

Abstract
Newborn screening (NBS) for phenylketonuria (PKU) which has a continuum of disease severities has been performed for more than 50 years. The screening method has undergone a continuous development with not only improvements of the positive predictive value but also identification of milder forms of the disease. With the introduction of genetic testing the confirmation of the diagnosis has improved. The Swedish NBS is centralized to one laboratory, which also performs confirmatory testing.
Here we present the results of NBS for PKU in Sweden during 1965–2014 describing an increase in diagnosed patients and a shift in the spectrum of phenylalanine hydroxylase (PAH) mutations towards an increasing heterogeneity. Milder mutations common in southern Europe and the Middle East together with lowering of the recall level for phenylalanine (Phe) have led to a shift towards milder phenotypes among the patients identified by the screening program. The inclusion of a Phe and tyrosine (Tyr) ratio as an additional marker has improved the positive predictive value to the present 0.92. Also discussed is what impact earlier sampling has had on the prediction of disease severity, concluding that the shift of age at sampling from 72 to 48 h does not increase the risk of missing patients in need of treatment.
Electronic supplementary material: The online version of this chapter (doi:10.1007/8904_2016_4) contains supplementary material, which is available to authorized users.
Keywords: Mutations, Neonatal screening, Newborn screening, Phenylalanine hydroxylase, Phenylketonuria
Introduction
Phenylketonuria (PKU; OMIM #261600) is caused by a deficiency of the liver enzyme phenylalanine hydroxylase (PAH; EC 1.14.16.1), which catalyses the conversion of phenylalanine (Phe) to tyrosine (Tyr) and requires tetrahydrobiopterin (BH4) as cofactor. Cofactor deficiencies constitute approximately 2% of patients with elevated Phe and need different treatment regimens (Blau et al. 2010).
Treatment of PKU from early infancy with a diet low in Phe and supplemented with Phe free amino acid mixture, vitamins and other trace elements results in normal development. Without early treatment neurological damage will occur. Neonatal screening for PKU is thus an important and successful intervention (Guthrie and Susi 1963).
The disease is inherited as an autosomal recessive and to date, 866 different mutations in the PAH gene have been described in the Human Gene Mutation Database (HGMD Professional 2016.1). Several of these cause less severe enzyme deficiency resulting in a continuum of severities of clinical disease.
The aim of the study here is to present the results of newborn screening (NBS) for PKU in Sweden 1965–2014. Since 1990 there has been an increase in the number of diagnosed patients and a shift of the mutation spectrum in the PAH gene due to a high rate of immigrants from non-Nordic countries which today represent 85% of Sweden’s 1,600,000 immigrants (http://www.scb.se).
Materials and Methods
Screening Methods and Cut Off
NBS for PKU started in Sweden in 1965, shortly after the first NBS programme was implemented in Massachusetts, USA (Guthrie and Susi 1963). The programme is voluntary and the coverage in Sweden has been >98% since 1972, and near 100% during the last four decades.
The Swedish neonatal screening is centralized to Centre for Inherited Metabolic Diseases, Karolinska University Hospital and the birth rate has been between 90,000 and 120,000 annually, presently being 116,000 infants per year. The confirmatory testing is performed by the same laboratory or at Sahlgrenska University Hospital, Gothenburg and Skånes University Hospital, Lund.
Blood specimens have been collected on filter paper cards Whatman™ 903 or Ahlstrom grade 226. From 1965 to 2008 the samples were collected at age 3–6 days. In 2008 new guidelines were implemented in preparation for extended tandem mass spectrometry (MS/MS) screening, resulting in sampling as soon as possible after 48 h of age.
The screening methods used in Sweden for elevated Phe levels have changed continuously since the Guthrie bacterial inhibition assay was introduced in 1965. At the same time the cut off value for Phe has been lowered and the ratio of Phe to Tyr has been included as a second marker (Table 1).
Table 1.
Incidences of PKU and MHP in relation to different cut offs and screening methods
| Year | Screening method | Second tier | Cut off Phe recall | Ratio Phe/Tyr | Screened newborns | PKU | MHP | Incidence PKU/MHP |
|---|---|---|---|---|---|---|---|---|
| 1965–1975 | Guthrie bacterial inhibition assay (BIA) | ≥360 μmol/L (6 mg%) | – | 945,523 | 50 | 6 | 1/16,900 | |
| 1976–1996 | Guthrie bacterial inhibition assay (BIA) | Ion exchange chromatography from 1978 to 1998 | ≥250 μmol/L (4 mg%) | – | 2,717,978 | 128 | 33 | 1/16,900 |
| 1997–2002 | Quantase phenylalanine kit (Shield Diagnostics) | No second tier from 1998 Tandem mass spectrometry in house method from 2002 |
≥250 μmol/L | – | ||||
| 2003–2004 | Quantase phenylalanine kit (Shield Diagnostics) | ≥180 μmol/L | ≥2.0 (2006-) | 1,305,706 | 67 | 30 | 1/13,500 | |
| 2005–2008 | Tandem mass spectrometry in house method | |||||||
| 2009–2014 | Tandem mass spectrometry non-derivatized kit PerkinElmer | |||||||
| 4,969,207 | 245 | 69 | 1/15,800 |
The project was approved by the Regional Ethical Committee of Stockholm.
Patients
Until December 31, 2014, a total of 4,969,207 newborns have been screened for PKU and 314 patients, 139 females and 175 males have been diagnosed, giving an incidence of 1/15,800 (Table 1). Four cases of BH4-deficiencies have also been identified; two with 6-pyruvoyl-tetrahydropetrin synthase deficiency, one with dihydropteridin reductase deficiency and one with GTP cyclohydrolase I deficiency. All four, except the one with GTP cyclohydrolase I deficiency, were caught in the screening programme. The patient with GTP cyclohydrolase I deficiency had a Phe value of 240 μmol/L in the screening sample taken on day four and the recall level was at that time 250 μmol/L.
The mutation study enrolled index cases from 279 families, 125 females and 154 males. Among these, 217 patients (193 index patients) were diagnosed when the recall level was Phe ≥360 μmol/L (1965–1975) or ≥250 μmol/L (1976–2002) and 97 patients (86 index patients) from 2003 when the recall level had been lowered to ≥180 μmol/L. From 2006 the ratio of Phe to Tyr ≥2.0 was an additional requirement. Of the 279 families, 32 have more than one child with PKU.
Also included were 33 immigrant/adoptive patients (14 females and 19 males) born between 1966 and 2011 and diagnosed either in their countries of birth or through the Swedish NBS program which is offered to immigrant and adoptive children under the age of 18 years on arrival in Sweden. From 2014 this was lowered to under the age of 8 years.
Genetic Analysis
Genetic analysis has been a part of the routine confirmatory testing of all PKU patients during the past 20 years. Patients born before genetic analyses became a standard procedure were genotyped retrospectively. Eighty-eight index cases born 1966–1985 were included in a previous study (Svensson et al. 1993). The mutation detection rate in that study was 73% and patients not fully genotyped were reanalysed when new methods became available.
Genetic analyses have not been performed in 9 (3.2%) out of the 279 index cases diagnosed by NBS and for another 4 (1.4%) only one mutation has been identified. Mutation analysis has not been requested for seven of the 33 immigrant/adoptive patients.
Genomic DNA was isolated from whole blood following standard procedures which have varied. Since routine genotyping has been in use, screening for mutations has been performed by denaturing gradient gel electrophoresis) or polymerase chain reaction amplification, the latter method most frequently used, followed by direct sequencing. Multiplex ligation-dependent probe amplification was performed to detect large genomic deletions in patients without identified mutations on one or both alleles. Confirmation by carrier analysis of parents was performed in more than 80% of the families.
Mutation nomenclature follows the guidelines and recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen) and novel mutations were validated using the program Mutalyzer (http://mutalyzer.nl/2.0/). Complementary DNA (cDNA) numbering commences from the ATG start codon, where +1 is the A of the ATG translation initiation codon (NM_000271.1).
Classification of Patients with MHP, Mild PKU and Classical PKU
Classification of patients with hyperphenylalaninemia (HPA) according to Phe levels is subjective and several classifications are used, from Phe values ≤360 to ≤1000 μmol/L (Hanley 2011).
We have chosen to define patients with at least one mutation known to be associated with non-PKU mild hyperphenylalaninemia (MHP) or having two mild mutations resulting in repeat Phe levels <500 μmol/L on a normal diet to be classified as MHP patients. PKU patients are classified as classical PKU when they have two null mutations or Phe levels ≥1200 μmol/L in the NBS sample. Patients in between are classified as having mild PKU.
Results and Discussion
Incidence of PKU/MHP in Sweden
During the period 1965 to December 2014, close to five million newborns were screened for PKU in Sweden. PKU was confirmed in 245 newborns and MHP in 69.
The merged incidence of MHP and PKU was 1/15 800, which is similar to what has been reported for many other European countries (Loeber 2007). The ratio of MHP to PKU was 1–3.5. Comparing the period 1965–1989 with 1990–2014, the incidence of PKU/MHP patients has increased from 1/18 300 to 1/14 200 equal to an increase from 5.5 to 7.1 per 100.000 born. This is almost solely due to an increase of patients diagnosed with MHP (Table 2).
Table 2.
Incidence of PKU and MHP during the two screening periods
| Year | Screened newborns | PKU | MHP | Incidence PKU/MHP |
|---|---|---|---|---|
| 1965–1989 | 2,322,536 | 111 | 16 | 1/18,300 |
| 1990–2014 | 2,646,671 | 134 | 53 | 1/14,200 |
| 1990–2014 a | 2,646,671 | 134 | 38 | 1/15,400 |
aIncidences re-calculated excluding 15 MHP patients recalled after lowering the cut off for Phe to ≥180 μmol/L
Screening Results and Recall Levels
The change in incidence shows the same pattern with an increase in the number of patients diagnosed with MHP when the PKU population is divided according to the different cut offs used for recalls in the neonatal screening (Table 1).
The infants recalled with Phe values <250 μmol/L are of special interest. Of the 15 infants in this cohort only one was sampled after 3 days of age, all the others were sampled at 2–3 days of age (Supplementary 1).
Six patients had Phe values <250 μmol/L also in the repeat samples taken at recall. Three additional infants had a Phe to Tyr ratio less than 2.0 in the sample taken at recall. All but two have at least one allele associated with MHP. Two of the patients were homozygous for p.Leu48Ser (c.143T>C) and p.Gly46Ser (c.136G>A), respectively. The former is known to give an inconsistent phenotype, whilst p.Gly46Ser is associated with a severe phenotype (Djordjevic et al. 2013; Eiken et al. 1996).
Taking into account the Phe values and Phe to Tyr ratios seen in the repeat tests of these 15 children, eight of them would not have been recalled as presumptive positive PKU/MHP cases. Their Phe values and Phe to Tyr ratios in the repeat dried blood samples (DBS) did not fulfil the criteria for recall (Phe ≥180 μmol/L and ratio Phe to Tyr at or above 2.0). All eight infants are compound heterozygous for known MHP mutations; p.Ser87Arg (c.261C>A), p.Asp145Val (c.434A>T), p.Arg176Leu (c.527G>T), p.Ala300Ser (c.898G>T), p.Ala322Gly (c.965C>G) and p.Ala403Val (c.1208C>T). When the incidence is re-calculated for the period 1990–2014 excluding the 15 MHP patients recalled with Phe values <250 μmol/L the incidence of MHP patients is still higher than for the period 1965–1989 (Table 2). Lowering the cut off thus cannot explain the increase of detected MHP-patients after 1989.
The increase of MHP patients has raised the question whether we now pick up and treat patients unnecessarily. In the minireview: “Non-PKU mild hyperphenylalaninemia (MHP) – the dilemma”, the author did not find any evidence for need of treatment to gain a normal outcome in this group of patients (Hanley 2011).
In Sweden we have seen an increase in the number of patients with a true positive screening test for PKU/MHP defined as carrying two mutations giving persistently elevated Phe values. Comparing the incidence according to the cut offs for Phe, no changes of the total incidence of true positive cases could be seen when the cut off was lowered from ≥360 to ≥250 μmol/L in 1976 (Table 1). From 2003 when the cut off was lowered to ≥180 μmol/L an increase similar to the one seen when the two periods 1965–1989 and 1990–2014 were compared was noted. It is important to have in mind that in 2006 the ratio of Phe to Tyr was added as an additional marker to better distinguish between infants with increased Phe levels due to PKU/MHP and infants with liver disease or having treatment with intravenous amino acid solutions.
Lowering the cut off to levels lower than 180 μmol/L would probably not improve the PKU screening. This is supported in an article from Italy (Trunzo et al. 2013), where they use a Phe cut off ≥120 μmol/L. Of the 30 infants included, 26 were recalled with Phe values 120–180 μmol/L. Eight of these patients were compound heterozygous or homozygous for MHP mutations, in another nine infants only one mutation was detected and in 6 infants no mutations could be found.
Phe Values in First and Second Sampling in Patients with MHP, Mild PKU and Classical PKU
Figure 1a shows the Phe values in true positive infants whose screening samples were collected at age 2–3 days in comparison with those whose samples were collected at age 4–5 days. Included are also the Phe values of the second DBS samples taken when the infants were recalled. The infants were recalled at ages ranging from 5 to 10 days.
Fig. 1.
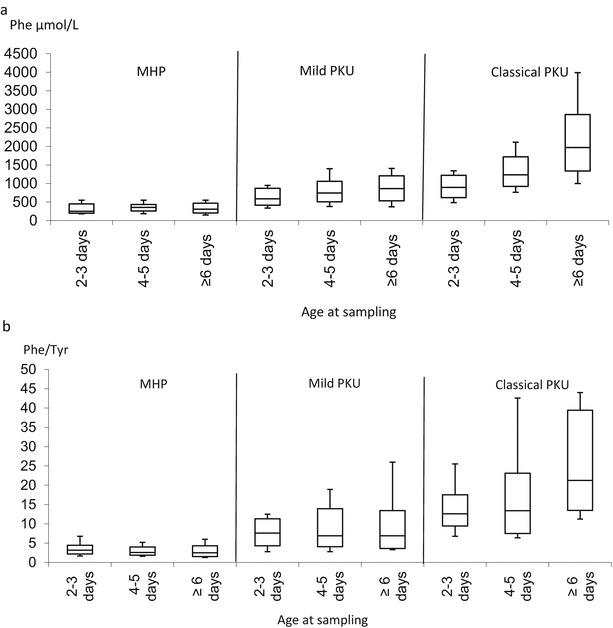
Phe values and Phe to Tyr ratios divided into different groups according to age at sampling. a Phe values in screening samples taken at ages 2–3, 4–5 and ≥6 days, divided into groups with MHP, mild PKU and classical PKU. b Phe to Tyr ratio in screening samples taken at ages 2–3, 4–5 and ≥6 days, divided into groups with MHP, mild PKU and classical PKU. The horizontal lines in the boxes and the lines outside the boxes indicate median and the minimum and maximum values, respectively. The top and bottom of each box represent the 90th and 10th percentiles, respectively
Phe values for the MHP group did not change with sampling at an older age. In the group with mild PKU there is an increase between sampling at age 2–3 days and age 4–5 days and later. This is also true in the group with classical PKU.
The ratio between Phe and Tyr was essentially unchanged between the ages at sampling in the MHP group and interestingly it did not change much between the ages at sampling in the group of mild PKU although it was higher in this group. In the group with classical PKU patients there was a clear influence of age at sampling (Fig. 1b).
Impact of Earlier Sampling
In 2008 the age at sampling was brought forward to as soon as possible after 48 h of age. Data from our laboratory shows that the mean age of sampling has decreased from 4.0 (year 2007) to 2.7 (year 2014) days. Earlier sampling may have an effect on the outcome of NBS for PKU since the Phe values may not have had time to reach the highest pre-treatment levels. In the MHP group no differences could be seen when comparing the Phe levels and Phe to Tyr ratios in the screening sample of the three age groups (Fig. 1a, b). In the mild PKU group an increase in the Phe values could be seen when the samples were taken at age 4–5 days compared with 2–3 days. This group seems to reach the highest pre-treatment values at age 4–5 days since there is no difference between samples taken at age ≥6 days and samples taken at age 4–5 days. The Phe to Tyr ratio did not change over time in this group.
Earlier sampling leads to lower Phe values in the first screening sample but does not necessarily lead to any disadvantages for infants with MHP since some who do not need treatment may go undetected. Another issue of earlier sampling is the risk of missing a woman who as an adult may have Phe levels detrimental for the foetus during pregnancy (Smith et al. 1990). A third factor to consider is the detection of infants with cofactor deficiencies. Some of these patients tend to have Phe levels in the MHP region when the neonatal screening sample is taken (Opladen et al. 2012).
Positive Predictive Value
The positive predictive value of the screening for PKU was 0.34 before the ratio of Phe to Tyr was added as a second marker. The majority of the false positives were premature or sick newborns on treatment with intravenous amino acid solutions. In 2006 when the ratio Phe to Tyr ≥2.0 was included, the positive predictive value increased to 0.92 much due to the fact that newborns treated with intravenous amino acid solutions could be sorted out (Table 3). The exclusion of these patients was further improved when the expanded MS/MS screening was implemented in 2010 making it possible to evaluate a total of 11 amino acids. Newborns on treatment with intravenous amino acids and a positive screening for PKU are no longer recalled as presumptive PKU cases; instead a new sample, taken 24 h after completed treatment with amino acids is requested by the laboratory.
Table 3.
Positive predictive value in the screening for PKU
| Years | Screened newborns | True positive | False positive | Positive predicted value | Ratio Phe/Tyr |
|---|---|---|---|---|---|
| 1965–2005 | 3,965,693 | 245 | 471 | 0.34 | No |
| 2006–2014 | 1,003,514 | 71 | 6 | 0.92 | Yes |
Mutation Analysis
The second major change that has had an impact on the increase in the number of MHP patients is the change of mutation spectrum. The mutations detected in patients born from 1990 show an increasing frequency of less severe mutations where MHP mutations represent 14.1% of all mutations compared to 6.9% before 1990. There has been simultaneously a decrease of the frequency of the two most common classical PKU mutations, p.Arg408Trp (c.1222C>T) and c.1315+1G>A, and the milder mutation, p.Tyr414Cys (c.1241A>G), previously associated with MHP but now considered a mild PKU mutation. The prevalence of the mutation p.Arg261Gln (c.782G>A), which is associated with less severe PKU as well as classical PKU, has increased (Fig. 2).
Fig. 2.
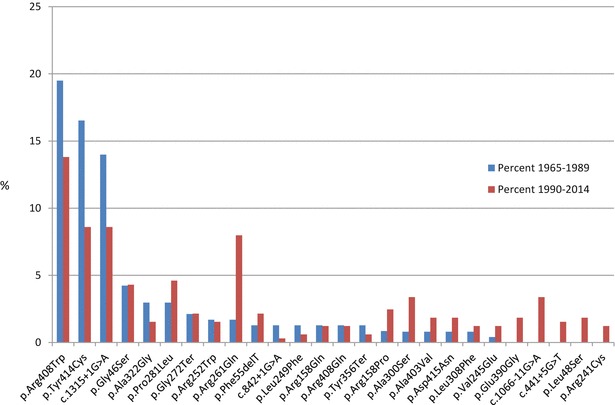
Mutations with a relative frequency >1.0% of all alleles in the NBS group for the periods 1965–1989 and 1990–2014
Mutations were detected in 536 out of 540 independent alleles (270 index patients) giving a mutation detection rate of 99.3%. For nine additional index patients no DNA was available and 26 of 33 immigrant/adoptive index cases were fully genotyped (52 alleles).
Ninety-four disease causing mutations were detected in the Swedish PKU/MHP-population: 62 missense mutations (66.0%), 16 intronic variations (17.0%), 13 deletions (13.8%) and 3 nonsense mutations (3.2%). A total of 17 of the 61 missense mutations were associated with MHP (Supplementary 2).
Five variants not described earlier in a peer-reviewed journal were detected. The missense variants p.Tyr77His (c.229T>C) and p.Asp143Val (c.428A>T) were run in the functional prediction programs PolyPhen-2, (http://genetics.bwh.harvard.edu/pph2), PROVEAN (http://provean.jcvi.org) and Mutation Taster (http://www.mutationtaster.org/) and both variants were predicted to be disease causing in all three programs. The intron variants c.843-13_843-10delTTCT, c.970-1G>T and c.1315+5G>A were run in Mutation Taster and were all three predicted disease causing. In Fruitfly (http://www.fruitfly.org/seq_tools/splice.html) one was predicted to cause an abolished splice site (c.970-1G>T) whilst the other two resulted in weakened splice sites (c.843-13_843-10delTTCT and c.1315+5G>A). Clinically the five variants are predicted to be associated with classical PKU; c.843-13_843-10delTTCT, c.970-1G>T and c.1315+5G>A, with mild PKU; p.Tyr77His (c.229T>C) and with MHP; p.Asp143Val (c.428A>T), respectively.
Among the 94 different PAH mutations found in the study group, 69 were found in the cohort identified during 1990–2014 as compared to 48 during 1965–1989 (Fig. 3). Only 32 of the 94 different mutations were found in both birth cohorts. Out of the 32 mutations, eight were also found in the immigrant/adoptive group. This further more confirms the increase of the heterogeneity of mutations detected in the PAH gene.
Fig. 3.
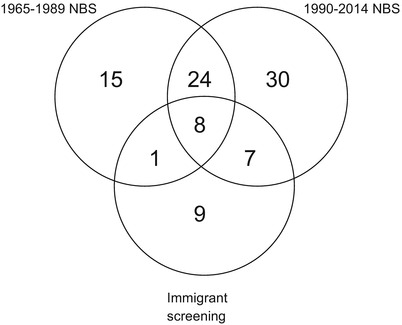
The Venn diagram describes the heterogeneity of mutations detected in the PAH-gene with only 32 mutations being common for the two NBS periods
Shown in Fig. 2 are mutations in the NBS group (index cases) with a relative frequency of >1.0% of all alleles for the periods 1965–1989 and 1990–2014, respectively. The three most frequent mutations, p.Arg408Trp, p.Tyr414Cys and c.1315+1A>G, are the same for both periods, although the mutations are occurring less frequently in the second period. Four well-known mutations associated with MHP; p.Ala300Ser, p.Ala403Val, p.Asp415Asn (c.1243G>A) and p.Glu390Gly (c.1169A>G) have added together been detected on 8.8% of all alleles during 1990–2014. Five mutations, p.Leu48Ser, c.441+5G>T, p.Arg241Cys, c.1066-11G>A and p.Glu390Gly which were not seen before 1990, are now detected at an increasing frequency. The two mutations showing the highest increase for the two periods are p.Arg261Gln (+6.2%) and c.1066-11G>A (+3.8%), both of which are common in the southern Europe (Zschocke 2003).
The three mutations with the highest frequency among the 26 immigrant/adoption cases were c.168+5G>C (9.6%), p.Arg408Trp (9.6%) and c.1066-11G>A (11.5%). Three of the novel variants were detected in this group, p.Tyr77His, c.970G-1G>T and c.843-13_843-10delTTCT.
In the total Swedish PKU population (NBS and immigration) more than 30 countries are represented. The major immigration influence on the mutation spectrum comes from Turkey, Iraq, Syria and Lebanon. The most frequently occurring mutations in these patients are p.Arg261Gln, p.Arg408Trp, c.1066-11G>A and p.Ala300Ser. All but p.Arg408Trp show an increased frequency also in patients detected by NBS after 1990.
The Change of Homozygosity
During 1965–1989 the two most prevalently occurring homozygous mutations in patients were the same as the mutations with the highest frequencies, p.Arg408Trp and p.Tyr414Cys. These two mutations occurred at the highest frequencies also in the most recent period but were only found in a homozygous state in one patient each. Instead the two most prevalent homozygous genotypes in this period were homozygosity for p.Arg261Gln and c.1066-11G>A. The percentage of patients being homozygous for a mutation was the same for the periods, 18.1% (21 patients) and 16.8% (27 patients), respectively. In the first period one homozygous patient had parents born abroad whilst in the second period 14 patients had parents born abroad. This change could probably be explained by the fact that the native Swedish population has become more movable during the past decades, reducing geographic isolation, whilst immigration from countries with a high rate of consanguineous marriages has become more common.
Conclusions
The Swedish spectrum of PAH mutations has during the past 25 years undergone a major change towards becoming more heterogeneous, reflecting the present structure of the Swedish population. Milder mutations common in southern Europe and in the Middle East together with lowering of the cut off values for Phe have led to a shift towards milder genotypes and phenotypes among the PKU patients detected by NBS. The inclusion of the Phe to Tyr ratio as an additional marker has led to an increase in positive predictive value and it also provides a tool to differentiate between mild and classical PKU. Lowering the cut off to Phe below 180 μmol/L most probably will increase the number of false positives and increase the number of newborns with MHP not needing treatment. The lower Phe value in the first screening sample due to earlier sampling age does not necessarily lead to any disadvantages. It does not lead to missed cases in need of treatment and enables children with very mild MHP to remain undiscovered.
This work was supported by grants from the Stockholm County Council and the Karolinska Institute Research Foundation.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
[INSERT CAPTION HERE] (XLSX 13 kb)
[INSERT CAPTION HERE] (XLSX 19 kb)
Concise Sentence
Since the start of NBS for PKU in Sweden due to the use of lower cut offs a major shift towards detecting patients with milder mutations and less severe disease are becoming more common in the Swedish population.
Contribution of Individual Authors
Experimental design and execution: AO and HB
Manuscript preparation: AO and UvD
Data analysis: AO, AW and UvD
Clinical investigation: AN and RZ
All authors have read and revised the final manuscript.
Ulrika von Döbeln serves as guarantor for the article, accepts full responsibility for the work, has access to the data and controls the decision to publish.
Conflict of Interest
The authors have nothing to declare.
Details of funding: Stockholm County Council and the Karolinska Institute Research Foundation.
The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Compliance with Ethics Guidelines
Genetic investigation of inborn errors of metabolism, approved by Regional Ethical Committee of Stockholm, 2008/351-31. Informed consent was not required since individual patients cannot be identified in the manuscript.
Contributor Information
Annika Ohlsson, Email: annika.ohlsson@ki.se.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376:1417–1427. doi: 10.1016/S0140-6736(10)60961-0. [DOI] [PubMed] [Google Scholar]
- Djordjevic M, Klaassen K, Sarajlija A, et al. Molecular genetics and genotype-based estimation of BH4-responsiveness in Serbian PKU patients: spotlight on phenotypic implications of p.L48S. JIMD Rep. 2013;9:49–58. doi: 10.1007/8904_2012_178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiken HG, Knappskog PM, Apold J, Flatmark T. PKU mutation G46S is associated with increased aggregation and degradation of the phenylalanine hydroxylase enzyme. Hum Mutat. 1996;7(3):228–238. doi: 10.1002/(SICI)1098-1004(1996)7:3<228::AID-HUMU7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:338–343. [PubMed] [Google Scholar]
- Hanley WB. Non-PKU mild hyperphenylalaninemia (MHP) – the dilemma. Mol Genet Metab. 2011;104(1–2):23–26. doi: 10.1016/j.ymgme.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Loeber JG. Neonatal screening in Europe; the situation in 2004. J Inherit Metab Dis. 2007;30(4):430–438. doi: 10.1007/s10545-007-0644-5. [DOI] [PubMed] [Google Scholar]
- Opladen T, Hoffmann GF, Blau N. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. J Inherit Metab Dis. 2012;35(6):963–973. doi: 10.1007/s10545-012-9506-x. [DOI] [PubMed] [Google Scholar]
- Smith I, Glossop J, Beasley M. Fetal damage due to maternal phenylketonuria: effects of dietary treatment and maternal phenylalanine concentrations around the time of conception (an interim report from the UK Phenylketonuria Register) J Inherit Metab Dis. 1990;13(4):651–657. doi: 10.1007/BF01799520. [DOI] [PubMed] [Google Scholar]
- Svensson E, von Döbeln U, Eisensmith RC, Hagenfeldt L, Woo SL. Relation between genotype and phenotype in Swedish phenylketonuria and hyperphenylalaninemia patients. Eur J Pediatr. 1993;152(2):132–139. doi: 10.1007/BF02072490. [DOI] [PubMed] [Google Scholar]
- Trunzo R, Santacroce R, D’Andrea G, et al. Mutation analysis in hyperphenylalaninemia patients from South Italy. Clin Biochem. 2013;46(18):1896–1898. doi: 10.1016/j.clinbiochem.2013.06.009. [DOI] [PubMed] [Google Scholar]
- Zschocke J. Phenylketonuria mutations in Europe. Hum Mutat. 2003;21:345–356. doi: 10.1002/humu.10192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[INSERT CAPTION HERE] (XLSX 13 kb)
[INSERT CAPTION HERE] (XLSX 19 kb)