

Abstract
Pulmonary alveolar proteinosis (PAP) is a potentially fatal complication of lysinuric protein intolerance (LPI), an inherited disorder of cationic amino acid transport. The patients often present with mild respiratory symptoms, which may rapidly progress to acute respiratory failure responding poorly to conventional treatment with steroids and bronchoalveolar lavations (BALs). The pathogenesis of PAP in LPI is still largely unclear. In previous studies, we have shown disturbances in the function and activity of alveolar macrophages of these patients, suggesting that increasing the activity and the number of macrophages by recombinant human GM-CSF (rhuGM-CSF) might be beneficial in this patient group.
Two LPI patients with complicated PAP were treated with experimental inhaled rhuGM-CSF (sargramostim) after poor response to maximal conventional therapy. BAL fluid and cell samples from one patient were studied with light microscopy and transmission electron microscopy.
Excellent response to therapy was observed in patient 1 with no compliance problems or side effects. Macrophages with myelin figure-like structures were seen in her BAL sample. Slight improvement of the pulmonary function was evident also in patient 2, but the role of sargramostim could not be properly evaluated due to the complicated clinical situation.
In conclusion, inhaled rhuGM-CSF might be of benefit in patients with LPI-associated PAP.
Keywords: Lysinuric protein intolerance, aminoaciduria, pulmonary alveolar proteinosis, sargramostim, granulocyte-macrophage colony-stimulating factor
Introduction
Lysinuric protein intolerance (LPI) is a rare inherited amino acid transport disorder leading to decreased plasma concentrations and increased urinary excretion of cationic amino acids (CAAs) arginine, ornithine and lysine by defective y+LAT1 transporter. The patients present with a variety of symptoms including failure to thrive, postprandial hyperammonaemia and haematological and immunological abnormalities, the exact mechanism of which is still unclear. The patients may remain asymptomatic for decades with a carefully planned low-protein diet and supplementation with low-dose oral l-citrulline and ammonia-scavenging drugs. Unfortunately, the current treatment regime does not seem to protect the patients from long-term complications including renal disease and pulmonary alveolar proteinosis (PAP), a rare condition characterized by accumulation of lipoproteinaceous material in the alveoli (Parto et al. 1993; Santamaria et al. 1996). Acute episodes of PAP may occur even in those patients with excellent treatment compliance and no previous respiratory symptoms. Unlike in idiopathic PAP, granulocyte-macrophage colony-stimulating factor (GM-CSF) antibodies have not been detected in the LPI patients, and the pathogenesis of PAP in LPI patients has thus remained unclear.
The LPI PAP patients often present initially with mild respiratory symptoms, which may progress rapidly and lead to pulmonary insufficiency and multi-organ failure. To date, the only treatment options, i.e. repeated bronchoalveolar lavations (BALs) and systemic corticosteroids, have shown limited effectiveness (Santamaria et al. 2004; Ceruti et al. 2007). During the last 10 years, four Finnish paediatric patients from our cohort of over 40 Finnish LPI patients have been diagnosed with acute PAP. Three of them died despite immediate management in an intensive care unit (Table 1). The incidence of PAP has been even larger in other LPI cohorts. Valimahamed-Mitha and colleagues have reported a series of 14 paediatric patients with LPI, out of which ten fulfilled the diagnostic criteria of PAP and six died of pulmonary failure (Valimahamed-Mitha et al. 2015). In a group of nine Italian LPI patients reported by Santamaria and colleagues, one patient died of respiratory insufficiency, and five other patients had signs of lung involvement when studied with high-resolution computed tomography (HRCT) imaging despite being asymptomatic at the time of the study (Santamaria et al. 1996).
Table 1.
Summary of the four Finnish paediatric patients with LPI-related PAP followed up at Turku University Hospital between years 1997 and 2014
| Age at diagnosis | Age at PAP | Presenting signs of PAP | Treatment for PAP | Result | |
|---|---|---|---|---|---|
| 1. | 1 year | 1. 12 years 2. 16 years |
1. Dyspnoea, dry cough 2. Poor exercise tolerance, progressive hypoxia |
1. Large-dose corticosteroids, BAL, supplementary oxygen, saline inhalations, salbutamol 2. BAL, peroral steroids and antibiotics, saline inhalations, sargramostim |
1. Partial recovery (decreased exercise tolerance, need for regular lung lavations) 2. Normal pulmonary function, able to participate to sports activities normally 1.5 years after the last episode of PAP |
| 2. | 1 year | 16 years | Fever, cough, epistaxis, haematemesis | Intravenous antibiotics, large-dose corticosteroids, ventilatory support | Multi-organ failure and death 2 days after admission to hospital |
| 3. | 3 months | 3 months | Dry cough, poor eating | Supplemental oxygen, corticosteroids, BAL | Died at the age of 12 months |
| 4. | 8 months | 4 years 8 months | Fever, fatigue, poor appetite, enlarged liver and spleen, enlarged lymph nodes | Intravenous antibiotics, large-dose corticosteroids, BAL | Died 10 days after admission to hospital |
Patient no. 1 is patient 1 described in the “Results” section
BAL bronchoalveolar lavation
Although the exact pathogenesis of PAP in LPI has been unclear, disturbances in the function and phagocytic activity of monocyte-derived macrophages have been demonstrated (Barilli et al. 2010, 2012; Kurko et al. 2015). Thus, accumulation of proteinous material into the lungs may be caused by insufficient clearance of proteins by poorly functioning alveolar macrophages. Inhaled granulocyte-macrophage colony-stimulating factor (GM-CSF) is used off-label in patients with idiopathic PAP with anti-GM-CSF antibodies to activate and attract monocyte-derived macrophages into the lungs. We hypothesized that increasing the activity and the number of alveolar macrophages in the alveolar fluid by recombinant human GM-CSF (rhuGM-CSF) inhalation could promote the resolution of PAP also in LPI. Barilli et al. (2010) have previously reported one Italian patient diagnosed with LPI-associated PAP at the age of 15 years, whose respiratory condition and CT showed marked improvement after rGM-CSF treatment. However, the authors were naturally unable to draw conclusions on the efficacy of rGM-CSF in LPI patients based on a single patient case.
Here, we describe two Finnish LPI patients, one child and one adult, with complicated PAP treated with experimental inhaled rhuGM-CSF (sargramostim, Leukine®, Genzyme) after already receiving maximal conventional therapy.
Patients and Methods
Patients
The research was conducted according to the principles of the Declaration of Helsinki. A written informed consent was obtained from the patients before the initiation of the experimental rhuGM-CSF treatment.
Methods
BAL Fluid and Cell Sample Collections
BAL fluid was collected routinely from patient 1. After Cyto-Tek and Cytospin cytocentrifugations of the BAL sample, routine Papanicolaou, May-Grünwald-Giemsa, Prussian blue and periodic acid-Schiff (PAS)-stained slides were prepared and studied under a light microscope. In addition, a part of the BAL fluid was filtered through a sterile gauze. The filtered cells were centrifuged 250g for 10 min, washed with ice-cold HBSS and suspended in the RPMI-1640 medium with a GlutaMAX supplement (Invitrogen Life Technologies, Carlsbad, CA, USA) and 10% FBS before the following experiments.
Transmission Electron Microscopy
The centrifuged cell pellet was fixed with 5% glutaraldehyde overnight, and osmium tetroxide was added to fix the sample for 2 h. The sample was then dehydrated with ethanol and embedded with propylenoxid in epoxy resin. Ultrathin sections contrasted with uranyl acetate and lead citrate were studied under the Jeol JEM-1400Plus transmission electron microscope (Jeol, Tokyo, Japan).
Histology
The cells for histopathological examination were fixed in 10% buffered formalin, centrifuged and pre-embedded in agar. Then, the agar blocks were further embedded in paraffin, and routine 4 μm thick histological sections were cut on slides. The sections were stained with haematoxylin and eosin, and PAS, and studied under a light microscope.
Results
In the BAL sample of patient 1, a total of 520 million cells per litre were detected. Of these, 55% were macrophages, 42% lymphocytes and the remaining 3% neutrophils. Cytological bronchoalveolar preparations showed macrophages which contained PAS-positive granules. Similar granules were also seen around the cells. In addition, the histological sections contained macrophages with PAS-positive granules (Fig. 1). In a sample studied with electron microscopy, several macrophages containing lysosomes as well as myelin figure-like structures were observed (Fig. 2).
Fig. 1.
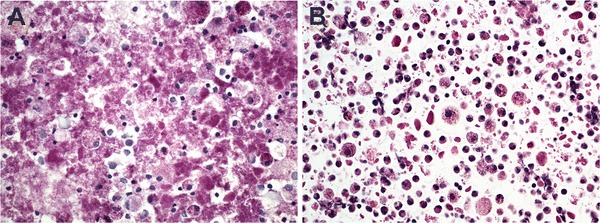
Bronchoalveolar lavage fluid with macrophages containing PAS-positive granules. The cells from the patient 1 were stained with periodic acid-Schiff and examined with a light microscope using a 400× enlargement (a). Microscope preparations of filtered and washed cells in an agar/paraffin block obtained from the patient 1 were stained with periodic acid-Schiff and examined with a light microscope using a 400× enlargement (b)
Fig. 2.
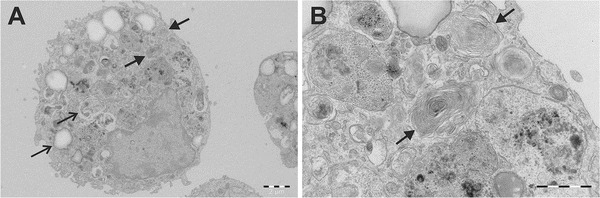
An alveolar macrophage containing lysosomes and myelin figures. The filtered and washed cells obtained from the bronchoalveolar lavage fluid of the patient 1 were examined with an electron microscope. A 3,000× enlargement shows lysosomes indicated by narrow arrows and myelin figures indicated by thick arrows (a). A 10,000× enlargement reveals more closely the myelin figure-like surfactant structures indicated by thick arrows (b)
Patient 1
The patient is a girl who was diagnosed with LPI at the age of 1 year after her older sister had been diagnosed with LPI. At that time, she had no clinical findings except for the characteristic urinary amino acid profile and elevated serum ferritin. Her treatment regime consisted of oral l-citrulline (100–200 mg/kg/day), sodium benzoate (125–250 mg/kg/day) and protein-restricted diet (1–1.2 g/g/day) supplemented with calcium carbonate, vitamins, l-lysine hydrochloride (13–17 mg/kg/day) and commercial carbohydrate energy supplements. She had mild growth retardation, mild proteinuria and recurrent urinary tract infections but was previously otherwise quite healthy and regularly participated in sports activities.
At the age of 12 years, she had an acute episode of dyspnoea and cough without symptoms of a respiratory infection. HRCT imaging suggested acute PAP. She was treated with repeated BALs and high-dose corticosteroids with a partial response but she remained dependent on supplemental oxygen. Hypertonic saline and salbutamol inhalations were not of significant benefit. After several large-volume BALs, her respiratory function gradually improved, and supplemental oxygen was discontinued 3 months after the initial episode. However, only a couple of months later, her pulmonary function started to worsen again and she developed dyspnoea and cough. A new BAL was performed. Three weeks later, the patient developed symptoms of pneumonia, and a PCR test performed on the lavation fluid was shown to be positive for human herpesvirus 6 (HHV-6) and Mycoplasma pneumoniae. She was treated in an intensive care unit with intravenous antibiotics and BALs which were of benefit, and her condition improved. However, regular BALs were continued as a prophylactic treatment.
During the following years, her condition remained relatively stable; she was able to participate in sports activities to some extent, and the glucocorticoid treatment was gradually weaned off. At the age of 16, her pulmonary function again decreased rapidly and she developed progressive dyspnoea and poor exercise tolerance. The response to high-dose corticosteroids was poor. HRCT revealed an acute episode of PAP. BAL was performed several times and surfactant was given to prevent atelectasis after the procedure. Chest physiotherapy was also initiated to improve lung function. After 6 months, her condition had not improved and she remained dependent on supplemental oxygen. Due to a poor overall prognosis, an experimental treatment with inhaled rhuGM-CSF (sargramostim) was offered.
The treatment started with 125 μg of rhuGM-CSF in 2 ml of saline given twice a day with PARI BOY nebulizer every other week for 12 weeks. Inhalation was well tolerated. Blood leucocyte counts and liver enzymes were followed up weekly, but no adverse effects were observed during the treatment. After no more than 2 weeks on rhuGM-CSF, her lung function had significantly improved and the radiographic findings had ameliorated (Figs. 3 and 4). After three courses (1 week each) with rhuGM-CSF, supplemental oxygen was discontinued and tapering off corticosteroids was initiated. Two years after the treatment, the pulmonary function has remained normal and no relapses have occurred. The patient is currently able to participate normally in sports activities and inhaled asthma medications have been discontinued. The respiratory function before and after the rhuGM-CSF treatment is summarized in Table 2.
Fig. 3.
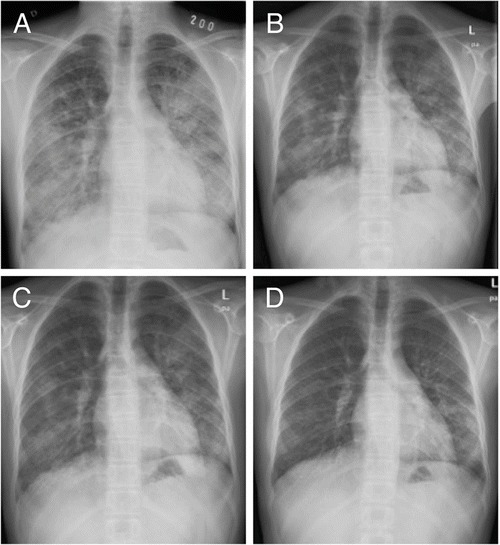
Chest radiographs of patient 1 (a) before treatment, (b) after the first cycle, (c) after the second cycle, and (d) after the last (6th) cycle of sargramostim
Fig. 4.
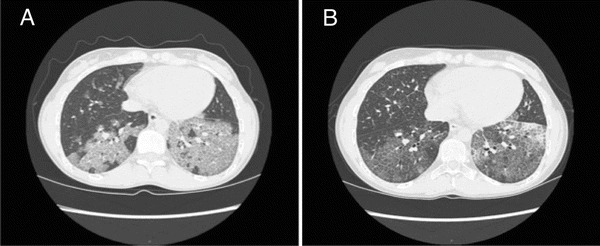
Computed tomography images of patient 1 (a) before treatment and (b) after the third cycle of sargramostim
Table 2.
Respiratory function of patient 1 before and after the sargramostim treatment
| Date | VC | VC %Ref | FVC (L) | FVC %Ref | FEV1 (L) | FEV1%Ref | FEV1/FVC | FEV1/FVC %Ref | PEF (L/min) | PEF %Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| Day −82 | 1.69 | 55 | 1.68 | 53 | 1.59 | 56 | 94 | 105 | 7.5 | 133 |
| Day −70 | 1.75 | 56 | 1.65 | 51 | 1.53 | 52 | 92.61 | 101 | 5.56 | 97 |
| Day −69 BAL | ||||||||||
| Day −34 | 1.56 | 51 | 1.52 | 48 | 1.39 | 49 | 92 | 102 | 6.06 | 107 |
| Day −28 | 1.64 | 53 | 1.62 | 51 | 1.5 | 52 | 92 | 103 | 5.78 | 102 |
| Day −27 BAL | ||||||||||
| Day −7 | 1.7 | 55 | 1.59 | 50 | 1.5 | 52 | 94 | 105 | 5.89 | 104 |
| Day 0 | 1.66 | 53 | 1.59 | 49 | 1.49 | 51 | 93.24 | 104 | 6.15 | 107 |
| Day +14 | 1.62 | 52 | 1.62 | 51 | 1.5 | 53 | 93 | 104 | 6.52 | 116 |
| Day +28 | 1.58 | 51 | 1.56 | 49 | 1.43 | 50 | 91 | 102 | 6.38 | 113 |
| Day +42 | 1.71 | 55 | 1.68 | 53 | 1.55 | 54 | 92 | 103 | 6.38 | 113 |
| Day +51 | 1.76 | 52 | 1.71 | 52 | 1.59 | 55 | 93.39 | 104 | 6.65 | 116 |
| Day +78 | 2.08 | 67 | 1.93 | 60 | 1.73 | 61 | 90 | 100 | 6.61 | 117 |
BAL bronchoalveolar lavation, VC vital capacity, FVC forced vital capacity, FEV1 forced expiratory volume in 1 s, PEF peak expiratory flow
Patient 2
The patient was a 58-year-old woman who was diagnosed with LPI in her early childhood. Her treatment regime consisted of a protein-restricted diet (about 1 g/kg/day) together with oral l-citrulline (about 2,500 mg/day), l-lysine (750 mg/day), sodium benzoate (2,000 mg/day) and calcium and vitamin supplements. At the age of 49 years, a statin therapy was initiated for combined hyperlipidaemia, and mild renal insufficiency was diagnosed at the age of 50 years. She also had a tendency to bruising and nosebleeds. At the age of 57 years, she complained of prolonged cough and dyspnoea. A BAL was performed twice, but the symptoms persisted and the patient also had an episode of pneumonia. Radiological findings were consistent with PAP. The patient was given oral and inhaled steroids without any effect. The patient became dependent on supplementary oxygen and was subsequently hospitalized. HRCT and pulmonary function tests could not be performed due to her poor general condition. Inhaled rhuGM-CSF treatment (125 μg) was initiated with the protocol described above, along with oral prednisolone (40 mg × 1), furosemide (40 mg × 1) and antibiotics (oral doximycin and intravenous moxifloxacin). The clinical condition of the patient improved enough for her to be released from hospital although still dependent on supplementary oxygen, and significant radiographic improvement was also observed (Fig. 5). Two weeks later, she was readmitted because of severe dyspnoea and elevated CRP (90 mg/L). A treatment with intravenous cefuroxime was initiated and the prednisolone dose was doubled to 40 mg × 2. A chest radiograph showed mildly increased alveolar opacity of the left lung. Five days after hospital admission, the patient developed severe epistaxis, which led to severe anaemia requiring red blood cell transfusions. After 2 weeks, the patient was again released from hospital as her CRP had normalized. The rhuGM-CSF dose was increased to 250 μg × 1, and inhaled corticosteroid was also initiated. With this regime, her condition seemed to remain stable. However, 2 months later she was again admitted to hospital due to severe epistaxis, and posterior tamponade was performed. Shortly afterwards, she was diagnosed with fulminant staphylococcal septicaemia, of which she subsequently died.
Fig. 5.
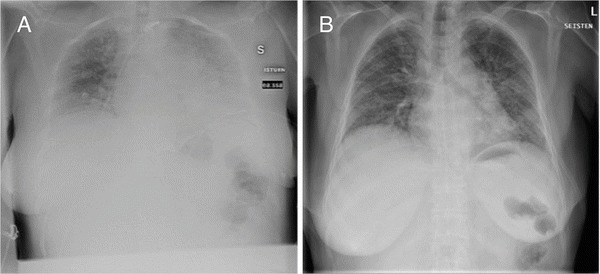
Chest radiographs of patient 2 before (a) and after (b) the first cycle of sargramostim
Discussion
Pulmonary alveolar proteinosis is a relatively poorly known clinical entity, which can develop with a variety of mechanisms including genetic defects of the GM-CSF protein or its receptor, neutralizing anti-GM-CSF antibodies, infections and malignancies (Campo et al. 2012). Patients with LPI are predisposed to secondary PAP, the prognosis of which seems to be especially poor in paediatric patients (Parto et al. 1993, 1994). The initial symptoms of PAP may be very subtle, making the diagnosis challenging. However, the disease can rapidly progress to a life-threatening multi-organ failure, and a careful monitoring of any LPI patient with dyspnoea, unexplained fatigue or fever in the absence of other signs of an infection is therefore warranted.
Toll-like receptor signalling, especially a response to viral DNA, has been shown to be impaired, and nitric oxide (NO) production decreased in LPI monocyte-derived macrophages reinforcing the role of macrophages in the development of secondary complications of LPI (Kurko et al. 2015). We hypothesized that, in contrast to the earlier suggestions by other groups (Sebastio et al. 2011; Ogier de Baulny et al. 2012), arginine reservoirs and the subsequent NO levels may actually be diminished in LPI macrophages as a result of reduced arginine influx by the defective y+LAT1 transporter, known to be the most important transporter of arginine in macrophages, including those of alveolar origin (Barilli et al. 2011; Rotoli et al. 2007). In addition, the defective CAA transport also results in impaired phagocytic activity of LPI macrophages (Barilli et al. 2012). This may, in turn, lead to an impaired clearance of phospholipoproteinaceous material from the alveoli. It is interesting that iNOS synthesizing NO from arginine is expressed in the airway epithelial cells where the gene encoding y+LAT1 is also expressed (Thomassen and Kavuru 2001; Rotoli et al. 2005). Therefore, NO synthesis in the LPI airway cells could be distorted affecting, for example, the production of cytokines in alveolar macrophages. Since NO is known to be involved in several inflammatory lung diseases (Thomassen and Kavuru 2001), it would be reasonable to measure NO levels in the airway aspirate or bronchoalveolar lavage fluid of the LPI patients with PAP.
In the study by Douda and colleagues, a supplementation with surfactant protein D (SP-D) and GM-CSF increased the uptake of protein and dying cells ex vivo, but GM-CSF increased the number of spontaneously generated granulomas (Douda et al. 2009). The authors therefore suggested that GM-CSF might not be a suitable mode of treatment for patients with LPI-associated PAP. The 2-year-old LPI patient in their report had large amounts of cholesterol, cholesterol crystals and lipid-laden macrophages in the airways, and the authors suggested that therapies aiming to decrease the amount of cholesterol in the airways might be beneficial instead. On the other hand, in the study by Ohashi and colleagues, the GM-CSF inhalation therapy improved lung clearance and decreased the amount of protein in BAL fluid in a patient with autoimmune alveolar proteinosis (Ohashi et al. 2012). Furthermore, in 2014, Yu and colleagues reported a patient with autoimmune PAP treated successfully with a combination of whole lung lavage and inhaled GM-CSF (Yu et al. 2014). It is therefore logical to assume that rhuGM-CSF might also be useful in patients with LPI-associated PAP. New, noninvasive therapeutic options would be warmly welcomed as the effectiveness of corticosteroids and whole lung lavage has proven to be inadequate in this patient group. Although repeated whole lung lavage has been shown to alleviate symptoms and reduce hypoxia in PAP, it does not actually stop the progression of the underlying disease (Gao et al. 2014). Also, general anaesthesia is required to perform the whole lung lavage, which may present a challenge due to the pre-existing respiratory failure.
Our experiences based on two patient cases together with the case previously reported by Barilli et al. (2010) suggest that inhaled rhuGM-CSF may be useful in LPI-associated pulmonary alveolar patients. In patient 1, an excellent response to therapy was observed with no compliance problems or side effects. In patient 2, the role of rhuGM-CSF could not be properly evaluated due to the complicated clinical situation, but slight improvement in the pulmonary function was observed also in this patient. However, comparing the outcomes is complicated by the marked age difference between the two patients.
In conclusion, the data on these two patients suggest that inhaled rhuGM-CSF might be of benefit in patients with LPI-associated PAP responding inadequately to high-dose corticosteroids and lung lavations. Such PAP often carries a dismal prognosis, especially in paediatric patients. However, further studies on this subject are warranted.
Take Home Message
Inhaled sargramostim induces resolution of pulmonary alveolar proteinosis associated with lysinuric protein intolerance.
Contributions
LT collected the clinical data. LT, JK and HA drafted the manuscript. JK and MT performed the laboratory analyses. HA performed the electron microscopy imaging and provided Figs. 1 and 2. JM, KN-S and HN supervised the study. KN-S, HN and HL were responsible for the treatment and follow-up of the patients and provided clinical data. HL planned the treatment protocol. All the authors commented on and approved the manuscript.
Guarantor
Laura Tanner
Compliance with Ethics Guidelines
Conflicts of Interest
Laura Tanner, Johanna Kurko, Maaria Tringham, Heikki Aho, Juha Mykkänen, Kirsti Näntö-Salonen, Harri Niinikoski and Heikki Lukkarinen declare that they have no conflict of interest.
Details of Funding
Johanna Kurko has received grants from the Päivikki and Sakari Sohlberg Foundation and the Magnus Ehrnrooth Foundation. The authors confirm independence from the sponsors; the content of the chapter has not been influenced by the sponsors.
The Ethics Committee of the Hospital District of Southwest Finland has approved the follow-up study concerning the pathogenesis of long-term complications of LPI and informed consent has been obtained from all the patients or their parents. A written informed consent was also obtained from the patients before the initiation of the experimental rhuGM-CSF treatment. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000.
Contributor Information
Laura M. Tanner, Email: lamaer@utu.fi
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Barilli A, Rotoli BM, Visigalli R, et al. In lysinuric protein intolerance system y+L activity is defective in monocytes and in GM-CSF-differentiated macrophages. Orphanet J Rare Dis. 2010;5:32. doi: 10.1186/1750-1172-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barilli A, Rotoli BM, Visigalli R, Bussolati O, Gazzola GC, Dall´Asta V. Arginine transport in human monocytic leukemia THP-1 cells during macrophage differentiation. J Leukoc Biol. 2011;90:293–303. doi: 10.1189/jlb.0910510. [DOI] [PubMed] [Google Scholar]
- Barilli A, Rotoli BM, Visigalli R, et al. Impaired phagocytosis in macrophages from patients affected by lysinuric protein intolerance. Mol Genet Metab. 2012;105:585–589. doi: 10.1016/j.ymgme.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Campo I, Kadija Z, Mariani F, et al. Pulmonary alveolar proteinosis: diagnostic and therapeutic challenges. Multidiscip Respir Med. 2012;7:4. doi: 10.1186/2049-6958-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti M, Rodi G, Stella GM, et al. Successful whole lung lavage in pulmonary alveolar proteinosis secondary to lysinuric protein intolerance: a case report. Orphanet J Rare Dis. 2007;2:14. doi: 10.1186/1750-1172-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douda DN, Farmakovski N, Dell S, Grasemann H, Palaniyar N. SP-D counteracts GM-CSF-mediated increase of granuloma formation by alveolar macrophages in lysinuric protein intolerance. Orphanet J Rare Dis. 2009;4:29. doi: 10.1186/1750-1172-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, GC L, Zhou XY, Yu Z, Wang HM, Bian T. Repeated whole-lung lavage for unremitting pulmonary alveolar proteinosis: an eight-year follow-up of a case. Genet Mol Res. 2014;13:6135–6141. doi: 10.4238/2014.August.7.29. [DOI] [PubMed] [Google Scholar]
- Kurko J, Vähä-Mäkilä M, Tringham M, et al. Dysfunction in macrophage toll-like receptor signalling caused by an inborn error of cationic amino acid transport. Mol Immunol. 2015;67:416–425. doi: 10.1016/j.molimm.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Ogier de Baulny H, Schiff M, Dionisi-Vici C. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol Genet Metab. 2012;106:12–17. doi: 10.1016/j.ymgme.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Sato A, Takada T, et al. Direct evidence that GM-CSF inhalation improves lung clearance in pulmonary alveolar proteinosis. Respir Med. 2012;106:284–293. doi: 10.1016/j.rmed.2011.10.019. [DOI] [PubMed] [Google Scholar]
- Parto K, Svedström E, Majurin ML, Härkönen R, Simell O. Pulmonary manifestations in lysinuric protein intolerance. Chest. 1993;104:1176–1182. doi: 10.1378/chest.104.4.1176. [DOI] [PubMed] [Google Scholar]
- Parto K, Kallajoki M, Aho H, Simell O. Pulmonary alveolar proteinosis and glomerulonephritis in lysinuric protein intolerance: case reports and autopsy findings of four pediatric patients. Hum Pathol. 1994;25:400–407. doi: 10.1016/0046-8177(94)90150-3. [DOI] [PubMed] [Google Scholar]
- Rotoli BM, Bussolati O, Sala R, Gazzola GC, Dall´asta V. The transport of cationic amino acids in human airway cells: expression of system y+L activity and transepithelial delivery of NOS inhibitors. FASEB J. 2005;19:810–812. doi: 10.1096/fj.04-2924fje. [DOI] [PubMed] [Google Scholar]
- Rotoli BM, Dall´Asta V, Barilli A, et al. Alveolar macrophages from normal subjects lack the NOS-related system y+ for arginine transport. Am J Respir Cell Mol Biol. 2007;37:105–112. doi: 10.1165/rcmb.2006-0262OC. [DOI] [PubMed] [Google Scholar]
- Santamaria F, Parenti G, Guidi G, et al. Early detection of lung involvement in lysinuric protein intolerance: role of high-resolution computed tomography and radioisotopic methods. Am J Respir Crit Care Med. 1996;153:731–735. doi: 10.1164/ajrccm.153.2.8564125. [DOI] [PubMed] [Google Scholar]
- Santamaria F, Brancaccio G, Parenti G, et al. Recurrent fatal pulmonary alveolar proteinosis after heart-lung transplantation in a child with lysinuric protein intolerance. J Pediatr. 2004;145:268–272. doi: 10.1016/j.jpeds.2004.04.047. [DOI] [PubMed] [Google Scholar]
- Sebastio G, Sperandeo MP, Andria G. Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am J Med Genet C Semin Med Genet. 2011;157:54–62. doi: 10.1002/ajmg.c.30287. [DOI] [PubMed] [Google Scholar]
- Thomassen MJ, Kavuru MS. Human alveolar macrophages and monocytes as a source and target for nitric oxide. Int Immunopharmacol. 2001;1:1479–1490. doi: 10.1016/S1567-5769(01)00092-3. [DOI] [PubMed] [Google Scholar]
- Valimahamed-Mitha S, Berteloot L, Ducoin H, Ottolenghi C, de Lonlay P, de Blic J. Lung involvement in children with lysinuric protein intolerance. J Inherit Metab Dis. 2015;38:257–263. doi: 10.1007/s10545-014-9777-5. [DOI] [PubMed] [Google Scholar]
- Yu H, Xf S, Wang Y, Xu Z, Huang H. Whole lung lavage combined with granulocyte-macrophage colony stimulating factor inhalation for an adult case of refractory alveolar proteinosis. BMC Pulm Med. 2014;14:87. doi: 10.1186/1471-2466-14-87. [DOI] [PMC free article] [PubMed] [Google Scholar]