

The miRNA complex with Argonaute protein recognizes and down-regulates target mRNA. The role of interorganellar interactions in controlling miRNA activity in mammalian macrophages infected with Leishmania is explored.
Abstract
MicroRNAs (miRNAs), the tiny regulatory RNAs, form complexes with Argonaute (Ago) proteins and inhibit gene expression in metazoan cells. While studying parasite-invaded macrophages, we identify a unique mode of gene regulation in which the parasite Leishmania donovani (Ld) causes mitochondrial depolarization, reduces mitochondrial dynamics, and restricts turnover of cellular microRNA ribonucleoprotein (miRNP) complexes in infected host cells. This leads to increased stability of miRNPs along with elevated levels of Ago2-bound cytokine mRNA in Ld-infected macrophages. Thus the increase of miRNP stability in Ld-infected cells curtails production of proinflammatory cytokines, which are otherwise detrimental for survival of the parasite within the infected macrophages. Loss of mitochondrial membrane potential is accompanied by reduced juxtaposition of endoplasmic reticulum (ER) and mitochondria as well as endosomes. This is likely coupled with enhanced sequestration and stabilization of ER- associated miRNPs observed in infected macrophage cells. Mitofusin 2 (Mfn2), a membrane protein implicated in ER–mitochondria tethering, also shows reduced expression in Ld-infected cells. A mitochondrial role in Ld-induced alteration of miRNA activity and stability is further corroborated by impaired compartmentalization and stabilization of miRNP components in Mfn2-depleted mammalian cells.
INTRODUCTION
MicroRNAs (miRNAs), 22- to 24-nucleotide-long noncoding RNAs, form microRNA ribonucleoprotein (miRNP) complexes with Argonaute (Ago) proteins and posttranscriptionally regulate diverse biological processes in metazoan cells. The miRNA-mediated gene repression process can occur via translation inhibition or mRNA degradation (Bartel, 2009). Recent studies in plants and in animals have allowed us to establish the association of miRNP complexes with endomembranes during miRNA biogenesis and repressive function (Kim et al., 2014; Barman and Bhattacharyya, 2015; Bose et al., 2017). In particular, the endoplasmic reticulum (ER) has been reported as the site for RNA-induced silencing complex (RISC) nucleation as well as translational repression, whereas RISC accumulation and its possible action have been associated with endosomes and multivesicular bodies (MVBs) (Gibbings et al., 2009; Lee et al., 2009; Li et al., 2013; Rogers and Chen, 2013; Stalder et al., 2013). Mitochondria, primarily considered as powerhouses of eukaryotic cells, show enrichment for a specific subset of small RNAs known as mito-miRs (Bandiera et al., 2011). In addition, the effect of mitochondrial membrane potential disruption on RNA interference and the dynamics of cytoplasmic RNA granules such as Processing-body (P-body) has been documented in mammalian cells (Kren et al., 2009; Huang et al., 2011a). Cumulatively, this evidence indicates a functional compartmentalization of miRNPs associated with various cellular organelles in space and time. What remains to be addressed is how these pools of miRNP complexes, located on different organelles, interact for efficient regulation of miRNA activity and turnover.
Defective miRNA-mediated repression of target RNA is associated with several human diseases. Moreover, specific changes in cellular miRNA profiles under altered cellular environments have also been reported (Mendell and Olson, 2012). Many pathogenic parasites are known to alter the miRNA-mediated repression in order to survive in host cells (Skalsky and Cullen, 2010; Huang et al., 2011b; Manzano-Roman and Siles-Lucas, 2012). Leishmania donovani (Ld) is a dimorphic pathogenic parasite that causes visceral leishmaniasis or “kala-azar” in vertebrates (Murray et al., 2005). Ld infects host macrophages upon its entry through sand fly bites (Desjardins and Descoteaux, 1998; Engwerda et al., 2004; Olivier et al., 2005). For efficient infection of macrophages, Ld down-regulates the proinflammatory response of host macrophages by inducing the expression of uncoupler protein 2 (Ucp2; Rousset et al., 2006; Basu Ball et al., 2011), a mitochondrial membrane protein that modulates mitochondrial membrane potential by uncoupling the electron transport chain (Skulachev, 1998).
Mitofusin (Mfn) 1 and Mfn 2 are mitochondrial dynamin–related GTPases. Global ablation of the Mfn1 or Mfn2 gene results in embryonic death. Mfn1 plays a role in mitochondrial docking and fusion (Chen et al., 2003; Koshiba et al., 2004). Mfn2, with its lower GTPase activity, is present on both mitochondria and ER membranes and plays a key role in controlling ER–mitochondria tethering, besides its well-established role in mitochondrial fusion (Chang et al., 2004; Ishihara et al., 2004; de Brito and Scorrano, 2008; Schrepfer and Scorrano, 2016).
Here we report how the pathogen Ld disrupts efficient miRNA turnover in infected host macrophages, possibly by causing mitochondrial depolarization via induced expression of Ucp2. Ucp2-mediated mitochondrial depolarization results in stabilization of host miRNPs and reduced proinflammatory cytokine production. Moreover, Ld down-regulates the protein Mfn2, a factor responsible for mitochondria–ER tethering, thereby possibly affecting mitochondria-ER-endosome interaction, and also modulates miRNA stabilization within mammalian cells. Thus we identified a unique mode of posttranscriptional gene regulation in mammalian cells by which mitochondria–ER interorganellar interaction may control stability of the miRNA–Ago complex. This process is affected by the pathogenic parasite Leishmania, which alters miRNA activity upon infection of host macrophage cells to restrict production of proinflammatory cytokines.
RESULTS
Ld invasion increases miRNP levels and activity in host macrophages
Ld is a pathogenic parasite that infects and resides within tissue macrophage. Entry of Ld within host macrophage is accompanied by an overall reduction of host proinflammatory response, which helps in establishing the infection. Most proinflammatory cytokine–encoding mRNAs either have predicted or established miRNA-binding sites in their 3′ untranslated regions (O’Neill et al., 2011). Therefore modulation of miRNA activity could restrict the expression of cytokines in mammalian macrophages. Does Ld affect miRNA activity in host macrophages? Modulation of miR-122 levels in Ld-infected murine liver has been reported. Reduction of miR-122 caused by the pathogen is essential for a reduction in cholesterol production that the parasite uses for its benefit. Of interest, during infection, an increased Ago2 protein level was observed in infected mouse liver, but the cause and consequence of that increase were not clear (Ghosh et al., 2013). With murine primary macrophage or RAW264.7 cells, we documented a similar increase of Ago2 protein level during Ld infection (Figure 1A). Gw182 protein, essential for miRNA-mediated gene repression, is also elevated in infected macrophages, whereas expression of Rck/p54 or DDX 6, a member of the dead box RNA helicase family, which is responsible for miRNA-mediated repression, is unchanged (Chu and Rana, 2006; Kedersha and Anderson, 2007; Figure 1B). In addition to the increased level of endogenous Ago2, elevated levels of exogenously expressed human Agos were observed in the infected cells (Figure 1C). Increased Ago2 level was accompanied by an increase in cellular levels of both endogenous and exogenously expressed miRNAs in Ld-infected cells. The miRNAs were quantified by quantitative real-time PCR (qRT-PCR) using specific Taqman probes against the miRNAs studied (Figure 1D and Supplemental Table S1). Furthermore, the increased miRNA also showed increased repressive activity, as shown by decreased expression (firefly normalized) of Renilla luciferase reporter having corresponding miRNA sites in Ld-infected cells (Supplemental Figure S1A). The miRISC isolated from Ld-infected cells also showed enhanced RISC cleavage activity per unit amount of Ago2 protein present in the in vitro RISC cleavage assays. The band intensities of immunoprecipitated Ago2 were quantified and used to normalize the cleaved product obtained in the RISC cleavage assays. The results suggest higher Ago2-associated miRNA content per unit of Ago2 protein isolated from the Ld-infected cells (Figure 1E). Similar observations occurred for an exogenously expressed small interfering RNA (siRNA) targeting Renilla reporter transcript (siRL), which showed higher slicer activity per unit amount of Ago2 isolated from the Ld-infected cells (Supplemental Figure S1, B and C). To confirm increased levels of Ago2-associated miRNAs in Ld-infected cells, we performed immunoprecipitation (IP) of Ago2 and subsequently quantified the associated miRNAs. Ago2 protein level was determined through quantification of immunoprecipitated Ago2 by Western blot followed by densitometry. The quantified immunoprecipitated Ago2 level was used to normalize the associated miRNA levels obtained by either densitometry from Northern blots or by qRT-PCR. The quantitative data revealed enhanced miRNA association per unit amount of Ago2 in both Ld-infected murine primary macrophage and RAW 264.7 cells (Figure 1, F and G).
FIGURE 1:
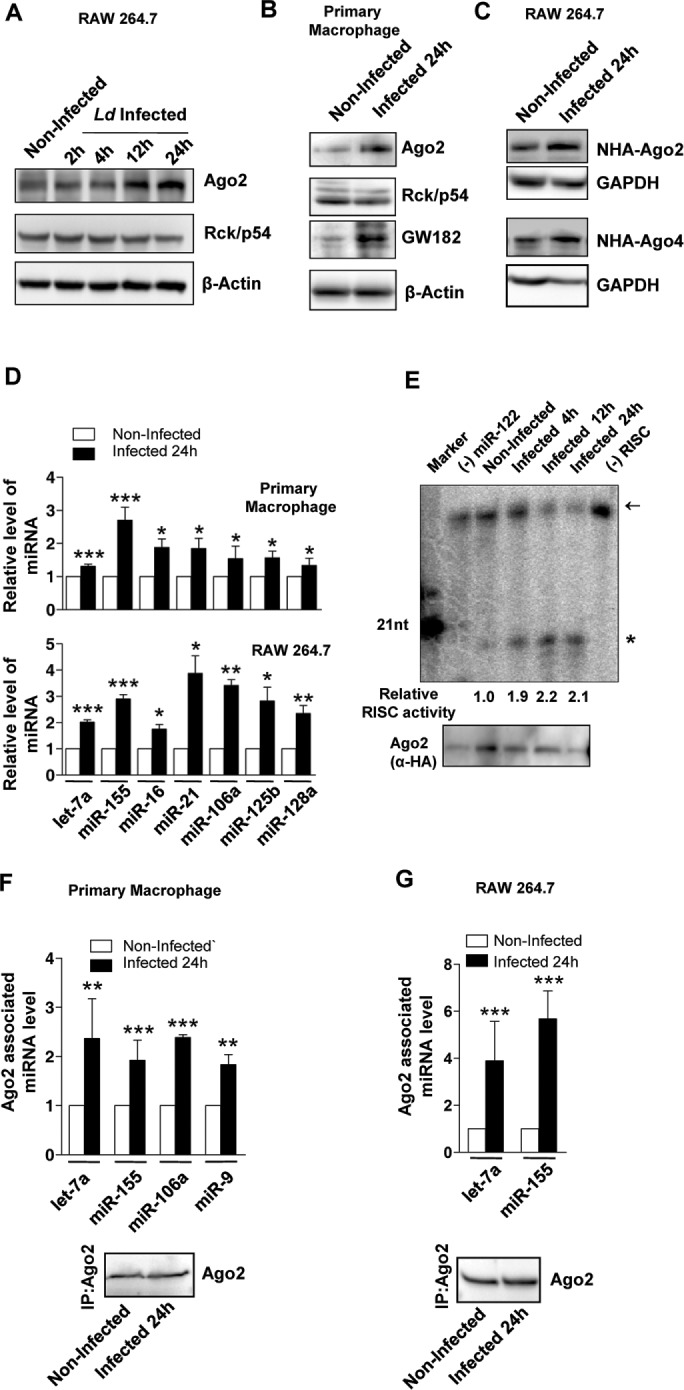
Increased level and activity of miRNPs in Ld-infected mammalian macrophages. (A, B) Representative images of Western blot analysis for indicated proteins in noninfected and Ld-infected cells at different times postinfection. β-Actin served as loading control. (C) Representative Western blot analysis of exogenously expressed Agos in noninfected and Ld-infected RAW 264.7 cells. Cells were infected for 24 h, and GAPDH served as loading control. (D) qRT-PCR–based relative quantification of endogenous miRNA levels on Ld infection in murine primary macrophage and RAW264.7 cells. Values were normalized against respective miRNA levels in noninfected cells. Mean and SEM from at least four independent experiments. (E) Representative autoradiogram showing variation in RISC cleavage activity associated with Ago2 isolated from naive and Ld-infected cells. In vitro RISC cleavage assay was done with isolated Ago2-miR-122 miRNPs from naive and infected RAW264.7 cells cotransfected with FH-Ago2 expression construct and a miR-122 expression plasmid, pmiR-122. The activities were measured and quantified in an in vitro RISC cleavage reaction using 5′-32P–labeled miR-122 target RNA as substrate. Values below the blot indicate relative RISC activity measured by estimating the cleaved band intensities against total (uncleaved + cleaved) intensity using densitometric analysis. Intensities were normalized to immunoprecipitated (IPed) FH-Ago2 levels. Asterisk indicates cleaved product. The arrow indicates uncleaved substrate. A 21-nucleotide-long, radiolabeled DNA oligo was used as size marker. Assay done with RISC isolated from cells not expressing miR-122 is designated as (−) miR-122, and assay carried out without added RISC is denoted as (−) RISC. Bottom, immunoprecipitated FH-Ago2 levels. The FH-Ago2 band intensities from this Western blot were used for calculating RISC cleavage activity per unit Ago2 or specific activity of miRISC, as given by the respective quantified values. (F, G) IP of endogenous Ago2 was performed in noninfected and 24-h Ld-infected cells. Changes in Ago2-associated miRNA levels on Ld infection in murine primary macrophage (F) and RAW264.7 cells (G). Quantification was done by qRT-PCR–based estimation. Values were normalized against amount of Ago2 immunoprecipitated and plotted against values of noninfected samples considered as unity. Representative blots showing immunoprecipitated endogenous Ago2 level used for normalization of respective miRNA levels. Mean and SEM from at least four independent experiments. All experiments were performed with Ld-infected RAW 264.7 or murine primary macrophage cells for indicated time. p values are calculated by Student’s t test; *p < 0.05, **p < 0.01, and ***p < 0.001.
Defective compartmentalization of miRNPs in Ld-infected cells
We sought to identify the association between subcellular fractions and enhanced level of miRNP in host macrophages. The permeabilization of cell membrane with digitonin, a detergent used for selective solubilization of cell membranes, and subsequent analysis of detergent-soluble and -insoluble fractions suggested association of the elevated Ago2 and miRNA levels with the digitonin-insoluble or membranous fraction of Ld-infected cells (Figure 2, A–C). This increased association of miRNPs with membranous fraction prompted us to identify the specific organelles with which the increased Ago2 and miRNA are associated. We used differential centrifugation on a continuous 3–30% OptiPrep gradient to separate different subcellular structures and organelles present in isotonic cell lysates of Ld-infected cells (Figure 2D). In infected cells a large fraction of Ago2 protein comigrated with fractions positive for the endosome/MVB marker HRS. The protein HRS or HGS is an ESCRT0 component present as peripheral membrane protein on the surface of early as well as late endosomes (EEs and LEs, respectively). Concurrently there was a relative decrease of Ago2 in calnexin (an ER marker)-enriched fractions of Ld-infected RAW264.7 cells (Figure 2E). Therefore the increase in cellular Ago2 can be predominantly attributed to this endosome-associated increase of Ago2 level. There can be a slight variation in Ago2 distribution between two different sets of an experiment. In our final observation, to minimize the effect due to these minor alterations in Ago2 distribution, we plotted relative Ago2 band intensities in individual fractions normalized against the total band intensities of Ago2 present in all 10 fractions for different experiments. This result further substantiated the changes in relative distribution of Ago2 protein present in individual fractions of an OptiPrep gradient upon infection (Figure 2F). Moreover, relative contents of different miRNAs associated with ER fractions (7–9) were elevated on Ld infection (Figure 2G). To ascertain that the enhanced endosomal association of Ago2 is due to specific interaction of Ago2 with endosomal membranes and not a cosedimentation artifact, we reisolated the pooled fractions 2–4, that is, endosomal marker–enriched fractions from the 3–30% OptiPrep gradient. Endosomal membrane–enriched fractions were further separated over a 5% density OptiPrep cushion so as to remove any comigrating, contaminating, nonassociating components. We used EEA1 as a peripheral endosomal membrane protein, Hrs as an endosomal membrane component along with Lamp1 as a lysosomal membrane–resident protein, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a cytosolic marker protein to check the purity of isolated membranes. We obtained EE/MVB-enriched fractions positive for LAMP1. This result is expected because a fraction of LE compartment is usually positive for LAMP1 due to progressive maturation of LEs into lysosomes. Moreover, there was no cytosolic contamination, which was evident from the absence of GAPDH after reisolation. To ascertain Ago2 association with EE/MVB fraction, we performed membrane IP assays using Ago2-specific antibody to pull down membranes associated with it in endosomal marker–enriched reisolated samples. We observed that Ago2 was predominantly associated with EE/MVB fractions, and these fractions were not positive for LAMP1. This interaction is specific: washing the immunoprecipitated fraction with 0.4% NP-40 detergent–containing IP buffer results in loss of Ago2 membrane association (Figure 2H). The result was extended to ascertain the increased association of Ago2 with EE/MVB compartments in Ld-infected RAW264.7 cells. Consistent with expectation, we observed increased association of Ago2 with endosomal fraction in Ld-infected cells (Figure 2H, right panel). However, NP-40 has an inhibitory effect on the efficiency of Ago2 IP, which might account for the relatively lower amount of immunoisolated Ago2 in NP-40–treated samples (Figure 2H, left panel).
FIGURE 2:

Differential distribution of miRNAs and Ago2 between ER and endosomal compartments in Ld-infected macrophages. (A) The fractionation process to get the digitonin-soluble supernatant (Sup.) and digitonin-insoluble (Pellet) fractions described in B and C. (B) Representative Western blot analysis of indicated proteins in digitonin-fractionated, Ld-infected and noninfected RAW 264.7 cells. Calnexin was used as membrane marker, and GAPDH served as cytosolic marker protein. (C) Quantitative estimation of relative miRNA level in membrane-enriched, digitonin-insoluble pellet fraction with respect to digitonin-soluble supernatant enriched for cytosolic components in noninfected and Ld-infected RAW 264.7 cells. Changes in miRNA content of individual pellet fraction are normalized against corresponding levels of miRNA in the supernatant. Mean and SEM from at least four independent experiments. (D) Procedure used to separate cellular organelles using iodixanol-based 3–30% OptiPrep gradients E–G. (E) Isotonic extracts of noninfected and Ld-infected cells were analyzed on an OptiPrep gradient (3–30%). Representative Western blots of indicated proteins were done with individual fractions. Ethidium bromide (EtBr)–stained RNA gel and Northern blot was used for detection of total RNA and let-7a miRNA, respectively. Position of ribosomal RNAs on EtBr-stained RNA gel is marked by arrows. (F) Levels of Ago2 (quantified by densitometry) in individual fractions of the OptiPrep gradients in E. Data were from more than two different experiments. Ago2 levels in individual fractions are normalized against the total Ago2 band intensity present in all the fractions. The resultant values are fitted on a scale of 0–10, where 10 indicates the sum of Ago2 density for all the fractions of an individual experiment. (G) Relative enrichment of miRNA in fractions enriched for ER or EE after Ld infection. Fractions 2–4 and 7–9 of OptiPrep gradients described in E were used for estimation of EE- and ER-associated RNA, respectively. The ER fraction from each set was normalized against corresponding EE levels. Mean and SEM from at least three independent experiments. (H) Membrane IP for endogenous Ago2 from reisolated endosomal-enriched fractions 2–4 of 3–30% OptiPrep gradient. Left, Presence of indicated proteins is shown by western blot in E. Total cell lysate is the postnuclear fraction from RAW 264.7 cells used for loading on the 3–30% OptiPrep gradient. Total EE-Input indicates the reisolated EE fraction used for IP. IgG immunoprecipitation was used as negative control to estimate nonspecific binding. NP-40 was used in the IP reaction to confirm the indirect interaction (organelle mediated) between Ago2 and EEA1. The IgG large fragment level was used to show effective IP reaction. Right, Estimation of endosomal membrane–interacting endogenous Ago2 with reisolated endosomes as described in H for RAW 264.7 macrophages infected with Ld. Densitometric analysis and relative quantification for both Ago2 and EEA1 band intensities from two different experiments are plotted. Experiments were performed with Ld -infected cells for indicated times. p values are calculated by Student’s t test; *p < 0.05, **p < 0.01, and ***p < 0.001.
Effect of Ld infection on ER–endosome and ER–mitochondria interaction in host macrophage
To decipher the link between differential spatial sequestration of miRNPs in ER and endosome with the changes in these organelles on Ld infection, we did microscopic and biochemical interaction studies on ER, endosomes, and mitochondria. Ld infection was associated with structural alteration of mitochondria in infected macrophages. The mitochondria became punctate and predominantly perinuclear on Ld infection of RAW 264.7 cells. We also observed altered ER–endosomes and ER–mitochondria colocalization but no discernible change in endosome–mitochondria interaction as revealed by the Manders and Pearson colocalization coefficients (Figure 3, A and B). Effects of these structural abnormalities were also reflected by altered dynamic behavior of the mitochondria. We observed a loss of movement and increase in fragmentation of mitochondria in live-cell dynamic studies of Ld-infected macrophages (Supplemental Figure S2A and Supplemental Videos S1 and S2). Furthermore, we performed organellar coIP assays in which we took the calnexin-enriched fractions 7–9 from the 3–30% OptiPrep gradients and reisolated them over a 15% cushion of OptiPrep density solution to remove any comigrating yet nonassociating organellar fractions such as nonassociated endosomal compartment with ER. In these experiments, we used EEA1 to immunoprecipitate early endosome and calnexin as an ER marker along with RCAS1, which served as a Golgi marker. Although Rcas1 is present in low amounts in the refractionated ER fractions, its interaction with EEA1-positive organelles is negligible, indicating that fractionation and IP were effective in removing ER-associated Golgi. GRP78, an ER luminal marker, was used to ascertain the integrity of ER used for this analysis (this ER luminal protein should otherwise be absent due to leakage of ER membrane that could happen during the isolation; Figure 3C). Infection of host macrophages with Ld resulted in reduced interaction of ER (Grp78) with endosomal membranes. These endosomal membranes were detected by either Eea1- or Alix-specific antibodies. Alix is a class E VPS protein involved in concentration and sorting of MVB cargo proteins and hence serves as a LE marker protein. This result supports our observation that interorganellar interaction is defective in infected macrophages. These results were quantified from three different organellar IP experiments (Figure 3D). Considering that miRNA and Ago2 exist as distinct miRNP pools on ER and endosomes, our data indicate that increased sequestration of miRNA on ER and Ago2 on EE/MVB in host macrophages might be due to altered ER–endosome interaction. The possible reduction in mitochondria–ER interaction and the distinct structural changes in mitochondria on Ld infection are noteworthy. Does this loss of mitochondrial interaction with ER–endosome axis play any role in miRNP stabilization?
FIGURE 3:

Altered ER–mitochondria and ER–endosome interaction in Ld-infected cells. (A) Representative frames with 3D surface reconstructions of ER-, EE-, and GP63-positive structures and mitochondria in noninfected and Ld-infected RAW264.7 cells. Cells expressing a mitochondrial-targeting variant of GFP (MT-GFP, green) and an ER-targeting variant of DsRed (pDsRed2-ER, red) were stained for endogenous EEA-1 (blue) marking the endosomal membranes. Ld was detected by indirect fluorescence against GP63 (magenta), an Ld-specific membrane protein. Right, boxed areas 10× zoomed. Scale bars, 10 μm. (B) Mean ± SEM from at least four independent experiments (10 cells/experiment) of interaction data for ER–mitochondria, mitochondria–EE, and ER-EE as in A. Manders and Pearson coefficients were used to depict the colocalization between organelles under consideration. (C) Representative picture of Western blot analysis of organelle-immunoprecipitated materials from the extract of 24-h Ld-infected RAW 264.7 cells. EEA1 was pulled down from reisolated ER-enriched fractions (7–9) of 3–30% OptiPrep gradient of infected cells for indicated proteins. Reisolated ER fraction was used for IP. IgG pull down is used as negative control to estimate nonspecific binding. NP-40 treatment reduces the amount of immunoprecipitated EEA1-associated calnexin and was used to confirm the indirect (organelle-level) interaction between EEA1 and calnexin. Right, in similar reaction to that with EEA1, Rab7-positive organelles were also isolated to confirm the absence of contaminating vesicles such as lysosomes positive for LAMP1. RCAS1 is used as Golgi body marker. Presence of GRP78, a luminal protein, confirms the association of intact ER with either EEA1- or LAMP1-positive organelles. (D) EEA1 immunoisolation from isotonic cell lysate was done from naive or Ld-infected RAW264.7 cells as labeled. For the quantification, the densitometric estimation of immunoisolated proteins from two independent experiments was plotted, with coimmunoprecipitated protein levels normalized against the level of immunoprecipitated EEA1 levels. For B, p values were calculated by Student’s t test; *p < 0.05, **p < 0.01, and ***p < 0.001.
Effect of mitochondrial depolarization on ER–endosome interaction in Ld-infected cells
Structural alteration of mitochondria into punctate structures is associated predominantly with loss of mitochondrial membrane potential (Chen et al., 2003). We also documented punctate mitochondria localization in perinuclear region and loss of mitochondrial membrane potential in RAW 264.7 cells infected with Ld (Figure 4A). Mitochondrial membrane potential was estimated using JC-1 staining–based flow cytometric analysis. JC-1 is a cationic carbocyanine dye that accumulates in mitochondria in a membrane potential–dependent manner. The dye exists as a monomer at low concentrations and yields green fluorescence in its monomeric form, detected at the fluorescein isothiocyanate channel (∼525 nm). At higher concentrations, the dye forms JC1 aggregates, which exhibit a broad excitation spectrum and an emission maximum at ∼590 nm. On infection by Ld, we observe a higher percentage (∼72%) of host cells with reduced red (∼590 nm) fluorescence, which signifies reduced mitochondrial membrane potential in Ld-infected macrophage cells (Figure 4, A and B). Up-regulated expression of Ucp2 and consequent decrease in mitochondrial membrane potential are linked with increased Ld pathogenesis (Carrion et al., 2013). Proteins like Ucp1 and Ucp2 are specific uncouplers of the mitochondrial electron transport chain. Unlike Ucp1, which is present only in brown adipose tissue of infants, Ucp2 is ubiquitously expressed (Rupprecht et al., 2010). Elevated expression of Ucp2 was previously shown to be responsible for mitochondrial depolarization in Ld-infected cells (Basu Ball et al., 2011; Carrion et al., 2013). We also observed elevated expression of Ucp2 in Ld-infected cells (Figure 4C). To ascertain whether Ucp2-mediated mitochondrial depolarization is responsible for the loss of ER–mitochondria interaction, we expressed FLAG-hemagglutinin (HA)–tagged Ucp2 (FH-Ucp2) in HeLa cells. We observed a subsequent loss of mitochondrial membrane potential, as well as changes in mitochondrial shape and size on FH-Ucp2 expression (Supplemental Figure S2, B and C; Fink et al., 2002; Krauss et al., 2002). Consistent with our observations in Ld-infected Raw 264.7 macrophages, we observed a concurrent reduction in mitochondria–ER and ER–endosome colocalization coefficients upon exogenous Ucp2 expression (Figure 4, D and E).
FIGURE 4:

Depolarization of mitochondria causes defective ER–endosome colocalization and alteration of miRNP levels in Ld-infected macrophage cells. (A) Representative surface reconstructions of mitochondria in noninfected and Ld-infected RAW264.7 cells. Cells expressing a mitochondrial-targeting variant of GFP (MT-GFP, green) were stained for GP63, an Ld-encoded metalloprotease (red). 4′,6-Diamidino-2-phenylindole (blue) was used to label the nuclei. Scale bars, 5 μm. (B) Representative plots using flow cytometry–based quantification of JC-1 staining of RAW264.7 cells in noninfected and Ld-infected conditions. Percentage of cells in quadrant 3 (Q3, low JC-1 aggregate) was measured and is indicated. (C) Western blot analysis of Ago2 and Ucp2 in noninfected and Ld-infected cells at different time of infection. β-Actin served as loading control. (D) Representative 3D surface reconstructions of ER, EE, and mitochondria in HeLa cells expressing FH-Ucp2. Cells were cotransfected with pDsRed2-ER (red) and pAcGFP1-Mito (green) plasmids. EE is labeled with endogenous EEA-1 (blue). Scale bars, 10 μm. Right, boxed areas 10× zoomed. (E) Mean ± SEM from at least three independent experiments (20 cells/experiment) of interaction data for ER–mitochondria, mitochondria–EE. and ER–EE as described in D. Manders and Pearson’s coefficients are used to indicate the changes in colocalization between different organelles. (F) Western blot analysis of indicated proteins in HeLa cells transfected with control or FLAG-HA-uncoupler protein 2 (FH-Ucp2)–expressing plasmids. β-Actin serves as loading control. (G) Relative repression level of a Renilla-based let-7a reporter with three imperfect let-7a binding sites in HeLa cells transfected with control or FH-Ucp2 expression plasmids. Coexpression of firefly luciferase was used as transfection control. Ratio of firefly normalized Renilla luciferase expression in cells transfected with RL-Con against that of RL-3xbulge-let-7a–transfected cells was used to calculate fold repression of the let-7a reporter. Mean and SEM from at least three independent experiments. (H) Representative Western blot analysis of Ago2 in HeLa cells treated with FCCP or oligomycin for 3 h. Dimethyl sulfoxide (DMSO)–treated cells served as control for FCCP treatment, and ethanol treatment was used as control for oligomycin-treated samples. β-Actin serves as loading control. (I) Estimation of miRNA levels in cells treated with FCCP and oligomycin. Values obtained from DMSO and ethanol were set as unity. U6 snRNA levels acted as endogenous control. Mean and SEM from at least three independent experiments. p values were calculated by Student’s t test; *p < 0.05, **p < 0.01, and ***p < 0.001.
Mitochondrial depolarization is sufficient to cause altered miRNA activity in mammalian cells
Mitochondrial membrane potential disruption is linked with down-regulation of RISC formation and delocalization of Ago2 from P-bodies (Huang et al., 2011a). Does mitochondrial membrane potential modulate miRNA activity in mammalian cells? To address this question, we expressed FH-Ucp2 in HeLa cells, which resulted in elevated levels of Ago2 and Gw182 proteins along with increased miRNA activity (Figure 4, F and G, and Supplemental Figure S3A). The increased miRNA activity and Ago2 accumulation in mitochondria depolarized cells expressing Ucp2 could be due to either membrane depolarization or depolarization-induced alteration in oxidative phosphorylation affecting cellular ATP content. Oligomycin treatment specifically blocks ATP production by inhibiting F0F1-ATPAse but does not alter mitochondrial membrane potential. p-Triflouromethoxyphenylhydrazone (FCCP) is an established uncoupler of mitochondrial membrane potential, and treatment of cells with FCCP depolarizes mitochondria without any major effect on cellular ATP content (Supplemental Figure S3, B and C). Of interest, only FCCP treatment and not oligomycin treatment affected miRNA and Ago2 levels in HeLa cells (Figure 4, H and I). Thus mitochondrial membrane depolarization is possibly responsible for miRNP accumulation in mammalian cells.
Mitochondrial depolarization is a prerequisite for Ld-mediated alteration of miRNA stability
Similar to Ld-infected macrophages, in FH-Ucp2–expressing cells, miRNAs and Ago2, the components of miRNPs, were found to become differentially associated with ER and endosomal fractions (Supplemental Figure S4, A and B). This increased population of miRNAs, which was primarily membrane associated, was observed to have higher Ago2 association in cells expressing FH-Ucp2 than with control cells (Figure 5A). Furthermore, the total cellular levels of proinflammatory cytokines such as interleukin-6 (IL-6), IL-1β, and tumor necrosis factor-α were decreased in Ld-infected macrophages (Figure 5B). Concurrently we found an elevated association of proinflammatory cytokine mRNAs such as IL-6 and IL-1β with Ago2 (Figure 5C). Moreover, the importance of Ucp2 in Ld-induced up-regulation of both miRNA activity and Ago2-miRNA association was confirmed when depletion of Ucp2 by siRNAs resulted in an impaired effect of Ld infection on the activity of miRNA and its association with Ago2 (Figure 5, D and E). In addition, Ucp2 depletion in Ld-infected cells rescued the enhanced sequestration of Ago2 in endosomal compartments (Supplemental Figure S4C and Figure 2E). Furthermore, Ucp2 depletion followed by Ld infection also reduced the enhanced level of cellular miRNAs (Supplemental Figure S4D and Figure 2C). To further validate our observations, we used a specific chemical blocker of Ucp2, genipin (Zhang et al., 2006; Parton et al., 2007). We found that 100 μM genipin added 4 h after Ld infection successfully reverted the decrease of mitochondrial membrane potential in Ld-infected cells (Supplemental Figure S5, A and B). Moreover, in addition to the rescue of Ld-mediated down-regulation of mitochondrial membrane potential, genipin treatment of Ld-infected RAW macrophages led to mitochondrial dynamics nearly similar to that of noninfected cells, as shown by efficient fluorescence recovery after photobleaching (FRAP) of Mito-green fluorescent protein (GFP; Supplemental Figure S5C and Supplemental Videos S5–S7). Furthermore, this concentration of genipin reduced Ucp2 and Ago2 protein levels, along with the cellular miRNA level in Ld-infected host cells, which was similar to the effect observed with Ucp2 depletion (Figure 5, F and G). Hence our claims are in accordance with previous observations of genipin-induced reduction of endogenous Ucp2 protein levels (Yu et al., 2015). Elevated levels of cellular miRNA can be attributed to either increased stability of miRNAs or their enhanced processing. To ascertain whether this increase in miRNA level in Ld -infected cells is due to increased stability, we checked the decay rate of exogenously expressed pre–miR-122 mimic in Ld-infected cells. Pre–miR-122 mimic was introduced into RAW 264.7 cells by transfection. Residual mature miR-122 levels were estimated over time in naive and Ld-infected macrophage cells. The residual level of mature miR-122 measured by TaqMan probe–based qRT-PCR was normalized with endogenous U6 levels. The final values for different time points were obtained by fitting the data for individual time points against the 0-h value (considered as 1) for respective data sets. We observed a slower decay rate of miR-122 in Ld-infected cells, indicating its enhanced stability, but we found no such change in the case of pre–miR-122. This effect on miR-122 stability was rescued on genipin treatment of Ld-infected cells (Figure 5H). Moreover, as observed previously, we also noted a reduction in Dicer1 protein level on Ld infection of Raw 264.7 macrophages, which is imperative for miRNA biogenesis from its precursor to mature form, in Ld-infected cells (Ghosh et al., 2013). This lends further support to our observation and clearly indicates that increased miRNA levels observed in Ld-infected RAW264.7 cells is primarily due to their increased stability and not because of altered miRNA processing. Furthermore, we also observed no change in either cytoplasmic cytochrome c or cleaved Parp levels in Ld-infected macrophages, which indicates that the observed changes are not due to apoptosis of Ld-infected macrophages (Figure 5I).
FIGURE 5:

Mitochondrial depolarization causes changes in miRNA stability in Ld-mediated cells. (A) Ago2-associated miRNA levels in HeLa cells expressing control or FH-Ucp2–encoding plasmid. Quantification of miRNA in Ago2-immunoprecipitated materials was done using by qRT-PCR. Values were normalized against amount of Ago2 immunoprecipitated. Representative Western blot for immunoprecipitated Ago2 level used for normalization is shown. For comparison, values corresponding to control cells were considered as unity. For estimation of Ago2-bound exogenous miR-122 levels, cells were transfected with 50 pmol of miR-122 mimic 24 h before the IP. Mean and SEM from at least three independent experiments. (B) Real-time quantitative estimation of relative mRNA level for different cytokines. Relative values in 24-h–infected samples were normalized and plotted against corresponding noninfected samples considered as unity. Mean and SEM from at least three independent experiments. (C) Ago2-associated cytokine mRNA levels in Ld-infected RAW 264.7 cells. Quantification of mRNA in Ago2-immunoprecipitated materials was done by qRT-PCR. Values were normalized against amount of Ago2 immunoprecipitated; representative Western blot for immunoprecipitated Ago2 level used for normalization is shown. For comparison, values corresponding to noninfected cells were considered as unity. (D) Effect of Ucp2 knockdown on in vivo activity of miR-155 and a siRNA (siRL) in Ld-infected RAW 264.7 cells. Relative repression level of Renilla-based miR-155 and siRL reporters in cells cotransfected with control siRNA (siCon) or siUcp2. For the miR-155 site containing RL reporter, RL-Con was used as control. Mean and SEM from at least three independent experiments. Also shown are the representative Western blot analyses for Ucp2, with β-actin as loading control. (E) Effect of Ucp2 knockdown on Ago2 association of miRNA. Associated miRNA levels were quantified by qRT-PCR. Cells were cotransfected with FH-Ago2 and specified siRNAs. Mean and SEM from at least three independent experiments along with representative blot for immunoprecipitated Ago2 level. Corresponding miRNA values were normalized with immunoprecipitated Ago2 levels from noninfected and Ld-infected cells 24 h postinfection. (F) Effects of Ucp2 depletion (siUcp2)– or blocking (genipin, different concentrations)-mediated rescue of Ago2 and Ucp2 protein levels in noninfected and Ld-infected cells upon 24 h of infection. Western blot analysis was performed for indicated proteins. β-Actin served as loading control. (G) Effect of genipin on endogenous miRNA levels on Ld infection in RAW264.7 cells estimated by qRT-PCR relative quantification. Values were normalized against respective miRNA levels in noninfected cells. Mean and SEM from at least three independent experiments. (H) Mature miR-122 decay rate estimated by qRT-PCR on Ld infection in RAW264.7 cells at different time points. Genipin was used at a concentration of 100 µM. RAW 264.7 cells were transfected with 50 pmol of pre-miR-122 at 48 h before addition of Ld. After 24 h of transfection, cells were subcultured and grown for another 24 h. Cells 48 h posttransfection were infected with Ld for indicated time periods. For each time point (8 and 24 h), the genipin addition was done 4 h postinfection with Ld, and RNA extraction was done at indicated time points. U6 snRNA levels served as endogenous control. Changes in Pre-miR-122 levels were calculated for 24-h samples, and relative changes are plotted for different conditions. (I) Western blot analysis of indicated proteins in noninfected and Ld-infected cells at 24 h of infection. GAPDH served as loading control. p values were calculated by Student’s t test. *p < 0.05, **p < 0.01, and ***p < 0.001.
Defective miRNP compartmentalization is associated with down-regulation of mitochondria–ER–associated protein Mfn2
Mitochondria–ER interaction might be connected to the effect we observed on miRNP machineries in Ld-infected cells. To elucidate this further, we checked the protein level expression of several factors that could modulate mitochondrial structural dynamics and its interaction with the ER. We determined the cellular levels of proteins responsible for mitochondrial fusion (Mfn1, Mfn2), mitochondrial fission (Dnm1L), and mitochondria-trafficking regulator molecules such as Miro (Rhot1 and Rhot2) and Milton (Trak1 and Trak2) in Ld-infected and naive cells (Frederick and Shaw, 2007; Tang, 2015; Schrepfer and Scorrano, 2016). We noted reduced expression of Mfn2 in Ld-infected macrophages (Figure 6, A and B), but, apart from mitofusins, we did not observe any significant change in protein level expression. Mfn1 and Mfn2 are both sensitive to mitochondrial membrane potential and thus expected to be affected as a result of mitochondrial depolarization on Ld infection. Mimicking the Ld-induced reduction of Mfn2, we used specific siRNAs to knock down Mfn2 and observed enhanced accumulation of Ago2 and Gw182 in HeLa cells (Figure 6C). Depletion of other mitochondrial dynamics–related factors had limited or no effect on Ago2 level (Supplemental Figure S6A).
FIGURE 6:

Loss of Mfn2 causes reduction in ER–endosome interaction and enhances accumulation of miRNPs on ER. (A, B) Representative blots indicating expression of Mfn2 in RAW164.7 cells infected with Ld for indicated time (A). Changes in the expression of other relevant factors were also done in 24-h–infected cells (B). β-Actin served as loading control. (C) Representative Western blot analysis of indicated proteins in HeLa cells transfected with siRNAs against Mfn2. siControl (siCon)-transfected cells were used as control. β-Actin level served as loading control. (D) Real-time quantification of miRNAs in HeLa cells transfected with specific siRNAs or siControl. Values were normalized to corresponding level of miRNAs in siControl (siCon)-transfected cells. Mean and SEM from at least three independent experiments. (E) Representative Western blot analysis of indicated proteins and Northern blotting of let-7a with fractions obtained from OptiPrep gradient (3–30%) of extracts from HeLa cells transfected with either control or Mfn2-specific siRNAs. Positions of the individual fractions enriched for different organelles as shown above. (F) Subcellular distribution of Ago2-associated endogenous let-7a in individual fractions of OptiPrep gradient as described in E. The let-7a associated with immunoprecipitated Ago2 was measured in individual fractions, normalized against immunoprecipitated Ago2 levels, and plotted. Representative Western blot depicting immunoprecipitated levels of endogenous Ago2 used for normalization of let-7a in corresponding fractions of the OptiPrep gradient. (G) Representative 3D surface reconstructions of ER, EE, and mitochondria in MEFs of indicated genotype. Cells were cotransfected with pDsRed2-ER (red) and pAcGFP1-Mito (green) plasmids. EE is labeled with endogenous EEA-1 (blue). Magenta indicates reconstructions of colocalization surface between ER and EE (Coloc. Surf.EE-ER). Yellow indicates colocalization of mitochondria with ER. Scale bars, 10 μm. Right, boxed areas 10× zoomed. Arrows depict relative EE–ER localization. (H) Mean and SEM from at least three independent experiments (15 cells/experiment) of interaction data for ER–mitochondria, mitochondria–EE, and ER–EE. Manders and Pearson’s coefficients were used to indicate the colocalization between different organelles. (I, J) Representative Western blot analysis of indicated proteins from membrane-immunoprecipitated samples. EEA-1 was pulled down from whole-cell lysate from different cell types. Knockdown of Mfn2 was achieved by siRNA specific to Mfn2, and control siRNA (siCon)-transfected cells were used as control (I). Wild-type or Mfn−/− MEFs were used in experiments. (J) Densitometric quantification of Western blot data was used to calculate the relative change in association between the organellar markers. Error bars indicate mean ± SEM (n ≥ 3). p values were calculated by Student’s t test; *p < 0.05, **p < 0.01, and ***p < 0.001.
Mfn2 was first reported as a mitofusin protein that, along with Mfn1, is found to be responsible for mitochondrial fusion, a phenomenon sensitive to mitochondrial membrane potential. Mfn2 has been found on the ER membrane, where it forms homodimers or heterodimers with Mfn2 or Mfn1 present on the surface of mitochondria and acts as a tethering molecule between mitochondria and ER (Chen et al., 2003; Koshiba et al., 2004; de Brito and Scorrano, 2008). In addition, prior studies demonstrated that only Mfn2 and not Mfn1 conditional knockout mice have functional defects in mitochondrial respiratory chain (Mourier et al., 2015). Compared to mitochondria, Mfn2 is enriched almost 14-fold in the MAM region of ER, where this elevated concentration of Mfn2 may play a role in the establishment of closely juxtaposed surfaces with mitochondria, which function in Ca2+ and lipid transfer (de Brito and Scorrano, 2008). Mfn2 was also found to be responsible for maintaining mitochondria–melanosome tethering (Daniele et al., 2014).
We found enhanced cellular miRNA levels in Mfn2-depleted cells. We also observed increased levels of a few miRNAs in Mfn1-depleted cells, but the effect was less substantial than with Mfn2 depletion (Figure 6D). In addition, in Mfn2-/- mouse embryonic fibroblasts (MEFs), increased miRNA repressive activity was observed for both let-7a and miR-122 Renilla-based reporters (Supplemental Figure S6B). Moreover, siMFN2 treatment of HeLa cells resulted in enhanced association of miRNAs with the ER fraction (Figure 6E). Furthermore, in both Mfn2-depleted and Mfn2-ablated cells, the accumulation of Ago2 was due to a specific increase in Ago2–miRNA interaction in ER-enriched membranous pools (Figure 6F and Supplemental Figure S6C). To explore how Mfn2 depletion might cause elevated accumulation of miRNAs on ER and Ago2 on EE/MVBs, we performed microscopic analysis to determine interorganellar interaction between ER, mitochondria, and endosomes. Prior reports also found that endosomes could interact with ER and this interaction was essential for maturation of endosomes from early to late stages (Eden et al., 2010; Rupprecht et al., 2010; West et al., 2011; Friedman et al., 2013; Rowland et al., 2014). On the basis of the Manders and Pearson coefficients, we documented reduced interorganellar colocalization of ER with both mitochondria and endosomes in Mfn2-ablated MEFs, which was similar to our observations in cells infected with Ld or expressing FH-Ucp2 (Figure 6, G and H). Furthermore, morphological changes in cellular organelles linked with Mfn2 ablation might affect the ER–mitochondria as well as ER–endosome pixel colocalization results.
Hence, to strengthen the conclusion and for normalization of these structural variation–mediated alterations in colocalization coefficients, we calculated a colocalization coefficient (CC) for mitochondria–ER and endosome–ER juxtapositioning. For CC determination, we calculated the surface of colocalization (Coloc. Surf.), that is, the total surface area of colocalization between the two organelles under consideration. The colocalized surface is the colocalized perimeter for the entire extent of pixel colocalization coverage (entire surface area of colocalization using a three-dimensional [3D] surface reconstruction). On the basis of these calculations, we noted the decrease in proximity of ER–mitochondria and ER–endosomes in Mfn2-ablated MEFs, as well as in Ld-infected macrophages or FH-Ucp2 over-expressed cells (Supplemental Table S2). We did not observe any increase in total surface area of either ER or mitochondria but instead noted a reduction in their overall surface area on Mfn2 ablation, as shown by Cosson et al. (2012) and Naon et al. (2016) (Figure 6, G and H, Supplemental Figure S6D, and Supplemental Table S2).
We also performed dynamic live-cell experiments for ER and endosome interaction in Mfn2-knockout MEFs. As expected, we observed reduced interaction between ER (ErDsred) and endosome (Endo GFP). The areas demarcating the EE-GFP–positive regions (white dotted areas) in the enlarged zones from representative frames suggest a morphological change and at the same time indicate loss of proximity between endosome and ER in Mfn2-ablated cells (Supplemental Figure S6E and Supplemental Videos S3 and S4). Furthermore, a possible reduction in membrane interaction between ER (positive for calnexin and GRP78) and endosomes (positive for Alix or EEA1) in Mfn2-depleted cells was supported by data from organelle pull-down assays (Figure 6, I and J). Of interest, genipin treatment of RAW cells infected with Ld rescued the loss of colocalization between ER–mitochondria and ER–endosome as ascertained by the Manders colocalization coefficient (Supplemental Figure S7, A–C). Thus Ld-mediated mitochondrial depolarization results in reduced physical juxtapositioning between ER and endosomes of host cells. Moreover, Mfn2-/- MEFs are sensitive to mitochondrial uncoupling and spontaneously lose mitochondrial membrane potential (Chen et al., 2003). These results support a possible role of Mfn2 in maintaining miRNP shuttling between ER- and endosome-associated organellar pools of miRNPs.
DISCUSSION
Here we report slow turnover of miRNPs in mitochondria-depolarized mammalian cells. This happens after infection of human macrophage cells with the unicellular protozoan parasite Ld, which, by inducing expression of Ucp2, depolarizes mitochondria and enhances the cellular level of miRNAs, as well as of key miRNA regulatory proteins, such as Ago2 and Gw182. Furthermore, increased miRNP content results in an elevated level of endogenous activity of miRISC. This overall increase in cellular miRNPs is primarily membrane associated. In particular, on Ld infection, Ago2 is increased in endosomal fractions comprising MVBs, whereas an increase in ER association occurs for miRNAs. Thus ER–endosomal interaction could act as a key responder to altered cellular needs and control gene expression posttranscriptionally by affecting miRNA activity in mammalian cells.
Only a handful of studies have reported the effect of Leishmania infection on host cell miRNAs (Ghosh et al., 2013; Kelada et al., 2013; Lemaire et al., 2013; Sahoo et al., 2014; Geraci et al., 2015). Most reports focused on Leishmania major as the model infective agent (Kelada et al., 2013; Lemaire et al., 2013), whereas few reported the effect of Ld infection on host miRNA (Geraci et al., 2015). These studies were conducted for shorter infection times, and none of them used Ld strain Ag83 (MAOM/IN/1083/AG83) for 24-h infection, for which the Ld-induced increase in Ucp2 protein level coincides with increased miRNA and Ago2 levels. In our study, the majority of data concerning miRNA levels of Ld-infected primary macrophages or RAW 264.7 cells indicate a global elevation in miRNA levels, although a few host miRNAs show a relatively lower level of elevation upon infection. This could be due to feedback regulation, substrate availability, or spatial sequestration of a specific miRNA, as discussed elsewhere (Bose et al., 2017).
The presence of at least one conserved miRNA-binding site and possible existence of numerous nonconserved sites suggest that the majority of human protein-coding genes are miRNA controlled. Thus it is not surprising that the biogenesis and function of miRNAs are tightly regulated. Hence loss of miRNA regulation as the cause for genesis and progression of human disorders has been a field of intense activity. miRNAs have been implicated in many diseases, including cancer, cardiovascular disorders, and many neurological and immunological maladies (Esteller, 2011; Im and Kenny, 2012; Lujambio and Lowe, 2012; Ha and Kim, 2014). Moreover, pathogens, including viruses (Skalsky and Cullen, 2010; tenOever, 2013), bacteria (Eulalio et al., 2012), and intracellular protozoan parasites such as Toxoplasma gondii (Zeiner and Boothroyd, 2010) can manipulate the miRNA network of infected host cells. This alteration in the host cell miRNA profile may indicate a subversion strategy used by the pathogen or a cellular defense mechanism. Recent studies indicate that miRNA biogenesis and turnover can be modulated depending on the organellar space acquired by the miRNP (Ha and Kim, 2014). Although organelle interaction–mediated regulation of miRNA biogenesis might be only a subset of the total cellular miRNA regulation process, if we consider the prevalence of organellar surfaces and their importance in various biological processes in a eukaryotic cell, it might turn out to be the major regulating factor. In this study, use of Ld helps us to unravel the underexplored field of miRNA organelle association and understand the fine tuning that takes place in this complex regulatory pathway due to mitochondrial membrane potential–mediated modulation of spatial dynamics between organelles.
Mitochondrial dysfunction affects turnover of existing miRNPs possibly by preventing their targeting to EE/MVBs. This targeting to EE/MVBs is associated with miRISC unloading and subsequent degradation of a fraction of unloaded Ago2 and miRNA. This cellular process is impaired on mitochondrial depolarization, which causes increased levels of Ago2 and miRNA in mammalian cells. We also found that the phosphorylated form of Ago2 is predominantly located in the EE/MVB fraction (unpublished data). Therefore it is also possible that phosphorylation at Tyr-529, which is known to inhibit miRNA binding of Ago2, may be responsible for increased sequestration of Ago2 on EE/MVBs (Rudel and Meister, 2008; Mazumder et al., 2013). The Ld parasite has evolved to target mitochondria, indirectly affecting miRNP biogenesis, and thus has devised an ingenious method of abrogating the proinflammatory response. This occurs due to enhanced repression of proinflammatory cytokines by preventing formation of new miRNPs in Ld-infected macrophage (Dogra et al., 2007; Gregory et al., 2008; Lemaire et al., 2013). Moreover, Mfn2 levels are exquisitely responsive to changes in mitochondrial membrane potential. On mitochondrial depolarization, PINK1 is stabilized, which recruits the E3-ubiquitin ligase parkin. Mfn2 (and Mfn1 as well) is a prime target of parkin, which undergoes polyubiquitination and subsequent degradation after loss of mitochondrial membrane potential (Wang et al., 2011; Hsieh et al., 2016). Thus the lower Mfn1 and Mfn2 levels observed on Ld infection could be explained by the loss of mitochondrial membrane potential. Therefore Mfn2 is necessary for maintaining mitochondrial membrane potential and is regulated by the latter. The fact that Mfn2-/- cells show a similar effect on miRNA stability and activity as observed in Ld-infected cells could be largely explained by this possible mechanism. In this context, it would be interesting to investigate further whether Mfn1/2 decrease upon Ld infection can be reverted by genipin. Furthermore, besides the mitochondrial membrane potential, several other components of the pathways regulating interaction of ER with EE/MVB and mitochondria are also likely candidates to affect miRNA activity under specific cellular needs.
The fate of the miRNA unloaded from Ago2 on MVBs is unknown. It has been postulated that lowered MVB targeting of miRNAs could increase their stability by reducing their exosomal export (Ghosh et al., 2015). It is possible that mitochondria depolarization might result in reduced interaction of ER with EE/MVBs (a process likely driven by mitochondria via an unknown mechanism), causing increased sequestration of miRNAs in the ER fraction. Moreover, the absence of Mfn2 protein, which is responsible for intracellular mitochondrial distribution in Mfn2-/- MEFs, also led to reduced ER–endosome association and increased miRNP level, as well as enhanced miRNA-mediated repression level. Therefore Mfn2-regulated ER–endosome tethering controls gene expression posttranscriptionally, possibly through modulation of miRNA stability in mammalian cells. Collectively this work suggests a possible role of interorganellar interaction in miRNA turnover through an exchange between ER- and MVB-located Ago2 pools, which could control miRNA activity in mammalian cells.
MATERIALS AND METHODS
Cell culture
All cell lines except RAW 264.7 cells and primary murine peritoneal macrophages were grown in DMEM supplemented with 2 mM l-glutamine and 10% heat-inactivated fetal calf serum (FCS). Wild-type (WT) and Mfn2-null MEFs and RAW 264.7, a murine macrophage-like cell line, were obtained from the American Type Culture Collection.
Primary murine peritoneal macrophages were elicited by 4% starch. A 1.5-ml amount of the solution was injected intraperitoneally in BALB/c mice, and peritoneal macrophages were isolated by lavage of the peritoneal cavity with cold 1× phosphate-buffered saline (PBS). Primary murine peritoneal macrophages and RAW 264.7 cells were cultured in RPMI 1640 supplemented with 2 mM l-glutamine and 10% heat-inactivated FCS. All cell culture reagents were from Life Technologies.
Parasite culture and infection
Ld strain AG83 (MAOM/IN/1083/AG83) was originally obtained from an Indian visceral leishmaniasis (“kala-azar”) patient and used to infect 4- to 6-wk-old golden hamsters with second-passage promastigotes (107 parasites/animal) by the intracardiac route. Amastigotes were isolated from Ld-infected golden hamsters and transformed to promastigotes. Ld was maintained until passages 2–4. Ld promastigotes were grown in M199 medium supplemented with 10% heat-inactivated FCS and used to infect RAW 264.7 or primary peritoneal macrophage cells in 1:20 ratio for all experiments.
Animal studies
Adult BALB/c mice and golden hamsters were obtained from the animal facility of the CSIR–Indian Institute of Chemical Biology. All experiments were performed according to the National Regulatory Guidelines issued by the Committee for the Purpose of Supervision of Experiments on Animals, Ministry of Environment and Forest, Government of India. All experiments involving animals were carried out with prior approval of the Institutional Animal Ethics Committee.
Cell transfection and treatments
For miRNA repression assays, 20 ng of Renilla luciferase (RL) reporter plasmids with 500 ng of firefly luciferase (FL) plasmid was cotransfected in a 24-well format. RL/FL values were measured after 48 h.
For target RNA or FH-Ucp2 overexpression experiments, 1 µg of RL reporter or FH-Ucp2 expression plasmid was transfected per well of a six-well plate. The siRNAs were transfected at a concentration of 50 pmol in the six-well format. Pre–miR122-mimic (Ambion, Thermo Scientific) was transfected at 10 pmol in a 12-well format with RNAiMAX (Life Technologies) according to the manufacturer’s protocol.
All plasmid transfections were carried out with Lipofectamine 2000 and siRNAs or mirVana hsa-miR-122 mimic (Life Technologies) with RNAiMAX (Life Technologies) according to the manufacturer’s instructions. All Dharmacon SMARTpool ON-TARGETplus used were purchased from GE Healthcare.
The indicated cell types were treated with genipin (100 µM; Sigma-Aldrich) after 4 h of administering Ld infection to a total of 24 h. FCCP (Clontech, 0.5 µM) and oligomycin (Clontech, 2.5 µM) treatments were done for the indicated time periods.
Luciferase-based repression assay and Western blot
Renilla and firefly luciferase activities were measured using a Dual-Luciferase Assay Kit (Promega) following the supplier’s protocol on a VICTOR X3 Plate Reader with injectors (PerkinElmer). Before measurement of luciferase activity, cells were lysed with the 1X Passive Lysis Buffer (Promega). FL-normalized RL expression levels for reporter and control were used to calculate fold repression as the ratio of control to reporter-normalized RL values.
Western blotting analysis of indicated proteins was performed as described previously (Pillai et al., 2005; Ghosh et al., 2015). Supplemental Table S3 lists antibodies used for Western blot and IP. Imaging of all Western blots was performed using an UVP BioImager 600 system equipped with VisionWorks Life Science software, version 6.80 (UVP). Densitometric analysis of Western blot protein band intensities was quantified using ImageJ software.
RNA isolation, Northern blotting, and real-time PCR analysis
Total RNA was isolated using TRIzol reagent (Life Technologies). Northern blotting of total cellular RNA (5-10 μg) was performed as described by Pillai et al. (2005). For detection, γ32P-labeled 22-nucleotide miRCURY complementary LNA probes for let-7a (Exiqon) or cDNA probe for siRL or U6 snRNA was used. Phosphorimaging of the blots was performed in a Cyclone Plus Storage Phosphor System (PerkinElmer), and ImageJ or OptiQuant software (PerkinElmer) was used for quantification.
For miRNA quantification by real-time PCR, TaqMan Universal PCR Master Mix (Applied Biosystems) and 25 ng of total RNA were used. An Applied Biosystems Taqman chemistry-based miRNA assay system was used. The U6 snRNA level was used as endogenous control. mRNA quantification was performed by preparing cDNA using random nonamers (Eurogentec Reverse Transcriptase Core Kit) and 100–200 ng of the total RNA. PCR was performed with gene-specific primers (Supplemental Table S4) using MESA GREEN pPCRMaster Mix Plus (Eurogentec). The 18S rRNA level was used as an endogenous control.
All miRNA quantification reactions were done in a QuantStudio 12K Flex Real-Time PCR System or Bio-Rad CFX96 Real-Time system. An Applied Biosystems 7500 Real Time System was used for mRNA quantification reactions. Cycles were set per manufacturer’s protocol. TaqMan-based Applied Biosystems primers were used for the amplification of different miRNAs as given in Supplemental Table S5.
Immunoprecipitation
For IP reactions, 6 × 106 cells (transfected with tagged protein expression vectors) were lysed in 1× lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM KCl, 5 mM MgCl2, and 1 mM dithiothreitol [DTT] with 1% Triton X-100, 40 U/ml RNase inhibitor [Fermentas], and 1× EDTA-free protease inhibitor cocktail [Roche]) for 30 min at 4°C. The lysates were clarified by centrifugation followed by incubation with antibody bound r-Protein G agarose beads obtained by preincubation of antibody to the r-Protein G agarose beads for 4 h at 4°C on a rotator. The beads were initially blocked with 5% bovine serum albumin (BSA) in 1× lysis buffer. The beads were separated into two parts. One half of the beads were boiled at 95°C for 10 min, centrifuged at 13,000 × g, and used for protein estimation, and the other half were used for RNA extraction. RNA estimations were done using real-time PCR as described previously. Protein estimation was done by Western blotting and quantified using ImageJ software, and these intensities were used to normalize the corresponding miRNA levels obtained from real-time analysis.
Cell fractionation
For digitonin fractionation, 1 × 106 RAW264.7 cells were lysed in digitonin lysis buffer (10 mM Tris-HCl, pH 7.5, 25 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 5 mM vanadyl ribonucleoside complex [Sigma-Aldrich], 1 mM DTT, 1× protease inhibitor cocktail [Roche], and 50 µg/ml digitonin [Calbiochem]) for 10 min at 4°C. The digitonin-insoluble fraction (pellet) was collected by spinning at 2500 × g for 5 min, followed by washing of the membrane pellet with the same lysis buffer without digitonin. RNA isolation from the fractions was done using acidic phenol:chloroform (5:1).
Cell fractionation using OptiPrep (Sigma-Aldrich) density gradient centrifugation was done as described previously (Mazumder et al., 2013; Ghosh et al., 2015). For Ago2 IP from fractionated samples, individual fractions as indicated were lysed in a 2× IP buffer with 0.9% Triton X-100 and 0.1% sodium deoxycholate for 30 min followed by IP with r-Protein G Agarose beads (Invitrogen, Thermo Scientific) as described earlier.
Membrane and organelle immunoprecipitation
Membrane and organelle IPs were performed as described previously with few variations (Stalder et al., 2013). For organelle IPs of anti-EEA1 with postnuclear supernatants, cells were incubated in hypotonic buffer for 10 min on ice. Cells (6 × 106) were transferred to extraction buffer. Cells were then lysed with Dounce homogenizer. Lysates were cleared for 5 min at 600 × g, and the supernatant was incubated with 5 μg of endogenous EEA1 antibody for 4 h at 4°C in extraction buffer. In all reactions, 25 μl of Protein G Agarose beads (Invitrogen) were preblocked with 5% (wt/vol) BSA in extraction buffer, and antibody–protein complexes were captured for 30 min at 4°C. Beads were washed three times with IP wash buffer (150 mM KCl, 4 mM MgCl2, 8.4 mM CaCl2, 10 mM ethylene glycol tetraacetic acid [EGTA], and 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]–NaOH, pH 7.2, supplemented with Complete EDTA-free protease inhibitor [Roche] and 0.5 mM DTT) with or without detergent (0.4% NP-40), and proteins were eluted by incubating the beads for 5 min at room temperature with 1× Laemmli buffer. Immunoglobulin G (IgG) binding was used to neglect any nonspecific binding.
For anti-EEA1 membrane IP done from OptiPrep gradient fractions, reisolation of either the EE/MVB-enriched fractions 2–4 or ER-enriched fractions 7–9 from 3–30% continuous OptiPrep density gradient was performed by diluting (4×) the fractions using gradient casting buffer. This was followed by loading the diluted fraction over a 5% (endosome) or 15% (ER) OptiPrep density solution, followed by ultracentrifugation at 370,000 × g in an SW61Ti Beckmann Coulter rotor for 30 min (endosome) to 60 min (ER) at 4°C. Pellet was resuspended and washed in extraction buffer (0.25 M sucrose, 78 mM KCl, 4 mM MgCl2, 8.4 mM CaCl2, 10 mM EGTA, and 50 mM HEPES-NaOH, pH 7.2, supplemented with Complete EDTA-free protease inhibitor and 0.5 mM DTT) and processed further for membrane or organelle IP. A 200-μl amount of the refractionated and resuspended pellet (total 400 μl) fraction (obtained from 1.8 × 107 cells) was used as input, and the IP was performed with 5 μg of antibody each. Membrane IP of endogenous Ago2 pool from reisolated pellet fraction was done in a similar manner as described using 10 μg of antibody and reisolated pool of the endosome-enriched fraction from the 3–30% OptiPrep gradient.
RISC cleavage assay
The RISC cleavage assay was performed as described previously (Mazumder et al., 2013).
Flow cytometry–based mitochondrial membrane potential estimation
JC-1 Dye (Life Technologies), a mitochondrial membrane potential probe (Life Technologies), was used to label cells according to the manufacturer’s protocol. Cells were harvested and incubated with 1 mM JC-1 in whole culture medium for 15 min at 37°C in the presence of 5% CO2. Cells were harvested and washed with 1× PBS and analyzed on a BD FACSCalibur instrument using the manufacturer’s protocol.
Tetramethylrhodamine methyl ester (TMRM), a cell-permeant, cationic, red-orange fluorescent dye that is readily sequestered by active mitochondria, was also used as a probe to detect mitochondria membrane potential. Cells were harvested and incubated with 20 nM TMRM in Hanks balanced salt solution for 15 min at 37°C in a CO2 incubator. Cells were harvested and washed with 1× PBS and analyzed on a BD FACSCalibur using the manufacturer’s protocol.
Immunofluorescence
Cells were grown on 18-mm round coverslips, transfected as indicated, and fixed after 24 h. For anti-HA, anti-EEA1 and anti-GP63 staining, cells were fixed for 30 min at room temperature in the dark with 4% ice-cold paraformaldehyde in PBS (pH 7.4). Cells were blocked and permeabilized by incubation with 3% BSA in PBS containing 0.1% Triton X-100 for 30 min at 25°C and subsequently incubated overnight at 4°C with anti-EEA1 (1:100), anti-GP63 (1:1000), or anti-HA (1:100) antibodies. Staining of the primary antibody was revealed after incubating for 60 min at room temperature with Alexa Fluor anti-rabbit, anti-mouse, or anti-goat secondary IgG conjugated to the appropriate fluorochrome.
Imaging
For fluorescence imaging, cells expressing pAcGFP1-Mito (Clontech) were imaged with an inverted Eclipse Ti Nikon microscope equipped with a Plan Apo VC 60×/1.40 oil objective equipped with a Nikon Qi1MC or QImaging-Rolera EMC2 camera.
For confocal live- or fixed-cell imaging, cells expressing pAcGFP1-Mito (Clontech), pAcGFP-Endo (Clontech), and pDsRed2-ER (Clontech) with requisite immunofluorescence were imaged in a Leica DMI6000 B inverted microscope equipped with Plan Apo 100×/1.40 oil objective (Leica TCS SP8 confocal system) or Olympus IX81 inverted microscope equipped with UApoN 100×/1.49 oil objective (Yokogawa CSU X1 spinning-disk confocal system using an IXON3 electron-multiplying charge coupled device [EMCCD] camera) and excited using the appropriate laser lines on a galvoscanner (Leica)- or peizoscanner (Olympus)-based motorized stage. FRAP analysis was performed on the Olympus IX81 inverted microscope equipped with UApoN 60×/1.49 oil objective (Yokogawa CSU X1 spinning-disk confocal system using an IXON3 EMCCD camera) equipped with an Andor FRAPPA unit. For confocal z-axis stacks, image stacks separated by 0.25 µm along the z-axis were acquired. Appropriate plug-ins of Imaris 7 (Bitplane) were used to perform 3D reconstruction and volume rendering of the stacks.
Postcapture image processing and organelle contact analysis
All images were surface rendered using Imaris 7. For mitochondria–ER, mitochondria–EE, and ER–EE interaction analysis, stacks were automatically thresholded after background subtraction using Imaris Coloc plug-in, 3D reconstructed, and volume rendered using Imaris Surpass plug-in. Interaction was quantified by Manders and Pearson colocalization coefficients using Coloc (Imaris; Zinchuk et al., 2007). For calculation of CCs, 3D surface-rendered images with colocalization channel constructed using autothresholding of Imaris Coloc plug-in were taken. This colocalization parameter was modified from Filadi et al. (2015) and Naon et al. (2016). This colocalization surface was used to construct the 3D surfaces, and Coloc. Surf. was calculated using Imaris surpass and vantage plug-in. Surface area was calculated using surpass plug-in for individual channels (i.e., Mito, ER, or EE). Total surface area was generated from the surface areas for individual organelles by normalizing it against total cellular surface area using a cell cytosolic marker such as actin or tubulin to stain the cell cytoplasm, hence outlining the cell periphery and defining the cellular volume. All images were processed with Adobe Photoshop CS5 for all linear adjustments and cropping.
Plasmids
RL-con, RL3xbulge-let-7a, RL-3xbulge-miR-122, pRL-per-let-7a, and firefly (FF) were kind gifts from Witold Filipowicz of FMI (Switzerland). To make RL-per-miR-155, annealed primers were inserted into the XbaI-NotI sites of pRL-per-let-7a after removing the let7a site by digestion with XbaI-NotI to get the construct:
miR155 S: 5′-GGCCGCTTAATGCTAATTGTGATAGGGGTC-3′
miR155 AS: 5′-CTAGACCCCTATCACAATTAGCATTAAGC-3′
To get WT FLAG-HA-Ucp2, murine Ucp2 was amplified from RAW264.7 cDNA using the following primers:
Ucp2 FP: 5′-CATGGTTGGTTTCAAGGCCACAG-3′
Ucp2 RP: 5′-CAGCATGGAGAGGCTCAGAAAG-3′
This amplified fragment was inserted in a TOPO-TA vector using TOPO TA Cloning Kit according to the manufacturer’s protocol. After screening of positive clones, the Ucp2 fragment was reamplified using primers containing NotI and EcoRI sites:
Ucp2 FP (with NotI site): 5′-AGCGGCCGCCAGTGTGCTGGTATTC-3′
Ucp2 RP (with EcoRI site): 5′-CTGCAGAATTCGCCCTTGGTCAGC-3′
The amplified fragment was then inserted in the NotI-EcoRI site in a FLAG-HA-AGO2 plasmid after removal of AGO2. FH-AGO2 was a kind gift from Tom Tuschl’s lab (The Rockefeller University).
The precursor miR-122 was cloned into the NheI and NotI sites of pTRE-tight BI vector (Clontech) to get the Tet-inducible precursor miR-122 plasmid (ip-miR-122). All other plasmids were described in previous studies (Chaussabel et al., 2003; Ghosh et al., 2015).
Statistics
All graphs and statistical analyses were generated in GraphPad Prism 5.00 (GraphPad, San Diego, CA). A two-sample Student’s t test was used for analysis. p < 0.05 was considered to be statistically significant and p > 0.05 not significant (ns). Error bars indicate mean ± SEM.
Supplementary Material
Acknowledgments
We thank Witold Filipowicz and Edouard Bertrand for clones and constructs. This work was supported by the Wellcome Trust Senior Research Fellowship Fund and Council of Scientific and Industrial Research Suprainstitutional Research Fund BeND (BSC 0116) to S.N.B. Y.C. received support from the Council of Scientific and Industrial Research, Government of India. We also acknowledge the support of High Risk High Reward Grant HRR/2016/000093 to S.N.B. from the Department of Science and Technology, Government of India.
Abbreviations used:
- CC
colocalization coefficient
- EE
early endosome
- ER
endoplasmic reticulum
- IP
immunoprecipitation
- Ld
Leishmania donovani
- miRNP
microRNA ribonucleoprotein
- MVB
multivesicular body
- RISC
RNA-induced silencing complex.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-06-0388) on May 24, 2017.
REFERENCES
- Bandiera S, Ruberg S, Girard M, Cagnard N, Hanein S, Chretien D, Munnich A, Lyonnet S, Henrion-Caude A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One. 2011;6:e20746. doi: 10.1371/journal.pone.0020746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman B, Bhattacharyya SN. mRNA targeting to endoplasmic reticulum precedes ago protein interaction and microRNA (miRNA)-mediated translation repression in mammalian cells. J Biol Chem. 2015;290:24650–24656. doi: 10.1074/jbc.C115.661868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu Ball W, Kar S, Mukherjee M, Chande AG, Mukhopadhyaya R, Das PK. Uncoupling protein 2 negatively regulates mitochondrial reactive oxygen species generation and induces phosphatase-mediated anti-inflammatory response in experimental visceral leishmaniasis. J Immunol. 2011;187:1322–1332. doi: 10.4049/jimmunol.1004237. [DOI] [PubMed] [Google Scholar]
- Bose M, Barman B, Goswami A, Bhattacharyya SN. Spatiotemporal uncoupling of microRNA-mediated translational repression and target RNA degradation controls microRNP recycling in mammalian cells. Mol Cell Biol. 2017;37:e00464-16. doi: 10.1128/MCB.00464-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrion J, Abengozar MA, Fernandez-Reyes M, Sanchez-Martin C, Rial E, Dominguez-Bernal G, Gonzalez-Barroso MM. UCP2 deficiency helps to restrict the pathogenesis of experimental cutaneous and visceral leishmaniosis in mice. PLoS Negl Trop Dis. 2013;7:e2077. doi: 10.1371/journal.pntd.0002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA, Xu C, Mason WS, Moloshok T, Bort R, et al. miR-122. a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- Chaussabel D, Semnani RT, McDowell MA, Sacks D, Sher A, Nutman TB. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood. 2003;102:672–681. doi: 10.1182/blood-2002-10-3232. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006;4:e210. doi: 10.1371/journal.pbio.0040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P, Marchetti A, Ravazzola M, Orci L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS One. 2012;7:e46293. doi: 10.1371/journal.pone.0046293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniele T, Hurbain I, Vago R, Casari G, Raposo G, Tacchetti C, Schiaffino MV. Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr Biol. 2014;24:393–403. doi: 10.1016/j.cub.2014.01.007. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Desjardins M, Descoteaux A. Survival strategies of Leishmania donovani in mammalian host macrophages. Res Immunol. 1998;149:689–692. doi: 10.1016/s0923-2494(99)80040-6. [DOI] [PubMed] [Google Scholar]
- Dogra N, Warburton C, McMaster WR. Leishmania major abrogates gamma interferon-induced gene expression in human macrophages from a global perspective. Infect Immun. 2007;75:3506–3515. doi: 10.1128/IAI.00277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden ER, White IJ, Tsapara A, Futter CE. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat Cell Biol. 2010;12:267–272. doi: 10.1038/ncb2026. [DOI] [PubMed] [Google Scholar]
- Engwerda CR, Ato M, Kaye PM. Macrophages. pathology and parasite persistence in experimental visceral leishmaniasis. Trends Parasitol. 2004;20:524–530. doi: 10.1016/j.pt.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Schulte L, Vogel J. The mammalian microRNA response to bacterial infections. RNA Biol. 2012;9:742–750. doi: 10.4161/rna.20018. [DOI] [PubMed] [Google Scholar]
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci USA. 2015;112:E2174–E2181. doi: 10.1073/pnas.1504880112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink BD, Hong YS, Mathahs MM, Scholz TD, Dillon JS, Sivitz WI. UCP2-dependent proton leak in isolated mammalian mitochondria. J Biol Chem. 2002;277:3918–3925. doi: 10.1074/jbc.M107955200. [DOI] [PubMed] [Google Scholar]
- Frederick RL, Shaw JM. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8:1668–1675. doi: 10.1111/j.1600-0854.2007.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Dibenedetto JR, West M, Rowland AA, Voeltz GK. Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol Biol Cell. 2013;24:1030–1040. doi: 10.1091/mbc.E12-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraci NS, Tan JC, McDowell MA. Characterization of microRNA expression profiles in Leishmania-infected human phagocytes. Parasite Immunol. 2015;37:43–51. doi: 10.1111/pim.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh J, Bose M, Roy S, Bhattacharyya SN. Leishmania donovani targets Dicer1 to downregulate miR-122. lower serum cholesterol. and facilitate murine liver infection. Cell Host Microbe. 2013;13:277–288. [Google Scholar]
- Ghosh S, Bose M, Ray A, Bhattacharyya SN. Polysome arrest restricts miRNA turnover by preventing exosomal export of miRNA in growth-retarded mammalian cells. Mol Biol Cell. 2015;26:1072–1083. doi: 10.1091/mbc.E14-11-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. 2009;11:1143–1149. doi: 10.1038/ncb1929. [DOI] [PubMed] [Google Scholar]
- Gregory DJ, Sladek R, Olivier M, Matlashewski G. Comparison of the effects of Leishmania major or Leishmania donovani infection on macrophage gene expression. Infect Immun. 2008;76:1186–1192. doi: 10.1128/IAI.01320-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schule B, Krainc D, Palmer TD, Wang X. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic parkinson’s disease. Cell Stem Cell. 2016;19:709–724. doi: 10.1016/j.stem.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Mollet S, Souquere S, Le Roy F, Ernoult-Lange M, Pierron G, Dautry F, Weil D. Mitochondria associate with P-bodies and modulate microRNA-mediated RNA interference. J Biol Chem. 2011a;286:24219–24230. doi: 10.1074/jbc.M111.240259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ. Biological functions of microRNAs: a review. J Physiol Biochem. 2011b;67:129–139. doi: 10.1007/s13105-010-0050-6. [DOI] [PubMed] [Google Scholar]
- Im HI, Kenny PJ. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012;35:325–334. doi: 10.1016/j.tins.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117:6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- Kelada S, Sethupathy P, Okoye IS, Kistasis E, Czieso S, White SD, Chou D, Martens C, Ricklefs SM, Virtaneva K, et al. miR-182 and miR-10a are key regulators of Treg specialisation and stability during Schistosome and Leishmania-associated inflammation. PLoS Pathog. 2013;9:e1003451. doi: 10.1371/journal.ppat.1003451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Maizel A, Chen X. Traffic into silence: endomembranes and post-transcriptional RNA silencing. EMBO J. 2014;33:968–980. doi: 10.1002/embj.201387262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Lowell BB. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc Natl Acad Sci USA. 2002;99:118–122. doi: 10.1073/pnas.012410699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kren BT, Wong PY, Sarver A, Zhang X, Zeng Y, Steer CJ. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 2009;6:65–72. doi: 10.4161/rna.6.1.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Pressman S, Andress AP, Kim K, White JL, Cassidy JJ, Li X, Lubell K, Lim do H, Cho IS, et al. Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol. 2009;11:1150–1156. doi: 10.1038/ncb1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire J, Mkannez G, Guerfali FZ, Gustin C, Attia H, Sghaier RM, Sysco C, Dellagi K, Laouini D, Renard P. MicroRNA expression profile in human macrophages in response to Leishmania major infection. PLoS Negl Trop Dis. 2013;7:e2478. doi: 10.1371/journal.pntd.0002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liu L, Zhuang X, Yu Y, Liu X, Cui X, Ji L, Pan Z, Cao X, Mo B, et al. MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell. 2013;153:562–574. doi: 10.1016/j.cell.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482:347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzano-Roman R, Siles-Lucas M. MicroRNAs in parasitic diseases: potential for diagnosis and targeting. Mol Biochem Parasitol. 2012;186:81–86. doi: 10.1016/j.molbiopara.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Mazumder A, Bose M, Chakraborty A, Chakrabarti S, Bhattacharyya SN. A transient reversal of miRNA-mediated repression controls macrophage activation. EMBO Rep. 2013;14:1008–1016. doi: 10.1038/embor.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier A, Motori E, Brandt T, Lagouge M, Atanassov I, Galinier A, Rappl G, Brodesser S, Hultenby K, Dieterich C, Larsson NG. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J Cell Biol. 2015;208:429–442. doi: 10.1083/jcb.201411100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366:1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernandez-Alvarez MI, et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci USA. 2016;113:11249–11254. doi: 10.1073/pnas.1606786113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Gregory DJ, Forget G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev. 2005;18:293–305. doi: 10.1128/CMR.18.2.293-305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 microRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- Rogers K, Chen X. Biogenesis. turnover. and mode of action of plant microRNAs. Plant Cell. 2013;25:2383–2399. [Google Scholar]
- Rousset S, Emre Y, Join-Lambert O, Hurtaud C, Ricquier D, Cassard-Doulcier AM. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine. 2006;35:135–142. doi: 10.1016/j.cyto.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK. ER contact sites define the position and timing of endosome fission. Cell. 2014;159:1027–1041. doi: 10.1016/j.cell.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel S, Meister G. Phosphorylation of Argonaute proteins: regulating gene regulators. Biochem J. 2008;413:e7–9. doi: 10.1042/BJ20081244. [DOI] [PubMed] [Google Scholar]
- Rupprecht A, Sokolenko EA, Beck V, Ninnemann O, Jaburek M, Trimbuch T, Klishin SS, Jezek P, Skulachev VP, Pohl EE. Role of the transmembrane potential in the membrane proton leak. Biophys J. 2010;98:1503–1511. doi: 10.1016/j.bpj.2009.12.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo GC, Ansari MY, Dikhit MR, Gupta N, Rana S, Das P. Computational identification of microRNA-like elements in Leishmania major. Microrna. 2014;2:225–230. doi: 10.2174/2211536602666131203232422. [DOI] [PubMed] [Google Scholar]
- Schrepfer E, Scorrano L. Mitofusins. from mitochondria to metabolism. Mol Cell. 2016;61:683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- Skalsky RL, Cullen BR. Viruses. microRNAs. and host interactions. Annu Rev Microbiol. 2010;64:123–141. [Google Scholar]
- Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta. 1998;1363:100–124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- Stalder L, Heusermann W, Sokol L, Trojer D, Wirz J, Hean J, Fritzsche A, Aeschimann F, Pfanzagl V, Basselet P, et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013;32:1115–1127. doi: 10.1038/emboj.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang BL. MIRO GTPases in mitochondrial transport, homeostasis and pathology. Cells. 2015;5 doi: 10.3390/cells5010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tenOever BR. RNA viruses and the host microRNA machinery. Nat Rev Microbiol. 2013;11:169–180. doi: 10.1038/nrmicro2971. [DOI] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M, Zurek N, Hoenger A, Voeltz GK. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J Cell Biol. 2011;193:333–346. doi: 10.1083/jcb.201011039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SX, Du CT, Chen W, Lei QQ, Li N, Qi S, Zhang XJ, Hu GQ, Deng XM, Han WY, Yang YJ. Genipin inhibits NLRP3 and NLRC4 inflammasome activation via autophagy suppression. Sci Rep. 2015;5:17935. doi: 10.1038/srep17935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiner GM, Boothroyd JC. Use of two novel approaches to discriminate between closely related host microRNAs that are manipulated by Toxoplasma gondii during infection. RNA. 2010;16:1268–1274. doi: 10.1261/rna.2069310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CY, Parton LE, Ye CP, Krauss S, Shen R, Lin CT, Porco JA, Jr, Lowell BB. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab. 2006;3:417–427. doi: 10.1016/j.cmet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Zinchuk V, Zinchuk O, Okada T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem Cytochem. 2007;40:101–111. doi: 10.1267/ahc.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.