody (H-95) (sc-67046, 1:500, Santa Cruz), rabbit anti-tissue inhibitor of metalloproteinase-1 (TIMP-1) (1:1000, Abcam) or goat anti-β-actin (1:500, Santa Cruz).
3.3. Primary cell culture of mouse CAMs and treatments
CAMs were isolated from CD38+/+ or CD38−/− mice as previously described (23) and also discussed in Supplementary material online. The expression of CD38 in CD38+/+ or CD38−/− CAMs was examined by Western blot. Cells were treated with TGF-β1 (20 nM, Sigma) for 24 h.
3.4. Confocal microscopic analysis
For confocal analysis, cultured CAMs were grown on glass coverslips, stimulated as indicated or left unstimulated, fixed in 4% paraformaldehyde in phosphate-buffer saline (PFA/PBS) for 15 min. After permeabilization with 0.1.% Triton X-100/PBS, the cells were rinsed with PBS and incubated overnight at 4°C with indicated primary antibodies: mouse anti-collagen 1 and rabbit anti-Lamp 1 or rabbit anti-LC3B (1:200, Cell Signaling). The samples were washed and incubated with Alexa-488- or Alexa-555-labeled secondary antibodies for 1 h at room temperature. The slides were then mounted and examined using sequentially scanning on a laser scanning confocal microscope (Fluoview FV1000, Olympus, Japan), with photos taken and the co-localization analyzed by the Image Pro Plus 6.0. software (Media Cybernetics, Bethesda, MD, USA). The summarized co-localization efficiency data was expressed as Pearson correlation coefficient (PCC) as described previously (24).
3.5. Statistics
Data are presented as means ± SEM. Significant differences in mean values between- and within-multiple groups were examined using analysis of variance (ANOVA), any significant difference revealed by this procedure were further investigated using Tuckey’s multiple-range test. Student’s t-test was used to detect significant difference between two groups. The statistical analysis was performed by the sigmastat 3.5. software (Systat Software, Chicago, IL, USA). P<0.0.5 was considered statistically significant.
4. RESULTS
4.1. Effects of CD38-deficiency on collagen deposition, atherosclerosis and autophagy in the coronary artery of mice
To test the significance of CD38 in collagen I deposition in vivo, we examined the collagen I content in coronary arterial walls of CD38+/+ and CD38−/− mice. Figure 1A showed that the coronary arterial wall in CD38−/− was significantly thickened by the WD treatment for 10 weeks, while that in CD38+/+ mice was unaffected by WD feeding. As shown in Figure 1B, collagen I staining around coronary arteries was much higher in CD38−/− than in CD38+/+ mice on either ND or WD treatment. The content of LC3B, an autophagosome marker, was also much higher in the coronary arterial wall of CD38−/− than of CD38+/+ mice on either ND or WD treatment (Figure 1C).
Figure 1.
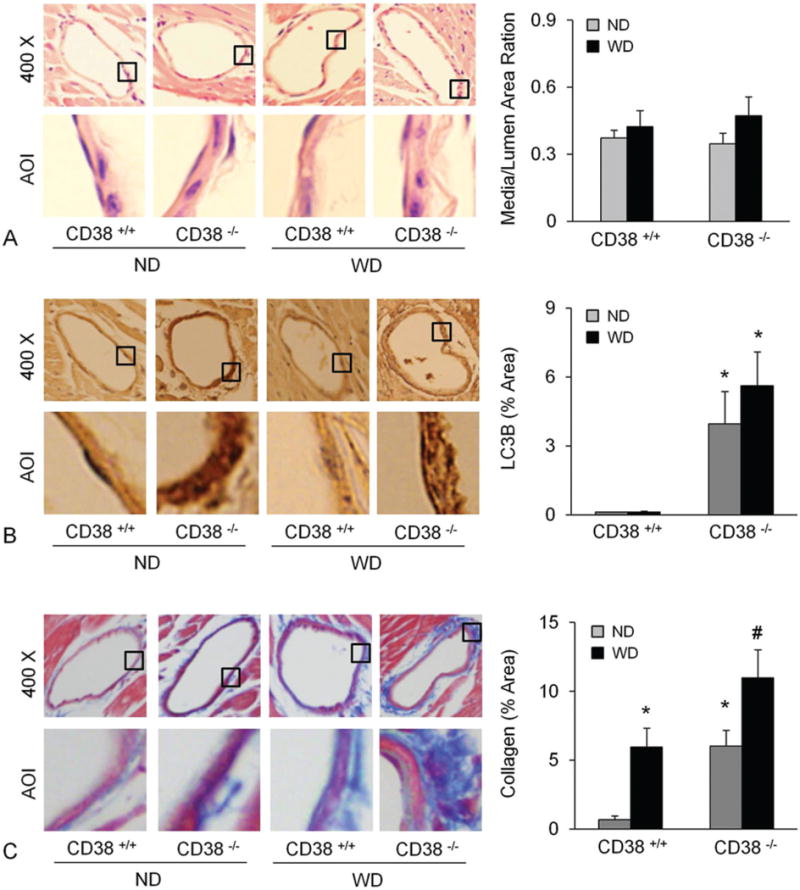
Deficiency of CD38 increased atherosclerosis, autophagy and collagen deposition in the coronary arterial wall. Arterial wall thickness (A and B), LC3B (C and D) and collagen I content (E and F) were determined in mice fed with a normal diet (ND) or a fat-enriched western diet for 10 weeks (WD). * p<0.0.5 vs. CD38+/+ mice.
4.2. Effect of CD38-deficiency on collagen I content in CAMs of CD38−/− and CD38+/+ mice
We next determined how CD38 deficiency affects the gene or protein expression of collagen I in primary cultured CAMs. In CAMs of CD38−/− mice, the protein expression of collagen I increased significantly as compared to that in CD38+/+ mice (Figure 2A,B). However, the mRNA abundance did not show significant difference between CD38+/+ and CD38−/− CAMs (Figure 2C) suggesting a postranscriptional regulation of collagen I by CD38.
Figure 2.
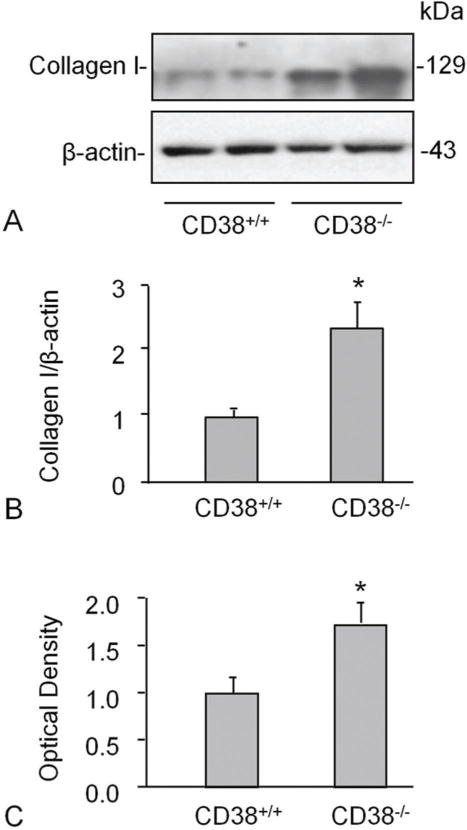
CD38-deficiency increased collagen I level in CAMs. Western blot studies show a higher expression of collagen in CD38−/− than CD38+/+ mouse CAMs. Shown are representative Western blot gel (A) and the quantitative analysis from 4 mice (B). mRNA abundance did not differ between CD38+/+ and CD38−/− CAMs (C) suggesting a postranscriptional regulation of collagen I. * P<0.0.5 vs. CD38+/+ group (n=4 mice).
4.3. Effect of autophagy inhibition on collagen I protein levels
CD38 could increase intracellular Ca2+ level via cADPR and NAADP, which finally leads to autophagy in CAMs. Autophagy has been proved being one of the main pathways of collagen I degradation (9). We therefore tried to Figure out whether autophagy mediates upregulation of collagen I protein in cells lacking CD38. We treated CAMs with 3-methyladenine (3-MA), an inhibitor of autophagosome formation, or bafilomycin (baf), an inhibitor of vacuolar type H+-ATPase (V-ATPase) and demonstrated that both treatments increased the levels of LC3B and collagen I significantly in both CD38+/+ and CD38−/− CAMs, but the former showed a markedly higher response than the latter (Figure 3A and D). The densitometric analysis showed that both 3-MA and Baf increased collagen levels in CAMs (B and E) which was accompanied by increased LC3B levels, an indicative of accumulated autophagosomes.
Figure 3.
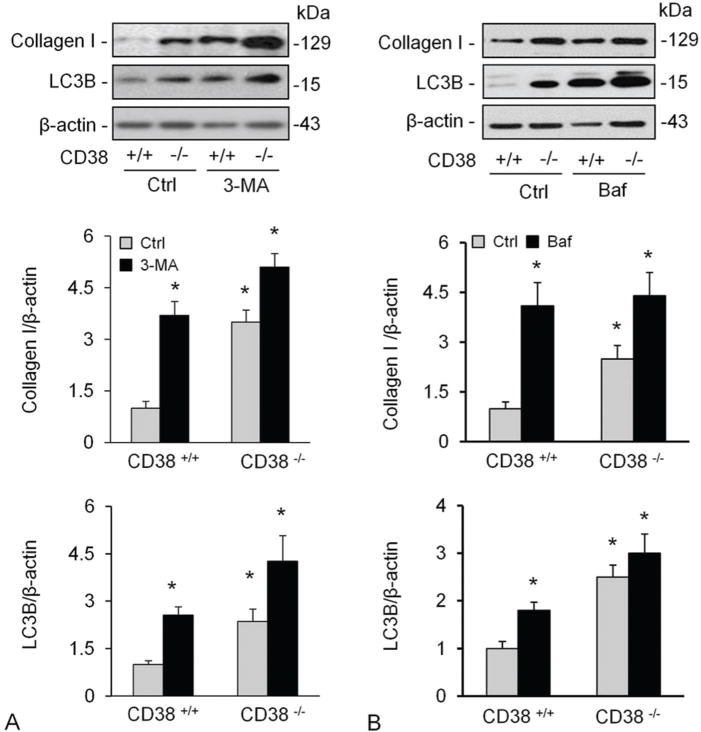
Inhibition of autophagy and lysosome functions elevated collagen I levels in CAMs. Treatment of CAMs with 3-MA (A), an inhibitor of autophagosome formation or bafilomycin (baf) (B), an inhibitor of vacuolar type H+-ATPase (V-ATPase). Shown are representative Western blot gel (A and D) and quantitative analysed data (n=4-5 mice). * P<0.0.5 compared to untreated CD38+/+ samples.
4.4. TGF-β induced more collagen I expression in CD38−/− than in CD38+/+ CAMs
TGF-β is a key regulator of matrix proteins production, which could elicit an increase of collagen I content in smooth muscle cells. After confirming the effect of CD38 on static collagen I production of CAMs, we tried to examine the role of CD38 in stimulation of collagen I production. Figure 4A and 4B shows that TGF-β increased the collagen I protein content significantly which was more pronounced in cells deficient for CD38. TGF-β did not alter mRNA abundance of collagen I (data not shown), again suggesting a posttranslational regulation of collagen I expression. Likewise, baf also increased the level of collagen I protein in both vehicle and TGF-β treated CAMs (Figure 4C and 4D).
Figure 4.
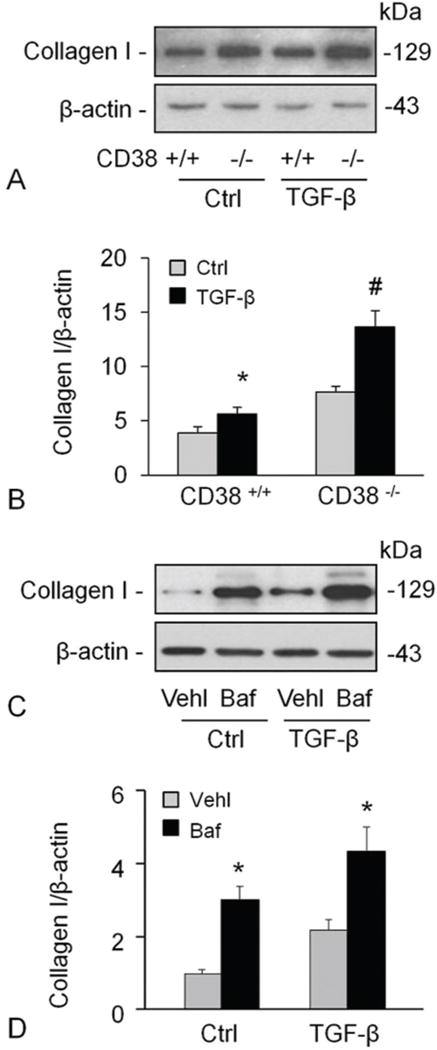
TGF-β-mediated induction of collagen I was promoted by CD38-deficiency or inhibition of lysosomal functions. TGF-β increased the collagen I protein content significantly, which was more markedly in cells with deficiency of CD38 gene (A, B). The effects of TGF-β on collagen I levels were enhanced by inhibition of lysosomal functions using Baf (C, D). Shown are representative Western blot gel (A and C) and the densitometric analysis from studies with CAMs from 4 mice (B and D). * P<0.0.5 compared to untreated CD38+/+ samples, # P<0.0.5 compared to TGF-β treated CD38+/+ samples.
4.5. Proteasome inhibition had no effect on collagen I accumulation of CAMs
Ubiquitin-proteasome system is another major nonlysosomal pathway for intracellular degradation of proteins. To analyze the function of the ubiquitin-proteasome system for collagen I deposition, we treated CAMs with MG132, a specific, potent proteasome inhibitor. However, MG132 treatment did not change the collagen I content in vehicle or TGF-β stimulated CAMs, but it indeed inhibited TGF-β-induced proteasome marker upregulation (Figure 5A–B). We also tested the effects of MMP inhibitor, batimastat (Bat) (ApexBio, 3.0. μg/ml pretreatment for 30 mins) on proteasome and collagen degradation and found that Bat had no effect on both proteasome and collagen-I level under control condition or during treatment of cells with TGF-β (Figure 5D–F).
Figure 5.
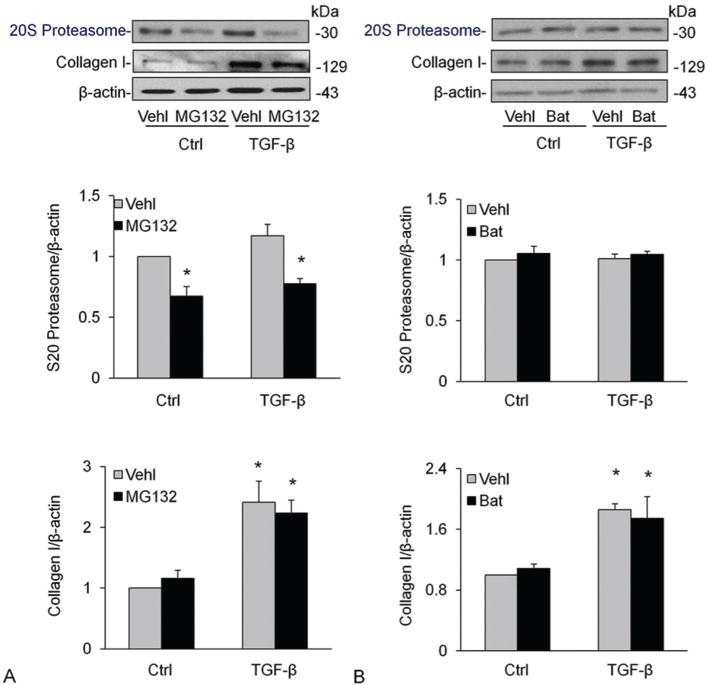
Effects of proteasome or MMP inhibition on proteasome and collagen I levels in CAMs. Treatment of CAMs with MG132, a specific, potent proteasome inhibitor (A-C) and MMP inhibitor, Bat (D-F) did not change the collagen I content in vehicle or TGF-β stimulated CAMs. Shown are representative Western blot gels (A and D) and the data from densitometric analysis (B, C, E, F) (n=4). * P<0.0.5 compared to vehicle-treated samples.
4.6. Effect of CD38-deficiency on MMP-9 or TIMP-1 of CAMs
MMPs are involved in the breakdown of ECM in physiological as well as in pathological processes. MMP-9/TIMP-1 has been previously reported to be involved in collagen I degradation (16). Thus, we tested whether CD38 expression regulates MMP and TIMP-1 expression in CAMs. However, TGF-β did not change the expression of MMP-9 or TIMP-1 in CAMs significantly and there was no remarked difference between CD38+/+ or CD38−/− CAMs on either basal or TGF-β stimulated expression of MMP-9 or TIMP-1 (Figure 6).
Figure 6.
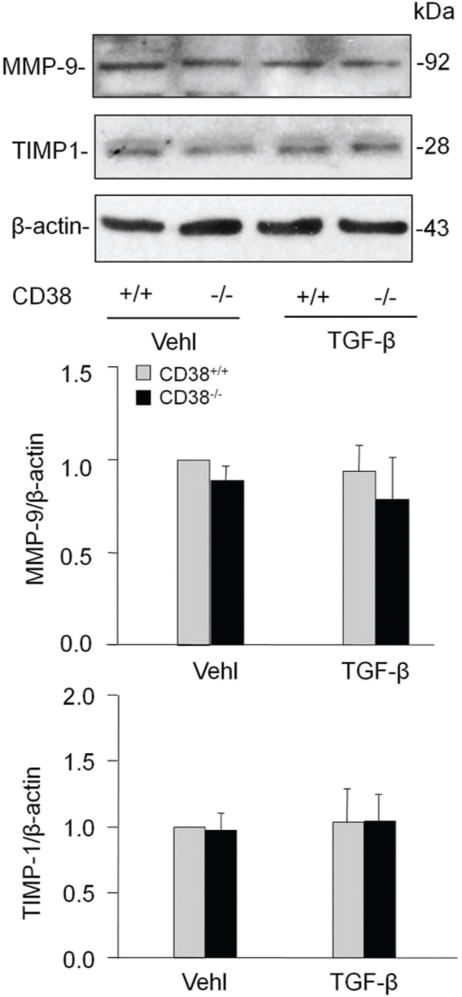
Effects of CD38 deficiency on matrix metalloprotein-9 (MMP-9) or tissue inhibitor of metalloproteinase-1 (TIMP-1) in CAMs. CD38 gene knockout did not significantly change the level of both MMP and TIMP-1 in CAMs with either vehicle or TGF-β treatment. Shown are representative Western blot gels (A) and the data from densitometric analysis (C and D) (n=4).
4.7. Increased collagen I accumulation mainly localizes to lysosomes and autophagosomes of CD38−/− CAMs
To clarify the location of collagen I accumulation, we examined whether collagen I co-localizes with Lamp1 or LC3B, specific markers of lysosomes and autophagosomes, respectively. The confocal microscopic analysis showed the co-localization of collagen I with either Lamp1 (Figure 7A and B) or LC3B (Figure 7C and D) which was unchanged by CD38 gene deficiency in control CAM. Upon TGF-β treatment, however, the colocalization of collagen I with either Lamp1 or LC3B increased significantly, which was further enhanced in CD38−/− CAMs.
Figure 7.
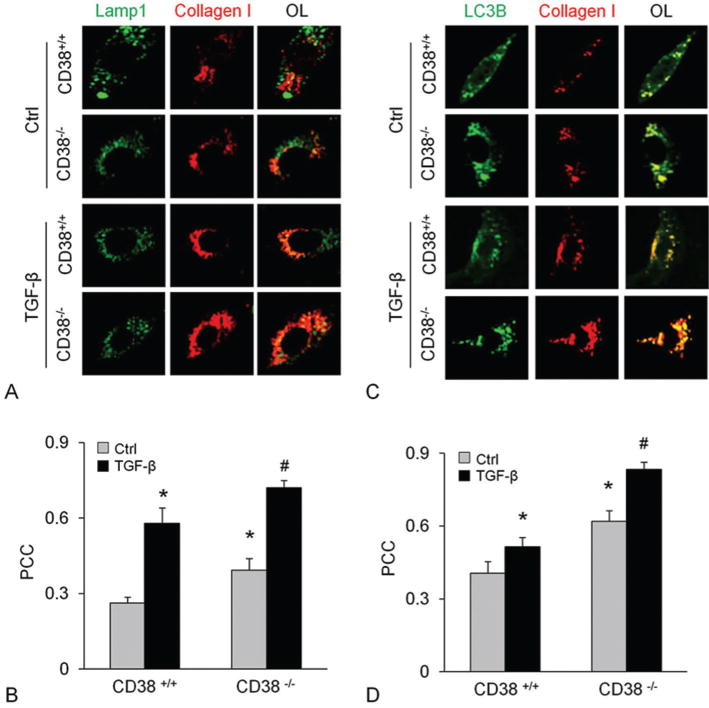
CD38-deficieny resulted in collagen I accumulation in lysosomes with autophagosomes in CAMs. Immunostainings of CAMs from CD38+/+ or CD38−/− cells for Lamp1 or LC3B with collagen showed enhanced colocalization of collagen I with Lamp1 (A and B) and LC3B (C and D) before and after TGF-β stimulation. Displayed are representative confocal microscopic images (A and C) and the quantitative analysis of colocalization coefficient in overlaid image (OL) (n=5) * P<0.0.5 compared to untreated CD38+/+ samples, # P<0.0.5 compared to TGF-β treated CD38+/+ samples.
5. DISCUSSION
The present study demonstrated that CD38-deficiency blocks collagen I degradation resulting in its deposition in CAMs. The accumulation of collagen I in CD38-deficient CAMs is mediated by an inhibition of autophagy in these cells.
Atherosclerosis starts with an inflammatory process that is stimulated by an increased uptake of LDL particles in the intimal space of the vessel wall. Various cytokines and growth factors then act on smooth muscle cells to proliferate and migrate the cell as well as produce ECM proteins including collagen (25). The formation of collagen is normally balanced by its degradation through mechanisms including autophagy (16), proteosome (26) and MMP-1 (27), the compromise of any of which might cause more collagen deposition. The present study demonstrates that CD38-deficiency enhanced collagen I deposition and atherosclerosis, in particular when the mice were fed with a fat-enriched WD. Simultaneously, autophagy was blocked with more LC3B present in Lamp1-positive vesicles. These results imply that under atherogenic stimulation the loss of CD38-expression causes a deregulation of autophagolysosome-formation reducing collagen I degradation and leading to deposition of collagen I around the coronary artery.
Our data further confirm that the accumulation of collagen I in CAMs of CD38−/− mice was due to impaired degradation, but not increased production of it. Using western blot and sirius red staining which is a specific morphologic method to detect different subtypes of collagen I (28), we proved the protein content of collagen I to be increased significantly in CD38−/− CAMs. Employing real time PCR, we found the mRNA abundance did not change when the CD38 gene was knocked out suggesting a posttranslational regulation of collagen I degradation by CD38. As mentioned above, CD38 gene products can induce Ca2+ mobilization in immunocytes contributing to cerebral injury after cardiac surgery, extracorporeal circulation, stoke, and temporary ischemia and reperfusion in the brain. Moreover, the CD38 immunopositive hepatic stellate cells (HSCs) were found correlated to fibrosis scores in patients with chronic hepatitis. Our data indicates CD38 expression in CAM may possess high capability to reduce the fibrogenesis and prevent the fibrosis of artery as compared to immunocytes and fibrocytes.
CD38-ADP-ribosylcyclase mediates signaling pathways, in particular the regulation of intracellular Ca2+ signaling, that have critical functions in the physiology and pathophysiology of coronary arteries (7, 29). The intracellular transport and fusion of lysosomes with autophagosome requires an increased cytosolic Ca2+-concentration (30, 31), a process that is regulated by CD38 in CAMs (14). Since autophagy mediates collagen I degradation not only in CAMs, but also in mesenchymal cell (32), hepeatic stellate cell (33), mesangial cell (16) and cardiac cell (34), we explored whether CD38-deficiency inhibits collagen I degradation through a defect in autophagy. We used 3-MA to block the formation of autophagosomes or baf to induce lysosomal dysfunction and demonstrated that the two inhibitors increased the protein content of collagen I in both CD38+/+ and CD38−/− CAMs with the extent much higher in the former. This indicates that CD38 at least partly regulates the collagen degradation by a suppression of autophagy.
In primary mouse mesangial cells, TGF-β acts as a potent inducer of fibrosis which promotes synthesis of collagen I. At the same time, it could also boost the autophagy mediated collagen I degradation. In TGF-β1 treated cell, inhibition of autophagy enhanced the collagen I deposition significantly (16, 35). We therefore tried to testify the effect of CD38-deficiency on TGF-β induced collagen I production and degradation in CAMs. Our results showed both CD38-deficiency and lysosomal dysfunction induced by baf increased collagen I protein content of CAMs. However, the increased collagen I mRNA level evoked by TGF-β did not change in CD38−/− CAMs (data not shown). These results suggested that CD38-deficiency or an inhibition of autophagy did not affect the synthesis of collagen I, but blocked its degradation resulting in the deposition of collagen I in CAMs.
Apart from autophagy, the ubiquitin-proteasome system and MMPs are two major non-lysosomal pathways for intracellular degradation of proteins (16, 36). According to recent studies on mechanisms of cardiovascular diseases (26), the ubiquitin-proteasome system is up-regulated by atherogenic stimuli promoting inflammation in endothelial and smooth muscle cells and thereby aggravating atherosclerosis damage. Thus, proteasome inhibition may attenuate collagen synthesis upon atherogenic stimulation and, therefore targeting the proteasome might be a new promising therapeutic strategy to prevent fibrosis and progression of organ failure (37, 38). However, cellular treatment with MG132, a specific inhibitor of ubiquitin-proteasome pathway (37, 38), did not induce any significant changes of collagen I protein content in both vehicle and TGF-β stimulated CAMs suggesting that the ubiquitin-proteasome pathway may not play a major role in collagen I deposition of CAMs. TGF-β treatment may also lead to kidney fibrosis via inhibition of MMPs in mesangial cell (16). However, we did not find any effect of CD38-deficiency on protein abundance of MMP-9 or TIMP-1, the endogenous inhibitor of MMP-9, and inhibition of MMP activity also had no effect on the collagen level in CAMs with or without treatment of TGF-β. These results indicate that in CAMs MMPs may not be a main pathway for collagen I degradation.
Finally, we employed morphological techniques to examine the distribution of the collagen I deposit inside CAMs. The results showed a significant co-localization of collagen I with either Lamp1 or LC3B strongly supporting the notion that CD38 regulates collagen I degradation through autophagy. This result is consistent with the report from Kim et al. who proved in primary cultured mouse kidney mesangial cells, TGF-β1 promoted collagen I protein expression was further enhanced by the treatment of baf. Under this condition, colocalization of collagen I with LC3B and Lamp1was also increased (16).
In summary, CD38 regulated collagen I degradation through the autophagic pathway. CD38-deficiency resulted in collagen I-deposition in CAMs mainly in lysosomes and autophagosomes. This process may result in coronary arterial fibrosis, a critical feature of atherosclerosis. Our data propose a novel way the degradation of collagen I is regulated which provide novel targets to prevent excessive collagen deposition in CAMs, a key feature of multiple pathological situations.
Supplementary Material
Acknowledgments
Jun-Xiang Bao, Qiu-Fang Zhang equally contributed as the first authors to this publication. This work was supported by the National Institutes of Health Grants (HL057244, HL075316, and HL091464 to P.-L.L.; HL122937 and HL122769 to Y.Z.).
References
- 1.Lan TH, Huang XQ, Tan HM. Vascular fibrosis in atherosclerosis. Cardiovasc Pathol. 2013;22(5):401–7. doi: 10.1016/j.carpath.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Rocnik EF, Chan BM, Pickering JG. Evidence for a role of collagen synthesis in arterial smooth muscle cell migration. J Clin Invest. 1998;101(9):1889–98. doi: 10.1172/JCI1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adiguzel E, Ahmad PJ, Franco C, Bendeck MP. Collagens in the progression and complications of atherosclerosis. Vasc Med. 2009;14(1):73–89. doi: 10.1177/1358863X08094801. [DOI] [PubMed] [Google Scholar]
- 4.Teggatz EG, Zhang G, Zhang AY, Yi F, Li N, Zou AP, Li PL. Role of cyclic ADP-ribose in Ca2+-induced Ca2+ release and vasoconstriction in small renal arteries. Microvasc Res. 2005;70(1–2):65–75. doi: 10.1016/j.mvr.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Lee HC. Structure and enzymatic functions of human CD38. Mol Med. 2006;12(11–12):317–23. doi: 10.2119/2006-00086.Lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ge ZD, Zhang DX, Chen YF, Yi FX, Zou AP, Campbell WB, Li PL. Cyclic ADP-ribose contributes to contraction and Ca2+ release by M1 muscarinic receptor activation in coronary arterial smooth muscle. J Vasc Res. 2003;40(1):28–36. doi: 10.1159/000068936. [DOI] [PubMed] [Google Scholar]
- 7.Zhang F, Zhang G, Zhang AY, Koeberl MJ, Wallander E, Li PL. Production of NAADP and its role in Ca2+ mobilization associated with lysosomes in coronary arterial myocytes. Am J Physiol Heart Circ Physiol. 2006;291(1):H274–82. doi: 10.1152/ajpheart.01064.2005. [DOI] [PubMed] [Google Scholar]
- 8.Thai TL, Arendshorst WJ. Mice lacking the ADP ribosyl cyclase CD38 exhibit attenuated renal vasoconstriction to angiotensin II, endothelin-1, and norepinephrine. Am J Physiol Renal Physiol. 2009;297(1):F169–76. doi: 10.1152/ajprenal.00079.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moroz VV, Salmina AB, Fursov AA, Mikhutkina SV, Linev KY, Mantorova NS, Shakhmaeva SV. Role of ADP-ribosyl cyclase in the pathogenesis of neurological disorders after coronary artery bypass surgery and experimental ischemia. Bull Exp Biol Med. 2009;147(5):570–2. doi: 10.1007/s10517-009-0580-5. [DOI] [PubMed] [Google Scholar]
- 10.Kolackova M, Kudlova MT, Lonsky V, Mand’ak J, Kunea P, Jankovicova K, Vlaskova D, Andrys C, Krejsek J. The expression of CD38 ADP-ribosyl cyclase ectoenzyme in immune cells of cardiac surgical patients. Acta Medica (Hradec Kralove) 2008;51(1):31–5. [PubMed] [Google Scholar]
- 11.Choe CU, Lardong K, Gelderblom M, Ludewig P, Leypoldt F, Koch-Nolte F, Gerloff C, Magnus T. CD38 exacerbates focal cytokine production, postischemic inflammation and brain injury after focal cerebral ischemia. PLoS One. 2011;6(5):e19046. doi: 10.1371/journal.pone.0019046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reyes LA, Boslett J, Varadharaj S, De Pascali F, Hemann C, Druhan LJ, Ambrosio G, El-Mahdy M, Zweier JL. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc Natl Acad Sci U S A. 2015;112(37):11648–53. doi: 10.1073/pnas.1505556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdeen SM, Olusi SO, Askar HA, Thalib L, Al-Azemi A, George S. The predictive value of CD38 positive hepatic stellate cell count for assessing disease activity and fibrosis in patients with chronic hepatitis. Acta Histochem. 2009;111(6):520–30. doi: 10.1016/j.acthis.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Xu M, Xia M, Li X, Boini KM, Wang M, Gulbins E, Ratz PH, Li PL. Defective autophagosome trafficking contributes to impaired autophagic flux in coronary arterial myocytes lacking CD38 gene. Cardiovasc Res. 2014;102(1):68–78. doi: 10.1093/cvr/cvu011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryter SW, Lee SJ, Smith A, Choi AM. Autophagy in vascular disease. Proc Am Thorac Soc. 2010;7(1):40–7. doi: 10.1513/pats.200909-100JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J Biol Chem. 2012;287(15):11677–88. doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He L, Livingston MJ, Dong Z. Autophagy in acute kidney injury and repair. Nephron Clin Pract. 2014;127(1–4):56–60. doi: 10.1159/000363677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Del Principe D, Lista P, Malorni W, Giammarioli AM. Fibroblast autophagy in fibrotic disorders. J Pathol. 2013;229(2):208–20. doi: 10.1002/path.4115. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Arroyo J, Sakagami M, Syed AA, Farkas L, Van Tassell B, Kraskauskas D, Mizuno S, Abbate A, Bogaard HJ, Byron PR, Voelkel NF. Iloprost reverses established fibrosis in experimental right ventricular failure. Eur Respir J. 2015;45(2):449–62. doi: 10.1183/09031936.00188013. [DOI] [PubMed] [Google Scholar]
- 20.Del Principe D, Vona R, Giordani L, Straface E, Giammarioli AM. Defective autophagy in fibroblasts may contribute to fibrogenesis in autoimmune processes. Curr Pharm Des. 2011;17(35):3878–87. doi: 10.2174/138161211798357791. [DOI] [PubMed] [Google Scholar]
- 21.LaRocca TJ, Gioscia-Ryan RA, Hearon CM, Jr, Seals DR. The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev. 2013;134(7–8):314–20. doi: 10.1016/j.mad.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bjursell M, Gerdin AK, Lelliott CJ, Egecioglu E, Elmgren A, Tornell J, Oscarsson J, Bohlooly YM. Acutely reduced locomotor activity is a major contributor to Western diet-induced obesity in mice. Am J Physiol Endocrinol Metab. 2008;294(2):E251–60. doi: 10.1152/ajpendo.00401.2007. [DOI] [PubMed] [Google Scholar]
- 23.Xu M, Zhang Y, Xia M, Li XX, Ritter JK, Zhang F, Li PL. NAD(P)H oxidase-dependent intracellular and extracellular O2*- production in coronary arterial myocytes from CD38 knockout mice. Free Radic Biol Med. 2012;52(2):357–65. doi: 10.1016/j.freeradbiomed.2011.10.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia M, Zhang C, Boini KM, Thacker AM, Li PL. Membrane raft-lysosome redox signalling platforms in coronary endothelial dysfunction induced by adipokine visfatin. Cardiovasc Res. 2011;89(2):401–9. doi: 10.1093/cvr/cvq286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strom A, Ahlqvist E, Franzen A, Heinegard D, Hultgardh-Nilsson A. Extracellular matrix components in atherosclerotic arteries of Apo E/LDL receptor deficient mice: an immunohistochemical study. Histol Histopathol. 2004;19(2):337–47. doi: 10.14670/HH-19.337. [DOI] [PubMed] [Google Scholar]
- 26.Marfella R, D’Amico M, Di Filippo C, Baldi A, Siniscalchi M, Sasso FC, Portoghese M, Carbonara O, Crescenzi B, Sangiuolo P, Nicoletti GF, Rossiello R, Ferraraccio F, Cacciapuoti F, Verza M, Coppola L, Rossi F, Paolisso G. Increased activity of the ubiquitin-proteasome system in patients with symptomatic carotid disease is associated with enhanced inflammation and may destabilize the atherosclerotic plaque: effects of rosiglitazone treatment. J Am Coll Cardiol. 2006;47(12):2444–55. doi: 10.1016/j.jacc.2006.01.073. [DOI] [PubMed] [Google Scholar]
- 27.McNulty M, Mahmud A, Spiers P, Feely J. Collagen type-I degradation is related to arterial stiffness in hypertensive and normotensive subjects. J Hum Hypertens. 2006;20(11):867–73. doi: 10.1038/sj.jhh.1002015. [DOI] [PubMed] [Google Scholar]
- 28.Veidal SS, Vassiliadis E, Bay-Jensen AC, Tougas G, Vainer B, Karsdal MA. Procollagen type I N-terminal propeptide (PINP) is a marker for fibrogenesis in bile duct ligation-induced fibrosis in rats. Fibrogenesis Tissue Repair. 2010;3(1):5. doi: 10.1186/1755-1536-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yue J, Wei W, Lam CM, Zhao YJ, Dong M, Zhang LR, Zhang LH, Lee HC. CD38/cADPR/Ca2+ pathway promotes cell proliferation and delays nerve growth factor-induced differentiation in PC12 cells. J Biol Chem. 2009;284(43):29335–42. doi: 10.1074/jbc.M109.049767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim HJ, Soyombo AA, Tjon-Kon-Sang S, So I, Muallem S. The Ca(2+) channel TRPML3 regulates membrane trafficking and autophagy. Traffic. 2009;10(8):1157–67. doi: 10.1111/j.1600-0854.2009.00924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirnikjoo B, Balasubramanian K, Schroit AJ. Mobilization of lysosomal calcium regulates the externalization of phosphatidylserine during apoptosis. J Biol Chem. 2009;284(11):6918–23. doi: 10.1074/jbc.M805288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hilscher M, Hernandez-Gea V, Friedman SL. Autophagy and mesenchymal cell fibrogenesis. Biochim Biophys Acta. 2012;1831(7):972–8. doi: 10.1016/j.bbadis.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thoen LF, Guimaraes EL, Grunsven LA. Autophagy: a new player in hepatic stellate cell activation. Autophagy. 2012;8(1):126–8. doi: 10.4161/auto.8.1.18105. [DOI] [PubMed] [Google Scholar]
- 34.Aranguiz-Urroz P, Canales J, Copaja M, Troncoso R, Vicencio JM, Carrillo C, Lara H, Lavandero S, Diaz-Araya G. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim Biophys Acta. 2011;1812(1):23–31. doi: 10.1016/j.bbadis.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Chin BY, Mohsenin A, Li SX, Choi AM, Choi ME. Stimulation of pro-alpha(1)(I) collagen by TGF-beta(1) in mesangial cells: role of the p38 MAPK pathway. Am J Physiol Renal Physiol. 2001;280(3):F495–504. doi: 10.1152/ajprenal.2001.280.3.F495. [DOI] [PubMed] [Google Scholar]
- 36.Marfella R, M DA, Di Filippo C, Siniscalchi M, Sasso FC, Ferraraccio F, Rossi F, Paolisso G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc Diabetol. 2007;6:35. doi: 10.1186/1475-2840-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goffin L, Seguin-Estevez Q, Alvarez M, Reith W, Chizzolini C. Transcriptional regulation of matrix metalloproteinase-1 and collagen 1A2 explains the anti-fibrotic effect exerted by proteasome inhibition in human dermal fibroblasts. Arthritis Res Ther. 2010;12(2):R73. doi: 10.1186/ar2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma Y, Chen Y, Yang Y, Chen B, Liu D, Xiong Z, Zhang C, Dong Y. Proteasome inhibition attenuates heart failure during the late stages of pressure overload through alterations in collagen expression. Biochem Pharmacol. 2013;85(2):223–33. doi: 10.1016/j.bcp.2012.10.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.