

Abstract
Background
Corticotropin-releasing factor (CRF) mediates anxiogenic responses by activating CRF1 receptors in limbic brain regions. Anxiety is further modulated by the endogenous cannabinoid (eCB) system that attenuates the synaptic effects of stress. In the amygdala, acute stress activates the enzymatic clearance of the eCB N-arachidonoylethanolamine (anandamide; AEA) via fatty acid amide hydrolase (FAAH), although it is unclear whether chronic stress induces maladaptive changes in amygdalar eCB signaling to promote anxiety. Here, we used genetically-selected Marchigian Sardinian P (msP) rats carrying an innate overexpression of CRF1 receptors to study the role of constitutive upregulation in CRF systems on amygdalar eCB function and persistent anxiety-like effects.
Methods
We applied behavioral, pharmacological, and biochemical methods to broadly characterize anxiety-like behaviors and amygdalar eCB clearance enzymes in msP versus non-selected Wistar rats. Subsequent studies examined the influence of dysregulated CRF and FAAH systems in altering excitatory transmission in the central amygdala (CeA).
Results
MsPs display an anxious phenotype accompanied by elevations in amygdalar FAAH activity and reduced dialysate AEA levels in the CeA. Elevations in CRF-CRF1 signaling dysregulate FAAH activity, and this genotypic difference is normalized with pharmacological blockade of CRF1 receptors. MsPs also exhibit elevated baseline glutamatergic transmission in the CeA, and dysregulated CRF-FAAH facilitates stress-induced increases in glutamatergic activity. Treatment with a FAAH inhibitor relieves sensitized glutamatergic responses in msPs and attenuates the anxiety-like phenotype.
Conclusions
Pathological anxiety and stress hyper-sensitivity are driven by constitutive increases in CRF1 signaling that dysregulate AEA signaling mechanisms and disable neuronal constraint of CeA glutamatergic synapses.
Keywords: central amygdala (CeA), endocannabinoid, anxiety, CRF1, glutamate, excitatory
Introduction
Acute stress mobilizes the peptide corticotropin-releasing factor (CRF) that orchestrates the endocrine, visceral, and behavioral responses to stress (1). Repeated induction of the CRF system contributes to maladaptive changes that promote negative affect and anxiety (2, 3). Although inextricably linked to the hypothalamic-pituitary-adrenal (HPA) axis, anxiety-like behavior is influenced by CRF type 1 (CRF1) receptor signaling in the amygdala and other limbic regions (4, 5). The amygdala receives sensory information in the lateral and basolateral regions, which relay to the central nucleus of the amygdala (CeA) serving as the primary integrator and distributor of stress-related information (6, 7). Neuronal activation of the medial CeA exerts bidirectional control of anxiety-like function that is influenced by glutamatergic signaling pathways (8). In this regard, acute stress and CRF administration increase CeA glutamatergic activity (9–12), and this effect is sensitized with prior stress experience (13). Dysregulated CRF-CRF1 signaling may therefore serve as a primer for inducing long-term neurological adaptations that influence amygdalar function and subsequent effects on stress and anxiety (3, 14–16).
Anxiety-like behavior is modulated by negative feedback systems that constrain the stress response. In this regard, the endogenous cannabinoid (eCB) system plays an important homeostatic role in the regulation of stress circuits. The primary eCBs, N-arachidonoylethanolamine (anandamide, AEA) and 2-arachidonoylglycerol (2-AG), are synthesized “on demand” in the postsynaptic neuron and act on cannabinoid type 1 (CB1) receptors in presynaptic terminals to suppress neurotransmitter release (17–20). Termination of eCB signaling occurs via enzymatic clearance by the serine hydrolases fatty acid amide hydrolase (FAAH) that degrades AEA, and monoacylglycerol lipase (MAGL) and α/β-hydrolase domain-containing 6 (ABHD6) that degrade 2-AG (21). Selective enzyme inhibitors reveal that eCB signaling by AEA and 2-AG produce distinct pharmacological profiles (22, 23) and mediate differential behavioral effects (24, 25) that may influence the development of anxiety-like disorders. In this regard, genetic polymorphisms in FAAH and MAGL are associated with disrupted limbic function (26, 27), and contribute to pathological changes in emotion- and reward-related processing (26, 28–30), post-traumatic stress disorder (31), and drug abuse (32, 33). Taken together, deficient eCB signaling may confer a critical loss in synaptic constraint of stress-related pathways that normally gate anxiety.
Stress exposure differentially alters eCB signaling in the amygdala that is associated with the HPA axis in distinct ways (16). For instance, acute stress rapidly increases amygdalar FAAH activity that is observed with decreases in AEA content, and these effects are blocked with a CRF1 antagonist (34). Although chronic stress induces similar responses (35), these effects vary with the nature of stress application that infer differences in amygdalar CRF-CRF1 signaling. To this end, genetically-selected Marchigian Sardinian P (msP) rats, originally developed for high alcohol intake, have co-segregated an anxiogenic-like phenotype that is consistent with aberrations in brain stress signaling. Specifically, msPs display point mutations in the Crhr1 locus that elevate gene transcript levels and receptor binding densities in amygdalar regions (36), enhance sensitivity to CRF1 antagonists (36, 37), and modulate stress responsivity and stress-coping performance (36, 38). Here, we utilized msP rats to explore the long-term consequences of dysregulated CRF1 signaling on amygdalar eCB function and anxiety-like measures. We implemented a broad-scale biochemical approach evaluating multiple eCB clearance enzymes in the amygdalar proteome, and subsequently identified a genotypic dysregulation in FAAH. We then applied behavioral, neurochemical, and electrophysiological approaches to examine the hypothesis that dysregulated CRF-FAAH mechanisms in the amygdala contribute to AEA signaling deficiencies and prime stress-reactive glutamatergic systems in the CeA to facilitate anxiety.
Methods and Materials
Animals
Adult male msP rats (300–550g) were bred at The Scripps Research Institute (TSRI) from a colony obtained from the University of Camerino (Italy). For genotypic comparisons, we used adult male Wistar rats (300–550g; Charles River, Raleigh, NC) from which the msP line was generated. Rats were group-housed on a 12h reverse light/dark cycle (lights off at 8:00 am) with food and water available ad libitum, and were handled for 3–5 days before experimentation. We conducted all procedures in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with the Institutional Animal Care and Use Committee (IACUC) policies of TSRI.
Chemicals and Drug Preparation
A list of chemical compounds, and the preparation methods used for the selective FAAH inhibitor PF-3845, the peptide CRF (human/rat), and the selective CRF1 receptor antagonist N,N-bis(2-methoxyethyl)-3-(4-methoxy-2-methylphenyl)-2,5-dimethylpyrazolo[1,5-a]pyrimidin-7-amine (MPZP) are provided in Supplementary Information.
Anxiety-like Behavioral Assessments
Elevated Plus Maze (EPM)
Rats were monitored for anxiety-like behavior using EPM procedures as previously described (39), but with minor modifications. Briefly, rats were habituated to a testing environment containing a standard EPM apparatus centered below a dimly-lit overhead lamp (see Supplemental Information). Rats were pretreated with doses of PF-3845 (3 or 10 mg/kg, i.p.), vehicle, or no injection and left undisturbed for 2h in an anteroom prior to EPM testing for open- and closed-arm activity (i.e., time/entries) for 5 min. A full assessment of performance is included in Supplemental Information (Tables S3, S4).
Novelty-induced Hypophagia
Rats were monitored for anxiety-like behavior using hypophagia procedures as previously described (40, 41). Briefly, rats were habituated to a quiet testing room illuminated by red light (see Supplemental Information). Bottles containing a palatable chocolate solution were presented for 30 min. Following acclimation, rats were monitored for baseline consummatory behaviors (i.e., latency to drink/overall intake). The next day, half of the rats from each genotype were pretreated with PF-3845 (10 mg/kg; i.p.) or vehicle, and left undisturbed for 2h prior to evaluation under novel testing conditions perceived to be stressful (i.e., lights on/new cages).
Biochemical Assessments of eCB Clearance Enzymes
Amygdalar Proteome
Amygdalar tissue was extracted bilaterally from a 2-mm coronal section with a 12 GA tissue extractor (Figure S1). Samples were snap-frozen in liquid nitrogen, and stored at −80°C. Membrane proteomes were processed in phosphate buffered saline as previously described (42).
Activity-based Protein Profiling (ABPP)
Amygdalar proteomes (1 mg/mL) were evaluated for eCB metabolic enzyme activity using ABPP as previously described (42). Details of the method are provided in Supplemental Information. Briefly, protein homogenates (50 μL) were incubated with a fluorophosphonate-rhodamine probe (1 μl, final concentration 1 μM) at 37°C for 30 min. Reactions were quenched with SDS-PAGE loading buffer (20 μL). Probe-labeled proteins were separated by SDS-PAGE (10% acrylamide) and visualized with a fluorescent scanner. Raw values of spectral counts were converted to reflect percentage change from Wistar controls.
FAAH Hydrolysis
A substrate hydrolysis assay was performed using liquid chromatography/triple quadrupole mass spectrometry as previously established (43). Briefly, amygdalar proteomes (50 μg/mL) were incubated with multiple concentrations of deuterated AEA standards (d4-AEA) to measure discrete timepoints of metabolic turnover into d4-ethanolamine (d4-EA; pmol/mg/min) (see Supplemental Information). The inverse of turnover values [1/(pmol/mg/min)] were then used to calculate the maximum velocity of enzymatic reaction (Vmax) and binding affinity (Km).
Pharmacological studies examined the role of CRF systems in modulating FAAH hydrolysis. To assess the relevance of CRF1 receptors across genotype, rats were given sub-chronic treatment with the selective CRF1 antagonist MPZP (10 mg/kg, 2 mL/kg, s.c., b.i.d.) or vehicle for 3 days, and sacrificed ~2h after the final dose. To examine the effects of acute CRF administration, Wistar rats were implanted unilaterally with 23 GA cannulas (PlasticsOne, Roanoke, VA) aimed ~1.5 mm above the lateral ventricles (from bregma AP: −0.8, ML: ±1.6, DV: −3.3, skull). One week later, rats were acclimated to injection procedures, and received CRF (1 μg) or vehicle infusions via a syringe pump dispensing 2 μL of solution in 2 min. Animals were sacrificed 30 min after injections.
Neurochemical assessments of eCBs and neurotransmitters
In-vivo Microdialysis
Rats were implanted unilaterally with guide cannulas aimed 1 mm above the CeA (from bregma AP: −2.3, ML: ±4.0, DV: −6.4, dura). One week later, microdialysis probes were inserted and secured into the guide cannulas. Dialysis and quantification of eCBs and neurotransmitters were performed in separate experiments as previously established (42, 44). Details are included in Supplemental Information.
Electrophysiological recordings
Slice Preparation
Coronal sections of the CeA (300–400 μm) were prepared using a Leica 1000S vibratome (Leica Biosystems, Buffalo Grove, IL) as previously described (12, 45). Slices were incubated with an interface configuration for 30–60 min, submerged, superfused, and equilibrated with 95% O2/5% CO2 artificial cerebral spinal fluid (aCSF) of the following composition: (in mM) 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 1.5 MgSO4-7H2O, 2.0 CaCl2, 24 NaHCO3, and 10 glucose.
Whole-Cell Voltage-Clamp Recordings
Recordings of spontaneous excitatory postsynaptic currents (sEPSCs) in 31 medial CeA neurons (n=16 from 4 msPs, n=15 from 4 Wistars) were made with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA), low-pass filtered at 2–5 kHz, digitized (Digidata 1440A; Axon Instruments), and stored on a computer using pClamp 10 software (Axon Instruments). The internal solution used was composed of (in mM): 145 K-gluconate; 5 EGTA; 5 MgCl2; 10 HEPES; 2 Na-ATP; 0.2 Na-GTP. Recordings (Vhold= −60mV) were performed in the presence of bicuculline (30 μM) and CGP55845A (1 μM). Drugs were dissolved in aCSF and applied by bath superfusion. sEPSCs were analyzed and visually confirmed based on a minimum time interval of 3–5 min and a minimum of 60 events using semi-automated, threshold-based mini detection software (Mini Analysis, Synaptosoft Inc., Fort Lee, NJ).
Intracellular Recording of Evoked Glutamatergic Responses
We recorded evoked excitatory postsynaptic potentials (eEPSPs) from 48 medial CeA neurons (n=24 from 13 msPs, n=24 from 14 Wistars) with sharp micropipettes filled with 3M KCl using discontinuous current-clamp by stimulating the adjacent basolateral amygdala (BLA) through a bipolar stimulating electrode. We held neurons near their resting membrane potential [Wistar: −77.4±1.0, msP: −81.4±0.9 mV]. Average input resistance by genotype was 157.1±9.5 MΩ for Wistar and 166.1±11.1 MΩ for msP. Data were acquired with an Axoclamp-2A preamplifier and stored for later analysis using pClamp software (Axon Instruments, Foster City, CA). Details of testing are included in Supplemental Information.
Statistics
For behavioral measures, genotype differences were examined using Student’s t-tests (unpaired, two-tailed), repeated measures analysis of variance (RM-ANOVA), or two-way ANOVAs followed by Bonferroni-corrected post hocs where appropriate. For enzymatic measures, genotype and/or drug differences were determined using non-linear regression analyses (d4-EA turnover), Student’s t-test (unpaired, two-tailed) or two-way ANOVAs (Vmax and Km) followed by post hoc assessments. Analyses of d4-EA turnover at each concentration of d4-AEA are provided in Supplemental Information (Table S2). For microdialysis measures, mean baseline levels were analyzed with a multivariate ANOVA (MANOVA) followed by RM-ANOVAs assessing genotypic differences, drug effects across time, and changes relative to baseline levels (Fisher’s PLSD). For electrophysiology studies, genotype and drug treatment were evaluated using t-tests or ANOVA as reported in each experiment. P<0.05 was considered statistically significant throughout.
Results
MsP rats display an innate anxiety-like phenotype
We characterized the anxious phenotype in msP rats using two models of anxiety-like behavior (Figure 1). In the EPM study, msPs displayed lower percentages of time in the open arms (Figure 1A) versus Wistars (t(10)=2.8, *p<0.05), albeit fewer closed-arm entries (Figure 1B), indicating reduced maze activity (t(10)=2.4, *p<0.05). In the hypophagia study, RM-ANOVA of latencies revealed a genotype x procedure interaction (F(1,18)=10.0, p<0.01). Relative to baseline, both groups exhibited increased latency (Figure 1C) during novelty stress (+p<0.05), although msPs displayed higher latencies than Wistars in either procedure (*p<0.05). Analysis of total consumption revealed a main effect of procedure (F(1,18)=38.9, +p<0.001), with decreased intake in both groups during novelty stress (Figure 1D), although msPs exhibited a further suppression of intake across procedures (F(1,18)=12.8, *p<0.01). The findings demonstrate an elevated anxiety-like phenotype in msPs.
Figure 1. MsP rats display an innate anxiety-like phenotype.

(A) Percentage of time spent on the open arm of the elevated plus maze (EPM) in non-selected Wistar (W, n=5) and msP (n=7) rats. (B) Total entries made into the closed arm of the EPM in rats from A. (C) Latency to drink from a palatable chocolate solution under baseline and novel environments in Wistars (n=9) and msPs (n=11). (D) Total volume of chocolate solution consumed in rats from C. Data expressed as mean ± SEM. Asterisks (*) denote significant genotype differences, whereas plus signs (+) denote significant differences from baseline conditions (p<0.05). A continuous line across groups denotes significant main effects.
Amygdalar FAAH activity is enhanced in msP rats
We examined genotypic differences in the functional state of amygdalar eCB signaling (Figure 2). ABPP labeling showed higher levels of active FAAH in msP (Figure 2B) than Wistars (t(17)=2.2, *p<0.05), with no difference in the 2-AG enzymes MAGL (t(17)=0.5, N.S.) or ABHD6 (t(17)=1.3, N.S.). Further assessment of FAAH activity using a substrate hydrolysis assay (Figure 2C) predicted higher d4-EA turnover (F(2,92)=3.8, *p<0.05) and Vmax rates (Figure 2D) in msPs than Wistars (t(14)=3.3, *p<0.05), whereas no differences in Km were observed (Wistar:0.93±0.06, msP:1.58±0.39 μM, t(14)=1.6, N.S.). To establish a functional role for genotypic differences in FAAH, in vivo microdialysis procedures examined eCB levels in the CeA, revealing lower baseline levels of AEA in msPs (Figure 2E) than Wistars (F(1,17)=4.6, *p<0.05). No differences were observed in CeA levels of 2-AG (Figure 2F) (F(1,17)=0.05, N.S.), or the ethanolamides palmitoyolethanolamine (PEA), or oleoylethanolamine (OEA) (Figure S2A, B). The findings show that msPs exhibit increased amygdalar FAAH activity and reduced AEA tone in the CeA.
Figure 2. Amygdalar FAAH activity is enhanced in msP rats.

(A) Representative gel image of serine hydrolase activity in amygdalar tissue collected from non-selected Wistar (W, n=10) and msP (M, n=9) rats. (B) Percentage of spectral counts in active site labeling of the endocannabinoid clearance enzymes fatty acid amide hydrolase (FAAH), monoacylglycerol lipase (MAGL), and α/β-hydrolase domain-containing 6 (ABHD6) in rats from A. (C) Substrate conversion of deuterated N-arachidonoylethanolamine (d4-AEA) into ethanolamine (d4-EA) in amygdalar tissue collected from Wistars (n=8) and msPs (n=8). (D) Velocity of reaction (Vmax) in rats from C. (E) Baseline dialysate concentration of N-arachidonoylethanolamine (anandamide, AEA) in the central nucleus of the amygdala (CeA) of Wistars (n=9) and msPs (n=10). (F) Baseline dialysate concentration of 2-arachidonoylglycerol (2-AG) in the CeA of rats from E. Data expressed as mean ± SEM. Asterisks (*) denote significant genotype differences (p<0.05).
Enhanced CRF1 receptor signaling drives elevated amygdalar FAAH activity
We examined whether CRF-CRF1 signaling contributes to genotypic differences in amygdalar FAAH activity (Figure 3). In the MPZP study, a two-way ANOVA of Vmax revealed a genotype x drug interaction (F(1,26)=7.9, p<0.01). While vehicle-treated msPs displayed increased FAAH hydrolysis versus Wistars (*p<0.05), MPZP (10 mg/kg) abolished the genotypic difference in d4-EA turnover (Figure 3A) (F(2,80)=0.6, N.S.), and reduced Vmax rates (Figure 3B) in msPs (+p<0.05) without altering Wistar responses. No significant changes in Km were observed (Table S1). Conversely, CRF administration (1 μg/2 μL) induces FAAH activity in Wistars that predicts higher d4-EA turnover (Figure 3C) (F(2,92)=13.3, *p<0.001) and Vmax rates (Figure 3D) versus vehicle controls (t(13)=2.2, *p<0.05) without altering Km (Vehicle:1.09±0.07, CRF:1.02±0.06 μM, t(13)=0.1, N.S.). Collectively, these findings establish a functional link between genotypic elevations in CRF-CRF1 signaling and increased amygdalar FAAH activity.
Figure 3. Enhanced CRF1 receptor signaling drives elevated amygdalar FAAH activity.

(A) Substrate conversion of deuterated N-arachidonoylethanolamine (d4-AEA) into ethanolamine (d4-EA) in amygdalar tissue collected from non-selected Wistar and msP rats treated with the corticotropin-releasing factor type 1 (CRF1) receptor blocker N,N-bis(2-methoxyethyl)-3-(4-methoxy-2-methylphenyl)-2,5-dimethylpyrazolo[1,5-a]pyrimidin-7-amine (MPZP, 10 mg/kg, 2 mL/kg, s.c., b.i.d., n=7 per genotype). (B) Velocity of reaction (Vmax) in Wistars and msPs treated with either MPZP (n=7 per genotype) or vehicle (VEH, n=8 per genotype). (C) Substrate conversion of d4-AEA into d4-EA in amygdalar tissue collected from Wistars receiving administration of the peptide hormone CRF (1 μg/2 μL, i.c.v., n=7) or VEH (n=8). (D) Vmax rates in rats from C. Data expressed as mean ± SEM. Asterisks (*) denote significant genotype differences (p<0.05), whereas plus signs (+) denote significant differences relative to vehicle controls (p<0.05).
Amygdalar glutamatergic transmission is enhanced in msP rats
Heightened activation of the amygdala facilitates anxiety-like behavior. To investigate the possibility of dysregulated amygdalar circuits, we characterized excitatory transmission in the rat CeA (Figure 4). Baseline levels of 10 different neurotransmitters were assessed using in vivo microdialysis. An omnibus MANOVA revealed a significant genotypic difference (F(10,7)=4.8, p<0.05), with follow-up analyses confirming a robust increase in msP glutamate levels (Figure 4A) versus Wistars (*p<0.05), whereas levels of GABA (Figure 4B) and other transmitters were comparable across genotype (Table 1).
Figure 4. Amygdalar glutamatergic transmission is enhanced in msP rats.
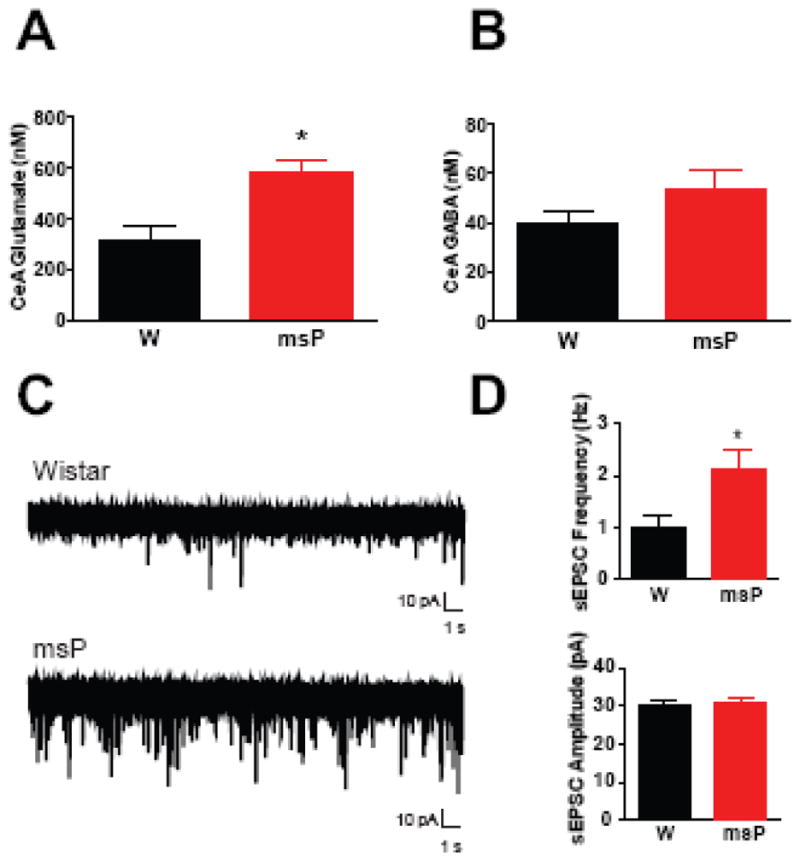
(A) Baseline dialysate concentrations of glutamate in the central nucleus of the amygdala (CeA) of non-selected Wistar (W, n = 9) and msP (n = 9) rats. (B) Baseline dialysate concentrations of gamma aminobutyric acid (GABA) in the CeA of rats from A. (C) Representative whole-cell patch-clamp recordings of spontaneous excitatory postsynaptic potentials (sEPSCs) from Wistars (top trace) and msPs (bottom trace). (D) Baseline frequency (top panel) and amplitude (bottom panel) of CeA sEPSCs in Wistars (n=15 neurons from 4 rats) and msPs (n=16 neurons from 4 rats). Data expressed as mean ± SEM. Asterisks (*) denote significant genotype differences (p<0.05).
Table 1. Neurotransmitter levels in the rat central amygdala.
Values reflect mean ± SEM baseline dialysate concentrations of a broad-scale analysis of neurotransmitters in the central nucleus of the amygdala of non-selected Wistar (n = 9) and msP (n = 9) rats. Asterisks (*) denote significant genotype differences (p<0.05).
| Baseline Dialysate (nM ± SEM) | Wistar | msP | Genotype Comparisons |
|---|---|---|---|
| Aspartate | 66.75 ± 11.54 | 121.8 ± 27.9 | F(1,16) = 3.3 |
| Dopamine | 0.27 ± 0.14 | 0.24 ± 0.04 | F(1,16) = 0.04 |
| GABA | 40.08 ± 4.53 | 53.48 ± 8.19 | F(1,16) = 2.1 |
| Glutamate | 317.8 ± 56.1 | 579.4 ± 50.2* | F(1,16) = 12.1* |
| Glutamine | 49.52 ± 2.72 | 49.25 ± 3.33 | F(1,16) = 0.01 |
| Glycine | 3419 ± 401 | 3611 ± 414 | F(1,16) = 0.1 |
| Norepinephrine | 0.60 ± 0.16 | 0.47 ± 0.06 | F(1,16) = 0.6 |
| Serine | 8056 ± 649 | 7872 ± 536 | F(1,16) = 0.1 |
| Serotonin | 0.09 ± 0.01 | 0.07 ± 0.01 | F(1,16) = 3.3 |
| Taurine | 2370 ± 212 | 2850 ± 389 | F(1,16) = 1.2 |
Given genotypic differences in CeA glutamate levels, we conducted whole-cell voltage-clamp recordings to characterize sEPSCs in 31 medial CeA neurons (n=16 from 4 msPs, n=15 from 4 Wistars). We found elevated baseline sEPSC frequency in msPs (Figure 4D, top panel) versus Wistars (t(14)=3.22, *p<0.05), supporting enhanced glutamate release in the CeA. There were no genotypic differences in baseline amplitude (Figure 4D, bottom panel) (t(29)=0.09, N.S.), rise (Wistar:1.7±0.1, msP:1.5±0.1 msec, t(29)=2.0, N.S.) or decay time (Wistar:1.3±0.1, msP:1.4±0.1 msec, t(29)=0.7, N.S.). The majority of glutamatergic terminals that innervate the CeA arise from the BLA (46, 47). To evaluate genotypic differences in glutamatergic signaling of this pathway, we recorded BLA-evoked EPSPs from 48 medial CeA neurons (n=24 from 13 msPs, n=24 from 14 Wistars). RM-ANOVA of eEPSP input-output curves (Figure S3A) revealed no significant differences in genotype, though there was a trend for higher responses in msPs. We did not observe genotypic differences in baseline paired-pulse facilitation ratios of eEPSPs (Figure S3B). These findings suggest that msPs display heightened glutamatergic activity in the CeA characterized by increased local spontaneous, but not BLA-evoked glutamate release.
FAAH inhibition alleviates stress-sensitive increases in glutamatergic transmission in msP rats
Enhanced glutamatergic tone in the amygdala may promote aberrant stress responsivity, and this may be influenced by FAAH. To examine the role of dysregulated FAAH in modulating stress-dependent changes in CeA glutamatergic transmission (Figure 5), the selective FAAH inhibitor PF-3845 was utilized for all in vivo and ex vivo studies. The specificity of PF-3845 is addressed in Supplemental Information.
Figure 5. FAAH inhibition alleviates stress-sensitive increases in glutamatergic transmission in msP rats.

(A) Percentage change in glutamatergic transmission in the central nucleus of the amygdala (CeA) across time (left panel) in which restraint stress (RES) was applied and measured for accumulative response (area under the curve, AUC, right panel) in msP rats pretreated with the fatty acid amide hydrolase (FAAH) inhibitor PF-3845 (10 mg/kg, i.p., n=9) or vehicle (VEH, n=7). (B) Percentage change in CeA glutamatergic transmission for similar procedures in non-selected Wistar (W) rats pretreated with PF-3845 (n=9) or VEH (n=7). (C) Representative whole-cell patch-clamp recordings of spontaneous excitatory postsynaptic potentials (sEPSCs) in msPs during control (top trace), superfusion of PF-3845 (1 μM, middle trace), and PF-3845 in combination with the peptide hormone corticotropin-releasing factor (CRF, 100 nM, bottom trace). (D) Baseline frequency of CeA sEPSCs following superfusion of CRF or CRF in the presence of PF-3845 in msPs (n=5–9 neurons from 4 rats, top panel) and Wistars (n=6–8 neurons from 4 rats, bottom panel). (E) Representative BLA-evoked excitatory postsynaptic potentials (EPSPs) from an msP CeA neuron (top panel) during control, PF-3845 application, and CRF co-applied with PF-3845. Histograms plotting the effects of CRF alone (n=8 neurons from 7 rats), PF-3845 alone (n=16 neurons from 9 rats) and CRF in the presence of PF-3845 (bottom panel, 11/16 neurons) in CeA neurons from msPs. (F) Representative BLA-evoked EPSPs from a Wistar CeA neuron (top panel) during control and CRF application. Histograms plotting the effects of CRF alone (n=9 neurons from 7 rats), PF-3845 alone (n=13 neurons from 7 rats) and CRF in the presence of PF-3845 (bottom panel, 10/13 neurons) in CeA neurons from Wistars. Data expressed as mean ± SEM. Asterisks (*) denote significant differences relative to PF-3845 treatment, whereas plus signs (+) denote significant differences relative to baseline levels (p<0.05).
To determine changes in CeA glutamatergic transmission during acute stress, glutamate release was assessed by in vivo microdialysis in restrained rats pretreated with PF-3845 (10 mg/kg) or vehicle. In msPs, RM-ANOVA of relative changes in glutamate levels revealed a drug x time interaction (F(9,126)=2.7, p<0.05). While vehicle-treated msPs displayed a robust increase in glutamatergic transmission (Figure 5A) during restraint stress (+p<0.05), PF-3845 abolished this response (*p<0.05). Conversely, in Wistars, there were no significant changes in CeA glutamatergic transmission (Figure 5B) across the timeline of restraint. PF-3845 mildly reduced glutamate levels (Figure S5) prior to stress induction, although the time-dependent decreases in this measure were comparable across genotype and did not produce baseline differences versus vehicle controls.
We evaluated the influence of CRF and FAAH on spontaneous glutamatergic responses in medial CeA neurons. Consistent with our previous work in msP neurons (12), CRF (100 nM) increased sEPSC frequency (Figure 5D, top panel) (139.0±16.2% of control, n=7 neurons from 4 rats). PF-3845 (1 μM) produced no changes in basal sEPSCs (102.1±8.3% of control, n=9 neurons from 4 rats), but significantly attenuated the CRF-induced increase (95.1±3.2% of control, n=5 neurons from 4 rats t(10)=2.2, *p<0.05). Conversely, in Wistar neurons, CRF produced opposing (increased or decreased) effects on sEPSC frequency (Figure 5D, bottom) (122.5±16.0% of control, n=8 neurons from 4 rats). PF-3845 did not significantly alter basal sEPSCs (101.4±3.1% of control, n=6 neurons from 4 rats, t(12)=1.1, N.S.), nor did the co-application of CRF alter this measure (111.2±7.4% of control, n=6 neurons from 4 rats). We next investigated the CRF-FAAH interaction on BLA-evoked CeA glutamatergic responses. CRF (100 nM) had no effect on eEPSP amplitude in msP neurons (Figure 5E) (101.9±6.03% of baseline, n=8 neurons from 7 rats, t(7)=0.32, N.S.). However, PF-3845 (1 μM) significantly reduced basal eEPSPs (by 15.64±3.65%, n=16 neurons from 9 rats, t(15)=4.29, +p<0.05) and this decrease persisted with the co-application of CRF (89.23±5.51% of baseline, 11/16 cells). Conversely, in Wistar neurons, CRF decreased eEPSPs (Figure 5F) (83.32 ± 3.36% of baseline, n=9 neurons from 7 rats, t(8)=4.96, +p<0.05), indicating that CRF inhibits BLA-evoked responses in the CeA. PF-3845 had no effect on basal eEPSPs (92.85±3.54% of baseline, n=13 neurons from 7 rats, t(12)=2.02, N.S.), although the co-application of CRF no longer decreased this measure (103.4±3.69% of baseline, 10/13 neurons, t(9)=0.92, N.S.). Our findings indicate that FAAH does not indiscriminately alter glutamatergic transmission, but rather modulates control of stress activation of these pathways. In this regard, msPs exhibit heightened sensitivity to stress-induced increases in glutamatergic transmission, and this effect is tempered with FAAH inhibition.
FAAH inhibition alleviates excessive anxiety-like behavior in msP rats
We examined whether FAAH inhibition alters the anxious phenotype in msPs (Figure 6). In the EPM study, a two-way ANOVA of open-arm time revealed a genotype x drug interaction (F(2,83)=3.2, p<0.05). While vehicle-treated msPs displayed lower percentages of time spent on the open arm (Figure 6A) versus Wistars (*p<0.01), PF-3845 (10 mg/kg) abolished this genotypic difference, and restored open-arm exploration in msPs (+p<0.01) without altering Wistar behavior. Analysis of closed-arm entries (Figure 6B) revealed that drug treatment did not alter this measure in either genotype (F(2,83)=0.8, N.S.), although msPs continued to display reduced maze activity (F(1,83)=29.3, p<0.001). In the hypophagia study, a two-way ANOVA of latencies revealed a genotype x drug interaction (F(1,33)=4.2, p<0.05) during novelty stress. Whereas vehicle-treated msPs displayed higher latencies (Figure 6C) than Wistars (*p<0.001), PF-3845 mitigated the genotypic difference (*p<0.01) and reduced latencies in msPs (+p<0.05) without altering Wistar behavior. Analysis of total consumption (Figure 6D) revealed similar findings in that drug treatment did not alter intake in either genotype (F(1,33)=0.02, N.S.), although msPs continued to display suppressed intake (F(1,33)=10.4, *p<0.01]. Collectively, these data suggest that FAAH inhibition alleviates excessive anxiety-like responses in msPs.
Figure 6. FAAH inhibition alleviates excessive anxiety-like behavior in msP rats.

(A) Percentage of time spent on the open arm of the elevated plus maze (EPM) in non-selected Wistar (W) and msP rats pretreated with doses of the fatty acid amide hydrolase (FAAH) inhibitor PF-3845 (3 or 10 mg/kg, i.p., n=12–15 per dose and genotype) or vehicle (VEH, n=19–20 per genotype). (B) Total entries made into the closed arm of the EPM in rats from A. (C) Latency to drink from a palatable chocolate solution during novelty stress in Wistars and msPs pretreated with PF-3845 (10 mg/kg, i.p., n=8–9 per genotype) or VEH (n=9–11 per genotype). (D) Total volume of chocolate solution consumed in rats from C. Data expressed as mean ± SEM. Asterisks (*) denote significant genotype differences, whereas plus signs (+) denote significant differences relative to vehicle controls (p<0.05). A continuous line across groups denotes significant main effects.
Discussion
Here, we report that relative to non-selected Wistar rats, msPs exhibit lower dialysate concentrations of AEA in the CeA in association with increases in amygdalar FAAH activity. Our findings are specific to AEA signaling mechanisms, as those related to 2-AG processing were comparable across genotype. Upregulated FAAH activity appears to derive from innate increases in CRF1 signaling, given that sub-chronic treatment with a CRF1 antagonist reduces genotypic differences in this measure. MsPs also exhibit higher CeA glutamate levels, increased spontaneous excitatory signaling, and greater stress-induced activation of glutamatergic signaling. FAAH inhibition attenuates the stress- and CRF-induced elevations in glutamatergic transmission, and modestly diminishes evoked responses in msPs. FAAH inhibition also reduced the anxious phenotype of msPs in two distinct models. Collectively, we suggest that prolonged CRF1 signaling induces long-term dysregulation of FAAH, the interaction of which primes stress-sensitive circuits in the CeA towards a hyper-excitable state to induce persistent anxiety-like effects.
The well documented increase in CRF1 signaling in msP rats (36, 48), together with the results showing that CRF1 receptor antagonism blocks elevated AEA hydrolysis suggest a constitutive interaction between CRF1 and FAAH that underlies anxiety-like pathologies. The findings are consistent with recent work showing that acute CRF1 activation rapidly mobilizes FAAH activity in association with reductions in amygdalar AEA content (34). Importantly, the increases in FAAH activity are sustained in rats displaying upregulated CRF1 systems that derived from phenotypic selection criteria of behavioral anxiety. Thus, we expand on the acute nature of the CRF1-FAAH interaction to provide a direct link between dysregulation in these mechanisms and persistent anxiety-like effects. Our findings are consistent with studies showing a similar pattern of AEA signaling dysfunction induced by sustained glucocorticoid exposure that increases amygdalar CRF signaling, as well as in mutant mice exhibiting forebrain overexpression of the CRF gene (49). Finally, whereas prior studies characterized CRF-FAAH interactions in the BLA, we have extended these observations to the CeA that serves as an integrative hub for processing and converting stress-related information into behavioral and physiologic responses (6). The regional distinction is noteworthy given recent evidence that anxiety-like behavior is influenced by AEA signaling in the BLA under conditions that elicit low, but not high states of emotional arousal (50). Although the amygdalar dissections performed here did not exclusively isolate the CeA region, the neurochemical/electrophysiological data clearly establish an important role of the CeA in CRF-FAAH interactions and resulting effects on stress-reactive systems that influence anxiety.
Our present findings reveal that acute stressors facilitate CeA glutamatergic transmission, and sensitivity to this response is relieved with FAAH inhibitors. Restraint stress stimulated in vivo CeA glutamate levels in msPs, whereas Wistars displayed a tempered and non-significant increase in this measure. Ex vivo studies complemented these results by showing that CRF-induced increases in spontaneous glutamate release were more frequent in CeA neurons from msP rats. Notably, our data support that dysregulated FAAH activity is a contributing factor to genotypic differences in CeA excitatory signaling (12). In this regard, FAAH co-localizes with CB1 receptors that are primarily restricted to glutamatergic synapses on pyramidal neurons (51–54) found in the medial and lateral CeA (55). All FAAH-positive cells co-express CRF1 labeling and gene expression, and mutant mice lacking the expression of CRF1 receptors on forebrain glutamatergic neurons display blunted reductions in amygdalar AEA content following restraint (34). The influence of stress on CeA excitability likely contributes to the anxious phenotype observed in msPs. Acute stress increases CeA glutamate levels after prolonged periods of restraint (9, 10), and intra-CeA treatments that indirectly enhance extracellular glutamate also increase anxiety-like symptoms in EPM and fear conditioning studies (56). Moreover, prior stress exposure primes the neurochemical responses to restraint stress, since fear-conditioned rats display sensitized CRF-induced increases in CeA glutamate levels (13).
We observed robust anxiety-like symptoms in msPs, and genotypic differences in these measures were attenuated with FAAH inhibition. Our evaluations of EPM and novelty-induced hypophagia characterized the anxiety-like phenotype in distinct ways. The EPM studies examined the rats’ willingness to actively explore open spaces, whereas hypophagia procedures examined consummatory behaviors under familiar versus novel contexts. PF-3845 selectively increased open arm exploration and reduced latencies to approach palatable substances in msPs, and these effects are closely related to the alleviation of anxiety-like function in rodents that are pre-stressed or exposed to aversive testing conditions (41, 57, 58). It is noteworthy that msPs continued to display suppressed motor activity and reduced chocolate intake despite the anxiolytic effects of PF-3845. These effects may relate to other known attributes of the msP line showing hypohedonic and depressive-like states (59), as well as innate tendencies for freezing that are resistant to pharmacological treatment (36, 38). The selectivity for which PF-3845 alleviates excessive anxiety may relate to the restoration of dysfunctional AEA-CB1 receptor signaling. Disrupted CB1 signaling is well known to facilitate anxiety (60–64), whereas CB1 agonists generally decrease this measure (61, 65–67). FAAH inhibition increases brain AEA levels and is observed to dampen anxiety-like responses under acute (30, 41) and chronic (35, 68, 69) stress conditions. The anxiolytic effects of FAAH inhibitors are thought to derive from CB1-mediated suppression of glutamatergic versus GABAergic signaling. Specifically, CB1 deletion on glutamate, but not GABA forebrain neurons reverses the anxiolytic effects produced by low-dose administration of a CB1 agonist (70). Relatedly, the anxiety-like effects of social defeat are reversed with a FAAH inhibitor and are associated with the dampening of spontaneous excitatory, but not inhibitory signaling in the mouse striatum (69). Taken together, our findings suggest that the anxiety-like phenotype in msPs results from increased FAAH activity allowing for enhanced stress activation of CeA glutamate systems that regulate anxiety.
The evidence of overactive FAAH in our studies represents a previously uncharacterized link between stress-promoting systems (i.e., CRF1 receptors) and in vivo dysregulation of amygdalar glutamatergic mechanisms. Accordingly, msP rats appear to be in a heightened state of vulnerability to the activation of stress mechanisms. We propose that the stress-sensitive features in msP rats relate to dysfunctional AEA signaling elements that regulate CeA glutamatergic transmission, and contribute to the etiopathology of anxiety. The evidence linking CRF1-FAAH dysfunction in amygdaloid circuitry with negative affective symptoms has translational value for recent work establishing a parallel between clinical symptoms of aberrant stress reactivity, anxiety disorders, and genomic variations in CRF1 and FAAH (26, 30, 71, 72). Moreover, our previous work relating the point mutations in the Crhr1 gene in msPs to aberrant stress responses in fear conditioning studies (36, 38) suggest a possible link between innately dysregulated eCB systems and pathological symptoms of post-traumatic stress disorder. At present, the source of the CRF1-FAAH interaction has not been determined. It is possible that second messenger systems linked to CRF1 receptor activation, such as cAMP/PKA and/or MAPK (ERK1/2) pathways (73), may differentially activate FAAH in msPs. However, we cannot rule out other possibilities including genotypic differences in local CRF-CRF1 neurocircuitry, synaptic function (12), AEA biosynthesis, and/or CRF1-eCB-glutamatergic mechanisms.
Supplementary Material
Acknowledgments
This work was supported by the National Institute on Alcohol Abuse and Alcoholism via the following mechanisms: P60-AA006420 (LHP and MR), R01-AA020404 (LHP), R01-AA021491 (MR), R37-AA017447 (MR), R01-AA015566 and a Research Supplement to Promote Diversity (LAN). We thank Drs. Benjamin Cravatt and Kim Janda for providing the fluorophosphonate-rhodamine chemical probe and N,N-bis(2-methoxyethyl)-3-(4-methoxy-2-methylphenyl)-2,5-dimethylpyrazolo[1,5-a]pyrimidin-7-amine (MPZP), respectively. We thank Dr. Joel Schlosberg for providing the FAAH inhibitor PF-3845. We also thank our colleagues David Stouffer, and Drs. Sarah A. Laredo and Michael Steinman for their technical and conceptual contributions. Finally, we thank Dr. George F. Koob for his insightful comments in the preparation of this manuscript. We humbly dedicate this manuscript in loving memory to our departed friend, colleague, and mentor Dr. Loren (Larry) Parsons, whose seminal contributions in the study of endocannabinoid signaling, stress, and drug addiction will be a driving force in the field for future generations.
Footnotes
Financial Disclosures
The authors of this manuscript report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- 2.Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets. 2006;5:453–479. doi: 10.2174/187152706777950684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Risbrough VB, Stein MB. Role of corticotropin releasing factor in anxiety disorders: a translational research perspective. Horm Behav. 2006;50:550–561. doi: 10.1016/j.yhbeh.2006.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P, et al. Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci. 2003;6:1100–1107. doi: 10.1038/nn1123. [DOI] [PubMed] [Google Scholar]
- 5.Koob GF, Heinrichs SC. A role for corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res. 1999;848:141–152. doi: 10.1016/s0006-8993(99)01991-5. [DOI] [PubMed] [Google Scholar]
- 6.Gilpin NW, Herman MA, Roberto M. The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol Psychiatry. 2015;77:859–869. doi: 10.1016/j.biopsych.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ressler KJ. Amygdala activity, fear, and anxiety: modulation by stress. Biol Psychiatry. 2010;67:1117–1119. doi: 10.1016/j.biopsych.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, et al. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reagan LP, Reznikov LR, Evans AN, Gabriel C, Mocaer E, Fadel JR. The antidepressant agomelatine inhibits stress-mediated changes in amino acid efflux in the rat hippocampus and amygdala. Brain Res. 2012;1466:91–98. doi: 10.1016/j.brainres.2012.05.039. [DOI] [PubMed] [Google Scholar]
- 10.Reznikov LR, Grillo CA, Piroli GG, Pasumarthi RK, Reagan LP, Fadel J. Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur J Neurosci. 2007;25:3109–3114. doi: 10.1111/j.1460-9568.2007.05560.x. [DOI] [PubMed] [Google Scholar]
- 11.Silberman Y, Winder DG. Corticotropin releasing factor and catecholamines enhance glutamatergic neurotransmission in the lateral subdivision of the central amygdala. Neuropharmacology. 2013;70:316–323. doi: 10.1016/j.neuropharm.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman MA, Varodayan FP, Oleata CS, Luu G, Kirson D, Heilig M, et al. Glutamatergic transmission in the central nucleus of the amygdala is selectively altered in Marchigian Sardinian alcohol-preferring rats: Alcohol and CRF effects. Neuropharmacology. 2016;102:21–31. doi: 10.1016/j.neuropharm.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skorzewska A, Bidzinski A, Hamed A, Lehner M, Turzynska D, Sobolewska A, et al. The effect of CRF and alpha-helical CRF((9–41)) on rat fear responses and amino acids release in the central nucleus of the amygdala. Neuropharmacology. 2009;57:148–156. doi: 10.1016/j.neuropharm.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 14.Radley J, Morilak D, Viau V, Campeau S. Chronic stress and brain plasticity: Mechanisms underlying adaptive and maladaptive changes and implications for stress-related CNS disorders. Neurosci Biobehav Rev. 2015;58:79–91. doi: 10.1016/j.neubiorev.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smoller JW. The Genetics of Stress-Related Disorders: PTSD, Depression, and Anxiety Disorders. Neuropsychopharmacology. 2016;41:297–319. doi: 10.1038/npp.2015.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morena M, Patel S, Bains JS, Hill MN. Neurobiological Interactions Between Stress and the Endocannabinoid System. Neuropsychopharmacology. 2016;41:80–102. doi: 10.1038/npp.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- 18.Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci. 2001;21:RC174. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 20.Diana MA, Marty A. Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE) Br J Pharmacol. 2004;142:9–19. doi: 10.1038/sj.bjp.0705726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiura T, Kondo S, Sukagawa A, Tonegawa T, Nakane S, Yamashita A, et al. N-arachidonoylethanolamine (anandamide), an endogenous cannabinoid receptor ligand, and related lipid molecules in the nervous tissues. J Lipid Mediat Cell Signal. 1996;14:51–56. doi: 10.1016/0929-7855(96)00508-1. [DOI] [PubMed] [Google Scholar]
- 23.Goodfellow CE, Glass M. Anandamide receptor signal transduction. Vitam Horm. 2009;81:79–110. doi: 10.1016/S0083-6729(09)81004-2. [DOI] [PubMed] [Google Scholar]
- 24.Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A. 2009;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hariri AR, Gorka A, Hyde LW, Kimak M, Halder I, Ducci F, et al. Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol Psychiatry. 2009;66:9–16. doi: 10.1016/j.biopsych.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC, et al. FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat Commun. 2015;6:6395. doi: 10.1038/ncomms7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carey CE, Agrawal A, Zhang B, Conley ED, Degenhardt L, Heath AC, et al. Monoacylglycerol lipase (MGLL) polymorphism rs604300 interacts with childhood adversity to predict cannabis dependence symptoms and amygdala habituation: Evidence from an endocannabinoid system-level analysis. J Abnorm Psychol. 2015;124:860–877. doi: 10.1037/abn0000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conzelmann A, Reif A, Jacob C, Weyers P, Lesch KP, Lutz B, et al. A polymorphism in the gene of the endocannabinoid-degrading enzyme FAAH (FAAH C385A) is associated with emotional-motivational reactivity. Psychopharmacology (Berl) 2012;224:573–579. doi: 10.1007/s00213-012-2785-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunduz-Cinar O, MacPherson KP, Cinar R, Gamble-George J, Sugden K, Williams B, et al. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol Psychiatry. 2013;18:813–823. doi: 10.1038/mp.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pardini M, Krueger F, Koenigs M, Raymont V, Hodgkinson C, Zoubak S, et al. Fatty-acid amide hydrolase polymorphisms and post-traumatic stress disorder after penetrating brain injury. Transl Psychiatry. 2012;2:e75. doi: 10.1038/tp.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sipe JC, Chiang K, Gerber AL, Beutler E, Cravatt BF. A missense mutation in human fatty acid amide hydrolase associated with problem drug use. Proc Natl Acad Sci U S A. 2002;99:8394–8399. doi: 10.1073/pnas.082235799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buhler KM, Huertas E, Echeverry-Alzate V, Gine E, Molto E, Montoliu L, et al. Risky alcohol consumption in young people is associated with the fatty acid amide hydrolase gene polymorphism C385A and affective rating of drug pictures. Mol Genet Genomics. 2014;289:279–289. doi: 10.1007/s00438-013-0809-x. [DOI] [PubMed] [Google Scholar]
- 34.Gray JM, Vecchiarelli HA, Morena M, Lee TT, Hermanson DJ, Kim AB, et al. Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci. 2015;35:3879–3892. doi: 10.1523/JNEUROSCI.2737-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill MN, Kumar SA, Filipski SB, Iverson M, Stuhr KL, Keith JM, et al. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol Psychiatry. 2013;18:1125–1135. doi: 10.1038/mp.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansson AC, Cippitelli A, Sommer WH, Fedeli A, Bjork K, Soverchia L, et al. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci U S A. 2006;103:15236–15241. doi: 10.1073/pnas.0604419103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ayanwuyi LO, Carvajal F, Lerma-Cabrera JM, Domi E, Bjork K, Ubaldi M, et al. Role of a genetic polymorphism in the corticotropin-releasing factor receptor 1 gene in alcohol drinking and seeking behaviors of marchigian sardinian alcohol-preferring rats. Front Psychiatry. 2013;4:23. doi: 10.3389/fpsyt.2013.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cippitelli A, Ayanwuyi LO, Barbier E, Domi E, Lerma-Cabrera JM, Carvajal F, et al. Polymorphism in the corticotropin-releasing factor receptor 1 (CRF1-R) gene plays a role in shaping the high anxious phenotype of Marchigian Sardinian alcohol-preferring (msP) rats. Psychopharmacology (Berl) 2015;232:1083–1093. doi: 10.1007/s00213-014-3743-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin D, Parsons LH. Anxiogenic-like effect of serotonin(1B) receptor stimulation in the rat elevated plus-maze. Pharmacol Biochem Behav. 2002;71:581–587. doi: 10.1016/s0091-3057(01)00712-2. [DOI] [PubMed] [Google Scholar]
- 40.Bechtholt AJ, Hill TE, Lucki I. Anxiolytic effect of serotonin depletion in the novelty-induced hypophagia test. Psychopharmacology (Berl) 2007;190:531–540. doi: 10.1007/s00213-006-0615-9. [DOI] [PubMed] [Google Scholar]
- 41.Bluett RJ, Gamble-George JC, Hermanson DJ, Hartley ND, Marnett LJ, Patel S. Central anandamide deficiency predicts stress-induced anxiety: behavioral reversal through endocannabinoid augmentation. Transl Psychiatry. 2014;4:e408. doi: 10.1038/tp.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buczynski MW, Herman MA, Hsu KL, Natividad LA, Irimia C, Polis IY, et al. Diacylglycerol lipase disinhibits VTA dopamine neurons during chronic nicotine exposure. Proc Natl Acad Sci U S A. 2016;113:1086–1091. doi: 10.1073/pnas.1522672113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rakers C, Zoerner AA, Engeli S, Batkai S, Jordan J, Tsikas D. Stable isotope liquid chromatography-tandem mass spectrometry assay for fatty acid amide hydrolase activity. Anal Biochem. 2012;421:699–705. doi: 10.1016/j.ab.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Buczynski MW, Polis IY, Parsons LH. The volitional nature of nicotine exposure alters anandamide and oleoylethanolamide levels in the ventral tegmental area. Neuropsychopharmacology. 2013;38:574–584. doi: 10.1038/npp.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herman MA, Kallupi M, Luu G, Oleata CS, Heilig M, Koob GF, et al. Enhanced GABAergic transmission in the central nucleus of the amygdala of genetically selected Marchigian Sardinian rats: alcohol and CRF effects. Neuropharmacology. 2013;67:337–348. doi: 10.1016/j.neuropharm.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pitkanen A, Amaral DG. Distribution of calbindin-D28k immunoreactivity in the monkey temporal lobe: the amygdaloid complex. J Comp Neurol. 1993;331:199–224. doi: 10.1002/cne.903310205. [DOI] [PubMed] [Google Scholar]
- 47.Veinante P, Freund-Mercier MJ. Intrinsic and extrinsic connections of the rat central extended amygdala: an in vivo electrophysiological study of the central amygdaloid nucleus. Brain Res. 1998;794:188–198. doi: 10.1016/s0006-8993(98)00228-5. [DOI] [PubMed] [Google Scholar]
- 48.Ciccocioppo R, Economidou D, Cippitelli A, Cucculelli M, Ubaldi M, Soverchia L, et al. Genetically selected Marchigian Sardinian alcohol-preferring (msP) rats: an animal model to study the neurobiology of alcoholism. Addict Biol. 2006;11:339–355. doi: 10.1111/j.1369-1600.2006.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gray JM, Wilson CD, Lee TT, Pittman QJ, Deussing JM, Hillard CJ, et al. Sustained glucocorticoid exposure recruits cortico-limbic CRH signaling to modulate endocannabinoid function. Psychoneuroendocrinology. 2016;66:151–158. doi: 10.1016/j.psyneuen.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morena M, Leitl KD, Vecchiarelli HA, Gray JM, Campolongo P, Hill MN. Emotional arousal state influences the ability of amygdalar endocannabinoid signaling to modulate anxiety. Neuropharmacology. 2016;111:59–69. doi: 10.1016/j.neuropharm.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 51.Egertova M, Cravatt BF, Elphick MR. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 52.Egertova M, Giang DK, Cravatt BF, Elphick MR. A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci. 1998;265:2081–2085. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, et al. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 54.Tsou K, Nogueron MI, Muthian S, Sanudo-Pena MC, Hillard CJ, Deutsch DG, et al. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett. 1998;254:137–140. doi: 10.1016/s0304-3940(98)00700-9. [DOI] [PubMed] [Google Scholar]
- 55.Ramikie TS, Nyilas R, Bluett RJ, Gamble-George JC, Hartley ND, Mackie K, et al. Multiple mechanistically distinct modes of endocannabinoid mobilization at central amygdala glutamatergic synapses. Neuron. 2014;81:1111–1125. doi: 10.1016/j.neuron.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.John CS, Sypek EI, Carlezon WA, Cohen BM, Ongur D, Bechtholt AJ. Blockade of the GLT-1 transporter in the central nucleus of the amygdala induces both anxiety and depressive-like symptoms. Neuropsychopharmacology. 2015;40:1700–1708. doi: 10.1038/npp.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haller J, Barna I, Barsvari B, Gyimesi Pelczer K, Yasar S, Panlilio LV, et al. Interactions between environmental aversiveness and the anxiolytic effects of enhanced cannabinoid signaling by FAAH inhibition in rats. Psychopharmacology (Berl) 2009;204:607–616. doi: 10.1007/s00213-009-1494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haller J, Goldberg SR, Pelczer KG, Aliczki M, Panlilio LV. The effects of anandamide signaling enhanced by the FAAH inhibitor URB597 on coping styles in rats. Psychopharmacology (Berl) 2013;230:353–362. doi: 10.1007/s00213-013-3161-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ciccocioppo R, Panocka I, Froldi R, Colombo G, Gessa GL, Massi M. Antidepressant-like effect of ethanol revealed in the forced swimming test in Sardinian alcohol-preferring rats. Psychopharmacology (Berl) 1999;144:151–157. doi: 10.1007/s002130050988. [DOI] [PubMed] [Google Scholar]
- 60.Haller J, Bakos N, Szirmay M, Ledent C, Freund TF. The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur J Neurosci. 2002;16:1395–1398. doi: 10.1046/j.1460-9568.2002.02192.x. [DOI] [PubMed] [Google Scholar]
- 61.Haller J, Matyas F, Soproni K, Varga B, Barsy B, Nemeth B, et al. Correlated species differences in the effects of cannabinoid ligands on anxiety and on GABAergic and glutamatergic synaptic transmission. Eur J Neurosci. 2007;25:2445–2456. doi: 10.1111/j.1460-9568.2007.05476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arevalo C, de Miguel R, Hernandez-Tristan R. Cannabinoid effects on anxiety-related behaviours and hypothalamic neurotransmitters. Pharmacol Biochem Behav. 2001;70:123–131. doi: 10.1016/s0091-3057(01)00578-0. [DOI] [PubMed] [Google Scholar]
- 63.Moreira FA, Crippa JA. The psychiatric side-effects of rimonabant. Rev Bras Psiquiatr. 2009;31:145–153. doi: 10.1590/s1516-44462009000200012. [DOI] [PubMed] [Google Scholar]
- 64.Haller J, Varga B, Ledent C, Barna I, Freund TF. Context-dependent effects of CB1 cannabinoid gene disruption on anxiety-like and social behaviour in mice. Eur J Neurosci. 2004;19:1906–1912. doi: 10.1111/j.1460-9568.2004.03293.x. [DOI] [PubMed] [Google Scholar]
- 65.Rubino T, Guidali C, Vigano D, Realini N, Valenti M, Massi P, et al. CB1 receptor stimulation in specific brain areas differently modulate anxiety-related behaviour. Neuropharmacology. 2008;54:151–160. doi: 10.1016/j.neuropharm.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 66.Patel S, Hillard CJ. Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: further evidence for an anxiolytic role for endogenous cannabinoid signaling. J Pharmacol Exp Ther. 2006;318:304–311. doi: 10.1124/jpet.106.101287. [DOI] [PubMed] [Google Scholar]
- 67.Moreira FA, Aguiar DC, Terzian AL, Guimaraes FS, Wotjak CT. Cannabinoid type 1 receptors and transient receptor potential vanilloid type 1 channels in fear and anxiety-two sides of one coin? Neuroscience. 2012;204:186–192. doi: 10.1016/j.neuroscience.2011.08.046. [DOI] [PubMed] [Google Scholar]
- 68.Lomazzo E, Bindila L, Remmers F, Lerner R, Schwitter C, Hoheisel U, et al. Therapeutic potential of inhibitors of endocannabinoid degradation for the treatment of stress-related hyperalgesia in an animal model of chronic pain. Neuropsychopharmacology. 2015;40:488–501. doi: 10.1038/npp.2014.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rossi S, De Chiara V, Musella A, Sacchetti L, Cantarella C, Castelli M, et al. Preservation of striatal cannabinoid CB1 receptor function correlates with the antianxiety effects of fatty acid amide hydrolase inhibition. Mol Pharmacol. 2010;78:260–268. doi: 10.1124/mol.110.064196. [DOI] [PubMed] [Google Scholar]
- 70.Rey AA, Purrio M, Viveros MP, Lutz B. Biphasic effects of cannabinoids in anxiety responses: CB1 and GABA(B) receptors in the balance of GABAergic and glutamatergic neurotransmission. Neuropsychopharmacology. 2012;37:2624–2634. doi: 10.1038/npp.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Demers CH, Drabant Conley E, Bogdan R, Hariri AR. Interactions between anandamide and corticotropin-releasing factor signaling modulate human amygdala function and risk for anxiety disorders: an imaging genetics strategy for modeling molecular interactions. Biol Psychiatry. 2016;80:356–362. doi: 10.1016/j.biopsych.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lazary J, Eszlari N, Juhasz G, Bagdy G. Genetically reduced FAAH activity may be a risk for the development of anxiety and depression in persons with repetitive childhood trauma. Eur Neuropsychopharmacol. 2016;26:1020–1028. doi: 10.1016/j.euroneuro.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 73.Grammatopoulos DK. Insights into mechanisms of corticotropin-releasing hormone receptor signal transduction. Br J Pharmacol. 2012;166:85–97. doi: 10.1111/j.1476-5381.2011.01631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.