

Hsp110 functions as both a nucleotide exchange factor and a protein molecular chaperone. A novel yeast Hsp110 mutant reveals that the ability to bind substrate proteins is not required for Hsp110 to support proteostasis under normal conditions but may enhance growth under chronic thermal stress.
Abstract
The highly conserved heat shock protein 70 (Hsp70) is a ubiquitous molecular chaperone essential for maintaining cellular protein homeostasis. The related protein Hsp110 (Sse1/Sse2 in Saccharomyces cerevisiae) functions as a nucleotide exchange factor (NEF) to regulate the protein folding activity of Hsp70. Hsp110/Sse1 also can prevent protein aggregation in vitro via its substrate-binding domain (SBD), but the cellular roles of this “holdase” activity are poorly defined. We generated and characterized an Sse1 mutant that separates, for the first time, its nucleotide exchange and substrate-binding functions. Sse1sbd retains nucleotide-binding and nucleotide exchange activities while exhibiting severe deficiencies in chaperone holdase activity for unfolded polypeptides. In contrast, we observed no effect of the SBD mutation in reconstituted disaggregation or refolding reactions in vitro. In vivo, Sse1sbd successfully heterodimerized with the yeast cytosolic Hsp70s Ssa and Ssb and promoted normal growth, with the exception of sensitivity to prolonged heat but not other proteotoxic stress. Moreover, Sse1sbd was fully competent to support Hsp90-dependent signaling through heterologously expressed glucocorticoid receptor and degradation of a permanently misfolded protein, two previously defined roles for Sse1. We conclude that despite conservation among eukaryotic homologues, chaperone holdase activity is not an obligate function in the Hsp110 family.
INTRODUCTION
Proteins must fold into a proper three-dimensional configuration, or native state, to execute their intended functions. Proteomic stressors such as exposure to harmful chemicals, oxidative stress, and aging can inhibit protein folding, disrupting protein homeostasis and resulting in cell death and human disease (Hartl et al., 2011). Misfolded proteins or amyloid aggregates contribute to the development or progression of neurodegenerative disorders: Alzheimer, Huntington, and Parkinson diseases are all fundamentally diseases of protein misfolding (Soto, 2003; Broadley and Hartl, 2009). Cell survival during and after stress conditions is promoted by molecular chaperones that optimize protein folding by stabilizing folding intermediates until native conformations have been obtained. The highly conserved Hsp70 chaperone is integral to protein biogenesis, quality control, and degradation of terminally misfolded proteins (Kampinga and Craig, 2010). The Hsp70 protein-folding cycle is ATP dependent and is regulated by cochaperones such as Hsp40s and nucleotide exchange factors (NEFs) that stimulate ATP hydrolysis and exchange, respectively (Mayer and Bukau, 2005; Mayer, 2013). The budding yeast Saccharomyces cerevisiae expresses three classes of cytosolic NEFs, all with human orthologues: the Hsp110-type proteins Sse1/Sse2, the HSPBP1-type protein Fes1, and the BAG-1–type protein Snl1 (Bracher and Verghese, 2015). SSE1 deletion results in slow growth and temperature sensitivity, whereas a combined deletion of SSE1 and SSE2 is lethal despite the presence of Fes1 and Snl1, suggesting a potentially unique role for the Hsp110 proteins (Trott et al., 2005; Raviol et al., 2006). The Hsp110 proteins are highly homologous to Hsp70, composed of an amino-terminal nucleotide-binding domain (NBD) and a substrate-binding domain (SBD), which is further subdivided into a β-sandwich domain and an α-helical “lid” domain (Liu and Hendrickson, 2007; Polier et al., 2008; Schuermann et al., 2008). Distinct from Hsp70, Sse1/2 bind ATP, which stabilizes the NBD, but catalytic activity (ATP hydrolysis) is not required to functionally complement the null mutant in vivo or accelerate Hsp70 nucleotide exchange in vitro (Shaner et al., 2005, 2006; Dragovic et al., 2006; Raviol et al., 2006; Andreasson et al., 2008a; Nillegoda and Bukau, 2015).
Whereas the NEF function of Hsp110/Sse is well established, possible biological roles for substrate binding by the SBD remain speculative. Crystal structures of the Hsp70-Sse1 complex depict the Hsp70 SBD in close proximity to the Sse1 β-domain, suggesting possible cooperative substrate binding (Polier et al., 2008; Schuermann et al., 2008). The Hsp110 SBD is structurally similar, but not identical, to that of Hsp70, and it is suggested that it binds peptides much like Hsp70 through interactions with both β-sheets and the connecting loops within the β-domain (Oh et al., 1997, 1999; Polier et al., 2010). Hsp110s are highly efficient at blocking aggregation of misfolded substrates in vitro (defined as “holdase” activity) and Sse1 possesses a unique peptide binding preference for regions enriched in aromatic amino acids relative to the yeast Hsp70, Ssa1 (Goeckeler et al., 2008; Xu et al., 2012). Although contributions to substrate selection and targeting to Hsp70 by Hsp40 cochaperones are established, it is unclear whether the holdase activity of Sse1 or other Hsp110 chaperones contributes to Hsp70-dependent functions in vivo (Johnson and Craig, 2001). Deletion mutagenesis to remove the Sse1 SBD is complicated by the fact that carboxyl-terminal deletions render the protein unstable that the α-helical domain is required for heterodimerization with Hsp70 (Shaner et al., 2005; Polier et al., 2008; Schuermann et al., 2008). Site-specific mutagenesis targeting residues in the Sse1 SBD modeled on the peptide-binding site of the bacterial Hsp70, DnaK, was likewise unsuccessful in significantly impairing holdase activity (Polier et al., 2008). Yeast cells lacking Sse1 are defective in folding of newly synthesized polypeptides and degradation of some misfolded proteins (Dragovic et al., 2006; Gowda et al., 2013; Abrams et al., 2014). However, overexpression of either Fes1 or a soluble, truncated mutant form of the normally endoplasmic reticulum-associated NEF Snl1, both of which lack demonstrated holdase activities, partially suppresses these phenotypes (Raviol et al., 2006; Mandal et al., 2010). In contrast, other NEFs cannot substitute for Hsp110 in protein disaggregation reactions, suggesting that Hsp110 possesses specific properties that could be linked to its unique SBD (Rampelt et al., 2012; Gao et al., 2015; Nillegoda and Bukau, 2015; Nillegoda et al., 2015).
We generated an Sse1 variant that separates, for the first time, the nucleotide exchange and substrate-binding functions of this chaperone. Multiple targeted single-residue substitutions in the β-sandwich region of the SBD were introduced to generate a novel mutant (Sse1sbd) that exhibits greatly reduced aggregation-preventing activity while retaining nucleotide-binding and Hsp70 nucleotide exchange potency. Strikingly, Sse1sbd was competent to restore growth to cells lacking SSE1 and/or SSE2, promote disaggregase activity in a reconstituted in vitro system, and support Hsp70-dependent signal transduction and protein degradation while exhibiting minor defects in stress resistance and protein quality control. The data presented here suggest that the substrate-binding function of Sse1, despite being conserved among the eukaryotic Hsp110 proteins, plays a minor role in maintaining protein homeostasis in the yeast system.
RESULTS
A novel SBD mutation specifically impairs Sse1 holdase activity
We generated a novel Sse1 SBD mutant based on previous structural studies (Polier et al., 2008; Xu et al., 2012) that indicated the region mutated could be within a putative peptide-binding site (Figure 1A). This putative substrate binding–defective mutant (Sse1sbd) includes four specific amino acid substitutions (L433A, N434P, F439L, and M441A) within the L3,4 region of the β-sandwich domain in Sse1. We first verified that the introduced mutations exclusively targeted substrate binding while maintaining proper nucleotide binding in the NBD. Using recombinant proteins purified from Escherichia coli (Supplemental Figure S1A), we measured ATP binding with fluorescently labeled nucleotide through fluorescence anisotropy. Compared with the wild-type protein, Sse1sbd bound FAM-ATP with approximately the same affinity (Kd = 12.1 ± 1.9 µM for Sse1sbd vs. 8.6 ± 1.4 µM for wild-type Sse1; Figure 1B). It was also essential that the mutant protein could still function as a nucleotide exchange factor (NEF) for Hsp70. We measured the exchange of α-32P-ATP loaded onto human Hsc70 (HSPA8) in the absence of NEF or in the presence of Sse1 or Sse1sbd and found no discernible difference in the accelerated exchange rates (Figure 1C). Together these results demonstrate that Sse1sbd retains critical nucleotide-binding and NEF features of the Hsp110 chaperone.
FIGURE 1:

A novel SBD mutant exhibits impaired chaperone holdase activity but retains Hsp70 nucleotide exchange capacity. (A) Crystal structure of the Sse1 β-domain, with amino acids selected for mutations highlighted in red (Xu et al., 2012). (B) Fluorescence anisotropy was performed with increasing concentrations of chaperone (Sse1 or Sse1sbd) binding fluorescently labeled ATP-FAM. (C) Nucleotide exchange activity assays using HSPA8 (Hsp70) prebound to α-32P-ATP in the presence or absence of Sse1. (D) Holdase experiments were conducted using chemically denatured FFL (200 nM) diluted into refolding buffer without chaperone (no chap), with Sse1 (400 nM), or with Sse1sbd (400 nM). FFL diluted into denaturing buffer instead of folding buffer was used as an aggregation control (denat). (E) Differential centrifugation analysis of FFL aggregation in the absence of chaperone or with Sse1 or Sse1sbd after a 30-min holdase assay. Samples were visualized by SDS–PAGE, followed by Coomassie stain, and scanning densitometry quantitation was performed to determine FFL aggregation under each condition. (F) Endpoint analysis of holdase experiments performed as in D, using denatured FFL with varying ratios of chaperone to substrate, quantified as fraction of total aggregation.
To assess whether substrate binding was impaired as predicted, we measured the ability of Sse1 and Sse1sbd to prevent the aggregation of chemically denatured firefly luciferase (FFL) using an established assay system (Garcia et al., 2016). Whereas wild-type Sse1 effectively reduced FFL aggregation relative to that observed in the absence of chaperone, the Sse1sbd protein was significantly impaired in aggregate prevention (Figure 1D). To verify that the spectrophotometric assays reflected substrate aggregation into insoluble material, we analyzed endpoint samples by differential centrifugation followed by SDS–PAGE and densitometry quantitation (Figure 1E). Sse1 maintained 72% of FFL in a soluble state after 30 min, whereas only 39% of FFL is soluble in the presence of Sse1sbd as the chaperone. Similar results were obtained with citrate synthase as the unfolded substrate (Supplemental Figure S1, B–E). Increasing the ratio of Sse1 to FFL allowed for better aggregate prevention, whereas increasing the ratio of Sse1sbd only mildly improved protection of the denatured substrate (Figure 1F). These data indicate that the novel Sse1sbd mutant is defective in its ability to passively chaperone unfolded proteins, whereas NEF function and nucleotide binding remain intact. However, we note that substrate binding, although significantly reduced, is not completely abolished.
Reduction in Sse1 substrate-binding capacity does not reduce disaggregation or refolding by the Hsp70/Hsp40/Hsp110 machine
Hsp110 boosts the aggregate solubilization activity of the Hsp70-based disaggregase machine in a manner that depends on its nucleotide exchange activity (Shorter, 2011; Rampelt et al., 2012; Gao et al., 2015; Nillegoda et al., 2015). We therefore tested whether the substrate-binding function of Sse1 is also required in this capacity. As a first step, we tested whether substrate binding by Sse1 was important for refolding of thermally denatured monomeric FFL. We heat denatured FFL in the presence of HSPA8 (hHsc70), DnaJB1 (hHsp40), and yeast Hsp26 (yHsp26) for 10 min at 42°C (Rampelt et al., 2012). The samples were shifted to 30°C, and a nucleotide regeneration system was added. Refolding of FFL was measured in the presence of no NEF, Sse1, Sse1sbd, or HSPH2 (hHsp110) as a control. It was previously established that yeast and human Hsp110s are functionally interchangeable (Rampelt et al., 2012). Sse1 and Sse1sbd were observed to aid Hsp70/Hsp40 equally in refolding FFL (Figure 2A). Next we tested whether the substrate-binding function of Sse1 might be necessary for the more difficult task of solubilizing aggregated FFL. FFL aggregates were formed by heat denaturation (15 min, 45°C) in the presence of Hsp26, and the aggregates were mixed with a cocktail of chaperones and cochaperones containing hHsc70, hHsp40, and no NEF, Sse1, Sse1sbd, or hHsp110. Again, substrate binding–deficient mutant Sse1sbd functioned with Hsp70/Hsp40 as effectively as the wild-type Sse1 or the hHsp110 control (Figure 2B). Because Sse1sbd is significantly reduced in substrate binding, we infer from these experiments that full Sse1 holdase activity is not obligatory for effective refolding or disaggregase activity of at least the model substrate FFL.
FIGURE 2:

Intact Sse1 SBD function is not obligate for the Hsp70/Hsp40/Hsp110 machine to disaggregate or refold substrates. (A) FFL refolding over time in the presence of different chaperone mixtures as indicated in the schematic and described in detail in Materials and Methods. (B) FFL disaggregation and refolding over time in the presence of the same chaperone mixtures shown in A, as indicated in the schematic and described in detail in Materials and Methods. Human proteins HSPA8, DnaJB1, and HSPH2 are labeled as Hsp70, Hsp40, and Hsp110, respectively, in the experimental schematic and as 70, 40, and 110 in the plots. Yield is calculated as the percentage reactivation of luciferase relative to activity before denaturation.
Sse1 substrate binding domain function is required during heat stress
Sse1 is a critical component of the protein quality control machineries. Indeed, sse1∆ cells demonstrate significant growth deficiencies, including temperature sensitivity, and sse1∆sse2∆ cells are inviable (Trott et al., 2005). Among known cytosolic NEFs, Sse1 and Sse2 are unique for possessing an ability to bind nonnative substrates, raising the possibility that this activity is important for protein quality control processes in vivo. To test this hypothesis, we began by determining the expression of Sse1sbd to ensure that the introduced mutations did not affect its stability in vivo. At 30 and 37°C, plasmid-borne Sse1, Sse1sbd, and a previously described NEF-defective mutant carrying the G233D mutation (here designated Sse1nbd) were expressed at similar levels, but both slightly higher than endogenous Sse1 (Figure 3, A and B, and Supplemental Figure S2, A and B). We also wanted to ensure that Sse1sbd retained interaction with the yeast cytosolic Hsp70s (Ssa and Ssb) to function as a NEF in vivo. All Sse1 proteins were expressed with a FLAG tag fused to the N-terminus, and coimmunoprecipitations were performed (Shaner et al., 2005). Sse1sbd was found to associate with the cytosolic Hsp70s, Ssa, and Ssb at both temperatures in a manner indistinguishable from wild-type Sse1 (Figure 3, C and D), whereas Sse1nbd failed to do so (Supplemental Figure S2, C and D).
FIGURE 3:

Sse1sbd stably interacts with endogenous yeast Hsp70 proteins and supports growth at normal but not heat shock temperatures. (A) Protein lysates from cells expressing the indicated SSE1 alleles and cultured at 30 or 37°C were analyzed by immunoblot to determine expression levels and stability. (B) Quantitative analysis of the immunoblots in A. (C) Coimmunoprecipitation experiments using FLAG-tagged Sse1 variants were performed to assess interactions with endogenous Ssa and Ssb proteins. Samples were analyzed via SDS–PAGE and Coomassie staining. Sse1; closed circle, Ssa/Ssb; open circle. (D) Quantitation of C. (E) Serial dilution plating of sse1Δ or sse1Δsse2Δ cells complemented with the indicated plasmid-expressed Sse1 alleles and cultured at the indicated temperatures. Wedges below images represent relative cell density.
Given that Sse1sbd displayed normal stability and retained Hsp70 interaction at both standard and heat shock temperatures, we next assessed the contribution of substrate binding to Sse1 functions in yeast. As previously mentioned, the sse1nbd allele contains the mutation G233D, which renders it unable to bind nucleotide, interact with Hsp70, or act as a NEF (Shaner et al., 2004, 2005; Dragovic et al., 2006). We compared the growth of sse1∆ cells expressing SSE1, sse1nbd, or sse1sbd under cold stress or heat stress. Whereas sse1sbd fully complements sse1∆ cells grown in optimal conditions and under cold stress, the mutant allele could not confer normal growth under heat stress (Figure 3E). This behavior contrasted with the inability of the sse1nbd allele to complement under any condition, suggesting that thermal stress may impose distinct requirements for Sse1 functions that include NEF and substrate holdase activities. To further probe this question and ask whether the presence of the closely related Sse2 protein masked growth defects of sse1sbd under non–heat shock conditions, we transformed sse1∆sse2∆ cells with sse1sbd- or SSE1-expressing plasmids using a plasmid shuffle technique (Trott et al., 2005). We again observed indistinguishable growth between the two alleles at 30°C, whereas sse1sbd was unable to maintain viability at 37°C (Figure 3E and Supplemental Figure S3A). Consistent with the phenotypes seen under thermal stress, cells grown in the presence of formamide, which acts as a general protein denaturant, exhibited phenotypes consistent with heat stress (Supplemental Figure S3B). Cells expressing the sse1nbd, or sse1sbd were hypersensitive to formamide, and this phenotype was augmented with combined heat stress. Of interest, exposing cells to ethanol stress at 30°C did not affect cells expressing sse1sbd, as they grew at least as well as cells expressing wild-type SSE1 (Supplemental Figure S3C; Trotter et al., 2002). These results suggest that despite being unnecessary for substrate refolding and disaggregation in vitro or resistance to other forms of proteotoxic stress, the Sse1 SBD and its holdase activity are important for cell physiology and survival under prolonged thermal stress. Previous work has established that defects in thermal tolerance in sse1∆ cells are linked to deficiencies in cell wall integrity rather than overall protein homeostasis and correspondingly remediated by growth in an osmotic support medium (Shaner et al., 2008). In keeping with this interpretation, addition of 1 M sorbitol suppressed the temperature sensitivity of sse1∆ and sse1sbd cells without altering the slow-growth phenotype of the sse1nbd mutant (Supplemental Figure S3D).
Reduction in Sse1 holdase activity results in mild proteotoxicity
We envisioned two possible explanations to account for the results obtained from the growth analyses with sse1sbd. One is that Sse1 substrate binding is important only during heat stress due to physiological insults that occur exclusively under those conditions. The second possibility is that the Sse1 SBD is functioning at all times to maintain proteostasis and is dispensable under normal growth conditions but is required to endure the increased burden on protein quality control systems during heat stress. To determine whether a nonfunctional Sse1 SBD has any effect on the proteome while cells are grown under optimal conditions, we assessed the activation of the heat shock response (HSR) as a proxy for disruption of proteostasis using an established HSE-lacZ reporter. To prevent possible variability from plasmid expression in these and subsequent experiments, we chose to directly integrate the SSE1 mutants into the yeast chromosome at the endogenous locus (Supplemental Table S1). It was previously shown that sse1∆ cells exhibit a twofold to fourfold elevated HSR, consistent with chronic proteostatic imbalance (Liu et al., 1999). We confirmed that cells expressing the NEF-defective allele sse1nbd also demonstrated an activated HSR (Figure 4A). Of interest, cells expressing sse1sbd exhibited modest activation of the HSR (∼1.8-fold) supporting the idea that the Sse1 SBD may play some role in proteome maintenance during nonstress conditions. Supplementation of the growth medium with 1 M sorbitol under nonstress conditions before determination of HSR status did not appreciably alter the general pattern observed in Figure 4A but decreased overall activation of the HSR in all strains tested by 20–30% (Supplemental Figure S3E). As a complementary approach, we assessed Hsp90 protein levels because expression of the two Hsp90-encoding genes HSC82 and HSP82 is under the exclusive transcriptional control of Hsf1 (Erkine et al., 1995; Solis et al., 2016). Using immunoblot analysis, we determined that the sse1∆, sse1nbd, sse1sbd cells exhibited a modest 1.5- to 3-fold increase in steady-state Hsp90 levels, in accordance with the HSR reporter activation results (Figure 4B). These data suggest that the holdase activity of Sse1 nominally contributes to proper functioning of the chaperone network under normal physiological conditions. In addition, we infer that the ability of sorbitol to rescue the 37°C growth defects of sse1 mutant strains without changing HSR activation status reflects a deficiency in cell wall integrity distinct from maintenance of protein quality control.
FIGURE 4:
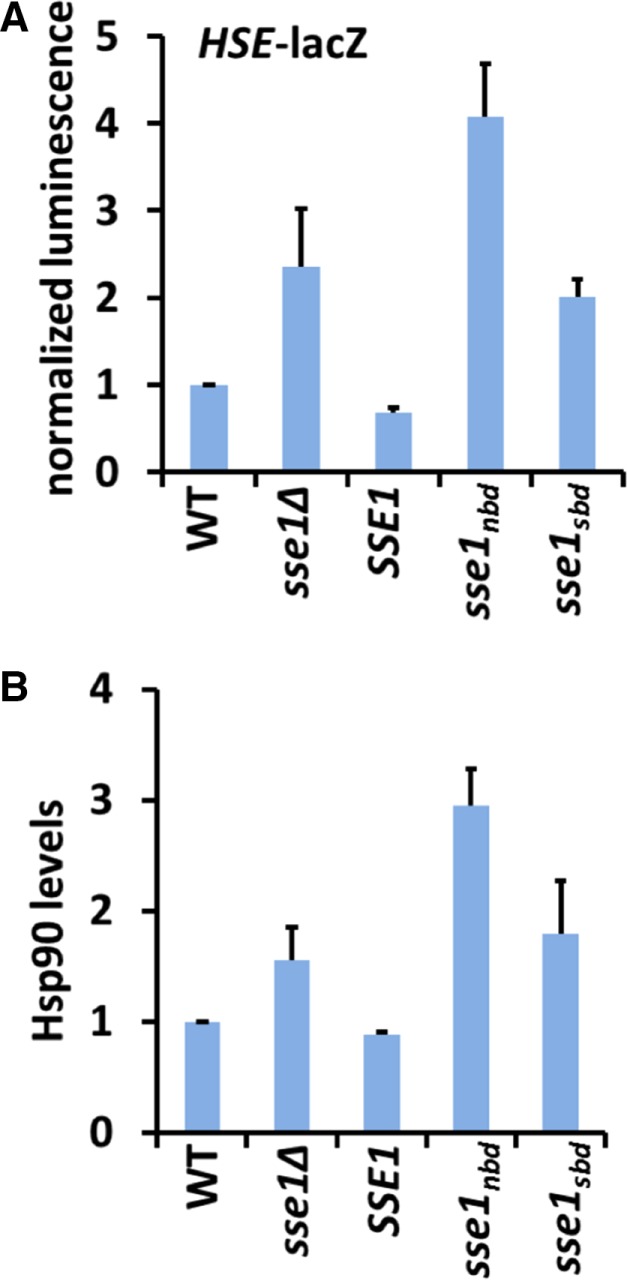
Reduced Sse1 holdase activity results in mild proteotoxicity. (A) β-Galactosidase activity assays from cells expressing the indicated SSE1 alleles integrated at the endogenous locus and expressing the HSF reporter pSSA3HSE-lacZ. (B) Protein lysates from cells grown to mid log phase were analyzed for Hsp90 protein levels by SDS–PAGE and immunoblot, quantified, and normalized to PGK load control. Experiments were repeated in triplicate, and error bars represent SD.
Reduction in Sse1 SBD function does not negatively affect Hsp90-based signal transduction or clearance of terminally misfolded proteins
In addition to general contributions to proteostasis, Sse1 supports signal transduction and functions of Hsp90 (Liu et al., 1999; Mandal et al., 2010). To assess whether this biological role required Sse1 to functionally interact with unfolded substrates, we used the maturation and activation of the mammalian glucocorticoid receptor (GR) in yeast cells as a benchmark of Hsp90 activity. β-Galactosidase activity was measured in cells coexpressing a glucocorticoid response element (GRE)–lacZ reporter and the different SSE1 alleles after activation of the GR via the synthetic hormone deoxycorticosterone (DOC; Figure 5A). Wild-type cells exhibited a robust response to DOC treatment, indicative of GR activation. Similarly, cells expressing sse1sbd were also able to activate the GR, whereas activation was abolished in sse1∆ and sse1nbd cells.
FIGURE 5:

Reduced SBD function does not impair Sse1-dependent biological processes. (A) Activation of the rat glucocorticoid receptor was measured via a lacZ reporter in the absence and presence of 10 µM DOC. (B) Degradation of CPYǂ-GFP after cycloheximide treatment analyzed using anti-GFP antibody. (C) Representative micrographs of the various SSE1 strains at 0 and 90 min after cycloheximide treatment to track CPYǂ-GFP aggregate clearance in the cell population. (D) Quantitation of the experiments in C; percentage calculated as aggregate-containing cells relative to time zero. (E) Representative micrographs of SSE1sse2∆ or sse1sbdsse2∆ strains, tracking CPYǂ-GFP aggregate clearance at the indicated time points. (F) Quantitation of the experiments in D; percentage calculated as aggregate-containing cells relative to time zero. All experiments were performed using cells expressing the indicated SSE1 alleles integrated at the endogenous locus.
Another established cellular role for Sse1 is its participation in the triage decision for Hsp70-mediated protein folding versus degradation, in which Sse1 is required for targeting terminally misfolded proteins to the proteasome for degradation (Mandal et al., 2010). Specifically, Sse1 stimulates ubiquitination and degradation of the model misfolded protein CPY‡–green fluorescent protein (GFP), an engineered variant of the vacuolar protease carboxypeptidase Y that lacks the endoplasmic reticulum (ER) signal sequence and is permanently misfolded (Heck et al., 2010; Abrams et al., 2014). We used CPY‡-GFP to assess whether Sse1 substrate binding was important for targeting terminally misfolded proteins for degradation. After treating cells with cycloheximide, we tracked the clearance of CPY‡-GFP in cells expressing SSE1, sse1ndb, or sse1sbd by immunoblot. We found that sse1nbd-expressing cells matched sse1∆ cells in their inability to clear the terminally misfolded protein after 2 h of cycloheximide chase (Figure 5B). In contrast, sse1sbd-expressing cells fully cleared CPY‡-GFP, indicating that the Sse1 SBD function is not required for targeting terminally misfolded proteins for degradation. In addition to immunoblot analysis, we assessed the amount of CPY‡-GFP aggregates forming in cells expressing the different Sse1 alleles and tracked their clearance over time using fluorescence microscopy. CPY‡-GFP aggregate clearance correlated precisely with protein clearance (Figure 5, C and D). To test whether Sse2 could be masking a substrate-binding role for Sse1 in protein degradation, we constructed SSE1sse2∆ and sse1sbdsse2∆ strains and tracked the ability of these cells to clear the CPY‡-GFP aggregates. Cells expressing the substrate binding–deficient mutant cleared the aggregates at the same rate as SSE1sse2∆ cells (Figure 5, E and F). Together these data strongly support the contention that Sse1 substrate binding is not required to support Hsp90 signaling activities or promote the degradation of terminally misfolded cytosolic proteins.
DISCUSSION
Among the three classes of cytosolic NEFs, Hsp110/Sse is the sole family demonstrated to possess holdase activity for unfolded proteins, yet no in vivo role has been exclusively attributed to this domain. To address this quandary, we generated a novel Sse1 allele that disrupts the ability of the chaperone to prevent aggregation, presumably via substrate binding and sequestration, while maintaining interaction with Hsp70 and NEF activity. Data from our laboratory and others strongly suggest that the yeast cytosolic Hsp110s, Sse1 and Sse2, play critical cellular roles in maintaining protein homeostasis during physiological and stress conditions. This interpretation is bolstered by the fact that sse1∆sse2∆ cells are inviable and, whereas overexpression of the other yeast NEFs can only partially complement growth phenotypes at 30°C, the complete absence of Hsp110 proteins can only be fully remedied by overexpression of either Sse1 or Sse2 (Mandal et al., 2010). In all cases studied, elimination of Hsp110/Sse NEF activity phenocopies the gene deletion, suggesting that, indeed, the NEF function is a primary, if not dominant, role for this class of chaperone. To determine which known Sse1 roles might additionally be affected by loss of Hsp110/Sse holdase activity, we tested refolding and disaggregation in vitro, responses to different proteotoxic stresses, signaling through Hsp90, and targeting of terminally misfolded cytosolic proteins for degradation. Strikingly, we did not observe a demonstrable role for Sse1 SBD function in the reconstituted luciferase refolding or disaggregation reactions, leading us to conclude that the holdase activity is dispensable for these activities. Similarly, Hsp90-dependent signaling and protein degradation was fully supported by the Sse1sbd mutant. This is in apparent contrast to a recent study that identified distinct sequences in two secretory pathway proteins, immunoglobulin γ1 heavy chain and NS-1 Κ light chain, which are preferentially bound by the ER homologue of Hsp110, Grp170, and, when eliminated, disrupt processing of these substrates (Behnke et al., 2016). Although these findings suggest a biological role for Grp170 substrate binding in human cells, these same regions within the substrates are recognized by the ER Hsp40 cochaperones ERdj4 and ERdj5, precluding a clear interpretation that holdase activity by Grp170 is specifically required. However, the finding that Grp170 and ERdj4/5 modulation of substrate processing can occur independently of the ER Hsp70 BiP also suggests that the passive holdase activity of these chaperones and, by extension, Sse1/Hsp110 affects proteome maintenance. This observation might explain the modest activation of the HSR we detect with the Sse1sbd mutant. It would be of interest to determine whether introduction of mutations in the Grp170 SBD analogous to those we demonstrated reduce substrate binding by Sse1 phenocopy elimination of the proposed target sequences in the secretory protein substrates. In addition, models have been proposed in which Hsp110 chaperones are competent to promote the folding of unfolded substrates when assisted by Hsp40 cochaperones in an ATP-dependent folding cycle, an activity that would presumably rely on the SBD (Mattoo et al., 2013). However, the ability of catalytically inactive SSE1 mutant alleles to fully support known Sse1-dependent activities challenges the biological relevance of this observation (Shaner et al., 2004; Raviol et al., 2006).
It is possible that the Hsp110/Sse SBD plays a (minor and perhaps redundant) role in protein folding events that is magnified under certain stress conditions. For example, we found that Sse1sbd was unable to serve as the sole Hsp110 allele under extended growth at 37°C or in the presence of formamide, the latter a phenotype that we and others have demonstrated to be functionally analogous to thermal stress (Hampsey, 1997; Trott and Morano, 2004). We cannot exclude, however, that these phenotypes are ultimately more tightly linked with cell wall integrity than protein homeostasis, an idea reinforced by the clear suppression of sse1 mutant phenotypes with sorbitol, an osmotic stabilizing agent (Shaner et al., 2008; Supplemental Figure S3D). Moreover, Sse1sbd was insensitive to proteotoxicity caused by chronic exposure to 8% ethanol, further challenging the notion that Hsp110/Sse holdase activity is a major contributor to protein homeostasis.
It is relevant to consider that the Sse1sbd mutant is not completely defective in substrate binding, retaining between 20 and 50% of its aggregation prevention potential in a substrate-specific manner. It is possible that a complete abrogation of substrate interaction is necessary to reveal more dramatic phenotypes in the different Sse1 functions tested. However, we attempted to generate a more severe holdase-defective mutant through additional targeted amino acid substitutions based on the work of Xu et al. (2012) but without success. Of importance, we observed nearly identical outcomes in multiple in vitro and in vivo assays that are highly dependent on Sse1 and sensitive to perturbations in its status. Tellingly, the recently described role for Hsp110/Sse as a critical component of the eukaryotic disaggregase machine provided a prime opportunity to answer the open question of whether substrate holding by this family of proteins contributed to the remarkable ability of the Hsp110•Hsp70•Hsp40 complex to extract and refold aggregated proteins. Our findings support the growing contention that Hsp110 NEF activity, not holdase activity, is the key accelerator of disaggregation in this context (Nillegoda and Bukau, 2015). Recent findings reveal that the yeast Hsp104-based disaggregation activity is coordinated in a manner that requires the interaction of Sse1 and Sse2 with Hsp70 (Kaimal et al., 2017). Efficient recruitment of Hsp104 to aggregates was found to require Sse1/2, and in turn recruitment of Sse1/2 was dependent on Ssa1/Hsp70, demonstrated by using the Hsp70 binding–defective SSE1 allele sse1-2,3 (Polier et al., 2008). However, Sse1/2 was also shown to be required for efficient Ssa1/Hsp70 association with aggregates, suggesting that Hsp110 may be limiting for generating sufficient cellular levels of Ssa1/Hsp70-ATP (Kaimal et al., 2017). Because the sse1-2,3 allele presumably retains substrate-binding function, it may be inferred that the Sse1 SBD is insufficient for recruitment to luciferase aggregates, consistent with our finding that disaggregation and clearance of CPYǂ-GFP aggregates proceeded unimpeded in cells expressing Sse1sbd.
Because the Sse1sbd mutant is not completely without substrate-binding capacity, we cannot yet formally exclude a role for substrate binding by Hsp110 chaperones in proteostasis. The passive holdase activity of Hsp110/Sse has been shown to promote the refolding of luciferase in yeast cytosol, likely by stabilizing the unfolded polypeptide and preventing its aggregation. This activity may also be compared with subtle interactions under certain conditions with the Sup35 prion in yeast, which appear to be independent of Sse1 NEF function (Brodsky et al., 1999; Sadlish et al., 2008). In both of the latter scenarios, the Sse1 holdase function is likely operating independently of Hsp70. It may be of interest to further probe potential contributions of Hsp110/Sse1 holdase activity in aggregate prevention for specific aggregation-prone substrates. For example, Hsp105α (HSPH1) in human cells is known to modulate cystic fibrosis transmembrane conductance regulator folding and processing (Saxena et al., 2012). Hsp110 suppresses the aggregation and associated toxicity of the mutant proteins that lead to amyotrophic lateral sclerosis and Alzheimer disease, respectively, when expressed in Caenorhabditis elegans and mice (Wang et al., 2009; Eroglu et al., 2010). Hsp110 has also been found to be an important modulator of neuronal degeneration caused by the expression of toxic polyglutamine proteins that model Huntington disease in the fly (Zhang et al., 2010; Kuo et al., 2013). Strikingly, Hsp110 can also ameliorate toxicity caused by the G85R variant of SOD1, a contributor to amyotrophic lateral sclerosis, significantly extending survival of SOD1G85R-YFP transgenic mice when overexpressed in motor neurons (Song et al., 2013; Nagy et al., 2016). The specific mechanisms by which Hsp110 prevents aggregation and disease progression in these model systems are unknown. Given the increasing significance of Hsp110 chaperones in modulation of proteotoxic aggregation, it will be important to define more precisely the features that contribute to such activities as a precursor to therapeutically manipulating the chaperone network to combat progression of protein-misfolding disorders.
MATERIALS AND METHODS
Strains, plasmids, and yeast culture
All yeast strains are derived from either BY4741 or W303 parent background (Supplemental Table S1). Yeast were propagated on standard rich (YPD; 1% yeast extract, 2% peptone, 2% glucose) or synthetic minimal complete media lacking amino acids for marker selection (Sunrise Science, San Diego, CA) at 30°C unless otherwise specified. Complementation of the lethal sse1∆sse2∆ mutant strain was conducted using a standard yeast plasmid shuffle technique with a URA3-based, SSE1-expressing plasmid and counterselection using 5-fluroorotic acid. Mutant sse1sbd was constructed via site-directed mutagenesis by PCR using the plasmid p413TEF-FLAG-SSE1 as a template. This SSE1 allele and the sse1nbd mutant (sse1-G233D; previously described in Shaner et al., 2004) were subcloned into the 413TEF vector using SpeI/XhoI restriction sites (Mumberg et al., 1995). For growth complementation and immunoprecipitation experiments, SSE1 alleles were expressed from the p413TEF plasmid. A FLAG epitope tag (DYKDDDDK) was added to the 5′ end of the SSE1 genes immediately after the start codon by using primers that included the FLAG-encoding, yeast-optimized sequence (GACTACAAGGACGACGATGACAAAATG). Strains expressing the various SSE1 alleles from the endogenous locus (YPL106C) were constructed by gene replacement. SSE1 amplicons were generated from plasmids containing a CYC1 terminator sequence using primers (5′-ATAACTCTGTCCTTGCCGT-3′) and (5′-TACTCTGTCAGAAACGGCCTGTACCGGCCGCAAATTAAAGCC-3′) to PCR amplify from nucleotide +35 relative to the ATG in SSE1 (forward primer) to the end of the CYC1 terminator with an overhang with homology to the LEU2 terminator (reverse primer). The LEU2 cassette was PCR amplified from plasmid DNA using a forward primer matching the CYC1 terminator (5′-GCTTTAATTTGCGGCCGGTACAGGCCGTTTCTGACAGAGTAAAATTCTTG) and a reverse primer with an overhang matching the endogenous SSE1 terminator (5′-AATCTTTTTTTAACTATACAGAGAAGATATTAGTATTTCACACCGCATATCG-3′). The two PCR amplicons were cotransformed into the BY4741 parent strain, and successful Leu+ double recombinants were selected. Individual clones were obtained and sequenced to verify correct integration, presence of desired mutations, and absence of additional nucleotide substitutions.
Hsf1 activity was measured with strains harboring plasmid pSSA3HSE-lacZ as described (Abrams et al., 2014). For experiments testing CPYǂ-GFP degradation, strains were constructed using pRH2081 (generous gift from Randy Hampton, University of California, San Diego), a plasmid that carries TDH3-driven CPY‡-GFP (Heck et al., 2010). The integrative plasmid was linearized using restriction endonuclease Van91I and transformed into indicated strains with Ura+ selection. For growth analysis, cells were grown to mid logarithmic phase and normalized to an OD600 of 1.0. Tenfold dilutions were plated on minimal medium and incubated as described in figure legends. Spot plates were imaged after 2–3 d of growth.
Protein purification
Purified firefly luciferase (Sigma L-9506) and citrate synthase (Sigma C-2360) were obtained from Sigma-Aldrich (St. Louis, MO). Sse1 was purified from E. coli BL21 (DE3) by metal affinity chromatography followed by size exclusion chromatography as described in Garcia et al. (2016). HSPA8 (hHsp70), DNAJB1 (hHsp40), and HSPH2 (hHsp110) were expressed in E. coli BL21 (DE3) with an N-terminal His6-Smt3 tag and purified by affinity chromatography using a Ni-IDA matrix and further subjected to size exclusion chromatography. The His6-Smt3 tag was cleaved with Ulp1 as published earlier (Andreasson et al., 2008b). Purified firefly luciferase (Sigma L-9506) and citrate synthase (Sigma C-2360) were obtained from Sigma-Aldrich.
Nucleotide-binding assay
Fluorescently labeled nucleotide, N6-(6-amino)hexyl-ATP-5-FAM (ATP-FAM; Jena Bioscience, Jena, Germany), was incubated at a concentration of 20 nM with increasing amounts of Sse1 chaperone in buffer (25 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 50 mM KCl, 5% glycerol) for 30 min at room temperature as described (Rauch and Gestwicki, 2014). Fluorescence polarization was measured (excitation λ = 485 nm, emission λ = 535 nm) using a SpectraMax M5 plate reader. Equilibrium binding constants were calculated using a saturation binding one-site equation via GraphPad Prism version 6 (GraphPad Software, La Jolla, CA).
Nucleotide exchange assay
The HSPA8 (Hsc70) protein was a generous gift from Betty Craig (University of Wisconsin, Madison, WI). HSPA8 (70 µg) was loaded with 100 µCi of α-32P-ATP in a total volume of 120 µl of complex buffer (25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]–KOH, pH 7.5, 100 mM KCl, 11 mM MgOAc, and 25 µM ATP) for 30 min at 4°C, and HSPA8–32P-ATP complex was obtained by centrifugation through a Microspin G-25 column (GE Healthcare, Chicago, IL). Labeled HSPA8 (7.8 μg) was incubated in the presence or absence of 5 μg of Sse1 or Sse1sbd at 30°C. At 0, 7, and 15 min, the HSPA8-Sse1 reactions were again passed over G-25 columns to separate from released nucleotide. Radiolabeled nucleotide that remained bound to HSPA8 was determined using a TRI-CARB 2900TR Liquid Scintillation Analyzer and normalized to counts obtained at time zero.
Protein aggregation assay
Substrate aggregation was measured in a Synergy MX Microplate Reader (BioTek, Winooski, VT) as described, with the following modifications (Garcia et al., 2016). Stock concentrations of firefly luciferase or citrate synthase were incubated in denaturing buffer (6 M guanidinium chloride, 5 mM dithiothreitol [DTT]) for 1 h at room temperature. In a 96-well, half-area, ultraviolet-transmissible plate (675801; Greiner Bio-One, Monroe, NC) refolding buffer alone (25 mM Tris-HCl, pH 7.5, 100 mM NaCl), varying concentrations of chaperone in refolding buffer, or denaturing buffer was preequilibrated at 25°C for 5 min, and baseline light scattering was determined. After equilibration, chemically denatured substrate was added to each sample at a final concentration of 200 nM into the refolding buffer to a final volume of 180 µl. The samples were mixed vigorously for 5 s, and aggregation was measured at 320 nm at 30-s intervals for 30 min. Changes in absorbance were calculated after subtracting baseline absorbance at time zero. To assess fractionation of protein into soluble and insoluble aggregates, samples (175 µl) were taken from the endpoint of the substrate aggregation experiments and subjected to centrifugation at 16,000 × g for 4 min. We recovered 170 µl as the supernatant or soluble fraction. The lower 5-µl fraction was considered the pellet or insoluble fraction, and volume was normalized to 170 µl with the addition of refolding buffer. We separated 30 µl of each fraction by 12% SDS–PAGE and stained them with Coomassie blue. Band densities were calculated using Image Studio Software (Li-Cor Biosciences, Lincoln, NE).
FFL refolding and disaggregation
Solubilization of aggregated luciferase was performed as described (Rampelt et al., 2012; Nillegoda et al., 2015). In brief, thermal aggregation was performed by incubating 0.02 μM of native luciferase with 0.1 μM of yHsp26 at 45°C for 15 min in refolding buffer (40 mM HEPES-KOH, pH 7.5, 50 mM KCl, 5 mM MgCl2, 2 mM DTT, 2 mM ATP, 10 μM bovine serum albumin) without the ATP-regenerating system in a water bath. The disaggregation reaction was started by adding 3 mM phosphoenolpyruvate and 20 ng/μl pyruvate kinase (ATP-regenerating system) with the indicated chaperone combinations at the concentrations 2 μM HSPA8 (hHsp70), 1 μM DNAJB1 (hHsp40), 0.2 μM HSPH2 (hHsp110), 0.2 μM Sse1 (yHsp110), and 0.2 μM Sse1sbd (yHsp110sbd), and shifting the reaction to 30°C. For the refolding-only assays, 0.02 μM luciferase was incubated with the indicated chaperone cocktails (2 μM HSPA8 [hHsp70], 1 μM DNAJB1 [hHsp40], HSPH2 0.2 μM [hHsp110], 0.2 μM Sse1 [yHsp110], 0.2 μM Sse1sbd [yHsp110sbd], and 0.1 μM yHsp26) and heat denatured at 42°C for 10 min in refolding buffer to generate thermally denatured luciferase (Nillegoda et al., 2015). Luciferase refolding was initiated by adding an ATP-regenerating system and shifting the reaction to 30°C. Luciferase reactivation was monitored at the indicated time points with a Lumat LB 9507 luminometer (Berthold Technologies) by transferring 1 μl of sample to 100 μl of assay buffer (25 mM glycylglycine, pH 7.4, 5 mM ATP, pH 7, 100 mM KCl, and 15 mM MgCl2) mixed with 100 μl of 0.25 mM luciferin.
Protein levels in vivo
Cultures were grown overnight and secondary cultures started and allowed to grow to an OD600 of 0.8, at which point cells were shifted to 37°C or maintained at 30°C for 6 h. Cells were collected and processed for protein lysates. Sse1 protein levels were detected by immunoblot using anti-Sse1 antiserum (generous gift from Jeff Brodsky, University of Pittsburgh, Pittsburgh, PA), and anti–phosphoglycerate kinase (PGK; Invitrogen, Carlsbad, CA) was used as a loading control. Band quantitation was performed using Image Studio Software, and Sse1 levels were normalized to the levels of PGK.
Immunoprecipitation and immunoblotting
Sse1 proteins were expressed with an N-terminal FLAG tag. Protein extracts were prepared from 30 ml of cultures grown at 30 or 37°C for 6 h. Protein lysates were incubated with 40 µl of M2 resin (Sigma-Aldrich) in TEGN (20 mM Tris-HCl, pH 7.9, 0.5 mM EDTA, 10% glycerol, 50 mM NaCl) at 4°C for 2 h. After washing with 4 ml of buffer, the resin was incubated with 40 µl of FLAG peptide for 25 min at room temperature to elute the FLAG-Sse1 complexes. Immunoprecipitated proteins were analyzed by SDS–PAGE and Coomassie stain. Band analysis was performed using Image Studio Software, and the coimmunoprecipitation efficiency of Hsp70 was calculated relative to the amount of Sse1 immunoprecipitated. Hsp90 levels were assessed by immunoblot using anti-Hsp90 (a generous gift from Avrom Caplan, CUNY, New York, NY) with anti-Sse1 and anti-PGK as internal controls. Band analysis was performed using Image Studio Software, and Hsp90 levels were normalized to PGK levels.
Glucocorticoid receptor activation
The various Sse1 strains were transformed with plasmids pCH-Flag-RatGR and pYRP-G2, expressing the glucocorticoid receptor protein and a GR-lacZ transcriptional reporter, respectively (Liu et al., 1999). Cells grown to mid logarithmic phase were treated with dimethyl sulfoxide (DMSO) only (–DOC) or 10 µM DOC in DMSO (+DOC) for 1.5 h. β-Galactosidase activity was measured by adding 50 µl of cell suspension at OD600 of 0.4 and 50 µl of Beta-Glo reagent (Promega, Madison, WI) and incubating for 30 min at 30°C, followed by luminescence detection using a Synergy MX Microplate Reader.
CPY‡-GFP degradation assay
To track the degradation of the CPY‡-GFP protein in vivo, cells were grown to mid logarithmic phase and treated with 100 µg/ml cycloheximide, and 10 ml of culture was collected at 0, 1, and 2 h. Denatured protein extracts were prepared using a glass bead lysis method with SUME buffer (1% SDS, 8M urea, 10 mM 3-(N-morpholino)propanesulfonic acid, pH 6.8, 10 mM EDTA). The CPY‡-GFP protein was detected by immunoblot using anti-GFP (Roche, Basel, Switzerland), and anti-PGK was used an internal control. In parallel experiments, CPY‡-GFP–expressing cells were collected immediately after treatment at 0, 45, and 90 min and visualized using an Olympus IX81-ZDC inverted microscope as described in Abrams and Morano (2013).
Statistical analysis
All experiments were performed at least in triplicate and quantitation shown as the mean, with error bars indicating SD as calculated in Microsoft Excel.
Supplementary Material
Acknowledgments
We thank Jason Gestwicki and Victoria Assimon (University of California, San Francisco) for help with the fluorescence anisotropy experiments and Elizabeth Craig (University of Wisconsin), Avrom Caplan (CUNY), Jeffrey Brodsky (University of Pittsburgh), and Randy Hampton (University of California, San Diego) for providing reagents. V.M.G. is supported by Grant F31GM113521 from the National Institute of General Medical Sciences/National Institutes of Health. This work was supported by Grant GM074696 from the National Institute of General Medical Sciences/National Institutes of Health (to K.A.M.) and the Deutsche Forschungsgemeinschaft (SFB1036, BU 617/19-3 to B.B.).
Abbreviations used:
- HSP
heat shock protein
- NBD
nucleotide-binding domain
- NEF
nucleotide exchange factor
- SBD
substrate-binding domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0070) on May 24, 2017.
REFERENCES
- Abrams JL, Morano KA. Coupled assays for monitoring protein refolding in Saccharomyces cerevisiae. J Vis Exp. 2013;77:e50432. doi: 10.3791/50432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams JL, Verghese J, Gibney PA, Morano KA. Hierarchical functional specificity of cytosolic heat shock protein 70 (Hsp70) nucleotide exchange factors in yeast. J Biol Chem. 2014;289:13155–13167. doi: 10.1074/jbc.M113.530014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson C, Fiaux J, Rampelt H, Druffel-Augustin S, Bukau B. Insights into the structural dynamics of the Hsp110-Hsp70 interaction reveal the mechanism for nucleotide exchange activity. Proc Natl Acad Sci USA. 2008a;105:16519–16524. doi: 10.1073/pnas.0804187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson C, Fiaux J, Rampelt H, Mayer MP, Bukau B. Hsp110 is a nucleotide-activated exchange factor for Hsp70. J Biol Chem. 2008b;283:8877–8884. doi: 10.1074/jbc.M710063200. [DOI] [PubMed] [Google Scholar]
- Behnke J, Mann MJ, Scruggs FL, Feige MJ, Hendershot LM. Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol Cell. 2016;63:739–752. doi: 10.1016/j.molcel.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracher A, Verghese J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front Mol Biosci. 2015;2:10. doi: 10.3389/fmolb.2015.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadley SA, Hartl FU. The role of molecular chaperones in human misfolding diseases. FEBS Lett. 2009;583:2647–2653. doi: 10.1016/j.febslet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB, McCracken AA. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem. 1999;274:3453–3460. doi: 10.1074/jbc.274.6.3453. [DOI] [PubMed] [Google Scholar]
- Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006;25:2519–2528. doi: 10.1038/sj.emboj.7601138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkine AM, Szent-Gyorgyi C, Simmons SF, Gross DS. The upstream sequences of the HSP82 and HSC82 genes of Saccharomyces cerevisiae: regulatory elements and nucleosome positioning motifs. Yeast. 1995;11:573–580. doi: 10.1002/yea.320110607. [DOI] [PubMed] [Google Scholar]
- Eroglu B, Moskophidis D, Mivechi NF. Loss of Hsp110 leads to age-dependent tau hyperphosphorylation and early accumulation of insoluble amyloid beta. Mol Cell Biol. 2010;30:4626–4643. doi: 10.1128/MCB.01493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Carroni M, Nussbaum-Krammer C, Mogk A, Nillegoda NB, Szlachcic A, Guilbride DL, Saibil HR, Mayer MP, Bukau B. Human Hsp70 disaggregase reverses Parkinson's-linked α-synuclein amyloid fibrils. Mol Cell. 2015;59:781–793. doi: 10.1016/j.molcel.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia VM, Rowlett VW, Margolin W, Morano KA. Semi-automated microplate monitoring of protein polymerization and aggregation. Anal Biochem. 2016;508:9–11. doi: 10.1016/j.ab.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeckeler JL, Petruso AP, Aguirre J, Clement CC, Chiosis G, Brodsky JL. The yeast Hsp110, Sse1p, exhibits high-affinity peptide binding. FEBS Lett. 2008;582:2393–2396. doi: 10.1016/j.febslet.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda NK, Kandasamy G, Froehlich MS, Dohmen RJ, Andreasson C. Hsp70 nucleotide exchange factor Fes1 is essential for ubiquitin-dependent degradation of misfolded cytosolic proteins. Proc Natl Acad Sci USA. 2013;110:5975–5980. doi: 10.1073/pnas.1216778110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampsey M. A review of phenotypes in Saccharomyces cerevisiae. Yeast. 1997;13:1099–1133. doi: 10.1002/(SICI)1097-0061(19970930)13:12<1099::AID-YEA177>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Craig EA. An essential role for the substrate-binding region of Hsp40s in Saccharomyces cerevisiae. J Cell Biol. 2001;152:851–856. doi: 10.1083/jcb.152.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimal JM, Kandasamy G, Gasser F, Andreasson C. Coordinated Hsp110 and Hsp104 activities power protein disaggregation in Saccharomyces cerevisiae. Mol Cell Biol. 2017;37:e00027–17. doi: 10.1128/MCB.00027-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The Hsp70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y, Ren S, Lao U, Edgar BA, Wang T. Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis. 2013;4:e833. doi: 10.1038/cddis.2013.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Hendrickson WA. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 2007;131:106–120. doi: 10.1016/j.cell.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XD, Morano KA, Thiele DJ. The yeast Hsp110 family member, Sse1, is an Hsp90 cochaperone. J Biol Chem. 1999;274:26654–26660. doi: 10.1074/jbc.274.38.26654. [DOI] [PubMed] [Google Scholar]
- Mandal AK, Gibney PA, Nillegoda NB, Theodoraki MA, Caplan AJ, Morano KA. Hsp110 chaperones control client fate determination in the Hsp70-Hsp90 chaperone system. Mol Biol Cell. 2010;21:1439–1448. doi: 10.1091/mbc.E09-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoo RU, Sharma SK, Priya S, Finka A, Goloubinoff P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J Biol Chem. 2013;288:21399–21411. doi: 10.1074/jbc.M113.479253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem Sci. 2013;38:507–514. doi: 10.1016/j.tibs.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- Nagy M, Fenton WA, Li D, Furtak K, Horwich AL. Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc Natl Acad Sci USA. 2016;113:5424–5428. doi: 10.1073/pnas.1604885113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Bukau B. Metazoan Hsp70-based protein disaggregases: emergence and mechanisms. Front Mol Biosci. 2015;2:57. doi: 10.3389/fmolb.2015.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, Arnsburg K, Gao X, Scior A, Aebersold R, et al. Crucial Hsp70 co-chaperone complex unlocks metazoan protein disaggregation. Nature. 2015;524:247–251. doi: 10.1038/nature14884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh HJ, Chen X, Subjeck JR. Hsp110 protects heat-denatured proteins and confers cellular thermoresistance. J Biol Chem. 1997;272:31636–31640. doi: 10.1074/jbc.272.50.31636. [DOI] [PubMed] [Google Scholar]
- Oh HJ, Easton D, Murawski M, Kaneko Y, Subjeck JR. The chaperoning activity of Hsp110. Identification of functional domains by use of targeted deletions. J Biol Chem. 1999;274:15712–15718. doi: 10.1074/jbc.274.22.15712. [DOI] [PubMed] [Google Scholar]
- Polier S, Dragovic Z, Hartl FU, Bracher A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell. 2008;133:1068–1079. doi: 10.1016/j.cell.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Polier S, Hartl FU, Bracher A. Interaction of the Hsp110 molecular chaperones from S. cerevisiae with substrate protein. J Mol Biol. 2010;401:696–707. doi: 10.1016/j.jmb.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012;31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch JN, Gestwicki JE. Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. J Biol Chem. 2014;289:1402–1414. doi: 10.1074/jbc.M113.521997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006;25:2510–2518. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadlish H, Rampelt H, Shorter J, Wegrzyn RD, Andreasson C, Lindquist S, Bukau B. Hsp110 chaperones regulate prion formation and propagation in S. cerevisiae by two discrete activities. PLoS One. 2008;3:e1763. doi: 10.1371/journal.pone.0001763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Banasavadi-Siddegowda YK, Fan Y, Bhattacharya S, Roy G, Giovannucci DR, Frizzell RA, Wang X. Human heat shock protein 105/110 kDa (Hsp105/110) regulates biogenesis and quality control of misfolded cystic fibrosis transmembrane conductance regulator at multiple levels. J Biol Chem. 2012;287:19158–19170. doi: 10.1074/jbc.M111.297580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuermann JP, Jiang J, Cuellar J, Llorca O, Wang L, Gimenez LE, Jin S, Taylor AB, Demeler B, Morano KA, et al. Structure of the Hsp110:Hsc70 nucleotide exchange machine. Mol Cell. 2008;31:232–243. doi: 10.1016/j.molcel.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner L, Gibney PA, Morano KA. The Hsp110 protein chaperone Sse1 is required for yeast cell wall integrity and morphogenesis. Curr Genet. 2008;54:1–11. doi: 10.1007/s00294-008-0193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner L, Sousa R, Morano KA. Characterization of hsp70 binding and nucleotide exchange by the yeast Hsp110 chaperone Sse1. Biochemistry. 2006;45:15075–15084. doi: 10.1021/bi061279k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner L, Trott A, Goeckeler JL, Brodsky JL, Morano KA. The function of the yeast molecular chaperone Sse1 is mechanistically distinct from the closely related hsp70 family. J Biol Chem. 2004;279:21992–22001. doi: 10.1074/jbc.M313739200. [DOI] [PubMed] [Google Scholar]
- Shaner L, Wegele H, Buchner J, Morano KA. The yeast Hsp110 Sse1 functionally interacts with the Hsp70 chaperones Ssa and Ssb. J Biol Chem. 2005;280:41262–41269. doi: 10.1074/jbc.M503614200. [DOI] [PubMed] [Google Scholar]
- Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One. 2011;6:e26319. doi: 10.1371/journal.pone.0026319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis EJ, Pandey JP, Zheng X, Jin DX, Gupta PB, Airoldi EM, Pincus D, Denic V. Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic proteostasis. Mol Cell. 2016;63:60–71. doi: 10.1016/j.molcel.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Nagy M, Ni W, Tyagi NK, Fenton WA, Lopez-Giraldez F, Overton JD, Horwich AL, Brady ST. Molecular chaperone Hsp110 rescues a vesicle transport defect produced by an ALS-associated mutant SOD1 protein in squid axoplasm. Proc Natl Acad Sci USA. 2013;110:5428–5433. doi: 10.1073/pnas.1303279110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- Trott A, Morano KA. SYM1 is the stress-induced Saccharomyces cerevisiae ortholog of the mammalian kidney disease gene Mpv17 and is required for ethanol metabolism and tolerance during heat shock. Eukaryot Cell. 2004;3:620–631. doi: 10.1128/EC.3.3.620-631.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott A, Shaner L, Morano KA. The molecular chaperone Sse1 and the growth control protein kinase Sch 9 collaborate to regulate protein kinase A activity in Saccharomyces cerevisiae. Genetics. 2005;170:1009–1021. doi: 10.1534/genetics.105.043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter EW, Kao CM, Berenfeld L, Botstein D, Petsko GA, Gray JV. Misfolded proteins are competent to mediate a subset of the responses to heat shock in Saccharomyces cerevisiae. J Biol Chem. 2002;277:44817–44825. doi: 10.1074/jbc.M204686200. [DOI] [PubMed] [Google Scholar]
- Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Sarbeng EB, Vorvis C, Kumar DP, Zhou L, Liu Q. Unique peptide substrate binding properties of 110-kDa heat-shock protein (Hsp110) determine its distinct chaperone activity. J Biol Chem. 2012;287:5661–5672. doi: 10.1074/jbc.M111.275057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Binari R, Zhou R, Perrimon N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics. 2010;184:1165–1179. doi: 10.1534/genetics.109.112516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.