

Abstract
Murine double minute 2 (Mdm2) is known to enhance the transactivation potential of human immunodeficiency virus (HIV-1) Tat protein by causing its ubiquitination. However, the regulation of Mdm2 during HIV-1 infection and its implications for viral replication have not been well studied. Here, we show that the Mdm2 protein level increases during HIV-1 infection and this effect is mediated by HIV-1 Tat protein. Tat appears to stabilise Mdm2 at the post-translational level by inducing its phosphorylation at serine-166 position through AKT. Although p53 is one of the key players for Mdm2 induction, Tat-mediated stabilisation of Mdm2 appears to be independent of p53. Moreover, the non-phosphorylatable mutant of Mdm2 (S166A) fails to interact with Tat and shows decreased half-life in the presence of Tat compared with wild-type Mdm2. Furthermore, the non-phosphorylatable mutant of Mdm2 (S166A) is unable to support HIV-1 replication. Thus, HIV-1 Tat appears to stabilise Mdm2, which in turn enhances Tat-mediated viral replication. This study highlights the importance of post-translational modifications of host cellular factors in HIV-1 replication and pathogenesis.
Keywords: HIV-1 replication, Mdm2, phosphorylation, Tat
Introduction
Human immunodeficiency virus (HIV-1) is known to cause acquired immunodeficiency syndrome (AIDS) by depleting the CD4+ T cell repertoire [1–4]. This virus depends completely on the host cell for its propagation. Apart from the structural genes, gag, pol and env, HIV-1 encodes two regulatory genes (Tat and Rev) and four accessory genes (Vpu, Vpr, Vif and Nef), which regulate viral infectivity and modulate pathogenesis [5]. HIV-1 exploits various cellular processes through either up-regulation or down-regulation of key cellular proteins for its propagation [6,7]. These modulations may be at the transcriptional, translational or post-translational level. HIV-1 Nef down-regulates CD4 and MHC proteins to evade innate immune recognition and to prevent superinfection. Similarly, HIV-1 Vif down-regulates APOBEC3G, Vpu down-regulates tetherin and Vpr degrades SAMHD1. In addition, HIV-1 Tat recruits cellular factors to increase the rate of viral transcription, and Rev is involved in the nuclear export of viral transcripts [8–12]. Similarly, cellular proteins also modulate viral pathogenesis [13–15]. For example, p53 and murine double minute 2 (Mdm2) contribute significantly to HIV-1 pathogenesis and are also manipulated by HIV-1 during its life cycle. Although Mdm2 is known to enhance Tat-mediated long terminal repeat (LTR) activation, its regulation has not yet been explored [16,17].
The Mdm2 (or Hdm2 in humans) gene was initially discovered in the spontaneously transformed mouse BALB/c cell line (3T3-DM), in which its expression was up-regulated 50-fold [18]. The over-expression of the oncogene Mdm2 can immortalise rodent primary fibroblasts and lead to the transformation of cultured cells [19]. Mdm2 serves diverse purposes; its up-regulation promotes survival of cells, whereas its down-regulation or inhibition can lead to apoptosis [20]. Like other proteins, Mdm2 is subject to post-translational modifications that may influence its functions; for example, phosphorylation by C-Abl at tyrosine-394 blocks the ability of Mdm2 to down-regulate p53 levels [21]. By contrast, S166 and S186 phosphorylation of Mdm2 positively influences its functions by stabilising it and promoting its nuclear localisation [22,23]. Under basal conditions, Mdm2 mediates p53 ubiquitination followed by its proteasomal degradation, whereas under genotoxic stress, ATM phosphorylates Mdm2 at the S395-position, which facilitates its interaction with p53 mRNA. This interaction stimulates p53 mRNA translation and suppresses Mdm2-E3 ligase activity for p53 [24,25]. In addition, p53 is known to be phosphorylated at other positions, which can either inhibit the Mdm2/p53 interaction or diminish Mdm2-E3 ligase activity for p53 [26,27]. DNA damage responses activate ATM (ataxia-telangiectasia mutated)/ATR (ATM-and Rad3-Related) and DNA-PK (DNA-dependent protein kinase), which leads to the phosphorylation of p53. This phosphorylation either stabilises p53 or inhibits its interaction with Mdm2. The ATM/ATR pathway participates in the activation of cell cycle checkpoints induced by DNA damage, whereas DNA-PK primarily acts during DNA repair [28]. DNA-PK-mediated phosphorylation of p53 at S15 disrupts the Mdm2-p53 complex in vivo and in vitro. An in vitro assay using DNA-PK shows phosphorylation of p53 at S15 and S37, which interferes with the ability of Mdm2 to inhibit p53 transactivation. These phosphorylation events are known to cause conformational changes in p53, which leads to p53 stabilisation during DNA damage [29,30]. ATM phosphorylates p53 at S15 in response to DNA damage, whereas ATR phosphorylates p53 at S15 and S37 during genotoxic stress [31–33]. Studies in severe combined immunodeficiency (SCID) mice with defective DNA-PK show that the cells are still able to induce p53 and undergo G1 arrest, suggesting that ATM and ATR kinases are mainly involved in the p53 S15 phosphorylation [34]. Similarly, casein kinase 1-mediated phosphorylation at S6, S9 and T18 and checkpoint kinase 1/2-mediated phosphorylation at S20 leads to p53 stabilisation [26,27]. RYBP has been reported to stabilise both p53 and Mdm2, but it inhibits Mdm2-mediated p53 ubiquitination [35]. Mdm2 also performs p53-independent functions, such as cell cycle control, cellular differentiation, cell fate determination, DNA repair and transcription [36].
Mdm2 has also been reported to facilitate the transcriptional activity of various viral genes and is known to be manipulated by viruses according to their requirements. In human papillomavirus infection, Mdm2 interacts with transcriptional regulator protein E2 and synergistically activates the type-16 promoter [37]. Epstein Barr virus (EBV) nuclear antigen 3C (EBNA3C) forms a trimeric complex with p53 and Mdm2, which enhances the ubiquitin ligase activity of Mdm2 for p53. This process favours the transformation and proliferation of EBV-infected cells [38]. Specific targeting of Mdm2 by simian virus 40 (SV40) prevents Mdm2-mediated degradation of p53 in SV40-transformed cells, and Mdm2 itself is known to be stabilised during this process [39,40]. Influenza A virus uses NS1 protein to activate the PI-3-kinase (PI3K)/AKT pathway, thereby blocking apoptosis to ensure the production of viral progeny and nuclear protein (NP1) in order to induce p53-dependent apoptosis [41,42]. Viral interferon regulatory factor 4 (vIRF4), in Kaposi's sarcoma-associated herpes virus (KHSV), deregulates Mdm2 to suppress p53 and escape cell cycle arrest and other immune responses [43].
In addition, Mdm2 appears to play a key role in HIV-1 pathogenesis through enhancing HIV-1 LTR activity by ubiquitinating Tat [16]. HIV-1 Tat and p53, on the other hand, show reciprocal modulation; p53 acts as a potent suppressor of Tat function while Tat protein inhibits the promoter activity of p53 [44]. Furthermore, Tat protein inhibits SIRT1, which results in the activation of the p53 pathway and a subsequent increase in the expression of genes that are a target of p53, namely, p21 and BAX [45]. Some reports also suggest a positive role for SIRT1 in HIV-1 replication through deacetylation of Tat [46]. Furthermore, Mdm2 has been reported to promote Vif degradation, thereby influencing the levels of APOBEC3G [47]. None of these studies, however, have addressed the mechanistic basis for the effect of Mdm2 levels on HIV-1 replication. Since Mdm2 is reported to have both positive and negative effects on HIV-1 pathogenesis, we were interested to study the regulation of Mdm2 during HIV-1 infection as well as the viral proteins involved in this process. Here, we report that HIV-1 Tat increased Mdm2 protein levels by stabilising it, thus creating a positive feedback loop between Tat and Mdm2. The Mdm2 S166 phospho-form increased after HIV-1 infection and the same was observed in Tat-transfected cells. Earlier reports have mainly suggested that Mdm2 functions only as a Tat transactivator, but here, we report for the first time that post-translational modification of Mdm2 by Tat is also important for HIV-1 replication.
Materials and methods
Cell culture and transfection
HEK-293T (Human Embryonic Kidney 293T cells), MCF-7 (Human breast adenocarcinoma cell line, p53 Wt), H1299 (non-small cell lung carcinoma, p53 null), HeLa (Human cervical adenocarcinoma cell line) were maintained in Dulbecco's Modified Eagle's Medium (DMEM; Himedia Laboratories) supplemented with 10% fetal bovine serum (Gibco, Invitrogen), 100 units penicillin, 0.1 mg streptomycin and 0.25 µg amphotericin B per ml at 37°C in the presence of 5% CO2 in a humidified incubator. MOLT-3 (T-lymphoblastoid cell line, p53 Wt), MOLT-3 p53 knockdown cells (MOLT-3 p53 kd), Jurkat E6.1 (T-lymphocyte) were cultured in RPMI (Himedia Laboratories) supplemented with 10% fetal bovine serum (Gibco, Invitrogen), 100 units penicillin, 0.1 mg streptomycin and 0.25 µg amphotericin B per ml at 37°C in the presence of 5% CO2 in a humidified incubator. Transfections were carried out using Lipofectamine 2000 (Invitrogen, U.S.A.) and Polyethyleneimine, Linear (MW 25 000, Polysciences Inc., U.S.A.) reagents using the manufacturers’ protocols.
shRNA-mediated p53 knockdaown
In total, 2 × 106 MOLT-3 cells were seeded in a 100 mm dish. After 24 h, these cells were transfected with either pBabe-U6-Puro plasmid expressing scrambled shRNA (5′-TTCTCCGAACGTGTCACGT-3′) or p53-specific shRNA (5′-GACTCCAGTGGTAATCTAC-3′) (20 µg/100 mm) using Lipofectamine 2000 (Invitrogen). Transfection was carried out as per the manufacturer's protocol. The cells were then grown in puromycin-rich media (5 µg/ml) for 24 h to select the shRNA (scrambled or p53-specific shRNA) expressing cells. The antibiotic selection was performed twice in order to obtain the pure population of puromycin-resistant cells. shRNA scrambled and p53-specific shRNA plasmids were provided by Dr. Sanjeev Das, NII, India [48].
siRNA-mediated knockdown of Mdm2
For Mdm2 knockdown, the siRNA duplex was designed and custom synthesised from Dharmacon, U.S.A. Mdm2-specific siRNA (5′-CGUACGCGGAAUACUUCGATT-3′) and control siRNA corresponding to luciferase (5′-CCACCUCACAGAUUCCAGCTT-3′) was obtained. HeLa and HEK-293T cells were transfected after every 24 h using Lipofectamine (Invitrogen) with 100 nM of siRNA and evaluated for Mdm2 protein levels. After 48 h of siRNA transfection, cells were transfected with the plasmid of interest using Lipofectamine (Invitrogen).
Plasmid constructs, proviral DNAs and recombinant proteins
Plasmid constructs (pCMV-Myc-Tat, pCMV-Myc-Rev, pCMV-Myc-Nef, pCMV-Myc-Vif, pCMV-Myc-Vpu and pCMV-Myc-Vpr) were made by cloning pNL4-3-derived genes in the pCMV-Myc plasmid from Clontech U.S.A., as described earlier [49]. pcDNA3.1, pcDNA3.1-Tat-HA, pBR322 SV40, pEBG and p-CMV-Myc3-Mdm2 were purchased from Addgene. pEGFP-Mdm2 Wt and pEGFP-Mdm2 S166A were a kind gift from Dr. Brian Hemmings, Friedrich Miescher-Institut, Switzerland. Renilla luciferase plasmid was a kind gift from Dr. Vivek Natrajan, IGIB, Delhi, India. pBabe-U6-shRNA (scrambled) puro and pBabe-U6-p53 shRNA puro were a kind gift from Dr. Sanjeev Das, National Institute of Immunology, New Delhi, India. HPV-18 construct was a kind gift from Dr. Lutz Gissmann, DKFZ, Heidelberg, Germany. HIV-1 Tat recombinant protein (Cat#2222) was obtained from the NIH-AIDS reagent programme.
Western blot analysis
HEK-293T, MCF-7, H1299 and HeLa cells were transfected with plasmids expressing the gene of interest for 36 h, then the cells were harvested and lysed in RIPA (Radioimmunoprecipitation assay) lysis buffer (1% NP-40, 20 mM TrisCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% sodium deoxycholate, 1 mM Na3VO4; pH 7.5,). MOLT-3 and Jurkat cells were similarly processed after infection. Protein estimation was carried out using BCA Protein Assay Kit (Pierce, Thermo Scientific, U.S.A.). An equal amount of protein was loaded on SDS-PAGE and was transferred to nitrocellulose membrane. The membranes were blocked with 5% non-fat dry milk (Himedia Laboratories, India) in 1× PBS (phosphate buffered saline; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) and washed three times with 1X PBS containing 0.1% Tween 20 (PBST). Membranes were then incubated with primary antibody at 4°C overnight, washed with 1× PBST and probed with horseradish peroxide (HRP)-conjugated secondary antibody. Blots were developed using ECL (Enhanced Chemiluminescence) reagent. The primary antibodies were anti-p53, anti-Mdm2, anti-Tubulin, anti-Tat, anti-Vif, anti-Rev (Santa Cruz Biotechnology), anti-S166 phosphorylated Mdm2, anti-S15 phosphorylated p53, anti-GAPDH (Cell Signalling Technology), anti-Myc, anti-HA (Clontech), anti-p24 (Cat No. 6457, NIH), anti-Vpu, anti-Vpr, anti-Nef (NIH AIDS Reagent programme, U.S.A.), anti-laminB2 (Proteintech group, Inc.), anti-SV40 VP1 (Abcam), and anti-HPV-18 E7 antibodies (Santa Cruz biotechnology). The secondary antibodies used were anti-rabbit/mouse/goat-HRP conjugated antibodies (Jackson Immuno Research).
Cycloheximide chase assay
To study the degradation kinetics of proteins, cycloheximide chase assay was performed. HEK-293T and H1299 cells were transfected with either 2 µg p-CMV-Myc3-Mdm2 alone or in presence of Myc-Tat for 36 h and then treated with cyclohexamide (CHX) (100 µg/ml; Sigma). Cell lysates were prepared at indicated time points, resolved on 10% SDS-PAGE followed by western blot analysis, as described above.
Real-time polymerase chain reaction (RT-PCR)
Real-time quantitative polymerase chain reaction (RT-PCR) is commonly used to detect mRNA expression level in the cell. MCF-7 cells were transfected with the desired plasmid for 36 h. Total RNA was isolated using Trizol reagent as described by the manufacturer's protocol (Invitrogen) and reverse transcribed to form complementary DNA (cDNA) using a cDNA synthesis kit (Promega). 1 µg of RNA was mixed with random primers and incubated at 70°C for 15 min and kept at 4°C. Reverse transcriptase was added to a reverse transcription mix containing 1× reaction buffer, MgCl2, dNTPs and rRNasin and it was incubated at 25°C for 5 min, 42°C for 1 h, 70°C for 15 min and then kept at 4°C. This cDNA was used for SYBR green-based RT-qPCR amplification of the Mdm2 gene using Mdm2 forward primer 5′-AAGCCTGGCTCTGTGGTAA-3′) and Mdm2 reverse primer (5′-TACACCAGCATCAAGATCCG-3′). Human β-actin was used as the control and amplified using the forward primer 5′-AGGCACCAGGGCGTGAT-3′, and the reverse primer 5′-GCCCACATAGGAATCCTTCTGAC-3′.
In vivo ubiquitination assay
In vivo ubiquitination assay was performed to detect ubiquitinated proteins in transfected mammalian cells. HEK-293T cells were grown in 35 mm dishes and co-transfected with 2 µg of pCMV-Myc3-Mdm2 and 1 µg His-Ub (6× Histidine-ubiquitin) for 36 h. After 36 h of transfection, 25 µM of MG132 (Sigma-Aldrich) was added and the cells were further incubated for 8 h. Thereafter, the cells were collected in 1× PBS and resuspended in 1 ml of lysis buffer (6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 10 mM imidazole; pH 8.0), sonicated, and centrifuged. Ni-NTA beads (50 µl) were added to the supernatant and the mixture was incubated at room temperature for 6 h while rotating. Subsequently, the beads were washed for 5 min at room temperature with 750 µl of each of the following buffers: buffer A (6 M guanidium-HCl, 0.1 M Na2HPO4/NaH2P04, 10 mM imidazole; pH 8.0); buffer TI (25 mM Tris, 20 mM imidazole; pH 6.8). After the last wash, ubiquitinated proteins were eluted by incubating the beads in 75 µl of buffer containing 200 mM imidazole, 5% SDS, 0.15 M Tris, 30% glycerol and 0.72 M β-mercaptoethanol at pH 6.7 for 20 min at room temperature. The eluates were mixed in a 1:1 ratio with 2× Laemmli buffer and resolved by SDS-PAGE followed by western blot analysis with the indicated antibodies.
Preparation of nuclear and cytoplasmic extracts
Nuclear and cytoplasmic extraction was performed using NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific). MCF-7 cells were transfected with the plasmids of interest and cells were harvested in 1× PBS after 36 h of transfection. Cells were centrifuged and ice cold CER I was added to the cell pellet, which was vortexed vigorously for 15 s in order to prepare the cell suspension and incubated on ice for 10 min. Ice-cold CER II was then added and vortexed for 5 s and incubated on ice for 1 min. It was again vortexed for 5 s and centrifuged for 5 min at 4°C. The supernatant was immediately transferred (cytoplasmic extract) to a clean pre-chilled tube and stored at −80°C. The insoluble (pellet) fraction containing nuclei was suspended in ice-cold NER and vortexed for 15 s; it was incubated on ice with vortexing for 15 s every 10 min, for a total of 40 min and centrifuged for 10 min at 4°C. The supernatant was immediately transferred (nuclear extract) to a clean pre-chilled tube at −80°C.
Luciferase assay
Luciferase assay was performed using Luciferase Reporter Assay kit (Promega, U.S.A.). MCF-7 cells were co-transfected with the firefly reporter construct and plasmids encoding the desired gene. After 24 h of post-transfection, cells were lysed in passive lysis buffer (Promega). Equal protein was taken for quantification of luciferase activity using the Luminometer (Tecan, Switzerland).
Immunoprecipitation assay
An immunoprecipitation assay was performed to study the protein–protein interaction. HEK-293T cells were transfected with plasmids encoding the proteins of interest. After 36 h of transfection, cells were harvested and lysed in CelLytic M (Cell Lysis Reagent, Sigma). Anti-HA antibody-conjugated agarose resin (Sigma) was added and kept overnight at 4°C on rotation. After incubation, the resin was pelleted and washed with IP buffer (Sigma). It was boiled in 2× SDS-PAGE loading buffer for 5 min and subjected to western blot analysis.
Statistical analysis
Results obtained are represented as mean ± standard error of the mean (s.e.m). P-values were calculated by a two-tailed t-test. Only values with P < 0.05 were considered to be significant.
Results
Mdm2 levels increase post-HIV-1 infection
To assess the levels of Mdm2 after HIV-1 infection, MOLT-3 and Jurkat cells were infected with VSVG-pseudotyped pNL4-3 virus at a multiplicity of infection (MOI) of 1 for different time periods and then the cells were harvested. The effect of HIV-1 infection on Mdm2 levels was assessed by western blot analysis. Mdm2 expression increased in a time-dependent manner in both the MOLT-3 and Jurkat cell lines and correlated well with the extent of HIV-1 replication (Figure 1a,b). To detect Mdm2 protein, we used monoclonal antibodies, which produced multiple bands in western blot analysis due to the presence of Mdm2 isoforms and their post-translational modifications. However, we showed the major Mdm2 isoform, which corresponds to a molecular weight of 90 kDa. This isoform of Mdm2 has been represented in most of the studies [50–52]. To investigate the viral gene involved in Mdm2 stabilisation, HEK-293T cells were co-transfected with Myc-Mdm2 along with individual viral gene constructs under the control of a CMV promoter (Tat, Rev, Vpu, Vpr, Vif and Nef). Mdm2 levels increased in the presence of Tat (∼1.5 fold), but not with other proteins (Figure 1c). As shown in Supplementary Figure S1, the identity of viral proteins was confirmed using specific antibodies. Since HEK-293T cells express SV40T antigen, it cannot be ruled out that the latter affects Mdm2 levels in this context. To eliminate this possibility, we co-transfected MCF-7 cells with Mdm2 and Tat in the presence or absence of SV40 proviral plasmid. Tat increased the expression of Mdm2 regardless of the presence of SV40 (Supplementary Figure S2a,b). When EGFP-Mdm2 was transfected in HEK-293T cells in the presence of HIV-1 Tat, these cells showed enhanced fluorescence (Figure 1d). This increase in EGFP-Mdm2 fluorescence was also validated in the presence of HA-Tat and another HA-tagged protein (HA-Rev) by western blot analysis using the pEBG vector as a transfection efficiency control. HA-Tat alone increased the expression of EGFP-Mdm2, while HA-Rev could not do so (Supplementary Figure S3). All of these experiments led to the conclusion that during HIV-1 infection, Mdm2 is potently up-regulated, and that HIV-1 Tat protein alone facilitates this phenomenon.
Figure 1. HIV-1 infection increases Mdm2 levels in a Tat-dependent manner.

(a) MOLT-3 cells were infected with VSVG-pseudotyped HIV-1 at an MOI (multiplicity of infection) of 1 for the indicated time periods. Cell lysates were prepared at 24 and 48 h post-infection, and western blot analysis was performed to assess total Mdm2 levels. GAPDH was used as a loading control. (b) Jurkat E6.1 cells were infected with VSVG-pseudotyped HIV-1 at MOI of 1 for 24 and 48 h. Cell lysates were prepared as described previously and western blot analysis of total Mdm2 was performed. GAPDH was used as a loading control. Bar diagrams represent the protein quantitation of the respective western blots using Image J software. (c) HEK-293T cells were co-transfected with p-CMV-Myc3-Mdm2 (2 µg) and one of the six viral regulatory/accessory gene constructs (2 µg each). Cells were collected 36 h post-transfection and subjected to western blot analysis. The bands of the viral proteins were obtained at their respective molecular sizes (Myc-Tat∼17, Myc-Rev∼19, Myc-Nef∼27, Myc-Vpr∼15, Myc-Vif∼23 and Myc-Vpu∼16; all in kDa). Tubulin was used as a loading control. (d) pEGFP-Mdm2 (3 µg) was transfected in HEK-293T cells either alone or with HA-Tat (1 µg) and the fluorescence of pEGFP-Mdm2 was assessed in the presence or absence of Tat. Expression of Tat was assessed by western blot analysis using anti-HA antibody with GAPDH as a loading control. The results are representative of three independent experiments. P-values were calculated by a two-tailed t-test (**P < 0.01).
HIV-1 Tat increases Mdm2 expression at the post-translational level
To investigate the mechanism of HIV-1 Tat-mediated Mdm2 increase, MCF-7 cells were transfected to express HA-Tat, and the levels of endogenous Mdm2 protein and mRNA were determined. Half of the cells from the same sample were used for RNA isolation using Trizol, followed by cDNA synthesis. Real-time qPCR, as described in materials and methods, was carried out using specific primers to analyse the effect of Tat on the mRNA levels of Mdm2. The mRNA levels of Mdm2 were found to be unaffected in HA-Tat-transfected cells compared with those in the control cells, whereas Mdm2 protein levels showed a significant increase compared with control cells (P < 0.01). These findings suggested that HIV-1 Tat regulates Mdm2 at the post-translational level (Figure 2a,b). The Mdm2 protein levels were also assessed in the presence and absence of the Tat protein in the presence of cycloheximide. Myc-Mdm2 was stabilised to a greater extent (by at least 4 h) in the presence of HIV-1 Tat (Figure 2c). Ubiquitination of Mdm2 was also assessed by transfecting a plasmid encoding Myc-Mdm2 along with His-Ub in the presence and absence of Tat. The proteasomal inhibitor MG132 was added, and, after 8 h of incubation, ubiquitinated species were detected by western blot analysis with c-Myc antibody after pull down of the ubiquitinated species using Ni-NTA beads. Consistent with the above results, Mdm2 ubiquitination decreased significantly in the presence of Tat, thus confirming its effect at the post-translational level (Figure 2d).
Figure 2. Tat increases Mdm2 levels at the post-translational level.

(a) MCF-7 cells were transfected with 0.5 µg and 1 µg HA-Tat and pcDNA 3.1 was used as a control DNA to normalise DNA concentration. Total Mdm2 was detected by western blot analysis. GAPDH was used as a loading control. Bar diagrams represent the protein quantitation of the respective western blots using Image J software. (b) In parallel, cells were analysed by real-time qPCR as described in the methods section. Mean value and standard deviation were calculated from 2dCt of three independent experiments. P-values were calculated by a two-tailed t-test (*P < 0.05, **P < 0.01; ns, not-significant). (c) HEK-293T cells were transfected with 2 µg of p-CMV-Myc3-Mdm2 either alone or in presence of Tat (1 µg). After 36 h of transfection, cycloheximide chase assay (CHX 100 µg/ml) was performed for 5 h to assess the half-life of Mdm2 in the presence or absence of HA-Tat. (d) HEK-293T cells were co-transfected with His-Ubiquitin (His-Ub) and either with Myc-Mdm2 alone or Myc-Mdm2 along with Myc-Tat. After 36 h, the cells were treated with MG132 for 8 h followed by lysis in denaturation buffer. Total ubiquitinated proteins were then pulled down using Ni-NTA beads and Mdm2 ubiquitination was assessed by western blot analysis with anti-Myc antibody. Bar diagrams represent the protein quantitation of respective western blots using Image J software. Data presented are representative (mean ± s.e.m.) of three independent experiments. P-values were calculated by a two-tailed t-test (*P < 0.05, **P < 0.01; ns, not significant).
Phosphorylation of Mdm2 is induced by HIV-1 Tat
Since the Mdm2 levels were increased at the post-translational level without any change in mRNA levels, we assessed the presence of serine-166 phosphorylated form of Mdm2, which is known to promote its stability. MOLT-3 cells were infected with VSVG-pseudotyped pNL4-3 virus. The cells were collected at different time points after infection and subjected to western blot analysis using S166 phospho-Mdm2 antibody. The MOLT-3 cells showed an increase in phospho-Mdm2 (S166) and total Mdm2 post-HIV-1 infection (Figure 3a). To detect p-Mdm2 protein levels, we used monoclonal antibodies, which produced multiple bands in western blot analysis due to the presence of Mdm2 isoforms and their post-translational modifications. However, we showed the major p-Mdm2 band, which corresponds to molecular weight of 90 kDa. This p-Mdm2 band has been represented in most of the studies [53–55]. Furthermore, in an over-expression system using HEK-293T cells, co-transfection of Myc-Mdm2 and HA-Tat resulted in an increase of total Mdm2 and phospho-Mdm2 (S166) levels (Figure 3b). A dose-dependent increase in endogenous phospho-Mdm2 (S166) and total Mdm2 was also observed in MCF-7 cells after transfection with HIV-1 Tat (Figure 3c). These results confirm that total Mdm2 and phospho-Mdm2 (S166) levels increase significantly in the presence of HIV-1 Tat and during HIV-1 infection.
Figure 3. Stabilisation of Mdm2 in the presence of HIV-1 Tat correlates with an increase in phospho-Mdm2 (S166).

(a) MOLT-3 cells were infected with VSVG-pseudotyped HIV-1 for the indicated time at an MOI of 1. Cell lysates were prepared and probed for total Mdm2 and phospho-Mdm2 (S166). GAPDH was used as a loading control. Band intensities of Mdm2, p-Mdm2 and GAPDH were measured by Image J software. Mdm2/GAPDH and p-Mdm2/GAPDH ratios were calculated for each sample. After normalisation, values of Mdm2 and p-Mdm2 for uninfected/control sample were assigned a value of 1 unit and compared with those of other samples to obtain the relative protein expression levels, as shown in the bar diagram. (b) HEK-293T cells were transfected with plasmids expressing Myc-Mdm2 (2 µg) alone or with HA-Tat (1 µg). Cells were collected 36 h post-transfection and subjected to western blot analysis for total Mdm2 and phospho-Mdm2 (S166). (c) MCF-7 cells were transfected with 0.5 µg and 1 µg of HA-Tat for 36 h. Endogenous Mdm2 and p-Mdm2 (S166) were detected using anti-Mdm2 and anti-phospho-Mdm2 (S166) antibodies, respectively. GAPDH was used as a loading control. Band intensities of Mdm2, p-Mdm2 and GAPDH were measured by Image J software. Mdm2/GAPDH and p-Mdm2/GAPDH ratios were calculated for each sample. After normalisation, values of Mdm2 and p-Mdm2 for uninfected/control sample were assigned a value of 1 unit and compared with those of other samples to obtain the relative protein expression levels, as shown in the bar diagram. The results are representative of three independent experiments. P-values were calculated by a two-tailed t-test (*P < 0.05, **P < 0.01).
HIV-1 Tat-mediated induction of Mdm2 levels is independent of p53
Since the levels of the Mdm2 protein increased during HIV-1 infection, we investigated the dependence of Tat-mediated Mdm2 increase on the p53 protein, which is known to induce Mdm2 expression. Surprisingly, the Mdm2 increase was accompanied by an increase in p53 during HIV-1 infection (Figure 4a). In HIV-1 infection, phospho-p53 (S15) is known to be up-regulated and this phosphorylation prevents p53 from Mdm2-mediated degradation [17,26,29,56]. Therefore, we also examined the levels of phospho-p53 (S15) during HIV-1 infection in MOLT-3 cells and found that both phospho-p53 (S15) and total p53 levels were up-regulated (Figure 4a). The strong induction of S15 phosphorylation of p53 is important for Bax up-regulation, which promotes HIV-1-mediated apoptosis. As shown in Supplementary Figure S4, HIV-1 Vpu increases total p53 and p-p53 (S15) levels by inhibiting β-TRCP1-mediated degradation of p53 [17]. DNA damage also up-regulates S15 phosphorylation of p53 during HIV-1 infection [57]. Since p53 is one of the major regulators of Mdm2 induction, in order to assess the p53 dependence of the HIV-1-mediated increase in Mdm2 levels, we used a MOLT-3 p53 knockdown cell line (MOLT-3 p53 kd), which was generated by transfecting p53 shRNA in these cells followed by puromycin (5 µg/ml) selection (Figure 4b). The MOLT-3 p53 kd cells also showed an increase in Mdm2 levels upon HIV-1 infection (Figure 4c). HIV-1 Tat-mediated increase in Mdm2 levels was also validated in H1299, a p53-null cell line. The H1299 cells were transfected with Myc-Mdm2 and HA-Tat. The cells were lysed 36 h post-transfection and subjected to western blot analysis. A significant increase in phospho-Mdm2 (S166) and total Mdm2 levels was observed, suggesting p53-independent induction of the Mdm2 by HIV-1 Tat (Figure 4d). Cycloheximide chase assay of Mdm2 was also performed in H1299 cells in the presence and absence of Tat. This assay also showed the enhanced stability (by at least 3 h) of Mdm2 in the presence of Tat, further supporting the fact that Tat-mediated Mdm2 stabilisation is independent of p53 (Figure 4e).
Figure 4. HIV-1 Tat-mediated Mdm2 increase is p53 independent.

(a) MOLT-3 cells were infected at an MOI of 1 with VSVG-pseudotyped HIV-1. The cells were collected at 12, 24 and 48 h post-infection and subjected to western blot analysis with anti-p53 and anti-phospho-p53 (S15) antibodies. GAPDH was used as a loading control. (b) MOLT-3 cells were transfected with shRNA scrambled (control) or p53-specific shRNA (20 µg/100 mm Petri dish) and selected using puromycin (5 µg/ml) as described in materials and methods section. The cells were collected after selection in puromycin-rich media and subjected to western blot analysis with p53 antibody. Bax was used as a positive control for the p53 knockdown experiment. GAPDH was used as a loading control. Band intensities of p53, p-p53 and GAPDH were measured by Image J software. p53/GAPDH and p-p53/GAPDH ratios were calculated for each sample. After normalisation, values of p53 and p-p53 for uninfected/control sample were assigned a value of 1 unit and compared with those of other samples in order to obtain the relative protein expression levels, as shown in the bar diagram. (c) MOLT-3 p53 kd cells were infected with VSVG-pseudotyped HIV-1 at an MOI of 1. The cells were collected at 12, 24 and 48 h post-infection and subjected to western blot analysis with anti-Mdm2 and anti-phospho-Mdm2 (S166) antibodies. GAPDH was used as a loading control. Band intensities of Mdm2, p-Mdm2 and GAPDH were measured by Image J software. Mdm2/GAPDH and p-Mdm2/GAPDH ratios were calculated for each sample. After normalisation, values of Mdm2 and p-Mdm2 for uninfected/control sample were assigned a value of 1 unit and compared with other samples in order to obtain the relative protein expression levels, as shown in the bar diagram. (d) H1299 p53-null cells were transfected with 2 µg of Myc-Mdm2 alone or in the presence of HA-Tat, and the cell lysates were collected 36 h post-transfection. Anti-Mdm2 and anti-phospho-Mdm2 (S166) antibodies were used for western blot analysis. (e) Cycloheximide chase assay was performed to assess the effect of Tat on Mdm2 stability in the absence of p53 using H1299 (p53-null) cells by treating the cells with 100 µg/ml of CHX 36 h post-transfection for the indicated time periods. The results are representative of four independent experiments. P-values were calculated by a two-tailed t-test (**P < 0.01).
Phosphorylation of Mdm2 promotes nuclear localisation
To assess the effect of HIV-1 Tat on the nuclear localisation of Mdm2, MCF-7 cells were transfected with HA-Tat. Nuclear and cytoplasmic extracts were prepared according to the manufacturer's protocol. Total Mdm2 and phospho-Mdm2 (S166) levels were assessed in both the cytosolic and nuclear fractions. In the cytosolic fraction, the increase in total Mdm2 and phospho-Mdm2 (S166) levels was less significant (Figure 5a,c), although the levels increased to some extent in the presence of Tat as expected. By contrast, in the nuclear fraction, the increase in Mdm2 and phospho-Mdm2 (S166) levels was significant in the presence of Tat (Figure 5b,c), compared with that in the cytosolic fraction. The significant increase in Mdm2 and phospho-Mdm2 (S166) indicates that Tat promotes nuclear localisation of Mdm2 by increasing its phosphorylation at S166 position, as described earlier [21,23].
Figure 5. HIV-1 Tat induces nuclear-cytoplasmic re-distribution of Mdm2.

(a) and (b) MCF-7 cells were transfected with 1 µg HA-Tat. Nuclear and cytoplasmic fractions were prepared 36 h post-transfection using NE-PER® Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Scientific). After preparing the fractions, western blot analysis was performed. LaminB2 was used as a loading control in the nuclear fraction whereas GAPDH was used as a loading control in the cytosolic fraction. (c) Relative protein expression in each fraction was calculated using Image J software. The results are representative of three independent experiments. P-values were calculated by a two-tailed t-test (*P < 0.05, **P < 0.01).
Mdm2 phosphorylation is important for HIV-1 replication
The effect of Mdm2 phosphorylation on HIV-1 replication was investigated using the wild-type Mdm2 and Mdm2 S166A. Co-transfection of pNL4-3 with either Mdm2 or Mdm2 S166A in HEK-293T and HeLa cells was performed and the cells were harvested 36 h post-transfection. p24 levels were assessed by western blot analysis. A significant increase in p24 levels was observed in the presence of wild-type Mdm2, whereas p24 levels in the presence of Mdm2 S166A were either marginally up-regulated or similar to those with pNL4-3 alone (control) (Figure 6a,b). This result shows that an increase in phosphorylation of Mdm2 at S166 is important for HIV-1 replication and that Mdm2 S166A does not support viral replication. We also validated the role of Mdm2 in HIV-1 replication with siRNA-mediated knock down of Mdm2 in HEK-293T and HeLa cells. Western blot analysis demonstrated a marked decrease in Mdm2 expression levels in these cells. The consequence of Mdm2 knockdown on viral replication was also analysed by transfecting Mdm2 kd cells and control siRNA-treated cells with proviral DNA of pNL4-3. p24 levels were assessed in both the control siRNA and Mdm2 siRNA-treated cells. Down-regulation of Mdm2 was found to lead to reduced virus production, as reported earlier [16] (Supplementary Figure S5). Since HeLa cells express HPV gene product HPV-18, in order to rule HPV-18's potential contribution to Tat-mediated Mdm2 up-regulation in our experimental system, we co-transfected MCF-7 cells with Mdm2 and Tat in the presence or absence of HPV-18 proviral plasmid. Tat increased the expression of Mdm2 irrespective of the presence or absence of HPV-18 (Supplementary Figure S2a,c).
Figure 6. S166A mutation abrogates Mdm2-mediated increase in HIV-1 replication by decreasing Mdm2 stability and its interaction with Tat.

(a) HeLa cells were transfected with pNL4-3 (1 µg) and EGFP-Mdm2 Wt (2 µg) or its mutant EGFP-Mdm2 S166A (2 µg), and p24 levels were assessed by western blot analysis. GAPDH was used as a loading control. (b) HEK-293T cells were transfected with pNL4-3 (500 ng) and Mdm2 Wt or Mdm2 S166A (3 µg each) and p24 levels were determined by western blot analysis. Bar diagrams represent the protein quantitation of respective western blots using Image J software. (c) MCF-7 cells were co-transfected with a LTR-luciferase construct (100 ng) along with the EGFP-Mdm2 Wt (3 µg), EGFP-Mdm2 S166A (3 µg) and HA-Tat (500 ng). pcDNA3.1 was used as a control DNA in order to normalise DNA concentration. LTR-luciferase acitivity was measured after 24 h using a luminometer. (d) HEK-293T cells were transfected with HA-Tat (1 µg) and EGFP-Mdm2 Wt (2 µg) or EGFP-Mdm2 S166A (2 µg). Cycloheximide chase assay was performed 36 h post-transfection after treatment of the cells with CHX (100 µg/ml) for the indicated time periods. (e) HEK-293T cells were transfected with HA-Tat (1 µg) and EGFP-Mdm2 Wt (2 µg) or EGFP-Mdm2 S166A (2 µg). Cell extracts were subjected to immuno-precipitation using anti-HA antibody-conjugated Protein A agarose beads (Sigma), followed by western blot analysis using anti-GFP antibody. The results are representative of three independent experiments. P-values were calculated by a two-tailed t-test (**P < 0.01; ns, not significant).
Since, HIV-1 p24 levels in the presence of Mdm2 S166A were comparable to those with pNL4-3 alone, we next investigated the role of Mdm2 S166A in Tat-mediated LTR promoter activity. In LTR-luciferase assay, Mdm2 S166A showed remarkably reduced LTR promoter activity compared with that of the wild-type Mdm2. This finding indicates the role of serine-166 phosphorylation of Mdm2 in the LTR promoter activity of Tat, which correlates with HIV-1 replication (Figure 6c).
S166A mutation of Mdm2 interferes with its stabilisation and interaction with Tat
The effect of S166A mutation on the stability of Mdm2 was investigated in the presence of HIV-1 Tat. Co-transfection of HIV-1 Tat with either wild-type Mdm2 or Mdm2 S166A was performed in HEK-293T cells. Cycloheximide chase assay was performed 36 h post-transfection to assess the stability of wild-type Mdm2 and Mdm2 S166A in the presence of Tat. Wild-type Mdm2 was found to be more stable in the presence of HIV-1 Tat, whereas Mdm2 S166A could not be stabilised (Figure 6d). Thus, S166 phosphorylation of Mdm2 is important for its Tat-mediated stabilisation.
The effect of S166A mutation of Mdm2 on its interaction with HIV-1 Tat was also investigated. An immuno-precipitation assay was performed using cell lysates transfected with HA-Tat, wild-type Mdm2 and Mdm2 S166A using anti-HA antibody conjugated beads. Wild-type Mdm2 showed strong interaction with Tat while Mdm2 S166A showed significantly decreased interaction with Tat as compared with wild-type Mdm2 (Figure 6e). The above results highlight the significance of S166 phosphorylation of Mdm2 because, in the presence of Tat, both Mdm2 stabilisation and the interaction of Mdm2 with Tat are compromised by the S166A mutation.
Involvement of AKT in Tat-mediated Mdm2 stabilisation
To assess the dependence of Tat-mediated Mdm2 stabilisation on AKT, Myc-Mdm2 was transfected alone or together with HA-Tat in HEK-293T cells. At 36 h post-transfection, the cells were treated with 4 µM AKT inhibitor for 8 h. The cells were then collected, lysed and subjected to western blot analysis. Total Mdm2 and phospho-Mdm2 (S166) levels decreased in the presence of AKT inhibitor (AKTi) compared with those of the control, thus showing the dependence of Tat-mediated stabilisation of Mdm2 on AKT (Figure 7a). Since AKT inhibition abrogated Tat-mediated Mdm2 up-regulation, we next investigated whether any components of the PI3K/AKT pathway were affected by Tat. HEK-293T and Jurkat T cells were serum starved for 36 h and then incubated with the Tat protein. Serum starvation was performed in order to rule out the influence of growth factors on the PI3K/AKT pathway. Both HEK-293T and Jurkat T cells showed an increase in p-AKT (S473) and p-Mdm2 (S166) levels (Figure 7b,c).
Figure 7. HIV-1 Tat stabilises Mdm2 through the PI3K/AKT pathway.

(a) HEK-293T cells were transfected with p-CMV-Myc3-Mdm2 (2 µg) alone or with HA-Tat (1 µg). At 36 h post-transfection, the cells were treated with AKT inhibitor (4 µM) for 8 h. Total Mdm2 and phospho-Mdm2 (S166) levels were determined by western blot analysis. (b) HEK-293T cells were serum starved for 36 h and then incubated with the Tat protein (10 ng/ml) for 30 min before harvesting the cells. The cells were then subjected to western blot analysis with anti-AKT, anti-phospho-AKT (S473), anti-Mdm2 and anti-phospho-Mdm2 (S166) antibodies. Total AKT was used as a loading control. (c) Jurkat T cells were serum starved for 36 h and incubated with the Tat protein (10 ng/ml) for the indicated time periods. The cells were collected and subjected to western blot analysis with anti-AKT, anti-phospho-AKT (S473), anti-Mdm2 and anti-phospho-Mdm2 (S166) antibodies. Total AKT was used as a loading control. Bar diagrams represent the protein quantitation of respective western blots using Image J software. The results are representative of three independent experiments. P-values were calculated by a two-tailed t-test (*P < 0.05, **P < 0.01).
Discussion
HIV-1 is known for its ability to exploit the host cellular machinery for its successful propagation. In the initial phase of its life cycle, HIV-1 needs healthy cells for replication and virus production. Apoptosis of host cells is required in the later stages to facilitate viral spread and to evade the immune response [58–61]. HIV-1 Tat protein is one of the early expressed proteins. It activates several cellular pathways and exerts apoptotic and anti-apoptotic effects, with a number of reports suggesting that it has a role in the activation of PI-3-kinase (PI3K)/AKT pathway [62–65]. This pathway has several important target proteins that promote cell survival either by inhibiting apoptotic factors or activating the anti-apoptotic factors. In addition, Mdm2 is a well-known downstream target of AKT [66,67]. AKT stabilises Mdm2 by causing its deubiquitination and it also promotes its nuclear localisation [22,23]. Direct modulation of Mdm2 in HIV-1 infection has not been reported. Therefore, we investigated the effect of HIV-1 infection on Mdm2. Mdm2 expression levels increased post-HIV-1 infection in both MOLT-3 and Jurkat T cells. Furthermore, HIV-1 Tat alone was responsible for increasing the Mdm2 levels in a co-transfection system. This observation was also confirmed by co-transfection of GFP-Mdm2 and Tat using fluorescence microscopy.
Since the Tat protein down-regulates p53 at the transcriptional level [44], we investigated whether Tat affects mRNA levels of Mdm2 using real-time qPCR analysis. In Tat-transfected MCF-7 cells, Mdm2 mRNA levels remained the same whereas expression of Mdm2 protein was up-regulated. This effect at the post-translational level was further validated by cycloheximide chase assay of Mdm2 in the presence of Tat in HEK-293T cells. We also performed an ubiquitination assay to determine the effect of Tat on the ubiquitination of Mdm2. HIV-1 Tat was found to decrease the levels of ubiquitinated Mdm2. To investigate the mechanism involved in Tat-mediated Mdm2 stabilisation, we assessed the phosphorylation status of Mdm2 in the presence of Tat. A number of phosphorylation sites are known to be present in Mdm2. Phosphorylation of Mdm2 at these sites serves diverse purposes such as cellular stress response (e.g., DNA damage) sub-cellular localisation, protein interactions and Mdm2 stabilisation [21,22]. Phosphorylation of Mdm2 at S166 is critical because it regulates the stability and localisation of Mdm2 [22,23]. We found that phospho-Mdm2 (S166) increases post-HIV-1 infection and also in the presence of Tat alone. This explains the post-translational regulation of Mdm2, which was also confirmed by the cycloheximide chase and the ubiquitination assay.
p53 levels are known to be up-regulated during HIV-1 infection and stabilisation of this protein by phosphorylation at the serine-15 residue is well documented [24,45,46]. Our laboratory has previously shown that phospho-p53 (S15) levels increase post-HIV-1 infection [17]. This contributes to HIV-1-directed apoptosis in the late stages of its life cycle. The data presented here also confirms this observation with both total p53 and phospho-p53 (S15) levels increasing upon HIV-1 infection. Since p53 is one of the crucial regulators of Mdm2 at the transcriptional level, we investigated the role of p53 in HIV-1 Tat-mediated stabilisation of Mdm2. p53 was knocked down in MOLT-3 cells followed by infection with HIV-1 virus. Total Mdm2 and p-Mdm2 (S166) levels increased upon HIV-1 infection of these cells. Mdm2 levels also increased in the presence of Tat in H1299 cells (a p53-null cell line). Furthermore, stabilisation of Mdm2 by Tat was also confirmed in H1299 cells by the cycloheximide chase assay. This showed that Tat-mediated Mdm2 stabilisation was independent of p53.
Mdm2 is known to enhance the transcriptional activity of Tat by causing its ubiquitination and thus promoting HIV-1 replication [16]; therefore, we investigated whether phospho-Mdm2 (S166) had any role in LTR transactivation and viral replication. A co-transfection based study in MCF-7 and HeLa cells clearly suggested that phosphorylation of Mdm2 at the serine-166 position increased HIV-1 replication, as shown by p24 levels. However, Mdm2 S166A showed significantly reduced LTR promoter activity compared with that of the wild-type Mdm2. The stabilisation of Mdm2 S166A by Tat was compromised and its interaction with Tat was significantly diminished, as illustrated in the Figure 6d,e. We also found that nuclear levels of total Mdm2 increased markedly in the presence of Tat. Thus, Mdm2 might alter the known functions of Tat in the nucleus. The results obtained with HEK-293T and HeLa cells need to be interpreted with caution since HEK-293T cells express SV40T antigen and HeLa cells harbour HPV-18 virus, both of which could influence the results. Hence, we separately investigated the effect of SV40 and HPV-18 on Tat-mediated Mdm2 stabilisation (Supplementary Figure S2) and we concluded that neither SV40 nor HPV-18 affected our results.
HIV-1 Tat-mediated stabilisation of Mdm2 was found to be dependent on the PI3K/AKT pathway as inhibition of AKT abrogated the effect of HIV-1 Tat on Mdm2. Tat protein was found to increase the levels of p-AKT and p-Mdm2 in both HEK-293T and Jurkat T cells. In summary, we show that the HIV-1 Tat protein potently stabilises Mdm2 through the PI3K/AKT pathway. Tat increases S166 phosphorylation of Mdm2, which leads to its stabilisation and phospho-Mdm2 (S166) in turn promotes HIV-1 gene expression and replication, thus creating a positive feedback loop between Mdm2 and Tat (Figure 8). Thus, the present study highlights the novel role of HIV-1 Tat-mediated post-translational modification in stabilising Mdm2, with significant implications for HIV-1 gene expression, replication and pathogenesis.
Figure 8. Mechanistic model showing HIV-1 Tat-mediated stabilisation of Mdm2.
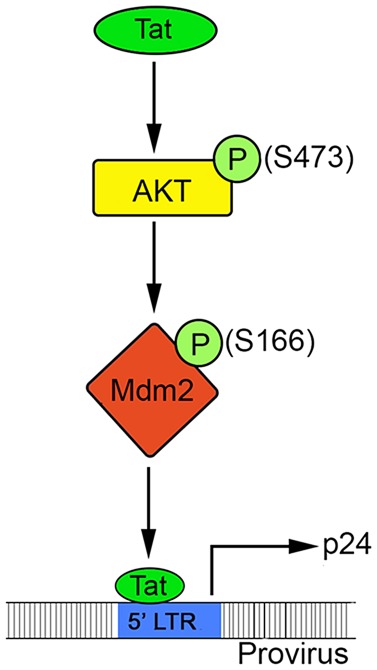
HIV-1 Tat protein is expressed in the initial phase of the HIV-1 life cycle and its activity is modulated by several cellular proteins. The present study shows that HIV-1 Tat protein induces phosphorylation of AKT at S473 position, which leads to the activation of AKT. Activated AKT in turn phosphorylates Mdm2 at serine-166, which is essential for stability of Mdm2 and its interaction with the Tat protein. Phosphorylated Mdm2 (S166) increases HIV-1 Tat-mediated transactivation of the viral promoter and hence HIV-1 replication. Thus, HIV-1 Tat and Mdm2 operate in a positive feedback manner resulting in the enhancement of viral replication. Arrows (→) indicate enhancement in activity.
Acknowledgements
We thank Brian Hemmings for the pEGFP-Mdm2 constructs, Sanjeev Das for the p53 shRNA constructs and Lutz Gissmann for the HPV-18 construct. We thank Amjad Ali and Sachin Verma for their support and guidance. We also thank Nancy Jayaraj and Lovely Princess Santos from Elsevier Language Editing Services and Samuel Kazer from Institute for Medical Engineering and Science, MIT for their help in manuscript editing. Several reagents were obtained from AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, MD, U.S.A.
Abbreviations
- AIDS
acquired immunodeficiency syndrome
- HIV
human immunodeficiency virus
- Kd
knockdown
- Mdm2
murine double minute 2
- RIPA
radioimmunoprecipitation assay
- Wt
wild-type
Author Contribution
Rameez Raja and Akhil Chandra Banerjea designed the research; Rameez Raja performed the experiments; Rameez Raja, Sneh Lata, Shubhendu Trivedi, Larance Ronsard analysed and interpreted the data; Rameez Raja, Larance Ronsard contributed to reagents/materials/analysis tools, Rameez Raja, Sneh Lata, Shubhendu Trivedi and Akhil Chandra Banerjea wrote the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the Department of Biotechnology (BT/PR1800/AGR/36/676/2011) and Indian Council of Medical Research (HIV/50/142/9-2011-ECD), Government of India to A.C.B. and NII, New Delhi. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
Supplementary Material
References
- 1.Connor R.I., Mohri H., Cao Y. and Ho D.D. (1993) Increased viral burden and cytopathicity correlate temporally with CD4+ T-lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected individuals. J. Virol. 67, 1772–1777 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevenson M. (2003) HIV-1 pathogenesis. Nat. Med. 9, 853–860 doi: 10.1038/nm0703-853 [DOI] [PubMed] [Google Scholar]
- 3.Terai C., Kornbluth R.S., Pauza C.D., Richman D.D. and Carson D.A. (1991) Apoptosis as a mechanism of cell death in cultured T lymphoblasts acutely infected with HIV-1. J. Clin. Invest. 87, 1710–1715 doi: 10.1172/JCI115188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss R.A. (1993) How does HIV cause AIDS? Science 260, 1273–1279 doi: 10.1126/science.8493571 [DOI] [PubMed] [Google Scholar]
- 5.Frankel A.D. and Young J.A.T. (1998) HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 67, 1–25 doi: 10.1146/annurev.biochem.67.1.1 [DOI] [PubMed] [Google Scholar]
- 6.Malim M.H. and Emerman M. (2008) HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microbe 3, 388–398 doi: 10.1016/j.chom.2008.04.008 [DOI] [PubMed] [Google Scholar]
- 7.Roulston A., Marcellus R.C. and Branton P.E. (2008) Viruses and apoptosis. Annu. Rev. Microbiol. 53, 577–628 doi: 10.1146/annurev.micro.53.1.577 [DOI] [PubMed] [Google Scholar]
- 8.Swanson C.M. and Malim M.H. (2008) Snapshot: HIV-1 proteins. Cell 133, 742 doi: 10.1016/j.cell.2008.05.005 [DOI] [PubMed] [Google Scholar]
- 9.Bieniasz P. D. (2009) The cell biology of HIV-1 virion genesis. Cell Host Microbe 5, 550–558 doi: 10.1016/j.chom.2009.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho D.D. and Bieniasz P.D. (2008) HIV-1 at 25. Cell 133, 561–565 doi: 10.1016/j.cell.2008.05.003 [DOI] [PubMed] [Google Scholar]
- 11.Rustagi A. and Gale M. (2014) Innate antiviral immune signaling, viral evasion and modulation by HIV-1. J. Mol. Biol. 426, 1161–1177 doi: 10.1016/j.jmb.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wojcechowskyj J.A., Didigu C.A., Lee J.Y., Parrish N.F., Sinha R., Hahn B.H. et al. (2013) Quantitative phosphoproteomics reveals extensive cellular reprogramming during HIV-1 entry. Cell Host Microbe 13, 613–623 doi: 10.1016/j.chom.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J. et al. (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926 doi: 10.1126/science.1152725 [DOI] [PubMed] [Google Scholar]
- 14.König R., Zhou Y., Elleder D., Diamond T.L., Bonamy G.M., Irelan J.T. et al. (2008) Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60 doi: 10.1016/j.cell.2008.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H., Xu M., Huang Q., Gates A.T., Zhang X.D., Castle J.C. et al. (2008) Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4, 495–504 doi: 10.1016/j.chom.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 16.Brès V., Kiernan R.E., Linares L.K., Chable-Bessia C., Plechakova O., Tréand C. et al. (2003) A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 5, 754–761 doi: 10.1038/ncb1023 [DOI] [PubMed] [Google Scholar]
- 17.Verma S., Ali A., Arora S. and Banerjea A.C. (2011) Inhibition of β-TrcP-dependent ubiquitination of p53 by HIV-1 Vpu promotes p53-mediated apoptosis in human T cells. Blood 117, 6600–6607 doi: 10.1182/blood-2011-01-333427 [DOI] [PubMed] [Google Scholar]
- 18.Fakharzadeh S.S., Trusko S.P. and George D.L. (1991) Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 10, 1565–1569 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haines D.S. (1997) The mdm2 proto-oncogene. Leuk. Lymphoma 26, 227–238 doi: 10.3109/10428199709051772 [DOI] [PubMed] [Google Scholar]
- 20.Haupt Y., Barak Y. and Oren M. (1996) Cell type-specific inhibition of p53-mediated apoptosis by mdm2. EMBO J. 15, 1596–1606 PMID: [PMC free article] [PubMed] [Google Scholar]
- 21.Meek D.W. and Knippschild U. (2003) Posttranslational modification of MDM2. Mol. Cancer Res. 1, 1017–1026 PMID: [PubMed] [Google Scholar]
- 22.Feng J., Tamaskovic R., Yang Z., Brazil D.P., Merlo A., Hess D. et al. (2004) Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 279, 35510–35517 doi: 10.1074/jbc.M404936200 [DOI] [PubMed] [Google Scholar]
- 23.Mayo L.D. and Donner D.B. (2001) A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl Acad. Sci. U.S.A. 98, 11598–11603 doi: 10.1073/pnas.181181198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gajjar M., Candeias M.M., Malbert-Colas L., Mazars A., Fujita J., Olivares-Illana V. et al. (2012) The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 21, 25–35 doi: 10.1016/j.ccr.2011.11.016 [DOI] [PubMed] [Google Scholar]
- 25.Ofir-Rosenfeld Y., Boggs K., Michael D., Kastan M.B. and Oren M. (2008) Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 32, 180–189 doi: 10.1016/j.molcel.2008.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bode A.M. and Dong Z. (2004) Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4, 793–805 doi: 10.1038/nrc1455 [DOI] [PubMed] [Google Scholar]
- 27.Chehab N.H., Malikzay A., Stavridi E.S. and Halazonetis T.D. (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl Acad. Sci. U.S.A. 96, 13777–13782 doi: 10.1073/pnas.96.24.13777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J., Yu Y., Hamrick H.E. and Duerksen-Hughes P.J. (2003) ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis 24, 1571–1580 doi: 10.1093/carcin/bgg137 [DOI] [PubMed] [Google Scholar]
- 29.Shieh S.-Y., Ikeda M., Taya Y. and Prives C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334 doi: 10.1016/S0092-8674(00)80416-X [DOI] [PubMed] [Google Scholar]
- 30.Lees-Miller S.P., Sakaguchi K., Ullrich S.J., Appella E. and Anderson C.W. (1992) Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol.Cell. Biol. 12, 5041–5049 doi: 10.1128/MCB.12.11.5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Canman C.E., Lim D.S., Cimprich K.A., Taya Y., Tamai K., Sakaguchi K. et al. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of P53. Science 281, 1677–1679 doi: 10.1126/science.281.5383.1677 [DOI] [PubMed] [Google Scholar]
- 32.Banin S., Moyal L., Shieh S., Taya Y., Anderson C.W., Chessa L. et al. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677 doi: 10.1126/science.281.5383.1674 [DOI] [PubMed] [Google Scholar]
- 33.Tibbetts R.S., Brumbaugh K.M., Williams J.M., Sarkaria J.N., Cliby W.A., Shieh S.-Y. et al. (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13, 152–157 doi: 10.1101/gad.13.2.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siliciano J.D., Canman C.E., Taya Y., Sakaguchi K., Appella E. and Kastan M.B. (1997) DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 11, 3471–3481 doi: 10.1101/gad.11.24.3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen D., Zhang J., Li M., Rayburn E.R., Wang H. and Zhang R. (2009) RYBP stabilizes p53 by modulating MDM2. EMBO Rep. 10, 166–172 doi: 10.1038/embor.2008.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ganguli G. and Wasylyk B. (2003) p53-independent functions of MDM2. Mol. Cancer Res. 1, 1027–1035 PMID: [PubMed] [Google Scholar]
- 37.Gammoh N., Gardiol D., Massimi P. and Banks L. (2009) The Mdm2 ubiquitin ligase enhances transcriptional activity of human papillomavirus E2. J. Virol. 83, 1538–1543 doi: 10.1128/JVI.01551-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saha A., Murakami M., Kumar P., Bajaj B., Sims K. and Robertson E.S. (2009) Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J. Virol. 83, 4652–4669 doi: 10.1128/JVI.02408-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henning W., Rohaly G., Kolzau T., Knippschild U., Maacke H. and Deppert W. (1997) MDM2 is a target of simian virus 40 in cellular transformation and during lytic infection. J. Virol. 71, 7609–7618 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hermannstädter A., Ziegler C., Kühl M., Deppert W. and Tolstonog G.V. (2009) Wild-type p53 enhances efficiency of simian virus 40 large-T-antigen-induced cellular transformation. J. Virol. 83, 10106–10118 doi: 10.1128/JVI.00174-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehrhardt C., Wolff T., Pleschka S., Planz O., Beermann W., Bode J.G. et al. (2007) Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 81, 3058–3067 doi: 10.1128/JVI.02082-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X., Deng X., Yan W., Zhu Z., Shen Y., Qiu Y. et al. (2012) Stabilization of p53 in influenza A virus-infected cells is associated with compromised MDM2-mediated ubiquitination of p53. J. Biol. Chem. 287, 18366–18375 doi: 10.1074/jbc.M111.335422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee H.-R., Toth Z., Shin Y.C., Lee J.-S., Chang H., Gu W. et al. (2009) Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J. Virol. 83, 6739–6747 doi: 10.1128/JVI.02353-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li C.J., Wang C., Friedman D.J. and Pardee A.B. (1995) Reciprocal modulations between p53 and Tat of human immunodeficiency virus type 1. Proc. Natl Acad. Sci. U.S.A. 92, 5461–5464 doi: 10.1073/pnas.92.12.5461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thakur B.K., Chandra A., Dittrich T., Welte K. and Chandra P. (2012) Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway. Biochem. Biophys. Res. Commun. 424, 245–250 doi: 10.1016/j.bbrc.2012.06.084 [DOI] [PubMed] [Google Scholar]
- 46.Pagans S., Pedal A., North B.J., Kaehlcke K., Marshall B.L., Dorr A. et al. (2005) SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 3, e41 doi: 10.1371/journal.pbio.0030041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Izumi T., Takaori-Kondo A., Shirakawa K., Higashitsuji H., Itoh K., Io K. et al. (2009) MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology 6, 1 doi: 10.1186/1742-4690-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sen N., Satija Y.K. and Das S. (2011) PGC-1α, a key modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell 44, 621–634 doi: 10.1016/j.molcel.2011.08.044 [DOI] [PubMed] [Google Scholar]
- 49.Arora S., Verma S. and Banerjea A.C. (2014) HIV-1 Vpr redirects host ubiquitination pathway. J. Virol. 88, 9141–9152 doi: 10.1128/JVI.00619-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng T.-H., and Cohen S.N. (2007) Human MDM2 isoforms translated differentially on constitutive versus p53-regulated transcripts have distinct functions in the p53/MDM2 and TSG101/MDM2 feedback control loops. Mol. Cell. Biol. 27, 111–119 doi: 10.1128/MCB.00235-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng T., Wang J., Zhao Y., Zhang C., Lin M., Wang X. et al. (2013) Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 4, 2996 doi: 10.1038/ncomms3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pochampally R., Fodera B., Chen L., Shao W., Levine E.A. and Chen J. (1998) A 60 kd MDM2 isoform is produced by caspase cleavage in non-apoptotic tumor cells. Oncogene 17, 2629–2636 doi: 10.1038/sj.onc.1202206 [DOI] [PubMed] [Google Scholar]
- 53.González E., Rother M., Kerr M.C., Al-Zeer M.A., Abu-Lubad M., Kessler M. et al. (2014) Chlamydia infection depends on a functional MDM2-p53 axis. Nat. Commun. 5, 5201 doi: 10.1038/ncomms6201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giovannini C., Minguzzi M., Baglioni M., Fornari F., Giannone F., Ravaioli M. et al. (2014) Suppression of p53 by Notch3 is mediated by Cyclin G1 and sustained by MDM2 and miR-221 axis in hepatocellular carcinoma. Oncotarget 5, 10607–10620 doi: 10.18632/oncotarget.2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malmlof M., Roudier E., Hogberg J. and Stenius U. (2007) MEK-ERK-mediated phosphorylation of Mdm2 at Ser-166 in hepatocytes: Mdm2 is activated in response to inhibited Akt signaling. J. Biol. Chem. 282, 2288–2296 doi: 10.1074/jbc.M604953200 [DOI] [PubMed] [Google Scholar]
- 56.Genini D., Sheeter D., Rought S., Zaunders J.J., Susin S.A., Kroemer G. et al. (2001) HIV induces lymphocyte apoptosis by a p53-initiated, mitochondrial-mediated mechanism. FASEB J. 15, 5–6 doi: 10.1096/fj.00-0336fje [DOI] [PubMed] [Google Scholar]
- 57.Perfettini J.-L., Nardacci R., Bourouba M., Subra F., Gros L., Séror C. et al. (2008) Critical involvement of the ATM-dependent DNA damage response in the apoptotic demise of HIV-1-elicited syncytia. PLoS ONE 3, e2458 doi: 10.1371/journal.pone.0002458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gougeon M.-L. (2005) Apoptosis as an HIV strategy to escape immune attack. Nat. Rev. Immunol. 3, 392–404 doi: 10.1038/nri1087 [DOI] [PubMed] [Google Scholar]
- 59.Hanauske-Abel H.M., Saxena D., Palumbo P.E., Hanauske A.R., Luchessi A.D., Cambiaghi T.D. et al. (2013) Drug-induced reactivation of apoptosis abrogates HIV-1 infection. PLoS ONE 8, e74414 doi: 10.1371/journal.pone.0074414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross T.M. (2001) Using death to one's advantage: HIV modulation of apoptosis. Leukemia 15, 332–341 doi: 10.1038/sj.leu.2402028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benedict C.A., Norris P.S. and Ware C.F. (2002) To kill or be killed: viral evasion of apoptosis. Nat. Immunol. 3, 1013–1018 doi: 10.1038/ni1102-1013 [DOI] [PubMed] [Google Scholar]
- 62.Chugh P., Bradel-Tretheway B., Monteiro-Filho C.M.R., Planelles V., Maggirwar S.B., Dewhurst S. et al. (2008) Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy. Retrovirology 5, 11 doi: 10.1186/1742-4690-5-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chugh P., Fan S., Planelles V., Maggirwar S.B., Dewhurst S. and Kim B. (2007) Infection of human immunodeficiency virus and intracellular viral Tat protein exert a pro-survival effect in a human microglial cell line. J. Mol. Biol. 366, 67–81 doi: 10.1016/j.jmb.2006.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deregibus M.C., Cantaluppi V., Doublier S., Brizzi M.F., Deambrosis I., Albini A. et al. (2002) HIV-1-Tat protein activates phosphatidylinositol 3-kinase/ AKT-dependent survival pathways in Kaposi's sarcoma cells. J. Biol. Chem. 277, 25195–25202 doi: 10.1074/jbc.M200921200 [DOI] [PubMed] [Google Scholar]
- 65.Borgatti P., Zauli G., Colamussi M.L., Gibellini D., Previati M., Cantley L.L. et al. (1997) Extracellular HIV-1 Tat protein activates phosphatidylinositol 3- and Akt/PKB kinases in CD4+ T lymphoblastoid Jurkat cells. Eur. J. Immunol. 27, 2805–2811 doi: 10.1002/eji.1830271110 [DOI] [PubMed] [Google Scholar]
- 66.Cooray S. (2004) The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 85, 1065–1076 doi: 10.1099/vir.0.19771-0 [DOI] [PubMed] [Google Scholar]
- 67.Manning B.D. and Cantley L.C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 doi: 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.