

Abstract
KRas4b is a small G-protein whose constitutively active oncogenic mutants are present in 90% of pancreatic cancers. Using fully post-translationally modified KRAS4b, we investigated the role of lipid identity in the recruitment of KRas4b to a membrane surface of defined composition. Application of a newly developed single frequency fluorescence anisotropy decay experiment to this system revealed that KRas4b has a significant binding preference for Nanodisc bilayers containing PIP2. We conducted molecular dynamics simulations to look for an origin of this specificity. In the case of membranes containing PIP2 the protein formed long-lived salt bridges with PIP2 head groups but not the monovalent DMPS, explaining the experimentally observed lipid specificity. Additionally, we report that PIP2 forms key contacts with Helix-4 on the catalytic domain of KRas4b that orient the protein in a manner expected to facilitate association with upstream and downstream signaling partners.
Keywords: KRas4b, Nanodisc, Cancer signaling, Lipid specificity, PIP2
Graphical abstract
Introduction
Ras proteins constitute an integral element of signal transduction where extracellular growth factors control various nuclear transcription events involved in cell division, proliferation, and apoptosis.[1]. The isoform KRas4b is mutated in 90% of pancreatic and 45% of colorectal adenocarcinomas [2], and mutants that activate KRas4b are considered “drivers” of the cancerous state. Intervention that either prevents KRas4b activation or returns the signaling cascade to its normal form could significantly improve cancer outcomes.[3] Ras activity is controlled, and signaling mediated by, critical protein-protein interactions. GTPase activating proteins (GAPs), such as p120, bind to activated Ras, dramatically increasing the rate of GTP hydrolysis thus returning the system to the inactive GDP bound state.[4] Guanine exchange factors (GEFs), such as Son of Sevenless (SOS), bind and effect the exchange of GDP for GTP, thus turning “on” KRas4b.[5] Most importantly, these multi-protein complexes all operate on a membrane surface which is a critical partner in signaling.[6]
KRas4b approaches and associates with the membrane surface via electrostatic interactions with the lysine rich hyper-variable region (HVR) and the catalytic domain, as well as through burial of the hydrophobic farnesyl anchor at the methylated C-terminus.[7] Despite the critical role of the membrane, there is incomplete knowledge as to the role of the bilayer composition in anchoring the protein to the membrane and the importance of specific lipid type in dictating the final orientation of KRas4b on the surface. To help address these lacunas, we use the Nanodisc system [8] and a new label-free means to quantitate the binding free energy of KRas4b to bilayers of defined composition. We use long-time molecular dynamics simulations to understand the approach of KRas4b to the membrane surface, identifying specific residues on the protein that interact with lipid components and determine the role of specific head group type in determining the final orientation. Here we utilized the Highly Mobile Membrane Mimetic (HMMM) model [9], implemented for broad distribution as a CHARMM-GUI plug-in [10], as it permits rapid calculation of the lateral lipid head group motion, allowing KRas4b to sample a greater number of possible protein/lipid contacts than would be possible in an all (lipid) atom simulation.
Materials and Methods
Materials
Phospholipids 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine (DMPS) and brain phosphatidylinositol 4,5 bisphosphate (PIP2) were purchased from Avanti Polar Lipids. Bis(2,2′-bipyridine)-4′methyl-4-carboxybipyridine-ruthenium N-succinimidyl ester was purchased from Sigma-Aldrich. Membrane scaffold proteins were expressed in E. coli BL21 DE3 and purified as previously described in detail.[11] The methods of Nanodisc self-assembly have been extensively documented in the literature.[12–14] Fully processed KRAS4b (farnesylated and methylated) was generously provided by Frederick National Laboratory for Cancer Research (FNLCR). Production methods for this protein have been recently described.[15]
Methods
MSP1D1 was labeled with a Ru(bpy)2(mcbpy)-NHS fluorophore in buffer containing 100 mM potassium phosphate and 50 mM NaCl at a pH of 7.4 at room temperature. A five-fold molar excess of dye dissolved in dimethyl sulfoxide was added dropwise to the MSP1D1 solution and stirred for 2 hours, at which point the reaction was quenched by addition of powdered Tris base to the solution. Excess dye was removed by gel filtration and the MSP concentration was quantified using “660 nm Protein Assay Reagent” (Pierce) using unlabeled MSP1D1 as a standard.
Fluorescence lifetime measurements were performed on an ISS (Urbana, IL) K2 phase fluorimeter using laser diode excitation at 486 nm. A long-pass 594 nm emission filter (Semrock) was positioned in the fluorimeter and frequency domain measurements were recorded by modulating the laser from 75 kHz to 6 MHz. In order to improve signal-to-noise, and reduce data acquisition times, the modulation frequency giving the highest signal-to-noise ratio is determined by performing multi-frequency anisotropy decay measurements during a complete titration of KRas4b binding to Nanodiscs. We followed the change in phase of the fluorescence emission at a single frequency of 262.3 kHz where binding of KRas4b to the disc manifests the largest signal. Each experiment contained 100 nM of Nanodiscs in a buffer containing 20mM HEPES, 150 mM NaCl, 5 mM MgCl2, and 1 mM EGTA at pH 7.3. The temperature controlled sample cell was maintained at 20°C with stirring. The difference in phase angle between horizontally and vertically polarized emission, ΔΦ, is used to monitor binding and was obtained as the average of at least three individual measurements with the signal averaged for 10s. The change in ΔΦ, ΔΔΦ, is proportional to the percent bound and is analyzed by the standard quadratic tight binding isotherm.
Molecular dynamics simulations were performed on the University of Illinois Campus Cluster, predominantly on the MCBSS node with 4 Graphic Processors and 28 CPUs. The crystal structure of GDP bound G12C KRas4B (pdb 4OBE) was used to generate full length KRas4b. The hypervariable region amino acids (residues 170 to 185) were added to the C-terminus and placed in a helical conformation based on secondary structure predictions [16], and the cysteine at position 12 was replaced with the wild-type glycine residue. Full Length KRas4b was solvated and equilibrated for 20 ns using NAMD 2.11 molecular Dynamics software package [17] and was later found to be nearly identical to the recently published full-length structure of KRas4b.[18]
Equilibrated KRas4B was farnesylated and placed 10 Å above a bilayer containing 120 lipids per leaflet. The lipid tails of DMPC, DMPS and PIP2 were modeled according to the HMMM.[9] The system was solvated and KCl added to a final concentration of 150 mM. The final system contained ~100,000 atoms and was energy minimized for 10,000 steps with fixed heavy atoms followed by three 25 ps simulations and then three 100 ps simulations while gradually reducing the harmonic constraints on the system at each step. Production runs were performed using NAMD 2.11 with an NPAT ensemble and the CHARMM36 force field.[17] Time steps were 2 fs and the particle mesh Ewald algorithm was used to calculate electrostatic forces.[19] The temperature was maintained at 303.15 K using Langevin dynamics with a coupling constant of 1 ps−1. Pressure was maintained at 1 bar using The Nose-Hoover Langevin Piston method using a piston period of 50 fs and a decay of 25 fs.[20,21] HMMM lipids were restrained using a weak harmonic potential of 0.05 kcal/mol/Å2 applied to the carbonyl carbons. Trajectory visualization and analysis was performed using the VMD software package.[22]
Results and Discussion
Binding of KRas4b to Nanodisc Membranes
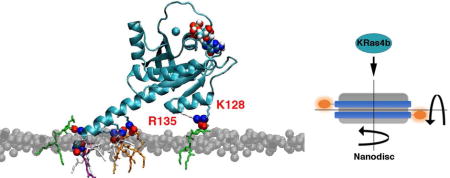
To realize a solution based assay to quantitate binding of KRas4b to the membrane without direct labeling of the target protein, we conjugated a long-lifetime Ru(bpy)2(mcbpy) fluorophore to the encircling membrane scaffold belt of the Nanodisc via N-hydroxysuccinimide chemistry (Figure 1, inset) and monitored the rotational motions of the Nanodisc. We used frequency domain anisotropy decay measurements to observe even small changes in the rotational correlation times of the Nanodisc as KRas4b molecules bind. At 20°C, the averaged rotational correlation time of DMPC Nanodiscs, in the absence of KRas4b, was measured to be ~70 nsec, which compares favorably to measurements obtained by NMR.[23,24] As KRas4b is added, a second, slower rotation becomes evident corresponding to formation of the KRas4b:Nanodisc complex. Titrations were performed using Nanodiscs of varying percentages of DMPC, and DMPS and PIP2. The dissociation constants derived from these experiments are summarized in Figure 1. The total formal charge on the Nanodisc is indicated in parentheses and corresponds to 77 lipids/leaflet and a charge of −1 for DMPS and −4 for PIP2 since the pH of the solution was held at 7.3. Clearly, the affinity of fully processed KRas4b for the membrane is significantly affected by changes in anionic lipid content. In the case of 50% DMPS, the measured dissociation constants decreased by approximately 2.8-fold compared to DMPC and is consistent with a recently published report using surface plasmon resonance. [15]
Figure 1.
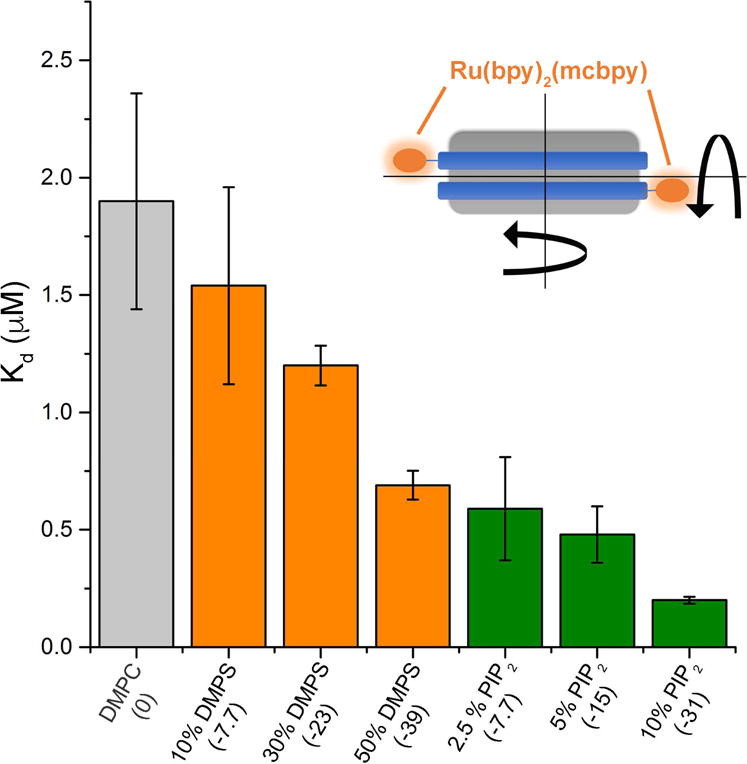
Dissociation constants of KRas4b when binding to Ru(bpy)2(mcbpy) labeled Nanodiscs of differing anionic lipid concentrations. The total formal bilayer charge is labeled in parenthesis. Error bars represent ± 1 standard deviation. Inset: Diagram of a Nanodisc with fluorescent label on each of the encircling membrane scaffold proteins.
Surprisingly, KRas4b bound substantially more tightly to membranes that included PIP2. Nanodiscs containing 2.5% PIP2 were found to bind as tightly to KRas4b as 50% DMPS Nanodiscs. At the highest concentration tested, 10% PIP2, the dissociation constant decreased nearly 10-fold relative to DMPC, despite having a less negative overall surface charge than 50% DMPS. Thus, we conclude that the affinity of KRas4b to anionic membranes is not solely determined by the total localized charge. Rather, our results are consistent with an important lipid specificity that favors multi-factorial contacts between protein and head-group such as that provided by PIP2.
Origin of Lipid Specificity in the Binding and Orientation of KRas4b on the Membrane Surface
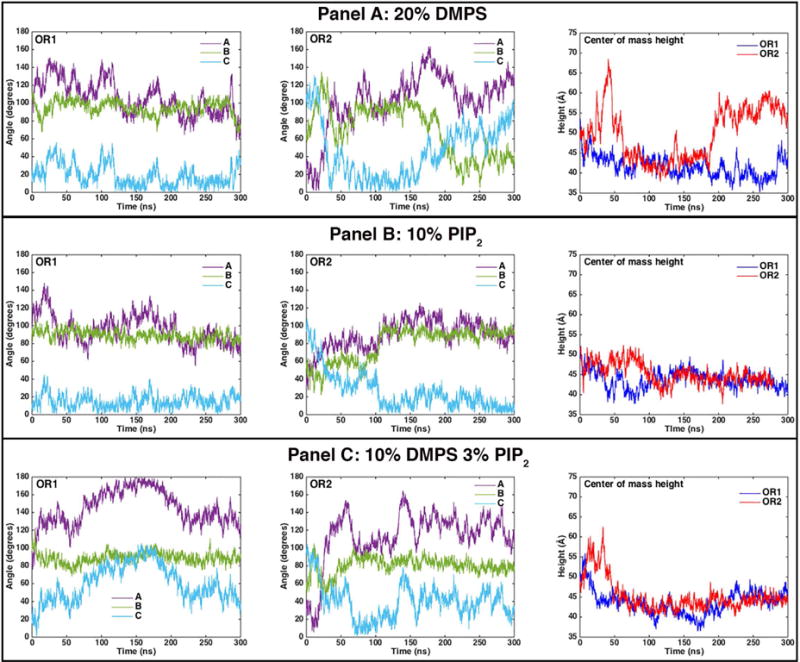
In order to understand the origins of the PIP2 specificity, we conducted longtime molecular dynamics simulations of the approach of KRas4b to a membrane surface and the burying of the hydrophobic farnesyl tail into the core of the bilayer. We first examined the approach of KRas4b to the membrane with different starting orientations of the protein relative to membrane bilayer (Figure 2). Orientation 1 (OR1) places the α-Helices 4 and 5 towards the membrane surface while Orientation 2 (OR2) is rotated 90 degrees about the axis of Helix-5 (HVR domain). To monitor changes in the orientation of KRas4b, we define the orthogonal vectors, A, B, and C between the carbonyl oxygen of L79, which lies near the center of mass of the catalytic domain, and three backbone atoms lying on or near the x axis (R97 alpha-carbon), y axis (F82 carbon), and z axis (T58 carbon) respectively. We report the angle of the vectors A, B, and C with respect to the bilayer normal (Figure 2, top panel) as a measure of the orientation of KRas4b relative to the membrane. In addition, as an indication of proximity, we measure the center-of-mass distance of the catalytic domain (CD), defined as residues 1 through 165, above the center of the bilayer. In all cases when the CD is in contact with the surface, the center-of-mass lies between 40 and 45 Å above the center of the bilayer. The simulations for membranes containing 20% DMPS, 10% PIP2, and a mixed system containing 10% DMPS and 3% PIP2 in a DMPC background, are described individually.
Figure 2.
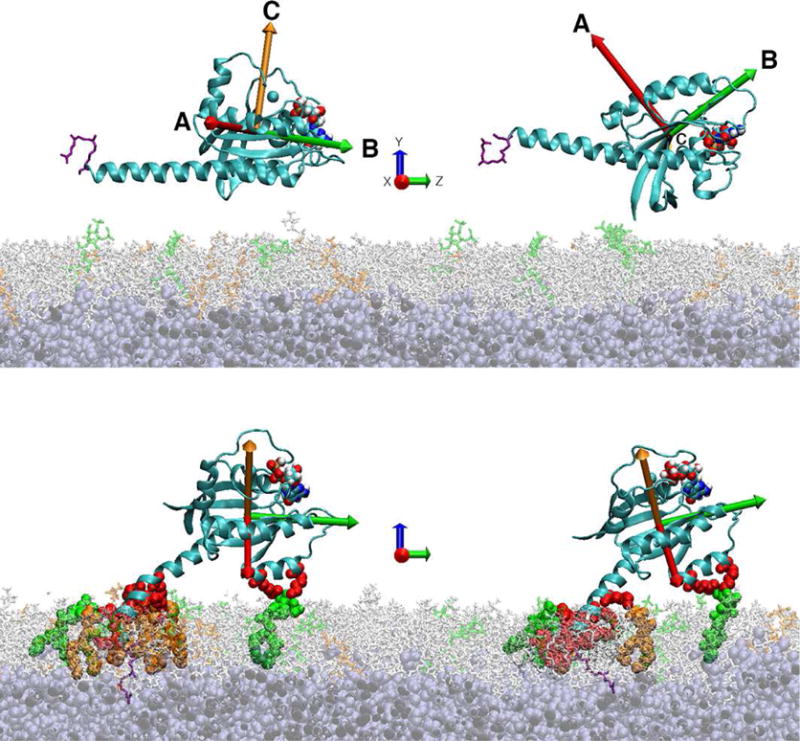
The top panel shows the starting orientations of KRas4b used in the simulations. Vectors A, B, and C are the axes used to define the protein orientation with respect to the membrane normal. The bottom panel shows the nearly equivalent final orientations of KRas4b on the membrane despite dramatically different starting orientations. The lipid bilayer contained 10% DMPS and 3% PIP2. PIP2 lipids are colored green, DMPS lipids are colored orange, DMPC is light gray, and the HMMM core is dark gray. Red colored residues show the formation of salt bridges with lipid head groups.
Simulations with a 20% DMPS Membrane
In simulations containing 20% DMPS and starting in OR1 (Figure 3, Panel A), we see that the orientation of the catalytic domain (CD) with respect to the bilayer rotates about vector B, giving rise to a decrease in the angle of vector A with the bilayer normal. Within the first 50 ns the catalytic domain approaches the surface and makes contact with the bilayer. Even though the KRas4b is associated with the membrane at 300 ns, the C-terminal farnesyl group remains exposed to solvent. In contrast, in OR2 the catalytic domain initially diffuses away from the surface and flips its orientation with respect to the bilayer before coming into contact with the membrane. Interestingly, at 150 ns, the orientation of the KRas4b catalytic domain is nearly identical to that seen at 300 ns of the OR1 simulation. This is followed by the dissociation of the CD from the surface and results in the KRas4b standing on end as evidenced by the large distance between the CD center-of-mass and the membrane.
Figure 3.
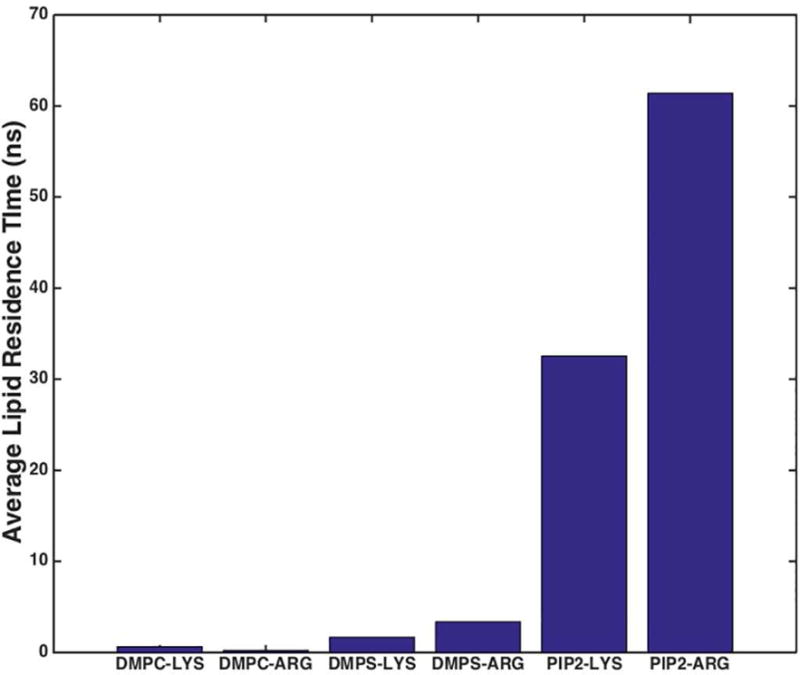
The angles of the orientation vectors A, B, and C with respect to the bilayer normal are shown as violet, green and light blue, respectively. Simulations run from 0 to 300 ns, with 20% DMPS (Panel A), 10% PIP2 (Panel B), and 10% DMPS + 3% PIP2 membranes (Panel C) for the starting orientations OR1 and OR2. The rightmost figure in each panel shows the distance of the KRas4b center-of-mass from the bilayer center for OR1 (dark blue) and OR2 (red) for each membrane composition. (See text)
Simulations with a 10% PIP2 Membrane
The approach and binding of KRas4b to bilayers containing 10% PIP2 differ from that seen in 20% DMPS bilayers. Both orientations bind the surface within the first 50 – 100 ns, and the KRas4b in the OR2 simulation once again flips over, resulting in both simulations having similar configurations to that of OR1 on 20% DMPS (Figure 3, Panel B). A significant difference is the absence of large-scale fluctuations in both orientation angle and the center-of-mass distance as compared to the DMPS simulations, which displayed a substantial amount of rotational mobility. This is due to a large number of long-lived salt bridges forming between PIP2 and the basic residues of the hypervariable region and the CD.
Simulations with a Mixed Bilayer 10% DMPS 3% PIP2
We also simulated a mixed bilayer system containing 10% DMPS and 3% PIP2 (Figure 3, Panel C). As observed in the previous simulations, membrane binding and reorientation occurs in the first 100 ns. These simulations converge to a nearly identical final configuration at 300 ns where the farnesyl group is buried within the hydrophobic core of the bilayer and stable salt bridges are formed between PIP2 and the basic residues of both the HVR and catalytic domains (Figure 2, bottom). A video of these simulations, showing the trajectories of both OR1 and OR2, is available in the supplemental material.
Importantly, in the final orientation stabilized by PIP2 contacts with cationic residues on Helix-4, the entire KRas4b molecule is in close proximity to the membrane with the GTP binding site oriented above the surface. In this position, the catalytic site is poised to interact with both upstream and downstream effector proteins as evidenced in the co-crystal structures of soluble Ras in the presence of SOS, p120, and the Ras Binding Domain of Raf Kinase.[4,5,25] In all cases access to their respective binding sites appears unencumbered by the membrane.
Lipid Residence Times: the Role of PIP2
Perhaps the most striking difference between the DMPS and PIP2 simulations is the substantial difference in residence time of lipid head groups interacting with cationic residues on the KRas4b protein. As seen in the bar graph (Figure 4), DMPS lipids make only transient contacts with the protein. These short-lived interactions persist an average of 2.2 ns, after which the monovalent salt bridge is broken and the lipid diffuses away. In sharp contrast, the PIP2:protein interactions are much more robust. In these simulations, formation of salt-bridges, defined as a nitrogen/oxygen distance of < 3.2 Å, resulted in long-lived contacts lasting on average 40 ns. The ability of PIP2 to form bifurcated salt bridges allows transition from one residue to another (eg. Lys-Arg or Arg-Arg) in a polybasic region without dissociation from the protein, thus promoting increased residence times. The PIP2:KRas4b interactions primarily occur in the highly charged HVR region, but are also observed between lysine and arginine residues in Helix-4 in the CD. In Figure 2 (bottom panel), these latter charge/charge interactions are clearly observed where a single PIP2 lipid forms a stable bifurcated contact with Lys128 and Arg135 that persists for hundreds of nanoseconds. These long-lived interactions are sufficient to explain the experimentally observed increase in affinity on PIP2 membranes.
Figure 4.
Contacts between a specific lipid and KRas4b amino acid type as a function of simulation time. Clearly evident is longer lived interactions with PIP2 verses DMPS.
Supplementary Material
Highlights.
New label-free method quantitates KRas4b binding to membranes
KRas4b binds preferentially to multi-factor anionic lipids
Molecular dynamics simulation visualizes the interaction of phospholipid head groups
Approach of KRas4b to the membrane, association and final conformation are visualized
Acknowledgments
This research was supported by a MIRA grant from the National Institutes of Health R35 GM118145. We especially acknowledge the partnership with the NCI RAS Initiative at the Frederick National Laboratory for Cancer Research under the guidance of Dr. Frank McCormick and Dr. David Heimbrook, including Drs. Andrew Stephen, William K Gillette, Dominic Espositio, and Carissa Grose. We also thank Drs. Randy Gapud, J-P Denson, Cheryl Habrukowich, Peter Frank, Shelley Perkins, Matt Drew at FNLCR who provided all of the KRas4b protein used in this work. We thank Dr. Ilia Denisov (University of Illinois) for numerous discussions and helpful suggestions.
Abbreviations
- CD
Catalytic Domain
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DMPS
1,2-dimyristoyl-sn-glycero-3-phospho-L-serine
- GAP
GTPase activating proteins
- GDP
Guanosine diphosphate
- GEF
Guanine exchange factor
- GTP
Guanosine triphosphate
- HMMM
Highly Mobile Membrane Mimetic
- HVR
Hypervariable Region
- MSP
Membrane Scaffold Protein
- PIP2
phosphatidylinositol 4,5 bisphosphate
- Ru(bpy)2(mcbpy)
Bis(2,2′-bipyridine)-4′methyl-4-carboxybipyridine-ruthenium
- SOS
Son of Sevenless
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: “it ain”t over “til it”s over’. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 2.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 3.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging Ras Back in the Ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 4.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, Wittinghofer A. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 5.Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature. 1998;394:337–343. doi: 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- 6.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 7.Jang H, Abraham SJ, Chavan TS, Hitchinson B, Khavrutskii L, Tarasova NI, Nussinov R, Gaponenko V. Mechanisms of Membrane Binding of Small GTPase K-Ras4B Farnesylated Hypervariable Region. J Biol Chem. 2015;290:9465–9477. doi: 10.1074/jbc.M114.620724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed Self-Assembly of Monodisperse Phospholipid Bilayer Nanodiscs with Controlled Size. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 9.Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA, Tajkhorshid E. Accelerating Membrane Insertion of Peripheral Proteins with a Novel Membrane Mimetic Model. Biophys J. 2012;102:2130–2139. doi: 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi Y, Cheng X, Lee J, Vermaas JV, Pogorelov TV, Tajkhorshid E, Park S, Klauda JB, Im W. CHARMM-GUI HMMM Builder for Membrane Simulations with the Highly Mobile Membrane-Mimetic Model. Biophys J. 2015;109:2012–2022. doi: 10.1016/j.bpj.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bayburt TH, Grinkova YV, Sligar SG. Self-Assembly of Discoidal Phospholipid Bilayer Nanoparticles with Membrane Scaffold Proteins. Nano Lett. 2002;2:853–856. [Google Scholar]
- 12.Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. Febs Lett. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG. Reconstitution of membrane proteins in phospholipid bilayer Nanodiscs. Methods Enzym. 2009;464:211–231. doi: 10.1016/S0076-6879(09)64011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luthra A, Gregory M, Grinkova YV, Denisov IG, Sligar SG. Nanodiscs in the studies of membrane-bound cytochrome P450 enzymes. Methods Mol Biol. 2013;987:115–127. doi: 10.1007/978-1-62703-321-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillette WK, Esposito D, Abreu Blanco M, Alexander P, Bindu L, Bittner C, Chertov O, Frank PH, Grose C, Jones JE, Meng Z, Perkins S, Van Q, Ghirlando R, Fivash M, Nissley DV, McCormick F, Holderfield M, Stephen AG. Farnesylated and methylated KRAS4b: high yield production of protein suitable for biophysical studies of prenylated protein-lipid interactions. Sci Rep. 2015;5:15916. doi: 10.1038/srep15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dharmaiah S, Bindu L, Tran TH, Gillette WK, Frank PH, Ghirlando R, Nissley DV, Esposito D, McCormick F, Stephen AG, Simanshu DK. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc Natl Acad Sci U S A. 2016;113:E6766–E6775. doi: 10.1073/pnas.1615316113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darden T, York D, Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 20.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. 1994;101:4177–4189. [Google Scholar]
- 21.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 22.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 23.Hagn F, Etzkorn M, Raschle T, Wagner G. Optimized Phospholipid Bilayer Nanodiscs Facilitate High- Resolution Structure Determination of Membrane Proteins. J Am Chem Soc. 2013;135:1919–1925. doi: 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puthenveetil R, Vinogradova O. Optimization of the design and preparation of nanoscale phospholipid bilayers for its application to solution NMR. Proteins Struct Funct Bioinforma. 2013;81:1222–1231. doi: 10.1002/prot.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter JC, Gurbani D, Ficarro SB, Carrasco Ma, Lim SM, Choi HG, Xie T, Marto Ja, Chen Z, Gray NS, Westover KD. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci U S A. 2014;111:8895–8900. doi: 10.1073/pnas.1404639111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.