

The ESCRT-I protein TSG101 can mitigate endoplasmic reticulum stress–mediated apoptosis involving caspase 4/12 caused by MGRN1 depletion, probably contributing to neurodegeneration. TSG101 associates with ALIX to prevent predisposition to apoptosis, whereas ALIX–ALG-2 interaction favors a cell death phenotype.
Abstract
ESCRT proteins are implicated in myriad cellular processes, including endosome formation, fusion of autophagosomes/amphisomes with lysosomes, and apoptosis. The role played by these proteins in either facilitating or protecting against apoptosis is unclear. In this study, while trying to understand how deficiency of Mahogunin RING finger 1 (MGRN1) affects cell viability, we uncovered a novel role for its interactor, the ESCRT-I protein TSG101: it directly participates in mitigating ER stress–mediated apoptosis. The association of TSG101 with ALIX prevents predisposition to apoptosis, whereas ALIX–ALG-2 interaction favors a death phenotype. Altered Ca2+ homeostasis in cells and a simultaneous increase in the protein levels of ALIX and ALG-2 are required to elicit apoptosis by activating ER stress–associated caspase 4/12. We further demonstrate that in the presence of membrane-associated, disease-causing prion protein CtmPrP, increased ALIX and ALG-2 levels are detected along with ER stress markers and associated caspases in transgenic brain lysates and cells. These effects were rescued by overexpression of TSG101. This is significant because MGRN1 deficiency is closely associated with neurodegeneration and prenatal and neonatal mortality, which could be due to excess cell death in selected brain regions or myocardial apoptosis during embryonic development.
INTRODUCTION
Endosomal sorting complexes required for transport (ESCRTs) are key regulatory proteins that guide formation and maturation of the endocytic compartments and recognize and sort ubiquitinated cargoes into them (Katzmann et al., 2001; Curtiss et al., 2007; Raiborg and Stenmark, 2009; Huotari and Helenius, 2011; Mayers and Audhya, 2012). Four complexes—ESCRT-0, -I, -II, and –III—along with several accessory components, in a well-synchronized sequence of events, sort endocytosed ubiquitinated proteins such as misfolded plasma membrane proteins, activated growth factors, and hormone and cytokine receptors in multivesicular bodies (MVBs; Hurley and Emr, 2006; Williams and Urbé, 2007; Hanson et al., 2009). Ubiquitinated cargo is captured by the ESCRT machinery in the endosomal pit, followed by deformation of endosomal membrane and breaking off of this invasion containing sorted cargoes. Ubiquitin-dependent cargo sorting was first identified in vacuolar protein sorting (VPS) mutants of Saccharomyces cerevisiae (Katzmann et al., 2001). ESCRT proteins are conserved across species, and their depletion causes impaired formation of intraluminal vesicles, abnormal genesis of MVBs, and inhibition of lysosomal degradation of proteins.
In addition, ESCRT proteins are implicated in maturation of autophagosomes (Lee et al., 2007; Manil-Segalén et al., 2012); they also affect fusion of vesicles with lysosomes to form autolysosomes (Luzio et al., 2007; Metcalf and Issacs, 2010). Inactivation of these proteins is associated with various neurodegenerative diseases, in which significant aberrations are detected in autophagic pathways (Rusten and Simonsen, 2008; Lee et al., 2009). In Drosophila melanogaster, null mutations in the ESCRT-I protein VPS28 causes accumulation of autophagosomes due to their blocked fusion with endolysosomes (Rusten et al., 2007). Recently monoubiquitinated tumor susceptibility gene 101 (TSG101), another ESCRT-I protein, was reported to potentiate fusion of amphisomes/MVBs and lysosomes (Majumder and Chakrabarti, 2015). Expression of mutant forms of the ESCRT-III protein charged multivesicular body protein 2B (CHMP2B) or depletion of CHMP4B in primary neurons leads to accumulation of autophagosomes as seen in frontotemporal dementia (Lee et al., 2009). Similarly, loss of function of the ESCRT-III component mSnf7-2 or mouse CHMP4B causes neuronal cell loss due to extensive accumulation of autophagosomes and multilamellar bodies.
Further, components of the ESCRT machinery are suggested to be a bridge between the endolysosomal system and cell death (Mahul-Mellier et al., 2006; Petiot et al., 2008; Rusten and Stenmark, 2009). Programmed cell death 6–interacting protein (PDCD6IP), also known as AIP1/apoptosis-linked gene 2 (ALG-2)–interacting protein X (ALIX), interacts with tumor susceptibility gene 101 (TSG101), an ESCRT-1 protein homologue of the yeast class E VPS protein complex ESCRT-III (Strack et al., 2003). ALIX was reported to be enriched in phagosomes (Garin et al., 2001) and exosomes (Théry et al., 2001). Group-specific antigen protein from membrane-containing viruses binds to ALIX and uses it in budding of the virus from the cell surface by recruiting the ESCRT machinery (Martin-Serrano et al., 2003, Strack et al., 2003, von Schwedler et al., 2003, Segura-Morales et al., 2005). Several observations also suggested that ALIX regulates endolysosomal pathway in multiple ways. It can interact with SRC homology 3 (SH3) domain–containing endophilins via its C-terminus, thereby inducing cytoplasmic vacuolization (Chatellard-Causse et al., 2002). The proline-rich domain of ALIX also interacts with Cbl-interacting protein of 85 kDa (CIN85)/SH3-encoding expressed in tumorigenic astrocytes/Ruk (Chen et al., 2000), a protein that in turn regulates endocytosis of ubiquitinated tyrosine receptors (Petrelli et al., 2002; Soubeyran et al., 2002). ALIX-CIN85 association sensitizes astrocytes to apoptosis in response to DNA damage (Chen et al., 2000). This also suggested a role of ALIX in regulating cell death.
In this context, the role of ALIX in controlling apoptosis was also reported via its interaction with the penta-EF-hand calcium-binding protein ALG-2 (Missotten et al., 1999; Vito et al., 1999), a product of the PDCD6 gene (Maki et al., 2011). Inhibiting ALG-2 expression blocked apoptosis in a T-cell line (Vito et al., 1996) without affecting caspase 3 activation (Lacana et al., 1997). However, death was not impaired in T-cells from ALG-2–deficient mice, suggesting that other proteins of similar function may replace ALG-2 (Jang et al., 2002). ALIX is also strongly up-regulated in neurons degenerating after kainate-induced epileptic seizures, demonstrating the involvement of this protein in cell death (Hemming et al., 2004). Further, ALIX mutants compromised in their binding to ALG-2 could not elicit death in neurons after K+ deprivation (Trioulier et al., 2004). These results suggested that the ALIX–ALG-2 complex could be linked to the effectors of cell death. Here we show that the presence of up-regulated protein levels of ALIX and ALG-2 and enrichment of cytosolic Ca2+ (e.g., by inducing ER stress) are simultaneously required to elicit predisposition to death. These events individually do not activate the downstream caspase cascade to eventually lead to apoptosis.
Mice deficient in the RING domain–containing E3 ligase Mahogunin RING finger 1 (MGRN1) have reduced embryonic viability and also develop adult-onset spongiform neurodegeneration (similar to Prion diseases; He et al., 2003). Embryonic lethality and enhanced mortality by weaning age in Mgrn1 mutants may be attributed to a battery of developmental defects (including heterotaxia and congenital heart defects) in mice (Cota et al., 2006; Jiao et al., 2009; Srivastava and Chakrabarti, 2014; McDowell et al., 2016). Studies implicate a role of MGRN1 in mitigating stress (Sun et al., 2007; Chhangani and Mishra, 2013, Mukherjee and Chakrabarti, 2016) and conversely suggest that lack of MGRN1 leads to up-regulating stress.
In this study in the course of seeking to understand the mechanism by which MGRN1 depletion predisposes cells to apoptosis, we unravel a unique mechanism by which the ESCRT-I protein, by virtue of its interaction with ALIX, can mitigate the effects of endoplasmic reticulum (ER) stress–mediated apoptosis. We show that whereas TSG101-ALIX interaction prevents predisposition to apoptosis, this effect is disrupted by deregulating the amount of cytosolic Ca2+ and simultaneously increasing the levels of ALG-2. Increased ALG-2 levels potentiate ALIX–ALG-2 interaction in the presence of high cytosolic Ca2+ and promote cell death. This is significant because CtmPrP-mediated induction of ER stress and associated increase in ALIX and ALG-2 levels are rescued by overexpression of TSG101, thus also making it pathologically relevant. Our analysis identifies a yet-unidentified role of TSG101 in which it directly participates in alleviating ER stress–mediated apoptosis.
RESULTS
Loss of MGRN1 predisposes cells to apoptosis
In HeLa cells, small interfering RNA (siRNA)–mediated depletion of MGRN1 resulted in the activation of a slew of caspases responsible for intrinsic pathway-mediated apoptosis. A consistent and significant approximately twofold increase in the ratio of cleaved to pro-form of the initiator (caspase 9 [CASP9]) and executioner (CASP3 and CASP7) caspases was detected in these cells compared with the controls (Figure 1, A and B). Comparable activation of these caspases was detected in SHSY5Y cells (Figure 1, C and D), suggesting a cell line–independent phenomenon. To avoid the off-target effects of gene silencing, we depleted MGRN1 in HeLa cells using two other independent siRNAs. Results indicated similar levels of CASP9 cleavage; this could be further rescued by exogenous expression of MGRN1–green fluorescent protein (GFP; (Supplemental Figure S1, A and B). No significant change was detected among the proapoptotic (B-cell/CLL lymphoma 2 [BCL2]–associated agonist of cell death [BAD] and BCL2-associated protein X [BAX]) or antiapoptotic (B-cell lymphoma-extra large [BCL-XL] and myeloid cell leukemia 1 [MCL1]) proteins of the BCL2 family or cleaved forms of CASP8 (associated with the extrinsic pathway) under similar conditions (Supplemental Figure S1, C and D). The fold change in cleaved:pro-CASP9 ratio was quantitatively lower than in ultraviolet (UV)-irradiated cells (Figure 1, E and F). UV treatment affected CASP9 activation to similar extent in untransfected cells and those with mock or MGRN1 siRNAs. During the time frame of the experiments, depletion of MGRN1 was not sufficient to induce death; instead, it predisposed cells to apoptosis. This was also reflected morphologically in the nuclei of cells stained with Hoechst 33342 (Figure 1G), in which an initial stage of nuclear condensation as opposed to total fragmentation was observed (Toné et al., 2007). Here the expression “nucleus” is used to describe shape of the Hoechst 33342–stained chromatin. Whereas UV-irradiated cells showed distinct nuclear fragmentation (nuclear volume, ∼68 µm3), the nuclei of MGRN1-depleted cells exhibited minor condensation. The nuclear volumes were ∼295 and ∼187 µm3 in mock and MGRN1 siRNA–treated cells, respectively (Figure 1, H and I). However, the number of condensed nuclei was significantly higher (approximately threefold) than in control cells. This led us to hypothesize that MGRN1 depletion was a contributing factor that predisposed cells to apoptosis without activation of the classic intrinsic or extrinsic pathways.
FIGURE 1:

Loss of MGRN1 leads to activation of caspases and causes nuclear condensation. (A) HeLa cells treated with mock or MGRN1 siRNAs were lysed and immunoblotted to check for caspases. Note an increase in active caspases with MGRN1 depletion. The levels of β-tubulin serve as loading controls. The blots are representative of at least five independent experiments. Efficiency of knockdown was checked using anti-MGRN1 antibody. (B) The immunoblots in A were analyzed for the fold change in caspase levels. Results from five independent experiments. *p ≤ 0.05 and **p ≤ 0.01 using Student’s t test. Error bars, ±SEM. (C) SHSY5Y cell lysates were analyzed as in A. (D) Histograms of immunoblots generated in C. Analyses from three independent experiments. *p ≤ 0.05 using Student’s t test. Error bars, ±SEM. (E) Untransfected HeLa cells or those transfected with mock or MGRN1 siRNA were either treated with UVB radiation (90 J/cm2 for 5 min) or left untreated. Cell lysates were immunoblotted against CASP9. Note ∼10-fold increase in cleaved CASP9 levels upon UV treatment, and ∼2.5 fold increase with MGRN1- depleted cells. The levels of GAPDH and MGRN1 serve as loading controls. (F) Histogram plotted with data from E. Analyses from six independent experiments. *p ≤ 0.05 using Student’s t test. NS, not significant (p = 0.3 and 0.1). Error bars, ±SEM. (G) HeLa cells treated with MGRN1 siRNA or mock siRNA or UV radiation were stained with Hoechst 33342 and imaged. Representative three-dimensional projection of single nucleus from each experimental condition to establish the imaging and analyses criteria used to define them. (H) Cells similarly treated as in G were imaged. Nuclear boundaries were analyzed using ImageJ. Note distinct nuclear fragmentation upon UV irradiation, whereas MGRN1 depletion led to nuclear condensation. Arrowhead, condensed nucleus. Scale bar, 10 µm. (I) Left, histogram plotting percentage of fragmented nuclei imaged in H. For each condition, 300 nuclei were analyzed from six independent experiments. **p ≤ 0.01 using Student’s t test. NS, not significant (p = 0.1). Inset, percentage of cells with condensed nuclei. Right, distribution of nuclear volume among the three experimental conditions for 70 randomly selected nuclei from the above set. The horizontal line in each box shows the median value, and the white plus sign inside each box is the mean. The upper and lower boundaries of individual boxes show the upper and lower quartiles, respectively; the whiskers are SDs. Outlier values are shown outside the whiskers.
Depletion of MGRN1 leads to onset of ER stress, perturbation in cytosolic calcium, and accumulation of ALIX–ALG-2
Recently we demonstrated up-regulation of autophagic proteins (Beclin1, p62, and LC3 II) upon functional depletion on MGRN1, which suggests the involvement of the genetically programmed and evolutionarily conserved cellular process of autophagy as the adaptive response to cellular stress (Majumder and Chakrabarti, 2015). We further reported that partial loss of MGRN1 function also led to blocked fusion between amphisomes/late endosomes and lysosomes, thus affecting lysosomal degradation. This in turn would result in cellular stress due to accumulation of undegraded cargo. In another study, we demonstrated that catalytic inactivation of MGRN1 led to a higher propensity to mitochondrial depolarization and mitophagy when treated with carbonyl cyanide m-chlorophenyl hydrazone to induce enrichment of cytosolic Ca2+ (Mukherjee and Chakrabarti, 2016). Third, it was reported that exposure of cells to a plethora of stresses up-regulated the mRNA levels of MGRN1. Conversely, overexpression of MGRN1 alleviated cellular stress (Chhangani and Mishra, 2013). Although these results implied that loss of MGRN1 would lead to cellular stress and eventually death, the exact mechanisms remained elusive.
Knockdown of MGRN1 led to ER expansion, a morphological manifestation of ER stress (Khaminets et al., 2015; Figure 2, A and B). Images of cells either left untreated or treated with 2 µM thapsigargin were used as representatives for normal and expanded ER, respectively (Supplemental Figure S2A). We also observed up-regulation of markers of ER stress, as detected by increased levels of phosphorylated PRKR-like endoplasmic reticulum kinase (PERK), binding immunoglobulin protein (BIP), C/EBP homologous protein (CHOP), and cleaved CASP4 compared with control cells (Figure 2, C and D). Treatment with two other independent siRNAs against MGRN1 yielded similar levels of CASP4 activation (Supplemental Figure S2, B and C). In addition, the levels of BCL2 were lower than in control cells (Supplemental Figure S2D). This is plausible because CHOP is known to negatively regulate BCL2 levels (McCullough et al., 2001; Oyadomari and Mori, 2004; Liu et al., 2014). In MGRN1 siRNA–treated cells, however, the enhanced levels of the stress markers were lower than when cells were treated with 2 µM thapsigargin, a drug that depletes the ER Ca2+ pool while enriching its cytosolic concentration. Treating MGRN1-depleted cells with 400 nM thapsigarin increased the levels of the ER stress markers to comparable levels as when control cells were treated with 2 µM drug. A titration for different concentrations of thapsigargin in HEK and HeLa cells showed that whereas 400 nM drug was adequate to up-regulate some of the ER stress markers, this amount was insufficient to elicit CASP9 cleavage (Supplemental Figure S2, E and F). When the stress is robust and persistent, CHOP levels are up-regulated, and, as a consequence, apoptosis occurs (Rutkowski et al., 2006). An increase in the total protein content at the ER was also detected in the microsome fractions isolated from MGRN1-knockdown cells compared with controls (Supplemental Figure 3, A and B). The total cell lysate or the nuclear/mitochondrial fraction had comparable protein amounts across both experimental conditions. Further, increased levels of cleaved or activated CASP4 were also detected in these cells compared with controls (Figure 2, C and D). CASP4 is localized to the ER membrane and is cleaved when cells are treated with ER stress–inducing reagents (Hitomi et al., 2004; Bian et al., 2009). This indicated a predisposition toward ER stress and apoptosis when MGRN1 was depleted. Cleaved CASP4 is in turn known to activate CASP9 (Yamamuro et al., 2011). In MGRN1-depleted cells, 400 nM thapsigargin treatment led to greater than twofold increase in cleaved CASP9 levels. This was partially rescued on exposure to the cytosolic Ca2+ chelator (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid [BAPTA]), either alone or with thapsigargin. However, in cells with mock siRNA, similar amounts of thapsigargin or BAPTA did not elicit alterations in the levels of activated CASP9 and CASP4 (Figure 2, E and F). Untransfected cells behaved like mock controls (Supplemental Figure 4, A and B). Free intracellular levels of Ca2+ were measured using Fura-2–acetoxymethyl-ester (FURA-2AM) in untreated control cells and in those treated with thapsigargin and/or BAPTA (Figure 2G). Knockdown of MGRN1 elevated the levels of free intracellular Ca2+ but to a lesser extent than treatment with thapsigargin.
FIGURE 2:

Depletion of MGRN1 perturbs the ER. (A) HeLa cells treated with mock or MGRN1 siRNAs transiently transfected with ER-GFP construct were either left untreated or treated with 400 nM thapsigargin (Tg) for 6 h; Tg, untransfected cells treated with 2 μM thapsigargin for 6 h. Cells were imaged to observe changes in ER morphology under the different conditions. Scale bar, 15 μm. Approximately 40 cells were imaged for each condition. (B) Histogram plotting percentage of total cell area occupied by the ER area imaged in A. Mean ± SD from three independent experiments. **p ≤ 0.01 using Student’s t test. Error bars, ±SEM. (C) Lysates from cells treated as in A were immunoblotted for markers of ER stress and CASP4. Western blot analyses showed activation of ER stress in MGRN1-depleted cells. Note that 400 nM Tg treatment in MGRN1-depleted cells elicits similar stress response as 2 μM Tg in untransfected cells. (D) Histogram plotting fold change of all proteins analyzed in C. Mean ± SD from three independent experiments. **p ≤ 0.01 and ***p ≤ 0.001 using Student’s t test. Error bars, ±SEM. (E) Mock or MGRN1 siRNA–transfected cells were left untreated or treated with Tg (400 nM for 6 h) alone or with BAPTA (75 μM for 6 h) in the indicated drug combinations. Lysates were immunoblotted using CASP9 and CASP4 antibody. The levels of GAPDH and MGRN1 serve as loading controls. (F) Histogram plotting fold change in CASP9 levels, analyzing data from E. Mean ± SD from three independent experiments. *p ≤ 0.05 using Student’s t test. Error bars, ±SEM. (G) HeLa cells with or without MGRN1 depletion were treated with Tg alone or with BAPTA in the indicated combinations or left untreated. FURA-2AM was loaded, and the cytosolic free Ca2+ concentration was measured from the ratio of fluorescence intensities obtained when samples were excited at 340 and 380 nm. Rmax and Rmin were calculated by Triton X-100 permeabilization of FURA-2AM–loaded cells and by subsequent treatment with ethylene glycol tetraacetic acid, respectively. The apparent Kd for FURA-2-Ca was taken as 224 nM. Mean ± SD for three independent experiments, with triplicates measured for each experiment. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 using Student’s t test. Note that the concentration of cytosolic free Ca2+ is significantly greater in MGRN1-depleted cells than in mock cells without any treatment.
ALG-2 and its interacting protein, ALIX, are implicated in ER stress–induced apoptosis, which alters Ca2+ homeostasis (Rao et al., 2004; Strappazzon et al., 2010). In MGRN1-depleted cells, protein levels of ALIX and ALG-2 were increased by approximately twofold compared with mock controls (Figure 3, A and B). Thapsigargin treatment further increased these levels, whereas addition of BAPTA partially but significantly rescued the amounts of ALIX and ALG-2 (Figure 3, C and D). The increase in ALIX and ALG-2 levels in thapsigargin-treated, MGRN1-depleted cells is probably due to stabilization of these proteins in the presence of very high cytosolic Ca2+, as was seen in mock siRNA–treated cells in the presence of 400 nM drug. We failed, however, to detect significant changes in the ALIX or ALG-2 mRNA level under similar experimental conditions (Figure 3E).
FIGURE 3:

MGRN1 depletion causes accumulation of ALIX–ALG-2. (A) Indicated cell lysates were analyzed by Western blots for the levels of ALIX and ALG-2 proteins. Increased ALIX and ALG-2 levels were observed in MGRN1-depleted samples. The levels of GAPDH and MGRN1 serve as loading controls. (B) Graphs plotted with data from A show analyses from six independent experiments. *p ≤ 0.05 and **p ≤ 0.01 using Student’s t test. Error bars, ±SEM. (C) Mock or MGRN1 siRNA–transfected cells were left untreated or treated with Tg (400 nM for 6 h) alone or with BAPTA (75 μM for 6 h). Note that immunoblotting with antibodies against ALIX and ALG-2 indicates a decrease in their protein levels with BAPTA treatment. The levels of GAPDH and MGRN1 serve as loading controls. (D) Histograms analyzing data from C. Mean ± SD from three independent experiments. *p ≤ 0.05 and **p ≤ 0.01 using Student’s t test. Error bars, ±SEM. (E) Total RNA was isolated from mock or MGRN1 siRNA–treated HeLa cells. Quantitative reverse transcription-PCR was performed using Syber Green and primers against ALIX, ALG-2, and GAPDH. Samples were present in triplicate. The 2-ΔΔCt values for each gene are plotted, with ΔCt calculated as the Ct of the gene minus the Ct of GAPDH in MGRN1-depleted cells. ΔΔCt was calculated as the ΔCt of MGRN1-knockdown samples minus that of the mock cells. The 2-ΔΔCt values do not differ significantly between mock- and MGRN1-depleted samples. Error bars indicate SD. NS, not significant (p = 0.1 and 0.15).
Lower levels of ALIX and ALG-2 proteins were maintained in cells primarily by autophagic degradation because an accumulation was detected after treatment with bafilomycin A1 (Supplemental Figure 4, C and D). Proteasomal degradation did not play a significant role in this process because the protein amounts did not change with addition of the proteasomal inhibitor MG132 (Supplemental Figure 4, E and F). Functional loss of MGRN1 is known to compromise autophagosomal degradation (Majumder and Chakrabarti, 2015).
Results presented so far suggest that compromised MGRN1 leads to ER stress–like conditions and also stabilization of ALIX and ALG-2 protein levels due to blocked autophagosomal degradation and altered calcium homeostasis.
Predisposition to apoptosis by MGRN1 depletion is rescued by the ESCRT-I protein TSG101
The ESCRT-I protein TSG101 was the first substrate identified as ubiquitinated by MGRN1 (Kim et al., 2007). More recently, we showed that overexpression of TSG101 and its monoubiquitination can rescue blocked degradation by the amphisomal-lysosomal and endolysosomal degradation pathways when MGRN1 is depleted (Majumder and Chakrabarti, 2015). Moreover, it has also been established that TSG101 and ALIX interact with each other, and this may involve the presence of ALG-2 (Strack et al., 2003; von Schwedler et al., 2003; Okumura et al., 2009). It was hence obvious to see whether TSG101 could rescue the predisposition-to-apoptosis phenotype. TSG101 overexpression reduced the amount of cleaved CASP9, CASP7, CASP3, and CASP4 generated when MGRN1 was depleted to comparable levels as in the mock control cells (Figure 4, A and B). A comparable decrease in the ratio cleaved:pro-forms of these caspases was detected in SHSY5Y cells (Figure 4, C and D), suggesting a cell line–independent phenomenon. TSG101 also rescued the cell-death phenotype, as assayed by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL; Figure 4E). This was also reflected morphologically in the nuclei of cells stained with Hoechst 33342. The nuclei of MGRN1-depleted cells exhibited minor but significant condensation similar to that detected previously. Overexpression of TSG101 in these cells partially rescued the phenotype; in other words, control and MGRN1-knockdown cells had morphologically similar nuclei without exhibiting nuclear condensation (Figure 5, A and B). The nuclear volumes were ∼323 and ∼281 µm3 in mock and MGRN1 siRNA–treated cells, respectively, when TSG101 was overexpressed. The expanded ER morphology due to MGRN1 knockdown was also rescued by overexpression of TSG101 (Figure 5, C and D).
FIGURE 4:

TSG101 rescues predisposition to apoptosis. (A) Lysates from HeLa cells treated with mock or MGRN1 siRNAs were either transiently transfected with empty vector or TSG101 and immunoblotted for proteins as indicated. Overexpression of HA-TSG101 rescued the depletion of MGRN1 phenotypes. The levels of TSG101, MGRN1, and GAPDH serve as loading controls. Arrowhead, HA-TSG101; arrow, endogenous TSG101. (B) Graphs plotted with data from A show analyses from five independent experiments. NS, not significant (p = 0.1, 0.56, and 0.69) using Student’s t test. Error bars, ±SEM. (C) SHSY5Y cells were treated as in A and probed for levels of indicated caspases. The levels of TSG101, MGRN1 and GAPDH serve as loading controls. Arrowhead, HA-TSG101; arrow, endogenous TSG101. (D) Histograms plotted with data from C show analyses from three independent experiments. NS, not significant (p = 0.1, 0.25, and 0.28) using Student’s t test. Error bars, ±SEM. (E) Mock and MGRN1 siRNA–treated HeLa cells were transiently transfected with TSG101 or empty vector; as control, untransfected cells were treated with UVB radiation (90 J/cm2 for 5 min). Cell death was observed by TUNEL assay. Note that MGRN1 siRNA–treated cells had more TUNEL-positive nuclei, which decreased with overexpression of TSG101. Approximately 30% of the cells were lysed to check levels of TSG101 and MGRN1; GAPDH served as loading control. TUNEL-positive nuclei are been marked by arrowheads. Scale bar, 50 μM. For the immunoblot, arrowhead indicates HA-TSG101, and arrow indicates endogenous TSG101.
FIGURE 5:
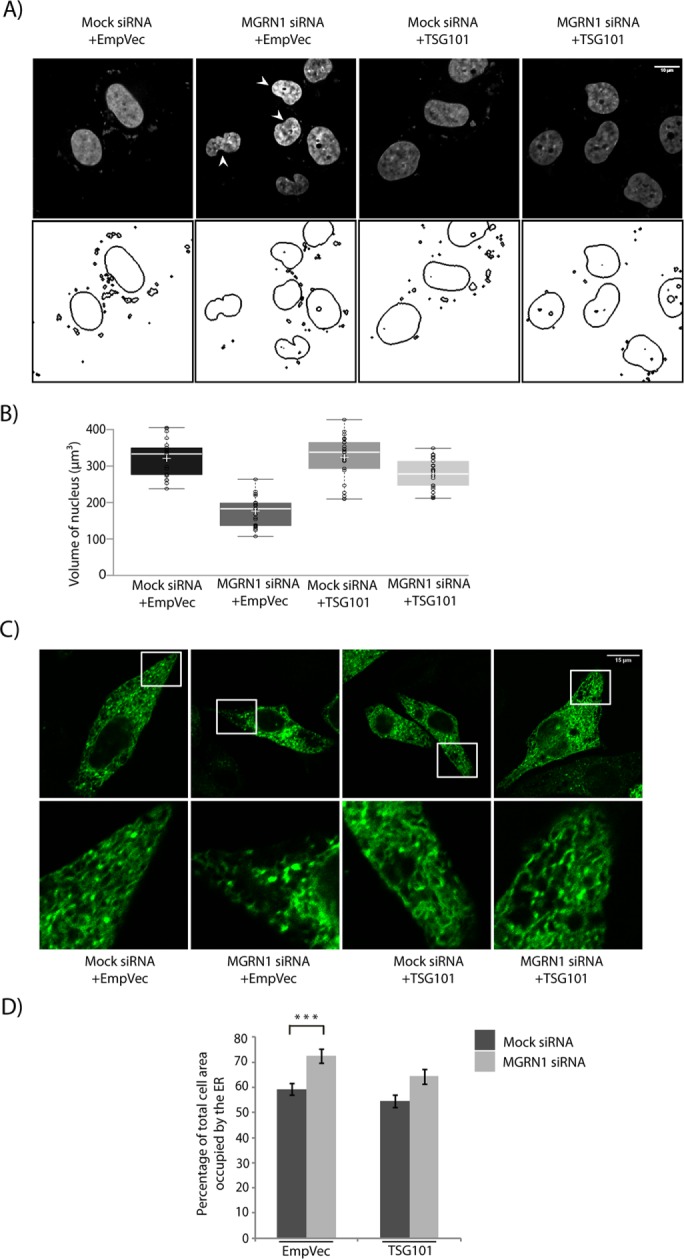
Nuclear condensation and alterations in ER morphology are reversed by TSG101. (A) HeLa cells treated with mock or MGRN1 siRNAs were transiently transfected with empty vector or TSG101, stained with Hoechst 33342, and imaged. Nuclear boundaries were analyzed using ImageJ. Note the significant rescue of nuclear condensation with HA-TSG101 overexpression. Condensed nuclei are marked by arrowheads. Scale bar, 10 µm. (B) Comparison of distribution of nuclear volume among the different experimental conditions. For each condition, 70 nuclei were analyzed from five independent experiments. The horizontal line in each box shows the median value, and the white plus sign inside each box is the mean. The upper and lower boundaries of individual boxes show upper and lower quartiles, respectively; the whiskers are SDs. (C) HeLa cells treated with mock or MGRN1 siRNAs were either transiently transfected with ER-GFP construct along with empty vector or TSG101. Cells were imaged to analyze perturbations in ER morphology. Note the recovery in ER morphology upon overexpression of HA-TSG101. Scale bar, 15 μm. (D) Histogram plotting percentage of ER area imaged in C. Mean ± SD from three independent experiments. ***p ≤ 0.001 using Student’s t test. Error bars, ±SEM.
Depletion of TSG101 recapitulates MGRN1 knockdown
In HeLa cells, siRNA-mediated depletion of TSG101 function resulted in enhanced cleavage and hence activation of CASP9, 7, 3, and 4 (Figure 6, A and B). To corroborate the effect of TSG101 knockdown on CASP9 and CASP4 cleavage, we also used two other independent siRNAs (Supplemental Figure S5, A and B). Consistent and significant ∼1.6-fold increase in the ratio cleaved:pro-form of these caspases was detected in TSG101-knockdown cells compared with the controls. The enhancement in active caspases was similar to that detected with the depletion of MGRN1 (Figure 1, A and B). Morphologically, the nuclei of TSG101-depleted cells stained with Hoechst 33342 showed minor condensation, similar to MGRN1 knockdown (Figure 6, C and D). UV-irradiated cells showed distinct nuclear fragmentation. The nuclear volumes were ∼268 and ∼193 µm3 in mock and TSG101 siRNA–treated cells, respectively. The ER morphology was also perturbed in TSG101-knockdown cells, similar to those with compromised MGRN1 function (Figure 6, E and F). Knockdown of TSG101 elevated the levels of free intracellular Ca2+ as measured with FURA-2AM (Figure 6G). This was similar to recent reports that depletion of some ESCRT proteins could deregulate Ca2+ homeostasis and elicit ER stress (Zhao et al., 2013; Oshima et al., 2016). This suggested that the destabilization of the Ca2+ homeostasis by depletion of either MGRN1 or TSG101 was primarily due to onset of ER stress.
FIGURE 6:

Loss of TSG101 phenocopies MGRN1 depletion. (A) Lysates from HeLa cells transiently transfected with mock or TSG101 siRNAs immunoblotted with indicated antibodies. Note that depletion of TSG101 predisposed cells to apoptosis, as assessed by the activation of the caspases. MGRN1 levels were unaltered across samples. Efficiency of knockdown was checked using anti-TSG101 antibody. The levels of β-tubulin serve as loading control. (B) Graphs plotted with data from A show analyses from three independent experiments. *p ≤ 0.05 and **p ≤ 0.01 using Student’s t test. Error bars, ±SEM. (C) HeLa cells treated with mock or TSG101 siRNAs were stained with Hoechst 33342 and imaged. Untransfected, UVB-irradiated (90 J/cm2 for 5 min) cells serve as apoptotic controls. Nuclear boundaries were obtained using ImageJ. Condensed nuclei are marked by arrowheads. Scale bar, 10 µm. (D) Distribution of nuclear volume among the different experimental conditions imaged in C. For each condition, 70 nuclei were analyzed from five independent experiments. The horizontal line in each box shows the median value, and the white plus sign inside each box is the mean. The upper and lower boundaries of individual boxes show the upper and lower quartiles, respectively; whiskers are SDs. (E) HeLa cells treated with mock or TSG101 siRNAs were transiently transfected with ER-GFP and imaged. Tg, cells treated with 2 μM Tg for 6 h and used as positive control for expanded ER morphology. Note that TSG101 and MGRN1 depletions have a similar effect on the ER structure. Approximately 35 cells were imaged for each condition. (F) Histogram plotting percentage of ER area imaged in E. Mean ± SD from three independent experiments. *p ≤ 0.05 using Student’s t test. Error bars, ±SEM. (G) Mock or TSG101-knockdown HeLa cells were loaded with FURA-2AM, and the cytosolic free Ca2+ concentration was measured using similar experimental conditions as in Figure 2G. Data represents mean ± SD for three independent experiments, with triplicates measured for each experiment. **p ≤ 0.01, Student’s t test.
Further, siRNA-mediated depletion of TSG101 also elevated the protein levels of ALIX and ALG-2 compared with mock controls (Figure 7, A and B). The extent of increase in these proteins was comparable to the depletion of MGRN1. TSG101 knockdown affects autophagosomal degradation (Filimonenko et al., 2007; Majumder and Chakrabarti, 2015). No significant changes in the ALIX or ALG-2 mRNA levels were detected under similar experimental conditions (Figure 7C). Whereas TSG101 and MGRN1 depletion phenotypically closely resembled each other, MGRN1 knockdown did not lead to down-regulation of TSG101 at the mRNA or protein level (Supplemental Figure 5, C and D).
FIGURE 7:

ALIX and ALG-2 accumulate when TSG101 is depleted. (A) Indicated cell lysates were analyzed by Western blots for the levels of ALIX and ALG-2 proteins. The levels of TSG101 and β-tubulin serve as loading controls. (B) Graphs plotted with data from A show analyses from thee independent experiments. *p ≤ 0.05, using Student’s t test. Error bars, ±SEM. (C) Quantitative reverse transcription-PCR was performed on samples generated from mock or TSG101- knockdown cells using similar primers and analyses as in Figure 3E. Error bars indicate SD. NS, not significant (p = 0.26 and 0.29) using Student’s t test.
Ubiquitination of TSG101 by MGRN1 maintains cell viability
We also checked whether the regulation of TSG101 function by MGRN1 could contribute to the activation of caspases. MGRN1-mediated ubiquitination of TSG101 was instrumental in regulating predisposition to apoptosis. For this, cells were transfected with MGRN1 mutants—either the RING-deleted (MGRN1ΔR, unable to ubiquitinate) or the strain with its PSAP motif altered (MGRN1(SRAP), with compromised ability to interact with TSG101; Figure 8A. Activation of the indicated caspases was affected. The levels of ALIX and ALG-2 were elevated, probably due to compromised fusion of autophagosomes/amphisomes with lysosomes, which eventually blocked lysosomal degradation (Majumder and Chakrabarti, 2015). Elevated levels of activated CASP9, ALIX, and ALG-2 upon depletion of MGRN1 could be reversed by exogenous expression of ubiquitin (Ub) or K0 Ub (a lysine-less Ub mutant promoting only monoubiquitination; Tan et al., 2008; Supplemental Figure 5E). Exogenous expression of hemagglutinin (HA)-TSG101 equalized the amounts of ubiquitinated TSG101 (functional protein) in mock- and MGRN1-depleted cells (Supplemental Figure 5F). MGRN1 depletion severely compromised ubiquitination of TSG101, as evident when endogenous TSG101 was immunoprecipitated and immunoblotted with α-ubiquitin antibody. Reduced TSG101 ubiquitination was evident even in HA-TSG101–overexpressed samples when immunoprecipitation was done with anti-TSG101 antibody. This indicated that the amount of functional endogenous TSG101 was less when MGRN1 was knocked down than with the control samples. However, such a difference in TSG101 ubiquitination was not obvious when immunoprecipitation was done with anti-HA antibody (Supplemental Figure 5F). TSG101 may be ubiquitinated by other E3 ligases, TAL and MDM2 (Amit et al., 2004; Cheng and Cohen, 2007). It cannot be completely ruled out that it is probably ubiquitinated by one of these ligases when MGRN1 is nonfunctional, as is evident in the brains of young Mgrn1-null mutant mice. Here ubiquitinated TSG101 is detected, although at relatively reduced levels (Jiao et al., 2009). It is reasonable to extrapolate that whereas MGRN1 modulates TSG101 function, this ESCRT-I protein is the key mediator in regulating cell viability in this scenario.
FIGURE 8:

Interaction between TSG101 and MGRN1 is pivotal for the rescue of the cell death phenotype. (A) Line diagram depicting interaction between TSG101 and MGRN1, motifs/domains responsible for TSG101 ubiquitination, and cellular fate due to this interaction and ubiquitination. HeLa cells transiently transfected with MGRN1, ΔR-MGRN1, or SRAP-MGRN1 constructs were treated with 400 nM Tg for 6 h, lysed, and immunoblotted against the indicated antibodies. The levels of GAPDH and MGRN1 serve as loading controls. (B) Line diagram of TSG101 and its mutants. Mock or MGRN1 siRNA–treated HeLa cells transiently transfected with the indicated HA-TSG101 constructs were lysed and immunoblotted against the indicated antibodies. The levels of GAPDH, MGRN1, and TSG101 serve as loading controls. (C) Histograms generated using data from B show analyses from three independent experiments. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. NS., not significant (p = 0.2, 0.34, and 0.28) using Student’s t test. Error bars, ±SEM.
The interaction between TSG101 and MGRN1 affected activation of the caspases. For this, mock- or MGRN1-depleted HeLa cells were transfected with TSG101 or its mutants that were compromised in their interaction with MGRN1. In the presence of TSG101, cleaved CASP9 levels, along with those of ALIX and ALG-2, were comparable between mock- and MGRN1-depleted cells (Figure 8, B and C). However, when cells expressed Δ ubiquitin E2 variant (ΔUEV)-TSG101 or M95R-TSG101, the levels of cleaved:pro-CASP9 significantly increased. ALIX and ALG-2 proteins were also increased in cells expressing TSG101 mutants. The TSG101 UEV domain binds specifically to P(S/T)AP sequences, but a mutation in the UEV domain abolishes this binding (Kim et al., 2007). These mutations also affect the interaction between TSG101 and ALIX (Pornillos et al., 2002; von Schwedler et al., 2003; Supplemental Figure 5G).
Taken together, the results so far suggest that TSG101 is an important molecular player in regulating predisposition of cells to ER stress–mediated apoptosis.
CASP4 activates CASP9 during ER stress
To establish a direct correlation between predisposition to apoptosis and ER stress, we examined the role of CASP4 activation in this process. Mock and MGRN1- or TSG101-knockdown HeLa cells were transfected with wild-type or mutant CASP4 constructs. CASP-C258A mutant is not cleaved or activated and hence is incapable of activating CASP9 (Pallepati and Averill-Bates, 2011; Yamamuro et al., 2011; Kajiwara et al., 2014). Here, although an increase in the levels of ALIX and ALG-2 were detected in the MGRN1- and TSG101-depleted samples, CASP9 was not cleaved, and nuclear condensation was not observed (Figure 9, A–C).
FIGURE 9:

CASP4 activates CASP9, but CASP4-C258A does not. (A) HeLa cells treated with the indicated siRNAs were transiently transfected with CASP4 or its CARD-domain mutant (CASP4-C258A). Cells were lysed 24 h posttransfection and treated with 200 nM Tg for 6 h and immunoblotted against the indicated antibodies. Note that loss of MGRN1 and TSG101 predisposed cells to apoptosis (as assessed by CASP9 cleavage) in the presence of CASP4 but not that of CASP4-C258A. ALIX and ALG-2 levels were elevated upon MGRN1 and TSG101 depletion, irrespective of CASP4 activity. The levels of TSG101, MGRN1, CASP4, and GAPDH serve as loading controls. Arrow, Myc-Pro-CASP4. (B) HeLa cells treated as in A were stained with Hoechst 33342 and imaged. Nuclear boundaries were obtained using ImageJ. Note that nuclear condensation closely follows the results of CASP9 cleavage. Boundaries of two random nuclei in each image are demarcated. Condensed nuclei are marked by arrowheads. Scale bar, 10 µm. (C) Histogram comparing the distribution of nuclear volume for the different experimental conditions in B. For each condition, 30 nuclei were analyzed from three independent experiments. The horizontal line in each box shows the median value, and the white plus sign inside each box is the mean. The upper and lower boundaries of individual boxes show the upper and lower quartiles, respectively; the whiskers are SDs.
Interaction between ALIX and ALG-2 predisposes to apoptosis but that of ALIX with TSG101 protects against cell death
ER stress–induced cell death is independent of Apaf-1 and cytochrome c molecules but recruits valosin-containing protein and ALG-2 (Rao et al., 2004). It was proposed that the ALIX-TSG101 binding is Ca2+ dependent and uses ALG-2 as an adaptor protein (Okumura et al., 2009; Sun et al., 2015). Further, it was also suggested that cell death as a result of cytosolic Ca2+ increase requires ALIX- and ALG-2–mediated CASP9 activation (Strappazzon et al., 2010). Our results so far indicate that when Ca2+ homeostasis is altered inside cells (such as due to depletion of MGRN1 or TSG101), CASP4 is cleaved. Simultaneous stabilization of the ALIX and ALG-2 levels due to blocked degradation at the lysosomes eventually leads to activation of CASP9. To understand the significance of each of these molecular players in regulating the phenotype of predisposition to apoptosis, we transfected cells with various combinations of TSG101 and ALIX constructs. The fold change in the levels of cleaved:pro-CASP9 increased in the presence of ALIX. Overexpression of TSG101 significantly reduced this (Figure 10, A and B). However, when ALIX-ΔTSG101 (a mutant of ALIX compromised in its binding with TSG101) was present, TSG101 could not rescue the phenotype, and a significantly high level of cleaved CASP9 was detected. Further, expressing ALIX–ΔALG-2 mutant did not elicit an increase in cleaved CASP9 in the presence of TSG101 or ΔUEV-TSG101. Our results also showed that the ALIX-TSG101 interaction was not drastically altered in the presence of ALIX–ΔALG-2 or when ALG-2 was depleted (Supplemental Figure 6, A–E). This association of ALIX with TSG101 under ALG-2–binding compromised conditions was verified in different cell lines. The presence of very low levels of functional ALG-2 enabling this interaction, however, could not be completely eliminated. Taken together, these results suggest that whereas interaction between ALIX and TSG101 rescued the apoptosis phenotype, the association between ALIX and ALG-2 supported predisposition to cell death. It has been suggested that under conditions of high cytosolic Ca2+, ALIX and ALG-2 interact to promote cell death (Missotten et al., 1999; Vito et al., 1999).
FIGURE 10:

ALIX-TSG101 interaction favors cell viability, whereas ALIX–ALG-2 interaction predisposes cell to death. (A) Lysates from HeLa cells transiently cotransfected with the indicated constructs were immunoblotted against the given antibodies. Note that CASP9 cleavage was not observed when ALIX and TSG101 could interact. The bands used to quantify the fold change of CASP9 activation are marked by arrows. ALG-2 levels were unaltered across samples. Levels of ALIX, TSG101, and GAPDH serve as loading controls. (B) Graph plotted with data from A shows analyses from three independent experiments. **p ≤ 0.01, using Student’s t test. Error bars, ±SEM. (C) HeLa cells transiently transfected with various ALIX constructs were left untreated or treated with Tg alone or with BAPTA. Cell lysates were analyzed by Western blots. (D) Histogram plotting results from C, using results from four independent experiments. *p ≤ 0.05 and **p ≤ 0.01, Student’s t test. Error bars, ±SEM. (E) MGRN1-depleted HeLa cells transfected with either empty vector or various ALIX constructs were lysed and immunoblotted for CASP9 cleavage. GAPDH serves as the loading control, and the expression of ALIX and its mutants were verified across different lysates. Arrowhead, FLAG-ALIX; arrow, endogenous ALIX. (F) Graph generated using results of E from three independent experiments. *p ≤ 0.05; NS, not significant (p = 0.11), using Student’s t test. Error bars, ±SEM. (G) HeLa cells transfected with mock or MGRN1 siRNAs were further knocked down with nontargeting or ALIX or ALG-2 siRNAs. Lysates were analyzed for CASP9 cleavage. Level of GAPDH serves as loading control; efficiency of knockdown was checked using antibodies against ALIX, ALG-2, and MGRN1. (H) Histogram plotted using results of G from three independent experiments. **p ≤ 0.01 and ***p ≤ 0.001 using Student’s t test. Error bars, ±SEM.
When ALIX or ALIX-ΔTSG101 was overexpressed in cells, a Ca2+-dependent increase in the levels of cleaved CASP9 was observed (Figure 10, C and D), probably due to stabilization of the ALIX–ALG-2 complex. No discernible Ca2+ dependence was detected when ALIX–ΔALG-2 or ALIX–ΔTSG101–ΔALG-2 was exogenously expressed in cells. To further understand why MGRN1 depletion resulted in predisposition to apoptosis, we transfected knockdown cells with ALIX and its mutants and assayed for the fold change in the levels of cleaved:pro-CASP9 (Figure 10, E and F). Here again, expression of ALIX and ALIX-ΔTSG101 led to increase in the amounts of cleaved CASP9, whereas ALIX–ΔALG-2 behaved like the control (with empty vector). Hence, absence of interaction between ALIX and TSG101 promoted cell death, whereas that of ALIX with ALG-2 had the opposite result. Under conditions of MGRN1 knockdown, depleting either ALIX or ALG-2 did not elicit an increase in the fold change in the level of cleaved:pro-CASP9 (Figure 10, G and H), again emphasizing that ALIX and ALG-2 interaction was crucial in guiding cells to apoptosis.
CtmPrP-mediated ER stress and predisposition to apoptosis are rescued by TSG101
The presence of the aberrant metabolic isoform of the ubiquitously expressed cell surface glycoprotein mammalian PrP, referred to as CtmPrP, is associated with elevated ER stress (Wang et al., 2011). Studies have shown that increased generation of CtmPrP by the expression of the naturally occurring human disease mutation (PrP(A117V)) leads to spongiform neurodegeneration in animal models (Hegde et al., 1998; Rane et al., 2010). Brain lysates from transgenic mice expressing CtmPrP (PrP(A117V)) showed increase in ER stress markers along with the levels of cleaved CASP9, ALIX, ALG-2, and ER stress–associated, murine-activated CASP12 compared with nontransgenic control (Figure 11, A and B). Induction of ER stress and activation of CASP12 have also been observed in the presence of the pathological form PrPSc (Hetz et al., 2003). In HeLa cells, expression of PrP(A117V) similarly resulted in the onset of ER stress and increased levels of cleaved CASP4, CASP9, ALIX, and ALG-2 (Figure 11, C and D). In addition, CtmPrP expression is implicated in depletion of MGRN1 in cells and selected brain regions (Chakrabarti and Hegde, 2009). Exogenously expressed TSG101 rescued the ER stress and predisposition-to-death phenotypes by restoring the levels of CHOP, activated caspases (CASP4 and CASP9), ALIX, and ALG-2 similar to the PrP controls (Figure 11, C and D). TSG101 further rescued the expanded ER morphology detected in cells expressing PrP(A117V) (Figure 11, E and F). Overexpressing PrP did not affect ER morphology. Here again, TSG101 partially reversed the effects of MGRN1 depletion and induction of ER stress in a more pathologically relevant situation. We can hypothesize that CtmPrP-mediated cytotoxicity involves close interplay among several ESCRT machinery proteins.
FIGURE 11:

TSG101 rescues CtmPrP-mediated ER stress. (A) Whole-brain lysates from transgenic mice were immunoblotted and analyzed for the indicated proteins. The levels of GAPDH serve as loading control. Note an increase in ER stress markers, active caspases (CASP12 and CASP9), and ALIX, and ALG-2 levels. (B) Graphs plotted with data from A show analyses from three independent experiments. **p ≤ 0.01 using Student’s t test. Error bars, ±SEM. (C) Lysates prepared from HeLa cells transiently cotransfected with wild-type PrP or CtmPrP-generating mutant (A117V) along with empty vector or TSG101 were immunoblotted against antibodies as indicated. The levels of PrP, TSG101, and GAPDH serve as loading controls. Note that overexpression of TSG101 rescues the levels of CHOP, active caspases (CASP4 and CASP9), ALIX, and ALG-2 comparable to the control. Arrowhead, HA-TSG101; arrow, endogenous TSG101. (D) Graphs plotted with data from C show analyses from three independent experiments. **p ≤ 0.01 and ***p ≤ 0.001 using Student’s t test; NS, not significant (p = 0.88, CHOP; p = 0.66, CASP4; p = 0.41, CASP9; p = 0.17, ALIX; and p = 0.67, ALG-2). Error bars, ±SEM. (E) HeLa cells transiently cotransfected with wild-type PrP or CtmPrP-generating mutant (A117V) along with empty vector or TSG101 and ER-GFP construct were imaged. Note that the ER morphology was rescued upon overexpresssion of HA-TSG101. ER-GFP is in green, PrP in blue, and HA-TSG101 in red. Scale bar, 15 µm. (F) Histogram plotting percentage of ER area imaged in E. Mean ± SD from three independent experiments. *p ≤ 0.05 using Student’s t test. Error bars, ±SEM.
DISCUSSION
This study elucidated the mechanism by which MGRN1 depletion predisposes various types of cells to apoptosis by up-regulating ER stress and using components of the ESCRT pathway. This activation of ER stress is not as robust as when cells were stimulated with 2 µM thapsigargin. ER stress–associated CASP4 is cleaved in MGRN1-depleted cells, and this effect is enhanced in the presence of 400 nM thapsigargin. In control cells, however, this amount of thapsigargin is not sufficient to elicit CASP4 or CASP9 cleavage, although the other markers of ER stress are increased. The levels of cleaved CASP9, ALIX, and ALG-2 are also higher in samples with MGRN1 depletion. Chelating away cytosolic Ca2+ partially restores the amounts of active CASP9, ALIX, and ALG-2 to the controls cells (mock siRNA–transfected or untransfected), suggesting that a minor insult can tilt the cellular balance toward apoptosis. A logically justified extrapolation would be that MGRN1 depletion predisposes cells to stress and hence apoptosis. The levels of ALIX and ALG-2 are increased due to impaired autophagosomal-lysosomal degradation. TSG101 depletion hence also stabilizes the levels of ALIX and ALG-2. At the same time, depletion of this ESCRT-I protein predisposes to ER stress. Increased levels of free cytosolic Ca2+ along with higher protein quantities of ALIX and ALG-2 favor predisposition to apoptosis when either MGRN1 or TSG101 is knocked down in cells. However, when TSG101 is overexpressed, autophagosomal degradation of ALIX and ALG-2 is restored, and their levels are not high. This abundance of TSG101 promotes interaction between ALIX and TSG101, thus protecting cells against apoptosis. Monoubiquitination of TSG101 is also able to mitigate the proapoptosis phenotype. Contrary to earlier studies, compromised interaction between ALIX and ALG-2 does not drastically affect binding between ALIX and TSG101. Impaired association of ALIX with TSG101 instead leads to activation of CASP9 (Figure 12). This study is significant because it further shows that ER stress induced by the disease-causing CtmPrP mutant (resulting in increased levels of ALIX and ALG-2) can be rescued by TSG101 overexpression. It is plausible to suggest that MGRN1 depletion is one of the contributing factors that predispose to apoptosis; any additional trigger would lead to final onset of the process.
FIGURE 12:

Schematic diagram summarizing the results. In healthy cells, when cytosolic Ca2+ is low, MGRN1-mediated ubiquitination of TSG101 promotes lysosomal degradation and helps maintain low levels of ALIX and ALG-2. MGRN1 depletion leads to ER stress and increase in cytosolic Ca2+, ALIX, and ALG-2 protein levels and facilitates the ALIX–ALG-2 interaction. ER stress–associated CASP4 is cleaved; this in turn activates CASP9 in the presence of high ALIX and ALG-2 levels. In the presence of excess TSG101, the concerted effect of multiple factors protects against apoptosis; lysosomal degradation resumes along with interaction with ALIX. Blocked pathways are indicated by red lines; EE, early endosome; MVB, multivesicular body; ER, endoplasmic reticulum; red dots, Ca2+; yellow ovals, ubiquitin.
Apoptosis is a normal cellular process and is tightly regulated during the lifespan of any organism. It plays an important role during embryonic development, leading to proper formation of various organs and structures. Deregulated apoptosis during embryogenesis is lethal for developing embryos (Brill et al., 1999). It is a highly regulated physiological process during normal heart development (Fisher et al., 2000; Barbosky et al., 2006; Sanchis et al., 2008). However, an increase in myocardial apoptosis during embryonic development results in cardiac dysfunction and congenital heart defects (Pexieder, 1975; Gittenberger-de Groot et al., 2005). Of interest, ventricular septal defects are among the most common congenital heart defects in children (Smith et al., 2015). Deficiency of endothelial nitric oxide synthase is implicated in increased cardiomyocyte apoptosis during embryonic heart development and congenital septal defects (Feng et al.; 2002). It is also speculated that prolonged ER stress in cardiac tissue ultimately triggers apoptosis (Groenendyk et al., 2010).
About 46–60% mortality by weaning age is found in mice homozygous for the null allele of MGRN1 (Cota et al., 2006). A significant majority of these deaths are due to complex congenital heart defects, including atrial and ventricular septal defects, thinning of the myocardium, right aortic arch, double-outlet right ventricle, unroofed coronary sinus, malposition of the great arteries, retroesophageal left subclavian artery, abnormal heart situs (mesocardia or dextrocardia), and/or pericardial and pleural effusion (Cota et al., 2006; Jiao et al., 2009). This raised the possibility that loss of expression of the ubiquitously expressed E3 ligase MGRN1 could also have a role in congenital heart defects by up-regulating ER stress.
The significance of MGRN1 in alleviating cellular stress was found in recent studies showing that functional loss of MGRN1 affected lysosomal degradation and enhanced propensity to mitochondrial depolarization and that its overexpression could abrogate the effects of a plethora of cellular stresses (Chhangani and Mishra, 2013; Majumder and Chakrabarti, 2015; Mukherjee and Chakrabarti, 2016). Increased presence of cytosolic Ca2+ along with the established markers of ER stress indicate that MGRN1 depletion indeed disrupts intracellular Ca2+ homeostasis and leads to onset of ER stress. A previous report demonstrated that ER stress–induced apoptosis involved CASP12 (mouse homologue of human CASP4) to activate CASP9 (Rao et al., 2002). We hence suggest that depletion of MGRN1 results in predisposition to apoptosis. We cannot exclude the possibility that prolonged ER stress may also involve mitochondria among other players in caspase activation and cell death.
Recent work suggests that ablation of the ESCRT-0 protein Hrs induces hippocampal neuronal cell loss in mouse models. Silencing of Hrs in cell culture systems results in impaired autophagic clearance and enhanced ER stress. This eventually culminates in apoptotic and neproptotic cell death by triggering c-Jun N-terminal kinase (JNK) activation (Oshima et al., 2016). In yeast cells, deletion of ESCRT components (Snf7, Snf8, Stp22, Vps20, Vps25, Vps28, or Vps36) activates calcium/calcineurin signaling. This leads to increased accumulation of intracellular Ca2+ in response to ER stress in these mutants and also altered calcium homeostasis (Zhao et al., 2013). Similarly, here we show that functional depletion of the ESCRT-I protein TSG101, previously shown to compromise autophagosomal-lysosomal degradation (Majumder and Chakrabarti, 2015), also induces ER stress, and this predisposes cells to apoptosis. MGRN1 depletion affects TSG101 function and perturbs lysosomal degradation. The present study finds an increase in the ER protein load in MGRN1-depleted samples, indicating onset of ER stress. Blocked clearance of lysosomal cargo could hence be a factor promoting ER stress and destabilizing cytosolic Ca2+, although the exact mechanism requires further study.
The Ca2+ binding to ALG-2 induces a conformational change that allows it to interact with the multifunctional adaptor protein ALIX (Vito et al., 1999). ALIX and ALG-2 act upstream of CASP9 activation after cytosolic Ca2+ increase (Strappazzon et al., 2010). Further, ALIX has also been shown to interact with TSG101 using ALG-2 as an adapter protein in a Ca2+-dependent manner (Okumura et al., 2009). This mediates sorting at MVBs (Sun et al., 2015). The association between ALIX and TSG101 may also be mediated through one of the adapter proteins involved in cytokinesis—CD2-associated protein (CD2AP), centrosomal protein 55 (CEP55), Rho-associated coiled-coil–containing protein kinase 1 (ROCK1), or IQ motif–containing GTPase-activating protein (IQGAP). CD2AP is best characterized as an adaptor protein that functions in various stages of endocytic protein trafficking and actin remodeling (Wolf and Stahl, 2003). Reductions in CD2AP levels affect MVB biogenesis (Kim et al., 2003), as well as abscission (Monzo et al., 2005). ALIX and TSG101 concentrate at centrosomes and are recruited to the midbodies of dividing cells via direct interactions with CEP55, IQGAP1, and ROCK1 (Morita et al., 2007). IQGAP1 and ROCK1 regulate cytoskeletal remodeling (Machesky, 1998; Matsumura, 2005) and possibly also during abscission (Adachi et al., 1997; Madaule et al., 1998; Skop et al., 2004). There is no experimental evidence that the interaction between ALIX and TSG101 involving CD2AP, CEP55, ROCK1, or IQGAP requires alteration of Ca2+ homeostasis. These results also suggest that ALIX-TSG101 interaction does not require ALG-2 as the adapter protein.
Here we provide the first evidence that whereas predisposing cells through ER stress mediates apoptosis, interaction between ALIX and ALG-2 and between ALIX and TSG101 are independent of each other but are both potentiated in the presence of cytosolic Ca2+. MGRN1 depletion results in onset of ER stress (and Ca2+ disrupted homeostasis) and simultaneously leads to increased protein levels of ALIX and ALG-2. These two events together would tilt the balance to ALIX–ALG-2 interaction and culminate in apoptosis. On the other hand, if TSG101 levels were exogenously increased, ALIX-TSG101 interaction would be more favored in protecting cells from activating caspases and initiating cell death. We speculate that blocked lysosomal degradation probably induces ER stress on one hand, and ER stress can then affect cytosolic Ca2+ levels. On the other hand, lack of lysosomal degradation also leads to accumulation of ALIX and ALG-2. We speculate that concerted action of these events ultimately culminates in cell death.
Finally, our study linking components of the ESCRT machinery via their involvement with MGRN1 may highlight a key event in regulating ER stress–mediated apoptosis that may guide early development of the cardiovascular system, and an excess of which translates into complex congenital heart defects, as well as lead to neurodegeneration in at least a subset of prion diseases.
MATERIALS AND METHODS
Constructs, antibodies, and reagents
MGRN1, MGRN1ΔR, PrP, PrP(A117V), and MGRN1-GFP constructs have been described (Chakrabarti and Hegde, 2009), as has MGRN1(SRAP) (Majumder and Chakrabarti, 2015). HA-tagged TSG101 was a gift of Juan S. Bonifacino (National Institutes of Health, Bethesda, MD); FLAG-tagged ALIX, ALIX–ΔALG-2, ALIX-ΔTSG101, and ALIX–ΔTSG101–ΔALG-2 were gifts of Rémy Sadoul (Grenoble Institute of Neurosciences, France); Myc-tagged CASP4 and CASP4-C258A were gifts of Hans-Dietmar Beer (University Hospital Zurich, Switzerland); ER-GFP construct was a gift of Erik L. Snapp (Janelia Research Campus); HA-tagged wild-type Ub was a gift of Rafael Mattera (National Institutes of Health, Bethesda, MD); and K0 Ub mutant was a gift of Kah-Leong Lim (Duke-NUS Medical School, Singapore). HA-ΔUEV-TSG101 was generated using standard cloning and mutation techniques, and HA-M95R-TSG101 was generated by standard site-directed mutagenesis methods.
Antibodies were from the following sources. Cell Signaling Technology: caspase 9 (9502P), cleaved caspase 9 (7237P), caspase 7 (9492P), cleaved caspase 7 (8438P), caspase 3 (9662P), cleaved caspase 3 (9664P), phospho-PERK (3179S), PERK (683P), CHOP (2895P), BIP (3177P), MCL1 (5453P), BCL-XL (2764P), BCL2 (2370P), and β-catenin (9562). Thermo Scientific: p62/SQSTM1 (PA5-20839). Sigma-Aldrich (St. Louis, MO): monoclonal anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) clone GAPDH-71.1 (G8795). Abcam: TSG101 (ab83), BAD (ab32445), ALG-2/PDCD6 (ab109181), ALIX (ab117600), β-tubulin (ab7792), caspase 4 (ab22687), caspase 8 p10 (ab2553), and BAX (ab7977). Novus: VDAC1 (15895) and Ub (NB300-130). The MGRN1, GFP, Myc, PrP, FLAG, TRAP-α, and HA antibodies were gifts of Ramanujan S. Hegde (MRC Laboratory of Molecular Biology, Cambridge, United Kingdom).
MG132, thapsigargin, FURA-2AM, bafilomycin A1, and BAPTA-AM were from Sigma Aldrich; Universal FastStart Syber Green Master (Rox) was from Roche; Hoechst 33342 was from Cell Signaling Technology; 4′,6-diamidino-2-phenylindole (DAPI) was from USB; and TRIzol reagent was from Invitrogen. Drug treatments were as follows: MG132 (25 µM, 6 h), bafilomycin A1 (300 nM, 6 h), thapsigargin (400 nM, 6 h; 200 nM, 6 h; 2 µM, 6 h), and BAPTA-AM (75 μM, 6 h). For thapsigargin titration experiments, drug treatments were done for 6 h. In situ BrdU-Red DNA Fragmentation (TUNEL) Assay Kit (ab66110) was from Abcam.
Cell culture and transfection
Cell lines used for the experiments were HeLa (human cervical cancer cell line), SHSY5Y (human neuroblastoma cell line), and HEK293T (human embryonic kidney cell line). Maintenance of cells in culture was as before (Srivastava and Chakrabarti, 2014). Briefly, cells were grown in 10% fetal bovine serum (FBS; Life Technologies, Grand Island, NY)/DMEM (Himedia, Mumbai, India) at 37°C and 5% CO2. SHSY5Y cells (a gift of Debashis Mukhopadhyay, Saha Institute of Nuclear Physics, Kolkata, India) and HEK293T cells (a gift of Subrata Banerjee, Saha Institute of Nuclear Physics, Kolkata, India) were grown under standard cell culture conditions.
For transfections of cells, Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used per the manufacturer’s instructions. At 24 h posttransfection, cells were lysed in suitable buffers.
All tissue culture plastic ware used for microscopy were from Nunc (Roskilde, Denmark), and bottom coverglass dishes were from SPL Life Sciences (Gyeonggi-do, South Korea).
siRNA-mediated knockdown
For siRNA-mediated knockdown, ON-TARGETplus SMARTpool siRNAs from Thermo Scientific (Dharmacon Products, Lafayette, CO) were used, consisting of a mixture of four individual siRNAs against MGRN1 (L-022620-00-0020), human TSG101 (L-003549-00-0005), human ALIX/PDCD6IP (L-004233-00-0005), human ALG-2/PDC6 (L-004440-00-0005), nontargeting siRNA (D-001810-01-20), and GFP (D-001300-01-20). Ambion Silencer Select Pre-designed (Inventoried) siRNAs (Life Technologies) consisting of single siRNAs were also used against human TSG101 (s14441 and s14439, referred to as TSG101 siRNA1 and siRNA2, respectively) and human MGRN1 (s23496 and s23495, referred to as MGRN1 siRNA1 and siRNA2, respectively). siRNAs were transfected using Lipofectamine 2000 following the manufacturer’s instructions.
Cells knocked down for 48 h were transfected with indicated constructs if required. They were used for experiments 20–24 h posttransfection.
Double siRNA-mediated knockdown was done by combining two SMARTpool siRNAs in equal amounts, keeping the total concentration the same, as recommended by the manufacturer.
UV treatment of cells
Subconfluent cells were washed with phosphate-buffered saline (PBS) and exposed to UV light. Vilber Lourmat Bio-Link BLX Crosslinker (BLX-E312), which emits UVB rays with an emission peak at 312 nm, was used for irradiating the cells in PBS. Cells were irradiated with a dose of 90 J/cm2 for 5 min. After this, PBS was discarded, and cells were maintained in fresh culture medium for 24 h at 37°C and 5% CO2. Cells were then harvested for biochemical analyses or used for imaging. Control cells were treated similarly without being exposed to UV.
Hoechst 3342 staining
After specific treatment, cells were fixed with 4% formaldehyde. They were then permeabilized using PBS/0.1% Triton X-100 (Sigma-Aldrich) for 25 min, followed by staining with a working concentration of 1µ g/ml Hoechst 33342. The samples were then imaged using a fluorescence microscope.
Western blotting and immunoprecipitation
The protocol for Western blotting was as described before (Srivastava and Chakrabarti, 2014). We used 7.5, 10, or 12% Tris-tricine gels for SDS–PAGE, followed by Western blotting.
For immunoprecipitation, cells were lysed in lysis buffer (142.5 mM KCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 0.2% NP-40, 50 μM CaCl2, pH 7.4) by passage through a 25-gauge needle 20 times. Unlysed cell debris was removed by spinning at 1500 × g. CaCl2 at 50 μM was used in most experiments, unless otherwise indicated. Immunoprecipitation was performed using standard protocols.
Quantification of Western blots was done using Bio-Rad Quantity One software. The data were tabulated in Microsoft Excel and used to generate the histograms. At least three independent experiments were performed, and band intensities were normalized to loading control. The p values were determined using Student’s t test.
Fold change in caspase activation is calculated as
 |
Microsome isolation protocol
Cells were lysed in microsomal isolation buffer containing 0.3 M sucrose and 10 mM HEPES (pH 7.0) by passage through a 25-gauge needle attached on a 1-ml syringe 30 times. This was centrifuged at 1500 × g to pellet unlysed cell debris. Nuclear and mitochondrial fractions were removed by centrifuging the supernatant at 12,000 × g. Microsomal fraction was enriched by centrifuging the supernatant at 43,000 rpm.
Quantitative PCR
After specific treatment, cells were harvested for immediate RNA extraction. Total RNA was prepared using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Quantitative PCR was used to compare the expression of human ALIX (forward primer, 5′-CTG ACA AAA TCA ATC GTG CC-3′; reverse primer, 5′-CC AAA GAC TGC TGT ACT GAC-3′), human ALG-2 (forward primer, 5′-TTC CAG AGG GTC GAT AAA GAC-3′; reverse primer, 5′-CGT GCG GAA GAC GTT CTG CCA-3′), and human TSG101 (forward primer, 5′-CCA AAT ACT TCC TAC ATG CCA-3′; reverse primer, 5′-ATC CCT ACT GGG ACC AAC AGT-3′) genes.
Determination of free intracellular Ca2+
After specific treatments, free intracellular Ca2+ was measured in cells as before (Mukherjee and Chakrabarti, 2016). Briefly, the cytosolic free Ca2+ concentration was measured using FURA-2AM. The ratio of fluorescence intensities was obtained for samples excited at 340 and 380 nm. Rmax and Rmin were calculated as previously described (Pérez et al., 1998; Grynkiewicz et al., 1985). The apparent Kd for FURA-2-Ca was taken as 224 nM. We used a Multiskan GO Microplate Spectrophotometer (Thermo Fisher Scientific).
Imaging and analysis
For fluorescence microscopy, cells were grown on coverslips or bottom-coverglass dishes and fixed after each experiment with 4% paraformaldehyde for 10 min. Cells were permeabilized with 0.1% saponin and 10% FBS in PBS for 1–2 h. Subsequently the coverslips/plates were incubated in a wet chamber with primary antibody solution overnight at 4°C. The next day, cells were washed with wash buffer containing 10% FBS in PBS and then incubated with secondary antibody solution for 1 h. Coverslips/dishes were finally washed three times with wash buffer and then observed under microscope.
Fluorescence imaging was performed using the Nikon A1R+ Ti-E with N-SIM and FCS microscope systems. For imaging, a 100×/1.4 numerical aperture (NA) oil immersion objective was used for Figures 1, G and H, 2A, 5, A and C, 6, C and E, 9B, and 11E and Supplemental Figure S2A, and a 40×/0.9 NA dry objective was used for Figure 4E. An Ar-ion laser was used for excitation of Hoechst 33342 and DAPI at 405 nm, with band pass (BP) 425–475 nm (emission filter 495LP), excitation of GFP at 488 nm, with BP 500–550 nm (emission filter 560LP), excitation of Alexa Fluor 546 at 561 nm, with BP 570–620 nm (emission filter 640LP), and excitation of Alexa Fluor 633 at 638 nm, with BP 663–738 nm (emission filter 640HP).
For quantitative analyses and comparisons between multiple samples, images were collected using identical excitation and detection settings. The detector gain settings were chosen to allow imaging of the desired cells within the linear range of the photomultiplier tube without saturating pixels, unless otherwise specified.
Image analysis was done using ImageJ software. Eight-bit images were converted to black and white using the threshold function; the free-hand selection tool was used to demarcate the nuclear perimeter (as indicated in the bottom parts of the figures). The volume of nuclei was measured using three-dimensional projections.
ER area measurements
HeLa cells were transfected with the indicated constructs along with ER-GFP (Snapp et al., 2006). After each experiment, cells were fixed and imaged. ER area was measured using ImageJ as described previously (Khaminets et al., 2015). In short, the background threshold was manually defined and set for all images. Borders of each cell were drawn, and the ER area was calculated; it is presented as a fraction of the total cell area.
TUNEL assay protocol
Mock or MGRN1 siRNA–treated HeLa cells were transiently transfected with an empty vector or TSG101-HA; untransfected cells were treated with UVB radiation (90 J/cm2 for 5 min). TUNEL staining was carried out as per manufacturer’s protocol. In short, cells were washed in 1× PBS thrice and fixed with 4% paraformaldehyde for 10 min. These were then permeabilized with 0.1% Triton X-100 in PBS for 10 min. The cells were incubated in the dark at 37°C with terminal deoxynucleotidyl transferase and BrdUTP for 1 h and then stained with anti–BrdU-red antibody for 30 min at room temperature. Cells were then counterstained with 0.1 µg/ml DAPI for 5 min and imaged under a fluorescence microscope.
In vivo ubiquitination assay
The in vivo ubiquitylation assay was performed as previously described (Srivastava and Chakrabarti, 2014). Mock- and MGRN1-depleted HeLa cells transfected with either empty vector or HA-TSG101 were lysed in immunoprecipitation buffer and immunoprecipitated under denaturing conditions with anti-HA or anti-TSG101 antibody. Ubiquitinated TSG101 was detected by immunoblotting with anti-Ub antibody.
Supplementary Material
Acknowledgments
We are grateful to R. Mattera, K.-L. Lim, J. S. Bonifacino, R. Sadoul, E. L. Snapp, and H.-D. Beer for constructs; S. Banerjee and D. Mukhopadhyay for cells; and R. S. Hegde for constructs, cells, and antibodies. We thank members of the Chakrabarti laboratory, especially D. Mookherjee and R. Mukherjee, for valuable support and help throughout the study. We thank P. Chakrabarti for helping in data acquisition on the Multiskan GO Microplate Spectrophotometer. This work was supported by the Integrative Biology on Omics Platform Project, intramural funding of the Department of Atomic Energy, Government of India.
Abbreviations used:
- AIP1
ALG-2–interacting protein 1
- ALG-2
apoptosis-linked gene-2
- ALIX
ALG-2–interacting protein X
- BAD
BCL2-associated agonist of cell death
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- BAX
BCL2-associated protein X
- BCL-XL
B-cell lymphoma–extra large
- BCL2
B-cell/CLL lymphoma 2
- BIP
binding immunoglobulin protein
- CASP
caspase
- CD2AP
CD2-associated protein
- CEP55
centrosomal protein 55
- CHMP
charged multivesicular body protein
- CHOP
C/EBP homologous protein
- CIN85
Cbl-interacting protein of 85 kDa
- DAPI
4′,6-diamidino-2-phenylindole
- ER
endoplasmic reticulum
- ESCRT
endosomal sorting complex required for transport
- FBS
fetal bovine serum
- Fura-2AM
Fura-2-acetoxymethyl ester
- GFP
green fluorescent protein
- HA
hemagglutinin
- IQGAP
IQ motif–containing GTPase-activating protein
- MCL1
myeloid cell leukemia 1
- MGRN1
Mahogunin RING finger 1
- MGRN1ΔR
Mahogunin RING finger 1 ΔRING mutant
- MVB
multivesicular body
- NA
numerical aperture
- PERK
PRKR-like endoplasmic reticulum kinase
- PBS
phosphate-buffered saline
- ROCK1
Rho-associated coiled-coil–containing protein kinase
- SH3
SRC homology 3 domain
- siRNA
small interfering RNA
- Tg
thapsigargin
- TSG101
tumor susceptibility gene 101
- UEV
ubiquitin E2 variant
- UV
ultraviolet
- VPS
vacuolar protein sorting.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-12-0855) on May 24, 2017.
REFERENCES
- Adachi H, Takahashi Y, Hasebe T, Shirouzu M, Yokoyama S, Sutoh K. Dictyostelium IQGAP-related protein specifically involved in the completion of cytokinesis. J Cell Biol. 1997;137:891–898. doi: 10.1083/jcb.137.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Yakir L, Katz M, Zwang Y, Marmor MD, Citri A, Shtiegman K, Alroy I, Tuvia S, Reiss Y, et al. Tal, a TSG101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004;18:1737–1752. doi: 10.1101/gad.294904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosky L, Lawrence DK, Karunamuni G, Wikenheiser JC, Doughman YQ, Visconti RP, Burch JB, Watanabe M. Apoptosis in the developing mouse heart. Dev Dyn. 2006;235:2592–2602. doi: 10.1002/dvdy.20885. [DOI] [PubMed] [Google Scholar]
- Bian ZM, Elner SG, Elner VM. Dual involvement of caspase-4 in inflammatory and ER stress-induced apoptotic responses in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2009;50:6006–6014. doi: 10.1167/iovs.09-3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill A, Torchinsky A, Carp H, Toder V. The role of apoptosis in normal and abnormal embryonic development. J Assist Reprod Genet. 1999;16:512–519. doi: 10.1023/A:1020541019347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti O, Hegde RS. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell. 2009;137:1136–1147. doi: 10.1016/j.cell.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatellard-Causse C, Blot B, Cristina N, Torch S, Missotten M, Sadoul R. Alix (ALG-2-interacting protein X), a protein involved in apoptosis, binds to endophilins and induces cytoplasmic vacuolization. J Biol Chem. 2002;277:29108–29115. doi: 10.1074/jbc.M204019200. [DOI] [PubMed] [Google Scholar]
- Chen B, Borinstein SC, Gillis J, Sykes VW, Bogler O. The glioma-associated protein SETA interacts with AIP1/Alix and ALG-2 and modulates apoptosis in astrocytes. J Biol Chem. 2000;275:19275–19281. doi: 10.1074/jbc.M908994199. [DOI] [PubMed] [Google Scholar]
- Cheng TH, Cohen SN. Human MDM2 isoforms translated differentially on constitutive versus p53-regulated transcripts have distinct functions in the p53/MDM2 and TSG101/MDM2 feedback control loops. Mol Cell Biol. 2007;27:111–119. doi: 10.1128/MCB.00235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhangani D, Mishra A. Mahogunin ring finger-1 (MGRN1) suppresses chaperone-associated misfolded protein aggregation and toxicity. Sci Rep. 2013;3:1972. doi: 10.1038/srep01972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota CD, Bagher P, Pelc P, Smith CO, Bodner CR, Gunn TM. Mice with mutations in Mahogunin ring finger‐1 (Mgrn1) exhibit abnormal patterning of the left–right axis. Dev Dyn. 2006;235:3438–3447. doi: 10.1002/dvdy.20992. [DOI] [PubMed] [Google Scholar]
- Curtiss M, Jones C, Babst M. Efficient cargo sorting by ESCRT-I and the subsequent release of ESCRT-I from multivesicular bodies requires the subunit Mvb12. Mol Biol Cell. 2007;18:636–645. doi: 10.1091/mbc.E06-07-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Song W, Lu X, Hamilton JA, Lei M, Peng T, Yee SP. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation. 2002;106:873–879. doi: 10.1161/01.cir.0000024114.82981.ea. [DOI] [PubMed] [Google Scholar]
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SA, Langille BL, Srivastava D. Apoptosis during cardiovascular development. Circ Res. 2000;87:856–864. doi: 10.1161/01.res.87.10.856. [DOI] [PubMed] [Google Scholar]
- Garin J, Diez R, Kieffer S, Dermine JF, Duclos S, Gagnon E, Sadoul R, Rondeau C, Desjardins M. The phagosome proteome insight into phagosome functions. J Cell Biol. 2001;152:165–180. doi: 10.1083/jcb.152.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Bartelings MM, Deruiter MC, Poelmann RE. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res. 2005;57:169–176. doi: 10.1203/01.PDR.0000148710.69159.61. [DOI] [PubMed] [Google Scholar]
- Groenendyk J, Sreenivasaiah PK, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107:1185–1197. doi: 10.1161/CIRCRESAHA.110.227033. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hanson PI, Shim S, Merrill SA. Cell biology of the ESCRT machinery. Curr Opin Cell Biol. 2009;21:568–574. doi: 10.1016/j.ceb.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Lu XY, Jolly AF, Eldridge AG, Watson SJ, Jackson PK, Barsh GS, Gunn TM. Spongiform degeneration in mahoganoid mutant mice. Science. 2003;299:710–712. doi: 10.1126/science.1079694. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- Hemming FJ, Fraboulet S, Blot B, Sadoul R. Early increase of apoptosis-linked gene-2 interacting protein X in areas of kainate-induced neurodegeneration. Neuroscience. 2004;123:887–895. doi: 10.1016/j.neuroscience.2003.10.036. [DOI] [PubMed] [Google Scholar]
- Hetz C, Russelakis‐Carneiro M, Maundrell K, Castilla J, Soto C. Caspase‐12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003;22:5435–5445. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Emr SD. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct. 2006;35:277–298. doi: 10.1146/annurev.biophys.35.040405.102126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang IK, Hu R, Lacaná E, D’Adamio L, Gu H. Apoptosis-linked gene 2-deficient mice exhibit normal T-cell development and function. Mol Cell Biol. 2002;22:4094–4100. doi: 10.1128/MCB.22.12.4094-4100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Sun K, Walker WP, Bagher P, Cota CD, Gunn TM. Abnormal regulation of TSG101 in mice with spongiform neurodegeneration. Biochim Biophys Acta. 2009;1792:1027–1035. doi: 10.1016/j.bbadis.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara Y, Schiff T, Voloudakis G, Sosa MA, Elder G, Bozdagi O, Buxbaum JD. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol. 2014;193:335–343. doi: 10.4049/jimmunol.1303424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 2001;106:145–155. doi: 10.1016/s0092-8674(01)00434-2. [DOI] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. doi: 10.1038/nature14498. [DOI] [PubMed] [Google Scholar]
- Kim BY, Olzmann JA, Barsh GS, Chin LS, Li L. Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol Biol Cell. 2007;18:1129–1142. doi: 10.1091/mbc.E06-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, Unanue ER, Shaw AS. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300:1298–1300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- Lacanà E, Ganjei JK, Vito P, D’Adamio L. Dissociation of apoptosis and activation of IL-1beta-converting enzyme/Ced-3 proteases by ALG-2 and the truncated Alzheimer’s gene ALG-3. J Immunol. 1997;158:5129–5135. [PubMed] [Google Scholar]
- Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- Lee JA, Liu L, Gao FB. Autophagy defects contribute to neurodegeneration induced by dysfunctional ESCRT-III. Autophagy. 2009;5:1070–1072. doi: 10.4161/auto.5.7.9823. [DOI] [PubMed] [Google Scholar]
- Liu K, Shi Y, Guo X, Wang S, Ouyang Y, Hao M, Liu D, Qiao L, Li N, Zheng J, Chen D. CHOP mediates ASPP2-induced autophagic apoptosis in hepatoma cells by releasing Beclin-1 from Bcl-2 and inducing nuclear translocation of Bcl-2. Cell Death Dis. 2014;5:e1323. doi: 10.1038/cddis.2014.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–632. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- Machesky LM. Cytokinesis: IQGAPs find a function. Curr Biol. 1998;8:R202–205. doi: 10.1016/s0960-9822(98)70125-3. [DOI] [PubMed] [Google Scholar]
- Madaule P, Eda M, Watanabe N, Fujisawa K, Matsuoka T, Bito H, Ishizaki T, Narumiya S. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature. 1998;394:491–494. doi: 10.1038/28873. [DOI] [PubMed] [Google Scholar]
- Mahul-Mellier AL, Hemming FJ, Blot B, Fraboulet S, Sadoul R. Alix, making a link between apoptosis-linked gene-2, the endosomal sorting complexes required for transport, and neuronal death in vivo. J Neurosci. 2006;26:542–549. doi: 10.1523/JNEUROSCI.3069-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder P, Chakrabarti O. Mahogunin regulates fusion between amphisomes/MVBs and lysosomes via ubiquitination of TSG101. Cell Death Dis. 2015;6:e1970. doi: 10.1038/cddis.2015.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki M, Suzuki H, Shibata H. Structure and function of ALG-2, a penta-EF-hand calcium-dependent adaptor protein. Sci China Life Sci. 2011;54:770–779. doi: 10.1007/s11427-011-4204-8. [DOI] [PubMed] [Google Scholar]
- Manil-Segalén M, Lefebvre C, Culetto E, Legouis R. Need an ESCRT for autophagosomal maturation. Commun Integr Biol. 2012;5:566–571. doi: 10.4161/cib.21522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Yaravoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci USA. 2003;100:12414–12419. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol. 2005;15:371–377. doi: 10.1016/j.tcb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Mayers JR, Audhya A. Vesicle formation within endosomes: an ESCRT marks the spot. Commun Integr Biol. 2012;5:50–56. doi: 10.4161/cib.18208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell GS, Lemire JM, Paré JF, Cammarata G, Lowery LA, Levin M. Conserved roles for cytoskeletal components in determining laterality. Integr Biol (Camb) 2016;8:267–286. doi: 10.1039/c5ib00281h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D, Isaacs AM. The role of ESCRT proteins in fusion events involving lysosomes, endosomes and autophagosomes. Biochem Soc Trans. 2010;38:1469–1473. doi: 10.1042/BST0381469. [DOI] [PubMed] [Google Scholar]
- Missotten M, Nichols A, Rieger K, Sadoul R. Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell Death Differ. 1999;6:124–129. doi: 10.1038/sj.cdd.4400456. [DOI] [PubMed] [Google Scholar]
- Monzo P, Gauthier NC, Keslair F, Loubat A, Field CM, Le Marchand-Brustel Y, Cormont M. Clues to CD2-associated protein involvement in cytokinesis. Mol Biol Cell. 2005;16:2891–2902. doi: 10.1091/mbc.E04-09-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R, Chakrabarti O. Ubiquitin-mediated regulation of the E3 ligase GP78 by MGRN1 in trans affects mitochondrial homeostasis. J Cell Sci. 2016;129:757–773. doi: 10.1242/jcs.176537. [DOI] [PubMed] [Google Scholar]
- Okumura M, Ichioka F, Kobayashi R, Suzuki H, Yoshida H, Shibata H, Maki M. Penta-EF-hand protein ALG-2 functions as a Ca 2+-dependent adaptor that bridges Alix and TSG101. Biochem Biophys Res Commun. 2009;386:237–241. doi: 10.1016/j.bbrc.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Oshima R, Hasegawa T, Tamai K, Sugeno N, Yoshida S, Kobayashi J, Kikuchi A, Baba T, Futatsugi A, Sato I, Satoh K. ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci Rep. 2016;6:24997. doi: 10.1038/srep24997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Pallepati P, Averill-Bates DA. Activation of ER stress and apoptosis by hydrogen peroxide in HeLa cells: protective role of mild heat preconditioning at 40 C. Biochim Biophys Acta. 2011;1813:1987–1999. doi: 10.1016/j.bbamcr.2011.07.021. [DOI] [PubMed] [Google Scholar]
- Pérez JF, Chemello ME, Liprandi F, Ruiz MC, Michelangeli F. Oncosis in MA104 cells is induced by rotavirus infection through an increase in intracellular Ca2+ concentration. Virology. 1998;252:17–27. doi: 10.1006/viro.1998.9433. [DOI] [PubMed] [Google Scholar]
- Petiot A, Strappazzon F, Chatellard-Causse C, Blot B, Torch S, Verna JM, Sadoul R. Alix differs from ESCRT proteins in the control of autophagy. Biochem Biophys Res Commun. 2008;375:63–68. doi: 10.1016/j.bbrc.2008.07.136. [DOI] [PubMed] [Google Scholar]
- Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S. The endophilin–CIN85–Cbl complex mediates ligand-dependent downregulation of c-Met. Nature. 2002;416:187–190. doi: 10.1038/416187a. [DOI] [PubMed] [Google Scholar]
- Pexieder T. Cell death in the morphogenesis and teratogenesis of the heart. Adv Anat Cell Biol. 1975;51:1–99. doi: 10.1007/978-3-642-66142-6. [DOI] [PubMed] [Google Scholar]
- Pornillos O, Alam SL, Rich RL, Myszka DG, Davis DR, Sundquist WI. Structure and functional interactions of the Tsg101 UEV domain. EMBO J. 2002;21:2397–2406. doi: 10.1093/emboj/21.10.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- Rane NS, Chakrabarti O, Feigenbaum L, Hegde RS. Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J Cell Biol. 2010;188:4515–4526. doi: 10.1083/jcb.200911115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- Rao RV, Poksay KS, Castro-Obregon S, Schilling B, Row RH, del Rio G, Gibson BW, Ellerby HM, Bredesen DE. Molecular components of a cell death pathway activated by endoplasmic reticulum stress. J Biol Chem. 2004;279:177–187. doi: 10.1074/jbc.M304490200. [DOI] [PubMed] [Google Scholar]
- Rusten TE, Simonsen A. ESCRT functions in autophagy and associated disease. Cell Cycle. 2008;7:1166–1172. doi: 10.4161/cc.7.9.5784. [DOI] [PubMed] [Google Scholar]
- Rusten TE, Stenmark H. How do ESCRT proteins control autophagy. J Cell Sci. 2009;122:2179–2183. doi: 10.1242/jcs.050021. [DOI] [PubMed] [Google Scholar]
- Rusten TE, Vaccari T, Lindmo K, Rodahl LM, Nezis IP, Sem-Jacobsen C, Wendler F, Vincent JP, Brech A, Bilder D, Stenmark H. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol. 2007;17:1817–1825. doi: 10.1016/j.cub.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis D, Llovera M, Ballester M, Comella JX. An alternative view of apoptosis in heart development and disease. Cardiovasc Res. 2008;77:448–451. doi: 10.1093/cvr/cvm074. [DOI] [PubMed] [Google Scholar]
- Segura-Morales C, Pescia C, Chatellard-Causse C, Sadoul R, Bertrand E, Basyuk E. Tsg101 and Alix interact with murine leukemia virus Gag and cooperate with Nedd4 ubiquitin ligases during budding. J Biol Chem. 2005;280:27004–27012. doi: 10.1074/jbc.M413735200. [DOI] [PubMed] [Google Scholar]
- Skop AR, Liu H, Yates J, Meyer BJ, Heald R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science. 2004;305:61–66. doi: 10.1126/science.1097931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Rajakaruna C, Caputo M, Emanueli C. MicroRNAs in congenital heart disease. Ann Transl Med. 2015;3, 333 doi: 10.3978/j.issn.2305-5839.2015.12.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp EL, Sharma A, Lippincott-Schwartz J, Hegde RS. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc Natl Acad Sci USA. 2006;103:6536–6541. doi: 10.1073/pnas.0510657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I. Cbl–CIN85–endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature. 2002;416:183–187. doi: 10.1038/416183a. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Chakrabarti O. Mahogunin-mediated α-tubulin ubiquitination via noncanonical K6 linkage regulates microtubule stability and mitotic spindle orientation. Cell Death Dis. 2014;5:e1064. doi: 10.1038/cddis.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Göttlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114:689–699. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- Strappazzon F, Torch S, Chatellard-Causse C, Petiot A, Thibert C, Blot B, Verna JM, Sadoul R. Alix is involved in caspase 9 activation during calcium-induced apoptosis. Biochem Biophys Res Commun. 2010;397:64–69. doi: 10.1016/j.bbrc.2010.05.062. [DOI] [PubMed] [Google Scholar]
- Sun K, Johnson BS, Gunn TM. Mitochondrial dysfunction precedes neurodegeneration in mahogunin (Mgrn1) mutant mice. Neurobiol Aging. 2007;28:1840–1852. doi: 10.1016/j.neurobiolaging.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhou X, Zhang W, Gallick GE, Kuang J. Unravelling the pivotal role of Alix in MVB sorting and silencing of the activated EGFR. Biochem J. 2015;466:475–487. doi: 10.1042/BJ20141156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- Théry C, Boussac M, Véron P, Ricciardi-Castagnoli P, Raposo G, Garin J, Amigorena S. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001;166:7309–7318. doi: 10.4049/jimmunol.166.12.7309. [DOI] [PubMed] [Google Scholar]
- Toné S, Sugimoto K, Tanda K, Suda T, Uehira K, Kanouchi H, Samejima K, Minatogawa Y, Earnshaw WC. Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp Cell Res. 2007;313:3635–3644. doi: 10.1016/j.yexcr.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trioulier Y, Torch S, Blot B, Cristina N, Chatellard-Causse C, Verna JM, Sadoul R. Alix, a protein regulating endosomal trafficking, is involved in neuronal death. J Biol Chem. 2004;279:2046–2052. doi: 10.1074/jbc.M309243200. [DOI] [PubMed] [Google Scholar]
- Vito P, Lacaná E, Adamio LD. Interfering with apoptosis: Ca2positive-binding protein ALG-2 and Alzheimer’s disease gene ALG-3. Science. 1996;271:521–525. doi: 10.1126/science.271.5248.521. [DOI] [PubMed] [Google Scholar]
- Vito P, Pellegrini L, Guiet C, D’Adamio L. Cloning of AIP1, a novel protein that associates with the apoptosis-linked gene ALG-2 in a Ca2+-dependent reaction. J Biol Chem. 1999;274:1533–1540. doi: 10.1074/jbc.274.3.1533. [DOI] [PubMed] [Google Scholar]
- Von Schwedler UK, Stuchell M, Müller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A. The protein network of HIV budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- Wang X, Shi Q, Xu K, Gao C, Chen C, Li XL, Wang GR, Tian C, Han J, Dong XP. Familial CJD associated PrP mutants within transmembrane region induced Ctm-PrP retention in ER and triggered apoptosis by ER stress in SH-SY5Y cells. PLoS One. 2011;6:e14602. doi: 10.1371/journal.pone.0014602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RL, Urbé S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8:355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- Wolf G, Stahl RA. CD2-associated protein and glomerular disease. Lancet. 2003;362:1746–1748. doi: 10.1016/S0140-6736(03)14856-8. [DOI] [PubMed] [Google Scholar]
- Yamamuro A, Kishino T, Ohshima Y, Yoshioka Y, Kimura T, Kasai A, Maeda S. Caspase-4 directly activates caspase-9 in endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells. J Pharmacol Sci. 2011;115:239–243. doi: 10.1254/jphs.10217SC. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Du J, Xiong B, Xu H, Jiang L. ESCRT components regulate the expression of the ER/Golgi calcium pump gene PMR1 through the Rim101/Nrg1 pathway in budding yeast. J Mol Cell Biol. 2013;5:336–344. doi: 10.1093/jmcb/mjt025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.