

Summary
Widespread resistance to first-line TB drugs is a major problem that will likely only be resolved through the development of new drugs with novel mechanisms of action. We have used structure-guided methods to develop a lead molecule that targets the thioesterase activity of polyketide synthase Pks13, an essential enzyme that forms mycolic acids, required for the cell wall of Mycobacterium tuberculosis. Our lead, TAM16, is a benzofuran class inhibitor of Pks13 with highly potent in vitro bactericidal activity against drug-susceptible and drug-resistant clinical isolates of M. tuberculosis. In multiple mouse models of TB infection, TAM16 showed in vivo efficacy equal to the first-line TB drug isoniazid, both as a monotherapy and in combination therapy with rifampicin. TAM16 has excellent pharmacological and safety profiles, and the frequency of resistance for TAM16 is ∼100-fold lower than INH, suggesting that it can be developed as a new antitubercular aimed at the acute infection.
PaperClip
Keywords: polyketide synthase, Pks13 thioesterase domain, crystal structure, benzofuran inhibitors, structure-based drug discovery, Mycobacterium tuberculosis
Graphical Abstract

Highlights
-
•
Structure-guided medicinal chemistry used to develop potent and safe lead molecule
-
•
First lead molecule reported that targets a polyketide synthase
-
•
TAM16 is active against clinical drug-resistant M. tuberculosis
-
•
Lower frequency of resistance than INH
A small molecule inhibitor of M. tuberculosis polyketide synthase shows strong efficacy in murine models of infection.
Introduction
Drug-resistance in Mycobacterium tuberculosis (Mtb) is a serious problem that threatens to worsen the global tuberculosis (TB) epidemic (World Health Organization, 2014). Although current six-month therapy for drug-susceptible TB can achieve a cure rate of >90%, the treatment of drug-resistant strains is more protracted (≥2 years) and involves the use of costly and less effective second-line drugs that have significant side effects (Zumla et al., 2013).
Isoniazid (INH) is a frontline TB drug that has been a mainstay of TB therapy since its introduction in 1952 (Bernstein et al., 1952). However, resistance occurs frequently with in vitro rates of about 1 in 10−5–10−6, which translates to high levels of clinical resistance ranging from 9.5% to 62%, based on geography and disease burden (Jenkins et al., 2011, World Health Organization, 2014). INH is a pro-drug that is activated by a catalase-peroxidase enzyme (KatG) to produce a radical that attacks nicotinamide adenine dinucleotide (NAD) to form a covalent adduct. This adduct inhibits the enoyl-ACP reductase, InhA (Rozwarski et al., 1998), an enzyme required for the synthesis of very long chain fatty acids that are used to form mycolic acids (Vilchèze et al., 2000). Because INH is activated by the non-essential KatG, resistance to INH often arises through loss-of-function mutations in the katG gene (Heym et al., 1995, Zhang et al., 1992). Indeed, the most common cause of INH resistance is the loss-of-function mutation KatG-S315T, which has been found in as many as 94% of INH-resistant and up to 82% of multidrug-resistant (MDR) Mtb clinical isolates (Torres et al., 2015). In addition, mutations in the inhA gene and its promoter region, i.e., the c-15t base change, further complicate the treatment of drug-resistant TB, conferring resistance not only to INH but also to the second-line TB drug ethionamide (ETH) (Banerjee et al., 1994), with reported frequencies of 35% and 55% in INH- and ETH-resistant clinical isolates, respectively (Vilcheze and Jacobs, 2014). After 65 years of use, the widespread and very high levels of INH resistance underscore the urgent clinical need for the development of alternative cell wall-active antibiotics for TB.
Mycolic acids are critical for viability and virulence of Mtb. Though these long-chain (C60–90) α-branched-β-hydroxylated fatty acids are primarily found esterified to the arabinogalactan-peptidoglycan cell wall core, they are also present as trehalose monomycolate and dimycolate esters in the cell envelope (Barry et al., 1998). In Mtb, mycolic acid biosynthesis occurs through the concerted action of more than 20 enzymes that are components of different multi-enzyme complexes (Takayama et al., 2005). Therefore, this pathway represents an important reservoir of novel targets for the development of new TB drugs, especially in the context of the emergence of drug resistance.
Polyketide synthases (PKS) are an important family of enzymes that have not been exploited as drug targets for any microbial pathogen. The Mtb H37Rv genome has about 24 PKS encoding genes (Cole et al., 1998). Genetic and biochemical studies have now linked most of the mycobacterial PKSs to participating in complex lipid biosynthestic pathways in Mtb (Chopra and Gokhale, 2009, Quadri, 2014). These PKS-derived lipid metabolites form essential components of the uniquely lipid-rich and complex cell wall of Mtb, which has been proposed as a means for it to survive under harsh conditions in host macrophages while also imparting an intrinsic resistance against many anti-microbial agents (Bhatt et al., 2007, Glickman et al., 2000).
In Mtb, Pks13 performs the final assembly step of mycolic acid synthesis, i.e., the Claisen-type condensation of a C26 α-alkyl branch and C40–60 meromycolate precursors (Portevin et al., 2004). It is comprised of five domains, including two acyl carrier protein domains, a β-ketoacyl-synthase, an acyltransferase, and a C-terminal thioesterase (TE) domain, that together contain all of the activities required for the condensation of two long-chain fatty acids. This activity has been shown to be essential both in vitro and in vivo (Portevin et al., 2004, Wilson et al., 2013). We recently discovered a small molecule that was active against Mtb H37Rv (TAM1; Figure 1A) and identified that Pks13 was the target through whole-genome sequencing and recombineering of the resistance mutations (Ioerger et al., 2013). In another study, a series of thiophenes were identified that kill Mtb by targeting the N-terminal ACPN domain of Pks13. Wilson et al., 2013, propose that the compounds function by blocking the interaction of ACPN with FadD32 protein, which transfers the meromycolyl chain. These results substantiate Pks13 as a druggable target for Mtb and highlight its potential for the development of new TB drugs that interfere with the critical pathway of mycolic acid synthesis.
Figure 1.

Novel Benzofurans Inhibit Pks13 Thioesetrase Domain
(A) Chemical structure of TAM1 highlighting the convention used for naming the substituent groups (P1, P2, P3, and P4) and numbering of the benzofuran ring. TAM1 inhibits the esterase activity of Pks13-TE with an IC50 = 0.26 ± 0.03 μM. The graph depicts percent activity relative to DMSO only control (mean ± SD).
(B) Overall view of the structure of the Pks13-TE-TAM1 complex showing structural features of the Pks13-TE domain. Catalytic residues His1699 and Ser1533 at the interface of the lid and core domains are shown as ball and sticks. TAM1 is shown as yellow sticks.
(C and D) Close-up views of inhibitor interactions show that benzofuran core of TAM1 (yellow sticks) wedges between Phe1670 and Asn1640 with its P3 group oriented toward the catalytic site. Hydrogen bonds are represented by dashed lines. Surface representation in (C) is colored by electrostatic potential (contoured at ± 5 kT/e, red for negative and blue for positive).
In this paper, we describe the structure-based development of a highly potent and very safe lead compound, TAM16 (Table 1), which targets Pks13. It is active against MDR and extensively drug-resistant (XDR) Mtb clinical strains in vitro, demonstrating a lack of cross-resistance with existing TB therapeutics. By inhibiting cell wall biosynthesis, it synergizes with other TB drugs, like rifampicin (RIF), likely by augmenting their penetration into Mtb. Importantly, in murine TB infection models, it demonstrates efficacy equal to INH. Furthermore, unlike INH, which shows a relative high frequency of resistance, TAM16 shows 100-fold lower frequency of resistance. These properties, combined with the excellent pharmacokinetic (PK) and toxicity profiles, will likely allow us to convert this lead into a first-line drug.
Table 1.
Preliminary SAR of TAM1 and Its Analogs
 | ||||||
|---|---|---|---|---|---|---|
| Compound | P1 | P2 | P3 | P4 | IC50 (μM)∗ | MIC (μM) |
| TAM1 | 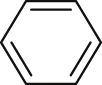 |
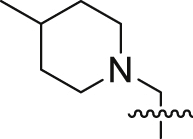 |
OH | 0.26 ± 0.03 | 2.3 | |
| TAM2 | 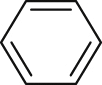 |
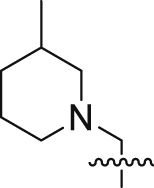 |
OH | 0.12 ± 0.02 | 4.4 | |
| TAM3 | 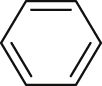 |
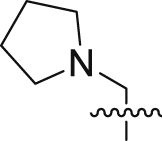 |
OH | 0.24 ± 0.02 | 4.1 | |
| TAM4 | 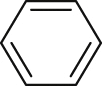 |
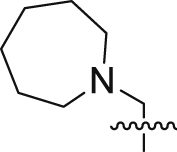 |
OH | 0.28 ± 0.03 | 4.6 | |
| TAM5 | 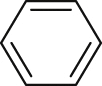 |
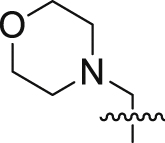 |
OH | 0.71 ± 0.05 | 13.3 | |
| TAM6 | 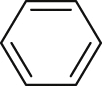 |
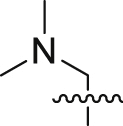 |
OH | 1.57 ± 0.15 | 7.3 | |
| TAM7 | 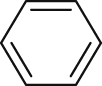 |
None | OH | 20 ± 1.9 | 20 | |
| TAM8 | 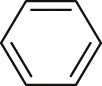 |
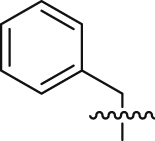 |
OH | 11.9 ± 2.3 | >40 | |
| TAM9 | 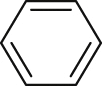 |
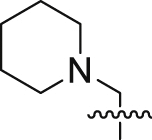 |
OH | 0.26 ± 0.04 | 0.4 | |
| TAM10 |  |
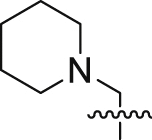 |
OH | 6.6 ± 0.7 | NI | |
| TAM11 |  |
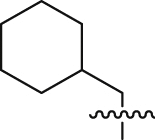 |
OH | 19.6 ± 1.4 | 5.2 | |
| TAM12 | 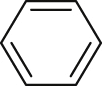 |
 |
 |
OH | 0.29 ± 0.01 | 0.2 |
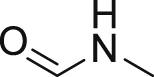
| TAM13 | 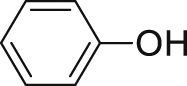 |
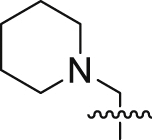 |
OH | 0.17 ± 0.02 | 1.2 | |
| TAM14 | 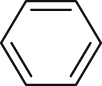 |
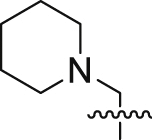 |
MeO | 35.8 ± 2.2 | ND | |
| TAM15 |  |
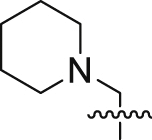 |
H | 2.0 ± 0.1 | 16 | |
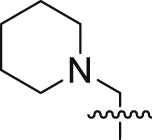
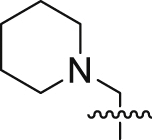
| TAM16 | 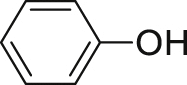 |
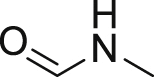 |
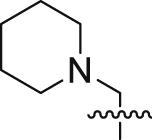 |
OH | 0.19 ± 0.01 | 0.09 |
| TAM17 | 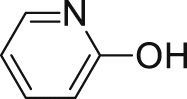 |
 |
OH | 0.36 ± 0.04 | 4 | |
| TAM18 | 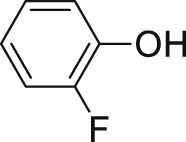 |
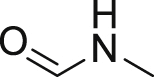 |
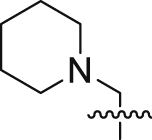 |
OH | 0.33 ± 0.03 | 0.5 |
| TAM19 | 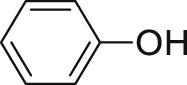 |
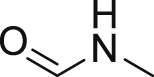 |
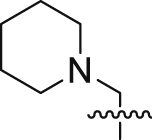 |
H, C6-OH | 0.57 ± 0.03 | 1 |
| TAM20 | 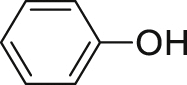 |
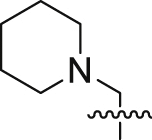 |
H, C6-OH | 0.45 ± 0.04 | 1.1 | |
| TAM21 | 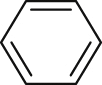 |
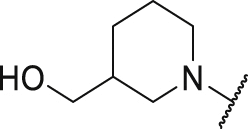 |
OH | 0.42 ± 0.04 | 10 | |
| TAM22 | 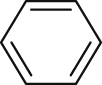 |
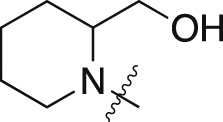 |
OH | >10 | >10 | |
| TAM23 | 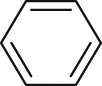 |
 |
OH | 0.38 ± 0.01 | 2.5 | |
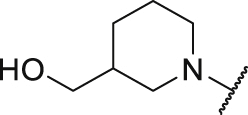
| TAM24 |  |
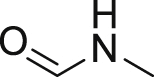 |
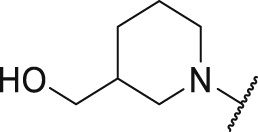 |
OH | 2.1 ± 0.3 | 0.5 |
IC50 values were determined using the Mtb Pks13-TE domain as described in the methods section. MIC values were determined for Mtb mc27000 in liquid medium in 96-well plates. MeO, methoxy; NI, no inhibition; ND, not determined.
Values are shown as mean ± SD of three independent measurements.
Results
TAM1 Inhibits Pks13 TE Domain Activity
Two laboratory-derived mutant strains resistant to TAM1 were found to harbor non-synonymous mutations, i.e., either D1607N or D1644G, both located in the TE domain of Pks13. To characterize the precise mechanism of action of TAM1 on the TE activity, a recombinant-expression plasmid was constructed to produce the domain for biochemical analysis. The pure recombinant protein, consisting of the TE domain of the Pks13 (Pks13-TE), was enzymatically active and produced diffraction-quality crystals complexed to TAM1.
An enzyme assay was developed for the TE activity of Pks13 using the fluorescent fatty acid ester, 4-methylumbelliferyl heptanoate (4-MUH) (Richardson and Smith, 2007). Pks13-TE was able to cleave the ester of 4-MUH, and kinetic analysis indicated a Michaelis constant (Km) ∼20 μM and kcat/Km ∼ 7.2 × 102 M−1 min−1 (Table S1). TAM1 inhibited the Pks13-TE activity with a half-maximal inhibitory concentration (IC50) of 0.26 μM (Figure 1A; Table S1).
TAM1 Blocks the Active Site of Pks13-TE
As a first step to structure-guided medicinal chemistry on the benzofuran inhibitor, we solved the crystal structure of Pks13-TE complexed with TAM1 and refined it to high resolution (2.0 Å; Table S2). The crystals contained two monomers in the crystallographic asymmetric unit (designated A and B). The overall structure of Pks13-TE consists of a core domain and a lid domain (Figure 1B). The larger core possesses a canonical α/β-hydrolase fold comprised of a central seven-stranded β sheet (β1–β7) flanked by four α helices (α1–α3 and α11) with the N-terminal β1 strand anti-parallel to other β strands (Nardini and Dijkstra, 1999). The lid domain (residues 1575–1645) is inserted between strands β5 and β6 and consists of four α helices, α4–α7, along with two short helices, α8 and α9, (residues 1665–1675) present between the strands β6 and β7 of the core domain. Based on the analysis with VAST server (Gibrat et al., 1996), the Pks13-TE lid domain appears to be relatively unique among TE structures reported to date.
The Pks13-TE active-site pocket is formed at the interface between the lid and core domains. The catalytic triad was identified to be Ser1533, Asp1560, and His1699, and the oxyanion hole is formed by the amide N-atoms of Leu1534 and Ala1477. Extending from the active site is a deep (∼30 Å) hydrophobic groove that spans the full length of the lid domain, with a total surface area of ∼1290 Å2 (Figure S1A). A similarly located surface groove (∼20 Å) in the α-helical lid domain of bovine palmitoyl-protein thioesterase 1 (PPT1, 15% identity with Pks13-TE), contained the substrate palmitic acid (Bellizzi et al., 2000). We observed unexpected electron density in this pocket of apo-Pks13-TE structure that could be built as an eight-carbon fragment of polypropylene glycol (PPG, C8O5), an additive in the crystallization buffer. The fragment is located in the fatty acyl chain-binding site based on the superimposition of Pks13-TE structure with the bovine PPT1 and human FAS TE (hFAS-TE) structures (Zhang et al., 2011). However, unlike the Pks13-TE acyl chain-binding site, the bovine PPT1 and hFAS-TE is not surface exposed. The PPG binding pocket is connected at the catalytic site to a series of tunnel-like regions that could also bind an acyl chain (Figure S1B). The more elaborate system of binding grooves and tunnels is likely related to the very long carbon chains (C80–90) that make up the mycolic acid precursor, compared to PPT1 and hFAS (Figure S1C).
Figure S1.

Structural Features of the Pks13-TE Crystal Structure, Related to Figure 1
(A) Surface representation of the Pks13-TE structure colored by electrostatic potential (contoured at ± 5 kT/e; red for negative and blue for positive) to illustrate the substrate binding groove (∼30 Å, double-headed yellow arrow) on the lid domain. The zoomed view shows the bound PPG fragment (cyan) in the catalytic pocked formed by residues Ser1533, His1699 and Asp1560 along with the residues of the oxyanion-hole (Leu1534 and Ala1477) rendered as sticks; catalytic water shown as red sphere. The 2Fo-Fc electron density map contoured at 1.2σ is shown for the bound PPG fragment. Hydrogen bond interactions are shown as black dashed lines.
(B) Predicted tunnels in Pks13-TE structure by CAVER analysis (Chovancova et al., 2012). The three potential tunnels are shown in pink, blue and green surface rendering. The largest of the tunnels (pink) opens onto the substrate binding surface groove and contained the bound PPG fragment.
(C) Docking of mycolic acid on Pks13-TE lid domain. A molecule of mycolic acid (shown as yellow sticks) was docked using Molsoft ICM-Pro software to determine a possible binding mode in the substrate binding groove. The zoomed view of the docking indicates that the surface groove can accommodate acyl chains of the mycolic acid precursor attached to the C-terminal ACP domain and position the thioester for cleavage near the catalytic Ser1533 residue.
TAM1 binds in the fatty acyl chain-binding groove at the entrance of the Pks13-TE active site, effectively blocking access of the substrate to the catalytic center of the enzyme (Figures 1B–1D). Several differences were seen between the apo- and TAM1-bound structures of Pks13-TE, the most significant being in the side chain of Phe1670. Phe1670 is located at the end of helix α8 directly adjacent to the acyl binding pocket. The phenyl ring of Phe1670 flips by about 80° in the TAM1 structure, compared to the apo protein, to form a slightly off-plane van der Waals stacking interaction with the furan ring (Figure S2A). The four different substituents attached to the benzofuran scaffold (P1, P2, P3, and P4; Figure 1A) were found to interact with residues that line the substrate-binding cleft. Indeed, most of the binding interactions occur between amino acids from helix α7 of the lid domain and the two supporting helices α8–α9 along with the loop that connects them to strand β6 of the core domain (Figures 1B and 1D). Overall, the structure showed that the phenyl group of TAM1 (P1) is solvent exposed; P2 ethyl ester is partially solvent exposed, while P3 piperidine and P4 OH are completely buried in Pks13-TE.
Figure S2.

Structural Changes in TAM1-Bound and D1607N-Mutant Pks13-TE Crystal Structures, Related to Figure 1
(A) Superimposition of Pks13-TE-TAM1 complex structure (purple) with Apo-Pks13-TE structure (yellow) shows that Phe1670 side chain (shown as stick) flips by ∼80° upon TAM1 binding. TAM1 interacting residues are shown in line representation in purple color, and the corresponding residues from Apo structure are shown as lines in yellow color. Catalytic residues (Ser1533 and His1699) are shown as ball and sticks. TAM1 is omitted for clarity of presentation.
(B) Superimposition of Pks13-TE-TAM1 complex structure (purple, TAM1 as yellow sticks) with the structure of D1607N mutant (pink) shows the conformational change in Arg1641 of the mutant due to disruption of ion pair interaction with Asp1607. In the wt Pks13-TE structure, Asp1607 carboxylate forms an ion pair interaction with the guanidinium of Arg1641 which helps stabilize the C-terminal end of helix α7. This allows Asp1644 to form hydrogen bond interaction (shown as dashed black line) with TAM1. The mutation D1607N breaks the ion pair interaction mediated anchoring of helix α7 that causes Asp1644 to move away by ∼3 Å (double-headed black arrow), consequently disrupting its interaction with TAM1.
The Pks13-TE structure shows that the D1644G mutation confers resistance through the loss of a direct H bond between a carboxylate oxygen and the P4 OH of TAM1. The crystal structure of the D1607N mutant (Figure S2B; Table S2) explained its more subtle effect on binding, through the loss of ion pair interaction between the carboxylate of Asp1607 on helix α6 and the guanidinium of Arg1641 (3 Å) located on the parallel helix, α7. Without this interaction, α7 shifts away from the substrate-binding groove by about 3 Å, moving Asp1644 out of hydrogen-bonding distance from the P4 OH of TAM1.
Structure-Based Development of TAM1 Analogs
TAM1 was modified using structure-guided approaches to improve the potency and pharmacological properties (Table 1; Figure 2; STAR Methods; Table S3). Briefly, the two major pharmacological liabilities of TAM1were the phenyl at P1, which was hydroxylated in mouse liver microsomes (MLMs) to yield primarily the 4-OH (Figure S3A), and the P2 ester, which was cleaved to the inactive acid in mouse serum. The structure of the Pks13-TE-TAM1 complex indicated that the side chain amide of Gln1633 was positioned in close proximity (∼4 Å) to the para-position of the P1 phenyl ring, suggesting that 4-OH form of the inhibitor would retain enzyme inhibitory activity. Indeed, the P1 4-OH-containing analog, TAM16, had greatly improved metabolic stability (Figure S3), showed significant improvement in enzyme potency, and exhibited >20-fold improvement in Mtb potency compared with TAM1 (Table 1). The structure also indicated that the bioisosteric replacement of the ester with a methyl amide that would be stable in serum would be tolerated in the binding pocket. Indeed, the methyl amide of TAM12 was stable and showed similar enzyme-inhibitory activity (IC50 0.3 μM) as the ethyl ester analog TAM9; however, it exhibited 2-fold better whole-cell activity (Table 1). Among P3 groups, the piperidine was the most potent of the P3 substituents synthesized. Structures of P3 substituted analogs showed that five-, six-, and seven-membered rings at P3 led to variations in the van der Waals and stacking interactions with the planar side chain of Tyr1674, and these interactions were abolished in TAM6 with acyclic dimethyl amine at P3. Thus, among the P3 analogs, the piperidine group was optimally located sandwiched between Tyr1663 and Tyr1674, and its protonated N appears to be a bifurcated hydrogen donor, forming an intra-molecular hydrogen bond with the P2 carbonyl oxygen (2.9 Å) and another with the side chain oxygen of Asn1640 (2.9 Å). We also explored alterations to the P4 OH group. All P4 analogs at C-5 showed a dramatic loss in enzyme potency presumably due to loss of hydrogen bond with the carboxylate of Asp1644. The resulting molecule, TAM16, was the most potent and stable inhibitor (IC50 0.19 μM, minimum inhibitory concentration [MIC] 0.09 μM) (Figures 2 and S3B).
Figure 2.

Structure-Guided Development of TAM16
Substitution of P1 and P2 groups in TAM16 with phenol and methyl amide, respectively, increased potency and metabolic stability. Calculated log(P), calculated log(partition coefficient); Mouse Cli., intrinsic clearance in mouse liver microsomes.
Figure S3.

Metabolic Stability of TAM1 Analogs, Related to Table 3
(A) Metabolic stability studies of TAM12 in mouse liver microsomes showed that TAM12 was hydroxylated at P1 phenyl. Graph represents mean values ± SD of two independent assays.
(B) Cartoon of the Pks13-TE-TAM16 complex structure showing hydrogen bond interaction between P1 4-OH of TAM16 (cyan) and the side chain carbonyl oxygen atom of Gln1633 (green). The gray mesh represents the 2mFo - DFc maximum-likelihood omit map, contoured at 1.2σ. Hydrogen bonds are shown as black dashed lines.
(C) Glucuronidation of TAM16 was assessed in mouse liver microsomes with UDPGA (5 μM for 60 min). At 0, 30 and 60 min, 100 μL aliquots of the reaction mixture were removed and placed in 100 μL of acetonitrile to terminate the reaction. Analysis by mass spectrometry for metabolite identification showed little metabolism of the parent compound (TAM16), the conjugate was barely detectable after 60 min incubation (peak A).
(D and E) Metabolic stability analysis of TAM16 incubated with glutathione and methoxylamine (50 μM for 180 min) in the presence of mouse plasma and HLMs for possible retro-Mannich metabolites or adducts. The parent compound was seen to decrease slightly over time and two metabolites were detected. For metabolite B the main ion see was at m/z 379 but a barely detectable ion was seen at m/z 397 indicating the metabolite may be due to oxidation which dehydrates readily in the MS. No trace of the quinone-methide or potential GSH adducts was seen in any of the samples.
a% of total peak area for parent and metabolite peaks of the proposed [M+H]+ ions at 60 min time point.
b% of total peak area for parent and metabolite peaks of the proposed [M+H]+ ions at 180 min time point.
Low Frequency of Resistance, No Cross-Resistance, and Combination Potential of TAM16
Resistant mutants emerged at frequencies of 7 × 10−7 and ∼8 × 10−8 at 10× and 20× MIC, which were ∼100-fold lower than the frequency observed for INH (1 × 10−6). Sequencing of two resistant mutants revealed point mutations in pks13 that resulted in a single non-synonymous amino acid substitution in Pks13 (D1644G or D1644Y), and they retained sensitivity to INH and RIF (MICs 0.2 and 0.04 μM, respectively).
We determined MIC values of TAM16 against 38 clinical Mtb strains representing a wide range of mutations covering all known molecular mechanisms involved in imparting resistance against first- and second-line TB drugs that form the core component of TB treatment regimen (Tables S4 and S5). TAM16 was highly active against pan-susceptible clinical isolates of Mtb (MICs 0.06–0.250 μM). Most importantly, TAM16 was potent against all MDR and XDR clinical isolates of Mtb evaluated (MIC range of 0.05–0.42 μM; Table 2). The lack of cross-resistance of TAM16 with current TB drugs shows the importance of developing new drugs with distinct modes of action.
Table 2.
TAM16 MIC Values for Mtb Strains with Different Drug-Susceptibility Profiles, Related to Tables S4 and S5
| Strain and resistance statusa | Number of strains | MIC90b range for multiple strains (μM) | Median MIC90 (μM) |
|---|---|---|---|
| H37Rv, fully susceptible lab strain | 1 | - | 0.125-0.25 |
| H37RvMa, fully susceptible lab strain | 1 | - | 0.125 |
| Mtb, fully susceptible clinical isolates | 12 | 0.060-0.250 | 0.100 |
| Mtb, poly-resistant clinical isolate | 1 | - | 0.420 |
| Mtb, MDR clinical isolates | 7 | 0.060-0.210 | 0.210 |
| Mtb, pre-XDR clinical isolates | 5 | 0.125-0.420 | 0.420 |
| Mtb, XDR clinical isolates | 5 | 0.125-0.250 | 0.125 |
| Mtb, INH mono-resistant, clinical | 6 | 0.050-0.125 | 0.100 |
| Mtb, RIF mono-resistant, clinical | 1 | - | 0.125 |
| Mtb, SM mono-resistant, clinical | 1 | - | 0.420 |
Poly-resistant, Mtb-strain resistant to isoniazid (INH), ethionamide, and streptomycin (SM); MDR, resistant to both INH and rifampicin (RIF), with or without resistance to other anti-TB drugs; pre-XDR, MDR strains with additional resistance to either a fluoroquinolone or an injectable but not both; XDR, MDR strains that are also resistant to any fluoroquinolone and to any of the three second-line injectables (amikacin, capreomycin, and kanamycin).
Detailed description of the strains with their drug-resistance phenotypes is given in Tables S4 and S5.
The lowest concentration of drug that inhibited growth of more than 90% of the bacterial population was considered to be the MIC90. The MIC90 values were determined using MGIT 960 system.
One of the advantages of drugs that target cell wall biosynthesis, like INH, is their ability to combine with other antibiotics to increase efficacy. We tested TAM16 in combination with INH, RIF, and EMB in vitro in two-drug combination studies by the combination index (CI) method (Chou, 2006) (wherein CI < 1, CI = 1, and CI > 1 indicate synergistic, additive, and antagonistic interactions, respectively). While TAM16 showed no synergistic activity with INH and EMB, the two other drugs that target cell wall, the combination of TAM16 with RIF, which targets the RNA polymerase, showed highly synergistic activity (CI = 0.55) against Mtb mc27000. Notably, at the combination ratio of 1:0.5, the MIC of RIF was improved ∼3-fold to 0.011 μM compared to RIF alone (MIC 0.35 μM), while the MIC of TAM16 improved by 5-fold (0.022 μM) compared to TAM16 alone (0.10 μM). This synergy is likely based on increased permeability of the bacterial cell wall leading to enhanced RIF accumulation. Indeed, two-drug combinations of TAM16 with antitubercular drugs TMC207 (a diarylquinoline compound that inhibits ATP synthesis) and streptomycin (SM, an aminoglycoside antibiotic that inhibits protein synthesis) also showed synergistic activity (CI ∼0.54 and 0.81, respectively). The combination of TAM16 and TMC207 (at a combination ratio of 1:0.5) improved the MIC of TAM16 in the presence of TMC207 by ∼6.5-fold to 0.016 μM, and the MIC of TMC207 in the presence of TAM16 was improved 2.6-fold to 0.005 μM compared to the MIC of TMC207 alone (MIC 0.013 μM). Similarly, the combination of TAM16 and SM (at a combination ratio of 1:2) was synergistic: it improved TAM16 MIC by ∼4-fold in the presence of SM, and the addition of TAM16 improved the MIC of SM ∼2-fold to 0.037 μM compared to the SM MIC when it was used alone (0.07 μM). Overall, these results are consistent with previously observed effects of cell wall inhibitors on improving the penetration of anti-Mtb drugs (Bhusal et al., 2005, Bosne-David et al., 2000, Lechartier et al., 2012).
Toxicology and Pharmacokinetic Properties of TAM16
TAM16 has excellent physicochemical, toxicological, and pharmacological properties (Table 3). It showed low plasma protein binding in both mouse and human plasma and exhibited very low clearance in MLM and HLMs (human liver microsomes) (CLint < 0.5 mL/min/g liver; Table 3), and the two phenolic OH groups were not significantly glucuronidated in the microsomal incubation assays (Figure S3C).
Table 3.
Physicochemical and Pharmacokinetic Properties of TAM16
| MW (g/mol) | 380.4 | |
|---|---|---|
| cLogP | 1.6 | |
| logD | 1.7 | |
| Lipophilic ligand efficiency | 5.1 | |
| H-bond donors | 3 | |
| H-bond acceptors | 4 | |
| TPSA (Å2) | 86 | |
| pKa | 9.95 | |
| Kinetic solubility (μM) | 74 | |
| (phosphate buffer, pH 7.4) | ||
| Plasma protein binding (%): | Mouse | 73 |
| Human | 72 | |
| Intrinsic clearance in liver microsomes | ||
| (CLint) (mL/min/g liver): | Mouse | <0.5 |
| Human | <0.5 | |
| CYP inhibition | No significant inhibition | |
| PK parameters: | ||
| Cmax (ng/mL) in plasma | 444 | |
| Tmax (hr) | 0.5 | |
| t1/2 (hr) in plasma | 1.0 | |
| AUC0-24 (ng.min/mL) (po) | 74,940 | |
| AUC0-24 (ng.min/mL) (iv) | 79,369 | |
| Clearance (mL/min per kg) | 37 | |
| Vss (L/kg) | 4.2 | |
| Oral bioavailability (F) (%) | 28 | |
Pharmacokinetic parameters were determined after administration of single oral (po) and intravenous (iv) doses of TAM16 at 10 mg/kg and 3 mg/kg, respectively, in female BALB/c mouse. Cmax, maximum concentration; Tmax, time to reach Cmax; t1/2, half-life; AUC, area under the concentration curve; Vss, volume of distribution at steady state; cLogP, calculated log(partition coefficient); Lipophilic ligand efficiency = pIC50 − cLogP; TPSA, total polar surface area.
See also Figures S3 and S4 and Table S6.
Pharmacokinetic studies in Swiss Webster mice were performed with TAM16 dosed by oral administration at 100 mg/kg twice daily. At this dose, maximum plasma concentration (Cmax) of TAM16 reached 3.6 μg/ml at 5 hr, and the AUC was 18.3 μg.hr/ml. At a single dose of TAM16, mouse exposure was higher than the MIC (34.3 ng/ml) for ∼12 hr. Pharmacokinetic parameters determined after a single oral and intravenous dose of 10 mg/kg and 3 mg/kg, respectively, in BALB/c mice showed that TAM16 had moderate total plasma clearance (37 mL/min per kg), a large volume of distribution (4.2 l/kg), good exposure, and oral bioavailability (28%) (Table 3; Figure S4).
Figure S4.

In Vivo Pharmacokinetics of TAM16, Related to Table 3
Mean blood concentration profiles of TAM16 following oral (p.o) and iv dose of 10 mg/kg and 3 mg/kg, respectively, in female BALB/c mouse. PK parameters (inset) were determined after the administration of a single dose (both p.o and iv) to mice. Cmax, maximum concentration; Tmax, time to reach Cmax, , half-life; AUC, area under the concentration curve; Vss, volume of distribution at steady state.
TAM16 was not cytotoxic to mammalian cells at concentrations up to 100 μM, and it was well tolerated in BALB/c mice at up to 300 mg/kg administered orally once daily for 3 days. Furthermore, TAM16 had no observable inhibitory activity against most of the major CYP isoforms (Table S6). hERG inhibition activity of TAM16 was evaluated by thallium (Tl+) flux assay (Schmalhofer et al., 2010) displaying an IC50 of 21 μM. Additional off-target activity screening against a broad panel of therapeutically relevant enzymes and receptors did not reveal any significant issues related to human safety. Moreover, in a two-strain Ames fluctuation assay for genotoxicity, the compound did not exhibit mutagenic potential in the two strains with or without S9.
For an antitubercular compound to be effective, it is essential that it achieves high exposure in the infected tissue consisting of macrophages and other immune cells in the human host. Cellular uptake studies using the cell line THP-1 as an in vitro macrophage model (Stokes and Doxsee, 1999) showed that, on average, cells maintained high (>70× MIC) concentrations of TAM16 (≥6.5 μM when treated with 10 μM compound) for up to 24 hr, indicating that the compound is freely permeable into the macrophage cytosol and can achieve high exposure. Indeed, TAM16 showed good activity in Mtb infected THP-1 cells.
Although TAM16 contains a Mannich substructure (C5OH C4 benzylamine scaffold), no adducts of TAM16 were formed in mouse plasma and HLMs upon incubation with glutathione and methoxylamine (Johansson et al., 2009) (Figures S3D and S3E). Moreover, TAM16 was stable after 4 weeks of incubation in vitro in low-pH buffered solutions (pH 3–5) at room temperature. Thus, taken together, the stability data suggests that the Mannich substructure in TAM16 does not represent a significant liability, consistent with previously reported stability of the piperidine adducts of hydroxyindoles (Monti and Castillo, 1970).
TAM16 Efficacy in Murine TB Infection Models
We evaluated TAM16 for in vivo efficacy in mouse models of acute and chronic TB infection. The TAM16 dosing regimens used in these animal studies were based on the maximum tolerated dose and pharmacokinetic studies. We first tested the in vivo activity of TAM16 using a mouse model of acute TB (Dutta et al., 2014), representing a population of actively multiplying Mtb in the host. BALB/c mice lungs were implanted with a very high inoculum of Mtb (∼4.4 log10 bacilli), which multiplied to a peak lung burden of ∼7.7 log10 CFU (colony-forming unit) 14 days after infection. Treatment of the mice was initiated 2 weeks post-infection with once-daily oral dosing of TAM16 (200 mg/kg) or INH (10 mg/kg). After 2 weeks of treatment, the TAM16 treated group showed a significant reduction in the lung CFU to ∼6.8 log10, which was indistinguishable from INH treatment, which reduced the lung CFU counts to ∼6.7 log10 (p > 0.05). Contrarily, all of the untreated control mice were moribund 1 week after treatment initiation due to uncontrolled bacillary growth in the lungs and were euthanized in accordance with institutional animal care regulations (Figure 3A). Histopathology of the mice post-treatment showed that TAM16 prevented the development of characteristic lung lesions (Figure S5).
Figure 3.

Efficacy of TAM16 in Mouse Models of TB
(A) In vivo activity of TAM16 against acute TB infection in immunocompetent BALB/c mice. Data represent mean M. tuberculosis burden (log10 CFU) in the lungs of mice (n = 5 per time point) expressed as mean ± SD. Week 0 indicates CFU counts in the lungs at treatment initiation (2 weeks after infection). Drugs were administered via oral gavage 5 days/week for 2 weeks. The mice in the untreated group were moribund after 3 weeks of infection and were euthanized in accordance with institutional animal care regulations. ∗p < 0.05 by Dunnett’s multiple comparison tests, as compared to the untreated (vehicle-only) control group; ns, no statistical significance.
(B) Efficacy of TAM16 in reducing M. tuberculosis burden in chronically infected immunocompetent mice after 4 weeks of treatment. Treatment was initiated 27 days after infection, and drugs were administered once daily via oral gavage for 5 days/week for 4 weeks. Data show the bacterial loads (mean log10 CFU ± SD) in the lungs and spleen of the infected mice (n = 5 per group). ∗p < 0.05 and ∗∗∗p < 0.001 by pairwise multiple comparison procedures (Tukey test); ns, no statistical significance.
(C) Efficacy of TAM16 administered in combination with anti-TB drug rifampicin (RIF) in chronically infected BALB/c mice. Treatment was initiated 28 days after infection, and drugs were administered once daily via oral gavage (5 d/wk) for 4 and 8 weeks. Data represent mean M. tuberculosis burden (log10 CFU) in the lungs of the infected mice (n = 6 for vehicle-only control group and n = 7 for each treatment group) after 4 and 8 weeks of therapy (mean ± SEM). In combination studies, TAM16 and isoniazid (INH) were administered 1 hr following prior administration of RIF. Dotted horizontal line indicates the limit of detection.
Figure S5.

Effect of TAM16 Treatment on Mice in Acute BALB/c Model, Related to Figure 3A
(A) Lung gross pathology images from untreated control (vehicle only) and treated mice. Untreated mice were moribund 1 week after treatment initiation (3 weeks post-infection) and were euthanized in accordance with institutional animal care regulations. INH, isoniazid.
(B and C) Effect of drug treatment on mean lung weights and, (C) mean body weights in M. tuberculosis infected mice (n = 5 per time point per group) after 2 weeks of treatment. Mice were infected on Day −13 and treatment was initiated on D0 (2 weeks after infection). Graphs represent mean values ± SD.
We next tested the in vivo activity of TAM16 in a chronic TB infection model in which lungs of BALB/c mice were aerosol-infected with Mtb using low-dose inocula, and treatment was initiated 4 weeks later when a steady-state infection was established at ∼7 log10 CFU per lung. After 4 weeks of treatment, mice treated with a once-daily oral dose of TAM16 (300 mg/kg) for 5 days per week showed a significant reduction in lung CFU counts by ∼0.9 log10 compared with the untreated control mice (p = 0.01) (Figure 3B). TAM16 treatment also significantly reduced the spleen bacterial burdens by ∼2.2 log10 CFU relative to that of the untreated control group (p < 0.001). Mice receiving INH (25 mg/kg) for 4 weeks showed a decrease in CFU counts by ∼1.1 log10 and ∼2.5 log10 in lungs and spleen, respectively, which again was statistically indistinguishable from the activity observed with TAM16 (p > 0.05) (Figure 3B).
Because of the synergy that was observed between TAM16 and RIF in the in vitro assays, we evaluated the drug combination of TAM16 and RIF in a mouse model representing a chronic TB infection. BALB/c mice were infected with a low-dose aerosol, and treatment was initiated 4 weeks post-infection with the combination of TAM16 (200 mg/kg) and RIF (10 mg/kg) in an 8-week study, where the combination was dosed orally once daily for 5 days per week. After 4 weeks of treatment, the combination of TAM16 and RIF resulted in a dramatic 2.54 log10 CFU reduction in the bacterial load in the mice lungs when compared to that of the vehicle-only control (p < 0.001) (Figure 3C). We saw a further reduction in bacterial load when the combination was continued for an additional 4 weeks with an impressive overall reduction of ∼3.9 log10 CFU in lungs over 8 weeks treatment when compared to the untreated control (p < 0.001). This was similar to the gold standard combination of INH and RIF that showed an efficacy of ∼4.1 log10 CFU reduction (p > 0.05) (Figure 3C; Table S7). Moreover, the combination of TAM16 and RIF was significantly more active than RIF alone, with an additional decrease of ∼1.5 log10 CFU counts after 8 weeks of treatment (p < 0.001). TAM16 as a single agent showed efficacy similar to RIF alone (CFU reduction of ∼1.9 log10 and ∼2.4 log10 in mice lungs, respectively) after 8 weeks of treatment (p > 0.05). In this study, no overt toxicity was observed in treated mice. Companion PK studies revealed a mean maximal drug concentration in plasma (Cmax ± SD) of 6.21 ± 1.5 μg/mL and 6.30 ± 2.3 μg/mL with trough concentrations of 1.91 ± 1.1 μg/mL and 1.58 ± 0.8 μg/mL for TAM16 30 min and 24 hr following oral administration alone, or 1 hr following prior RIF administration when sampled at steady state (week 8 of dosing), respectively. Thus, taken together, the efficacy data indicates that TAM16 has potent in vivo activity equivalent to INH, which is the most bactericidal first-line TB drug in current use, and highlights its potential for further pre-clinical development.
Discussion
INH, an inhibitor of the mycolic acid synthesis, has been a mainstay of TB therapy for decades. Although, in general, INH is well tolerated, it has several limitations. Because it is a pro-drug whose activation requires an Mtb enzyme that is not essential, resistance arises frequently through loss-of-function mutations in the activator KatG. Thus, a compound that targets mycolic acid synthesis and is not a pro-drug, such as the Pks13-targeting TAM16 developed in this study, should prove to be a better alternative to INH. Indeed, the frequency of resistance is about 100-fold lower for TAM16 as compared to INH. Though several other such compounds, such as InhA inhibitors, that do not require activation have been described (Guardia et al., 2016, Manjunatha et al., 2015, Pan and Tonge, 2012), none have advanced to the point where they can easily be compared with INH in animal studies, unlike TAM16.
TAM16 is a novel benzofuran class lead molecule with excellent drug-like properties and favorable pharmacokinetic and safety profiles. It exhibits potent in vivo efficacy in both acute and chronic mouse TB models when administered orally as a single dose, which is a highly desirable feature for a new TB therapeutic as it simplifies dosing regimen, encouraging patient compliance. However, the primary advantages of TAM16 are the novel target and the fact that it is not a pro-drug. This unique mode of action means that there will be no pre-existing resistance in clinical strains. Our studies verify that TAM16 is potent against multidrug-resistant and extensively drug-resistant Mtb strains.
One of the advantages of drugs that target mycobacterial cell wall biosynthesis is their potential to combine with other antibiotics to increase efficacy by improving their penetration. This makes compounds such as TAM16, which probably selectively kills the most rapidly growing bacteria in the population, producing rather rapid clearance initially with a more modest effect later in infection, very useful parts of a combination drug regimen. TAM16 combines well with RIF, a drug that kills different subpopulations of bacteria, suggesting that it is also likely to synergize with other drugs that have different modes of action. Indeed, TAM16 also showed synergistic activity in combination with other TB drugs like ATP synthesis inhibitor TMC207 and protein synthesis inhibitor streptomycin. Thus, TAM16 has suitable attributes for inclusion into either the current or any newly developed combination therapy for TB treatment. Overall, the results presented here demonstrate that TAM16 represents a promising candidate as a replacement of INH and validate Pks13 as a drug target in Mtb.
Structure-guided drug development has proven to be a powerful approach to producing new agents but has had limited application in the process of developing new antimycobacterial compounds (Lechartier et al., 2014, Zuniga et al., 2015). These findings re-emphasize the utility of structure-guided approaches for antimycobacterials when starting with compounds that have good whole cell and low toxicity. Whole-genome sequencing of resistant mutants for target identification can quickly provide a detailed understanding of the mechanism of action of new compounds with antibiotic activity as a path to turning these promising molecules into drugs.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| M. tuberculosis | Johns Hopkins University | H37Rv |
| BL21(DE3)pLysS | Novagen | Cat# 70236-3 |
| M. tuberculosis H37Rv | ATCC | ATCC 27294 |
| Clinical M. tuberculosis isolates, see Table S5 | Stellenbosch University: in-house culture bank | N/A |
| Clinical M. tuberculosis isolates, see Table S5 | Strain Collection at Mycobacteriology Laboratory of the Institute of Medical Microbiology, University of Zurich | N/A |
| M. tuberculosis mc2-7000 | Texas A&M University: in-house frozen stock (Sambandamurthy et al., 2006) | N/A |
| Mycobacterium tuberculosis Erdman | Trudeau Mycobacterial Culture Collection | TMC#107 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat# P8139 |
| D-Pantothenic acid | Sigma-Aldrich | Cat# P5155 |
| Resazurin | Sigma-Aldrich | Cat# R7017 |
| 4-Methylumbelliferyl heptanoate (4-MUH) | Sigma-Aldrich | Cat# M2514 |
| Ammonium sulfate | Sigma-Aldrich | Cat# A4418 |
| Polypropylene glycol P-400 | Sigma-Aldrich | Cat# 81350 |
| Fomblin | Sigma-Aldrich | Cat# 317950 |
| Polyethylene glycol 400 (PEG 400) | Hampton Research | Cat# HR2-603 |
| BTC-AM dye | ThermoFisher Scientific | Cat# B6791 |
| Uridine di-phosphoglucuronic acid (UDPGA) | Sigma-Aldrich | Cat# U6751 |
| β-Nicotinamide adenine dinucleotide 2′-phosphate reduced (NADPH) | Sigma-Aldrich | Cat# N1630 |
| β-Nicotinamide adenine dinucleotide (NADP) | Sigma-Aldrich | Cat# N3886 |
| Propranolol | Sigma-Aldrich | Cat# PO884 |
| Glucose-6-phosphate Dehydrogenase (from Baker’s yeast S. cerevisiae) | Sigma-Aldrich | Cat# G7877 |
| Ethoxyresorufin (ER) | Sigma-Aldrich | Cat# E3763 |
| 3-Cyano-7-Ethoxycoumarin (CEC) | Sigma-Aldrich | Cat# UC455 |
| Miconazole nitrate | Sigma-Aldrich | Cat# M3512 |
| D-Glucose-6-phosphate sodium salt | Sigma-Aldrich | Cat# G7879 |
| 7-methoxy-4-(trifluoromethyl)-coumarin (MFC) | Cypex | Cat# CYP517 |
| 7-methoxy-4-(aminomethyl)-coumarin (MAMC) | Cypex | Cat# CYP515 |
| Diethoxyfluorescein (DEF) | Cypex | Cat# CYP531 |
| 7-Benzyloxyquinoline (7BQ) | Cypex | Cat# CYP512 |
| Benzoquinone | TCI America | Cat# B0089 |
| ethyl 3-oxo-3-phenylpropanoate | Combi-Blocks | Cat# QA-0159 |
| ethyl 3-(4-methoxyphenyl)-3-oxopropanoate | TCI America | Cat# M1380 |
| Piperidine | Sigma-Aldrich | Cat# 411027 |
| Methylamine solution | Sigma-Aldrich | Cat# 395056 |
| Oxalyl chloride | Sigma-Aldrich | Cat# 221015 |
| Copper(II) trifluoromethanesulfonate | Sigma-Aldrich | Cat# 283673 |
| 2,5-Dihydroxybenzaldehyde | Combi-Blocks | Cat# OR-1145 |
| 3-methoxy-5-methylphenol | TCI America | Cat# M0895 |
| Palladium(II) acetate | Sigma-Aldrich | Cat# 205869 |
| Boron tribromide | Sigma-Aldrich | Cat# 230367 |
| Trifluoromethanesulfonic anhydride | Sigma-Aldrich | Cat# 176176 |
| Diethyl azodicarboxylate solution | Sigma-Aldrich | Cat# 563110 |
| Phenylmagnesium bromide | Sigma-Aldrich | Cat# 331376 |
| ethyl 5-hydroxy-4-[(4-methyl-1-piperidinyl)methyl]-2-phenyl-1-benzofuran-3-carboxylate (TAM1) | ChemBridge Corporation | ID# 6238794 |
| ethyl 5-hydroxy-4-[(3-methyl-1-piperidinyl)methyl]-2-phenyl-1-benzofuran-3-carboxylate (TAM2) | ChemBridge Corporation | ID# 6241075 |
| ethyl 5-hydroxy-2-phenyl-4-(1-pyrrolidinylmethyl)-1-benzofuran-3-carboxylate hydrochloride (TAM3) | ChemBridge Corporation | ID# 6238866 |
| ethyl 4-(1-azepanylmethyl)-5-hydroxy-2-phenyl-1-benzofuran-3-carboxylate hydrochloride (TAM4) | ChemBridge Corporation | ID# 6240697 |
| ethyl 5-hydroxy-4-(4-morpholinylmethyl)-2-phenyl-1-benzofuran-3-carboxylate (TAM5) | ChemBridge Corporation | ID# 6240924 |
| ethyl 4-[(dimethylamino)methyl]-5-hydroxy-2-phenyl-1-benzofuran-3-carboxylate (TAM6) | ChemBridge Corporation | ID# 5169001 |
| Ethyl 5-hydroxy-2-phenylbenzofuran-3-carboxylate (TAM7) | This paper | N/A |
| Ethyl 4-benzyl-5-hydroxy-2-phenylbenzofuran-3-carboxylate (TAM8) | This paper | N/A |
| Ethyl 5-hydroxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (TAM9) | This paper | N/A |
| 5-hydroxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylic acid (TAM10) | This paper | N/A |
| Ethyl 4-(cyclohexylmethyl)-5-hydroxy-2-phenylbenzofuran-3-carboxylate (TAM11) | This paper | N/A |
| 5-hydroxy-N-methyl-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (TAM12) | This paper | N/A |
| Ethyl 5-hydroxy-2-(4-hydroxyphenyl)-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (TAM13) | This paper | N/A |
| Ethyl 5-methoxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (TAM14) | This paper | N/A |
| Ethyl 2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (TAM15) | This paper | N/A |
| 5-hydroxy-2-(4-hydroxyphenyl)-N-methyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (TAM16) | This paper | N/A |
| Ethyl 5-hydroxy-2-(6-hydroxypyridin-3-yl)-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (TAM17) | This paper | N/A |
| 2-(3-fluoro-4-hydroxyphenyl)-5-hydroxy-N-methyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (TAM18) | This paper | N/A |
| 6-hydroxy-2-(4-hydroxyphenyl)-N-methyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (TAM19) | This paper | N/A |
| Ethyl 6-hydroxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (TAM20) | This paper | N/A |
| Ethyl 5-hydroxy-4-((3-(hydroxymethyl)piperidin-1-yl)methyl)-2-phenylbenzofuran-3-carboxylate (TAM21) | This paper | N/A |
| Ethyl 5-hydroxy-4-((2-(hydroxymethyl)piperidin-1-yl)methyl)-2-phenylbenzofuran-3-carboxylate (TAM22) | This paper | N/A |
| Ethyl 5-hydroxy-4-((3-(hydroxymethyl)piperidin-1-yl)methyl)-2-(4-hydroxyphenyl)benzofuran-3-carboxylate (TAM23) | This paper | N/A |
| 5-Hydroxy-4-((3-(hydroxymethyl)piperidin-1-yl)methyl)-N-methyl-2-phenylbenzofuran-3-carboxamide (TAM24) | This paper | N/A |
| Critical Commercial Assays | ||
| PCR Mycoplasma Test kit | Life Technologies | Cat# 409010 |
| QuikChange II Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200523 |
| Innovative Grade US Origin Mouse Plasma CD-1 | Innovative Research | Cat# IGMS-N |
| Mouse Microsomes (CD-1) | Life Technologies | Cat# MSMC-PL |
| Human liver microsomes (HLM) - Female CD1 | Xenotech | Lot# 910255 |
| Human CYP1A2 + human CYP reductase | Cypex | Cat# CYP/EZ001 |
| Human CYP2C9 + human CYP reductase + purified human cytochrome b5 | Cypex | Cat# CYP/EZ037 |
| Human CYP2C19 + human CYP reductase | Cypex | Cat# CYP/EZ008 |
| Human CYP2D6 + human CYP reductase | Cypex | Cat# CYP/EZ007 |
| Human CYP3A4 + human CYP reductase + purified human cytochrome b5 | Cypex | Cat# CYP/EZ005 |
| Membrane protein (control) | Cypex | Cat# CYP/EZ003 |
| Safety Profiling | AbbVie, IL, USA | N/A |
| Deposited Data | ||
| Pks13-TE domain (apo) structure | This paper | PDB: 5V3W |
| Pks13-TE:TAM1 complex structure | This paper | PDB: 5V3X |
| Pks13-TE:TAM16 complex structure | This paper | PDB: 5V3Y |
| Pks13-TE:D1607N mutant structure | This paper | PDB: 5V3Z |
| Pks13-TE:TAM6 complex structure | This paper | PDB: 5V40 |
| Pks13-TE:TAM5 complex structure | This paper | PDB: 5V41 |
| Pks13-TE:TAM3 complex structure | This paper | PDB: 5V42 |
| Experimental Models: Cell Lines | ||
| Human Dermal Fibroblast | ATCC | ATCC PCS-201-010 |
| THP-1 cells | ATCC | ATCC TIB-202 |
| hERG T-REx-CHO Cell line | ThermoFisher Scientific | Cat# K1237 |
| Experimental Models: Organisms/Strains | ||
| Mouse: ND4 Swiss Webster outbred | Envigo, Indianapolis, IN | Strain code: 032 |
| Mouse: BALB/c AnNCrl | Charles River laboratories, Wilmington, MA | Strain code: 028 |
| Oligonucleotides | ||
| LIC Primer: wt Pks13-TE Forward: TACTTCCAATCCAATGCCCAGATCGATGGGTTCGTCCGCAC | This paper | N/A |
| LIC Primer: wt Pks13-TE Reverse: TTATCCACTTCCAATGTTATCACTGCTTGCCTACCTCACTTGTTCG | This paper | N/A |
| Primer: Site directed mutagenesis Pks13-TE:D1607N Forward: GAGGAGCTCGACAACGAGGGCCAGG | This paper | N/A |
| Primer: Site directed mutagenesis Pks13-TE:D1607N Reverse: CCTGGCCCTCGTTGTCGAGCTCCTC | This paper | N/A |
| Primer: Site directed mutagenesis Pks13-TE:D1644G Forward: CCGGGCGATCGGCACCGCCCAGA | This paper | N/A |
| Primer: Site directed mutagenesis Pks13-TE:D1644G Reverse: TCTGGGCGGTGCCGATCGCCCGG | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pMCSG-19b | Midwest Center for Structural Genomics (http://bioinformatics.anl.gov/mcsg/technologies/vectors.html) | N/A |
| Pks13-TE-pMCSG-19b | This paper | N/A |
| Pks13-TE:D1607N_pMCSG-19b | This paper | N/A |
| Pks13-TE:D1644G_pMCSG-19b | This paper | N/A |
| Software and Algorithms | ||
| Masslynx 4.1 | Waters | N/A |
| MetaboLynx | Waters | N/A |
| HKL2000 | Otwinowski and Minor, 1997 | N/A |
| CCP4 suite | http://www.ccp4.ac.uk/ | N/A |
| Phenix Suite | Adams et al., 2010 | N/A |
| UCSF Chimera | Pettersen et al., 2004 | N/A |
| Coot | Emsley and Cowtan, 2004 | N/A |
| PyMOL 1.4 | Schrodinger, LLC | N/A |
| Prism 4.0, 5.0 | GraphPad Software | N/A |
| IN Cell Investigator image analysis software for IN CELL ANALYZER 2000 | GE Life Sciences | N/A |
| CompuSyn software | http://www.combosyn.com/ | N/A |
| SigmaPlot 11.0 | Jandel Corporation | N/A |
| CAVER 3.0 PyMOL plugin | http://www.caver.cz | N/A |
| Molsoft ICM-Pro 3.8 | http://www.molsoft.com | N/A |
| Other | ||
| Rapid Equilibrium Dialysis (RED) Device Single Use Plate with Inserts, 8K MWCO | ThermoFisher Scientific | Cat# 90006 |
| HiLoad 16/600 Superdex 200 pg Column | GE Life Sciences | Cat# 28989335 |
| Rifampin | Sigma-Aldrich | Cat# R3501-5G |
| Rifampicin | Sigma-Aldrich | Cat# R3502 |
| Ethambutol | Sigma-Aldrich | Cat# E4630 |
| Streptomycin | Sigma-Aldrich | Cat# S6501 |
| Isoniazid | Sigma-Aldrich | Cat# I3377 |
| Verapamil | Sigma-Aldrich | Cat# V4629 |
| Penicillin-Streptomycin (10,000 U/mL | Life technologies | Cat# 15140122 |
| DMEM, high glucose, GlutaMAX Supplement | Life technologies | Cat# 10566-016 |
| Fetal Bovine Serum | Corning | Cat# 35-015-CV |
| Gentamycin | GIBCO | Cat# 15710064 |
| Carbenicillin | Caisson Labs | Cat# C033-100GM |
| Chloramphenicol | GoldBio | Cat# C-105-100 |
| Warfarin PESTANAL, analytical standard | Sigma-Aldrich | Cat# 45706 |
| Tyloxapol | Sigma-Aldrich | Cat# T8671 |
| BBL Middlebrook 7H11 Selective Agar | Becton Dickinson | Cat# 8801671 |
| Difco Middlebrook 7H10 agar | Becton Dickinson | Cat# 262710 |
| Difco Middlebrook 7H9 broth | Becton Dickinson | Cat# 271310 |
| Middlebrook OADC enrichment | Becton Dickinson | Cat# 212351/212240 |
| BACTEC MGIT 960 tubes (7 ml) | Becton Dickinson | Cat# 245122 |
| Bedaquiline (TMC207) | Advanced ChemBlocks Inc | Cat# 10288, Lot# 10355 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James C. Sacchettini (sacchett@tamu.edu). Texas A&M University requires that a material transfer agreement (MTA) be signed for transfer of materials. Small amounts of compounds synthesized for this study will be made available as reference standards when a sufficient supply is available.
Experimental Model and Subject Details
Animal studies
All animal experiments described in this manuscript followed protocols approved by the respective Institutional Animal Care and Use Committee’s at Colorado State University, Johns Hopkins University and Texas A&M University.
Pharmacokinetic studies in mice
Swiss-Webster female mice (∼20 g each) (Envigo, Indianapolis, IN) were used in the PK study. The formulation of TAM16 was prepared in 90% canola oil and 10% DMSO. Mice were dosed at 100 mg/kg by oral gavage at 0 hr and 4 hr time points in 0.1 mL aliquots to yield a final dose per day of 200 mg/kg. Three mice were used per time point. The mice were anesthetized with ketamine-xyalzine and ∼0.1 mL of blood was drawn for survival bleeds at 0.5, 1.5, 5 hr, time points, and ∼0.3 mL of blood was drawn for terminal bleeds at 4, 8 and 12 hr time points. Blood was collected from the brachial region. The blood samples were centrifuged (5,000 x g, 15 min) for plasma separation. 500 μL methanol containing 0.1% formic acid was then added to a 50 μL serum aliquot to precipitate the protein. The methanol precipitation step was performed twice to ensure maximum extraction of TAM16 from the serum. After removing the precipitate by centrifugation, the clear supernatant was evaporated to dryness. The dry samples were then reconstituted with 50 μL methanol and subjected to LC-MS analysis on a micrOTOF-Q II mass spectrometer (Bruker Daltonics Inc.) hyphenated with an Agilent 1200 Infinity series HPLC with temperature controlled autosampler and photodiode array detector. 2.1 X 100 mm Atlantis T3, 5 μm C18 HPLC column (Waters) was used in the analysis at a flow rate of 0.5 ml.min-1. The mobile phase consisted of water with 0.1% formic acid as Solvent line A and acetonitrile with 0.1% formic acid as solvent line B. The gradient conditions were maintained as follows: 90% A, 10% B to 100% B in 8 min; 100% B held for 4 min; 100% B back to 90% A, 10% B in 2 min; 90% A, 10% B held for 3 min. The injection volume of the analyte was 10 μL and MS was operated in the positive mode with electrospray ionization at source. Mass spectra were monitored in the range of (m/z) 50 to (m/z) 1000.
Efficacy studies in mice
BALB/c acute TB infection mouse model
For this study, mice (Charles River Labs, Wilmington, MA) were maintained under specific pathogen-free conditions and fed water and chow ad libitum. For drug preparation, isoniazid (INH, Sigma) was dissolved in sterile distilled water. A dosing solution of 1 mg/ml was prepared weekly and kept at 4°C. TAM16 was suspended in vehicle of canola oil and 10% DMSO and stored at 4°C for up to 1 week. A total of 70 female BALB/c mice (7-8 weeks-old) were aerosol-infected with M. tuberculosis H37Rv using the Inhalation Exposure System (Glas-Col Inc., Terre Haute, IN) calibrated to deliver ∼104 (high-inoculum) colony-forming units (CFU) per mouse lung. After aerosol infection, mice were blindly randomized into treatment groups: no drug (negative control) and drug treated (positive control), and were treated daily via oral gavage administered in 0.2 mL 5 days per week for 2 weeks at 200 mg/kg once daily. Five mice from each group were sacrificed on the day after infection, on the day of treatment initiation (Day 0), and on day 14 after treatment to determine the numbers of CFU implanted in the lungs, pretreatment baseline CFU counts and the CFU counts after 14 days of treatment, respectively. Animal body weights were recorded at the time of sacrifice. The lungs of sacrificed mice were homogenized in 2.5 mL PBS. Lung homogenates were plated on selective 7H11 plates (containing cycloheximide (50 μg/ml), carbenicillin (100 μg/ml), polymyxin B (200 U/ml), and trimethoprim (20 μg/ml)) for CFU enumeration. Statistical analysis was done on CFU data derived from 3 to 5 mice per group. Log-transformed CFU were used to calculate means and standard deviations. Comparisons of data among experimental groups were performed by Student’s t test. Group means were compared by one-way analysis of variance (ANOVA) with Dunnett’s post-test (Day 0 or untreated controls versus treatment groups) or Bonferroni comparison (all treatment groups), using GraphPad Prism version 4 (GraphPad, San Diego, CA). Values of p < 0.05 were considered to be statistically significant.
BALB/c chronic TB infection mouse model
In this study, 6- to 8-week-old female specific-pathogen-free immunocompetent BALB/c mice (Charles River, Wilmington, MA) were infected via a low-dose aerosol exposure to M. tuberculosis Erdman in a Middlebrook aerosol generation device (Glas-Col Inc., Terre Haute, IN). One day post aerosol infection, three mice were sacrificed to verify the uptake of an average of about 100 CFU of bacteria per mouse. Following infection, the mice were randomly divided into treatment groups. Negative control mice remained untreated. Positive control mice received INH (at 25 mg/kg of body weight). Each group consisted of five mice at each time point. Treatment was started day 27 post aerosol and continued for 4 weeks. Five infected mice were killed at the start of treatment as pretreatment controls. Drugs were administered in canola oil, 5 days per week by oral gavage. To determine drug efficacies, mice from each treatment group were sacrificed after 4 weeks of treatment. The mice were humanely euthanized by CO2 inhalation. The spleens and left lung lobes were aseptically removed and disrupted in a tissue homogenizer. The number of viable organisms was determined by serial dilution of the homogenates on nutrient Middlebrook 7H11 agar plates (GIBCO BRL, Gaithersburg, MD). The plates were incubated at 37°C in ambient air for 4 weeks prior to determine the total number of culturable mycobacteria per organ. For statistical analysis, the number of culturable mycobacteria were converted to logarithms, which were then evaluated by a one-way analysis of variance, followed by a multiple-comparison analysis of variance by a one-way Tukey test (SigmaPlot software program). Differences were considered significant at the 95% level of confidence.
Combination studies in mouse model of chronic TB
Six- to 8-week-old female specific-pathogen-free immunocompetent BALB/c (Charles River, Wilmington, MA) were infected via a low-dose aerosol exposure to M. tuberculosis Erdman (∼50-100 bacilli/mouse) using the Glas-Col Inhalation Exposure System. One day post aerosol infection, five mice were sacrificed to determine bacterial uptake. Following infection, the mice were randomly divided into treatment groups of six mice each. Negative control mice remained untreated. At Day 28 post-infection 6 mice were sacrificed to determine bacterial load in the lungs at the start of therapy. Therapy administered via oral gavage, was started day 28 post-aerosol infection and continued for 4 and 8 weeks. Drugs were administered by gavage daily, for 5 days a week (Mon-Fri), in a volume of 200 μl/animal/drug. For animals receiving two drugs, RIF was dosed first and then at least 1 hr later, the second drug was administered. To determine drug efficacies, six mice from each group (untreated, vehicle, TAM16, RIF, INH and combination treatment mice) were sacrificed after 4 and 8 weeks of treatment following a three day drug washout period after the last day of dosing. For the companion plasma PK studies (plasma collected at both Cmax/Cmin), test bleeds were performed via the submandibular route using a GoldenRod lancet during last week of treatment (week 8) from n = 3 mice from TAM16 only and TAM16+RIF treatment groups. For statistical analysis, the viable CFU counts were converted to logarithms, which were then evaluated by a one-way ANOVA followed by a pairwise multiple comparison using the Dunnett’s test (SigmaPlot software program). Differences were considered significant at the 95% level of confidence.
Cell lines and bacterial strains
Human Dermal Fibroblast (HDF) cells were purchased from ATCC (catalog number PCS-201-010) and were cultured in DMEM (Lonza) media supplemented with 10% fetal bovine serum (Lonza) and penicillin/streptomycin (Lonza). THP-1 cells were purchased from ATCC (catalog number TIB-202) and were differentiated with PMA (100 nM) for 3 days prior to infection studies. For assaying hERG activity, an inducible hERG T-REx™-CHO Cell line was purchased by AbbVie from ThermoFisher (catalog number K1237). Cell lines obtained from ATCC were tested for mycoplasma contamination by PCR Mycoplasma test kit (MD Bioproducts). Purchased cell lines were not further authenticated.
BL21(DE3)pLysS competent E. coli cells were from Novagen (catalog number 70236-3). M. tuberculosis mc27000 (Sambandamurthy et al., 2006) was obtained from in-house frozen stock (−80°C) at Texas A&M University. To obtain starter culture, a 1 mL aliquot of frozen cells was thawed and cultured in 7H9 media supplemented with OADC (Middlebrook), 0.05% Tyloxapol (Sigma), and 25 μg/ml pantothenate for 6-7 days to an OD600 of 1–1.5. Clinical M. tuberculosis isolates were selected from the culture collections at the Department of Molecular Biology and Human Genetics, Stellenbosch University, South Africa, and at the Mycobacteriology Laboratory of the Institute of Medical Microbiology, University of Zurich, Switzerland (for a list of strains and mutations see Table S5). The identity of the isolates was determined by IS6110 restriction fragment length polymorphism (van Embden et al., 1993) and their phylogenetic lineages were assigned by spoligotyping (Kamerbeek et al., 1997, Streicher et al., 2007). Whole genome sequencing showed that all of the drug-susceptible isolates lacked high confidence resistant markers which suggests susceptibility against the conventional first- and second- line drugs.
Method Details
Cloning and overexpression of Mtb Pks13 TE domain constructs
The TE domain construct corresponding to the predicted TE domain in Mtb pks13 gene (Rv3800c) was made by PCR from the Mtb H37Rv genomic DNA as the template (forward primer: 5′ – TAC TTC CAA TCC AAT GCC CAG ATC GAT GGG TTC GTC CGC AC – 3′, reverse primer: 5′ – TTA TCC ACT TCC AAT GTT ATC ACT GCT TGC CTA CCT CAC TTG TTC G – 3′). The amplified DNA fragments were incorporated into the pMCSG-19b vector by ligation independent cloning (LIC) to yield TEV protease cleavable N-terminal His6-tagged TE domain construct (Donnelly et al., 2006). The Pks13-TE-pMCSG-19b vector was transformed into E. coli BL21(DE3)pLysS cells (Novagen) and the transformed cells were grown at 37°C in LB media containing carbenicillin (100 μg/ml) and chloramphenicol (34 μg/ml) to an OD600 of 0.6. Expression of TE construct was induced with 0.5 mM IPTG, and cells were harvested after 16 hr of growth at 20°C. The D1607N and D1644G mutants of Pks13 TE domain were constructed using the QuikChange II site-directed mutagenesis kit (Agilent Technologies). The mutations were confirmed by DNA sequencing. Mutant plasmids were transformed into E. coli BL21(DE3)pLysS cells, and mutant proteins were expressed by induction with 0.5 mM IPTG at 20°C for 18 hr.
Purification of Pks13 TE domain
The harvested cells were resuspended in the lysis buffer (50 mM Tris-HCl pH 8.0, 0.5 M NaCl, 10% (v/v) glycerol, 1 mM β-mercaptoethanol (BME) and DNase) and lysed by French press. The resulting cell extract was clarified by centrifugation (15,000 x g) for 1 hr at 4°C. The cleared supernatant was loaded onto a Ni-affinity column and the His-tagged TE domain constructs were eluted with a linear gradient of 10-250 mM imidazole in 20 mM Tris-HCl, pH 8.0 and 0.5 M NaCl. The peak fractions were pooled and the His-tag was cleaved by overnight incubation with TEV protease in dialysis buffer (20 mM Tris-HCl pH 8.0, 10% (v/v) glycerol and 1 mM DTT). The TEV cleaved protein was passed through Ni-column to remove any uncleaved His-tagged protein using 20 mM Tris-HCl (pH 8.0) with 100 mM NaCl and 1 mM BME. His-tag cleaved protein eluted in the flow-through and was concentrated for loading onto a Superdex-200 gel filtration column (GE Healthcare). The 283 residue long TE domain starting from residue 1451 in full length Pks13 (referred to as Pks13-TE in this paper) eluted under a single peak as a monomer (∼32 kDa) from the gel filtration column and was > 95% pure as observed by SDS-PAGE. The purified protein was concentrated to 20-25 mg/ml, flash-frozen and stored at −80°C. The TE domain mutants were purified using the same protocol as for the wild-type Pks13-TE domain constructs. Both the mutants and the wt Pks13-TE domain protein constructs have the amino acids SNA from the TEV cleavage site appended to the N terminus.
Crystallization and soaking with ligands
Initial screening for crystallization conditions for the Pks13-TE domain was done using sitting drop method using 1 μL of purified protein (15-20 mg/ml) and 1 μL of crystallization buffer from the well solution. The Pks13-TE crystals were obtained in crystallization buffer containing 0.1 M Tris-HCl, pH 8.5 and 2.0-1.8 M ammonium sulfate as precipitant. Crystals were further optimized by using polypropylene glycol P-400 as an additive at 2%–5% (v/v) in the original condition. To obtain Pks13-TE-inhibitor complex crystals, soaking of the inhibitors was done by transferring apo-Pks13-TE crystals into a drop consisting of 0.1 M Tris-HCl, pH 8.5 and 2-2.2 M ammonium sulfate with 1-2.5 mM inhibitor added from a DMSO stock keeping the final DMSO concentration at < 5%, and incubated at 18°C for 4-48 hr. Crystals of the Pks13-TE:D1607N mutant were obtained by sitting drop method at 18°C. The crystallization drops contained an equal volume of the protein solution (15-20 mg/ml) and mother liquor (0.1 M HEPES, pH 7.5, 2%–4% (v/v) PEG 400, and 1.8-2 M ammonium sulfate), and the diffraction quality crystals were obtained within 2 weeks.
Data collection and processing
For diffraction data collection the crystals were cryo-protected using Fomblin (Sigma) and flash frozen in liquid nitrogen. High resolution data was collected at wavelengths of 0.98 – 1.03 Å on the beamlines 19-ID and 23-ID at the Advanced Photon Source (APS) of the Argonne National Laboratory, Chicago, IL, USA. All the datasets were processed and scaled with HKL2000 (Otwinowski and Minor, 1997). Analysis of the integrated and scaled data by Xprep (Sheldrick, 2008) indicated that Pks13-TE crystallized in P21212 space group. Solvent content analysis in CCP4 suite indicated the presence of two molecules (VM 2.16, VS 43.2%) in the asymmetric unit (Matthews, 1968).
Determination of Pks13-TE structures and model refinement
The structure of the Pks13-TE domain was solved by molecular replacement method (MR) using E. coli EntF (PDB: 3tej) structure, as search model. A single MR solution was obtained using Phenix AutoMR (Adams et al., 2010) which was input into the AutoBuild wizard to generate the initial model for apo-Pks13-TE. The initial model was improved by further manual rebuilding in COOT (Emsley and Cowtan, 2004). The final model was obtained after iterative cycles of model building and Phenix refinement with simulated annealing yielding a 1.72 Å resolution apo-Pks13-TE model with Rcryst of 16.9% and an Rfree of 20.1% with good stereochemistry (Table S2). The final refined apo-model has two chains, designated A and B, a fragment of additive PPG P400 and 471 water molecules in the asymmetric unit. The crystal structures Pks13-TE-inhibitor complex structures, as well as the D1607N mutant structures were refined with simulated annealing (start temperature 5000 K, Phenix). Inspection of electron density maps showed clear |Fo-Fc| positive difference density for the ligands which were fit into the density using Ligandfit routine in Phenix (Terwilliger et al., 2006). The ligand model and geometry restraint files were created in ELBOW BUILDER of the Phenix suite (Moriarty et al., 2009). Iterative cycles of model building and NCS-restrained maximum likelihood refinement with simulated annealing yielded high quality models for Pks13-TE-inhibitor complexes (Tables S2 and S3). In all of the structures > 98% of residues are placed in the favored region of the Ramachandran plot (MolProbity, Chen et al., 2010). Figures of the structures were made with UCSF Chimera package (Pettersen et al., 2004) and PyMOL Molecular Graphics System version 1.4.1 (Schrodinger, LLC). Structural analysis of Apo Pks13-TE for the identification of tunnels and channels was done using CAVER 3.0 PyMol plugin (Chovancova et al., 2012). Electrostatic surface potentials were calculated using APBS (Baker et al., 2001) and displayed using the APBS plugin for PyMOL. Atomic coordinates and structure factors for the reported crystal structures (Tables S2 and S3) have been deposited with the Protein Data Bank under accession codes: PDB: 5V3W (Apo Pks13-TE), PDB: 5V3X (Pks13-TE:TAM1), PDB: 5V3Y (Pks13-TE:TAM16), PDB: 5V3Z (Pks13-TE(D1607N)), PDB: 5V40 (Pks13-TE:TAM6), PDB: 5V41 (Pks13-TE:TAM5), and PDB: 5V42 (Pks13-TE:TAM3).
Enzyme assay
Activity of Pks13-TE was assessed using 4-methylumbelliferyl heptanoate (4-MUH, Sigma) as a fluorogenic substrate in a 96-well plate format (Richardson and Smith, 2007). To make initial velocity measurements, Pks13-TE (1 μM) in 0.1 M Tris-HCl, pH 7 buffer was incubated with different concentrations of 4-MUH (2-150 μM in DMSO) in a 100 μL reaction volume, and the fluorescence of the hydrolyzed product 4-methylumbelliferone was read (excitation at 355 nm and emission at 460 nm) (PolarStar Omega plate reader BMG Labtech) at 5-10 min intervals over 80-120 min. The reaction rate was observed to be linear in the measured range. 4-MUH in buffer alone was included as a control to quantify its background hydrolysis. Data points were plotted as an average of triplicates and each experiment was repeated twice independently. The initial velocity data was curve fit to Michaelis-Menten equation by nonlinear regression using Prism software (GraphPad) to determine the kinetic parameters Km and Vmax. The assay and data analysis for Pks13-TE mutants was done the same way as that for the wild-type protein with the 4-MUH concentration varying from 2 to 300 μM.
IC50 determination
To determine the potency of TAM1 and its analogs against wt Pks13-TE, the compounds were tested at concentrations ranging from 0.012 to 20 μM in a 96-well plate format. The reaction mix contained 0.1 μM Pks13-TE in 0.1 M Tris-HCl, pH 7 buffer with 1 μL of each dilution of the compound or DMSO in a total volume of 99 μL. The reaction was initiated by addition of 1 μL of 2 mM 4-MUH in DMSO (20 μM final concentration) to the reaction mix. Initial velocity data was obtained by monitoring increase in the fluorescence due to hydrolysis of the substrate using PolarStar Omega plate reader at 10 min intervals over 110 min. The data points were collected in triplicate and the averaged value was used to generate concentration-response plots for TAM1 and its analogs. The IC50 value for each compound was obtained by nonlinear regression curve fitting of a four-parameter variable slope equation to the dose-response data using Prism software. The IC50 values of TAM1 for Pks13-TE mutants were determined in the same way as that for wt Pks13-TE, using inhibitor concentration range of 0.04 to 40 μM.
Whole cell activity and cytotoxicity testing
Whole cell testing for determining MIC was done using Alamar blue assay in 96-well plates (Franzblau et al., 1998). Mtb mc2-7000 cells were grown to an OD600 of 1–1.5. The cells were then diluted into testing media (7H9 media with 0.2% dextrose, 0.085% NaCl, 0.05% Tyloxapol, and 25 μg/ml pantothenate) to an OD600 of 0.01 and dispensed into testing plates at 196 μL per well. Then the compounds were added (4 μL) as a 2-fold serial dilution in DMSO (2% DMSO final in each well). The test plates also had a DMSO only control and a Rifampicin control. The plates were incubated with shaking at 37°C for 6 days and then stained with resazurin (Sigma) for an additional 2 days at 37°C. After staining the fluorescence of reduced resazurin was read (λEx = 544 nm, λEm = 590 nm) using PolarStar Omega plate reader. The fluorescence data were plotted as percent growth inhibition against the compound concentration and curve fitting was done by nonlinear regression using Prism software. Minimum inhibitory concentration (MIC) values, defined as the concentration giving 50% growth inhibition, were determined from the fitted curves.
Compounds were tested for toxicity by the Human Dermal Fibroblast (HDF) cytotoxicity assay. The cells were cultured in DMEM (Lonza) media supplemented with 10% fetal bovine serum (Lonza) and penicillin/streptomycin (Lonza). For setting the cytotoxicity assay, compound stocks were serially diluted starting from the highest concentration of 100 μL in phosphate buffered saline (PBS) plus 10% DMSO. On the day of assay, HDF cells were trypsinized, counted and resuspended at a concentration of 64,000 cells/ml in the media. Cells were plated, overlaid with the compound serial dilutions and incubated at 37°C. After 48 hr, resazurin dye was added and the assay plates were cultured for another 24 hr. The next day the absorbance of the resazurin was measured on a microplate reader (BMG Labtech) to assess cell death.
THP-1 Drug Efficacy Assay
THP-1 monocytes were differentiated with PMA (100nM) for 3 days prior to infection and seeded at 40k cells/well in a 96-well dish coated with 0.1% gelatin. Mtb mc27000 cells constitutively expressing fluorescent reporter mCherry were incubated with the THP-1 for 2 hr, washed gently, and incubated with gentamycin (10 μg/mL) for 2 hr to kill extracellular Mtb mc27000. Cells were then washed and incubated with drug (1% DMSO final). Fluorescence was measured on days 0 and 5 post infection using a GE IN Cell Analyzer 2000 at 20x magnification and analyzed using the complementary GE software suite. Fold change in fluorescence was normalized to rifabutin (10 μM) as the positive control. All experiments were performed with technical triplicates and were repeated six times independently.
Isolation of resistant mutants and whole-genome sequencing
For the isolation of TAM16 resistant mutants, ∼109 Mtb mc27000 cells were plated onto replicate 7H10 plates containing either 0.9 μM (10x MIC) or 1.8 μM (20x MIC) of TAM16. In addition, serial dilutions of Mtb mc27000 culture were plated out on 7H10 plates containing no TAM16. Plates were screened for the appearance of resistant mutants after 3-4 weeks of incubation at 37°C, and the frequencies of resistance were determined by dividing the CFUs on TAM16 containing plates by the CFUs on TAM16-free plates. Genomic DNA from the mutants was extracted by CTAB-lysozyme method as described previously (Larsen et al., 2007) and subjected to whole-genome sequencing for identification of single nucleotide polymorphisms (Ioerger et al., 2013).
In vitro activity of compounds against clinical M. tuberculosis isolates
The Mycobacterial Growth Indicator Tube drug susceptibility testing system (MGIT 960 DST) was used to determine the MICs of TAM16 against drug-susceptible and drug-resistant clinical M. tuberculosis isolates with diverse genetic background.
MGIT DST at University of Zurich
Drug susceptibility was tested using the proportion method and MGIT equipped with EpiCenter TB eXiST as described previously (Springer et al., 2009). In brief, bacterial suspensions were prepared from MGIT subcultures. TAM16 was dissolved in DMSO and two-fold dilution series in DMSO were prepared. 0.08 mL of each drug dilution was inoculated into the MGIT vial (final DMSO concentration 1%) together with 0.5 mL of the bacterial suspension. For preparation of the drug-free growth control tube, the bacterial suspension was diluted 1:100 with sterile saline solution, and then 0.5 mL was inoculated into the MGIT vial together with 0.08 mL DMSO. Results were interpreted as follows: at the time the growth unit (GU) value for the drug-free control was > 400, the strain was categorized as susceptible when the GU of the drug-containing tube was < 100 and as resistant, if the GU was ≥ 100. The MIC of each strain was defined at the lowest drug concentration that was categorized as sensitive per the definition above. The epidemiological cut-off (ECOFF) value is the MIC value identifying the upper limit for the wild-type type-population.
MGIT DST at Stellenbosch University
Bacterial suspensions were prepared from frozen stock (−80°C) in MGIT 960 medium. These cultures were then sub-cultured and grown at 37°C for 2 additional days after the MGIT tube became positive before they were used as inocula (∼1 × 105 CFU/ml). The compounds were dissolved in 100% DMSO to obtain stock solutions of 1680 mM. Stock solutions were further diluted (1:20) to a concentration of 84 mM in 50% DMSO followed by serial 2-fold dilutions ranging from 84 mM to 1.3 mM. From each dilution, 0.1 mL was transferred into MGIT tubes containing 7.0 mL modified Middlebrook 7H9 broth base supplemented with 0.8 mL OADC. The tubes were then inoculated with 0.5 mL of the test organisms for final 2-fold drug concentrations ranging from 1.0 to 0.015 mM. A drug-free 1:100 diluted inoculum was included per drug set as control to indicate 1% growth according to the proportion method. Results were based on a threshold growth unit (GU) reading of 400 by the drug-free control at 37°C in MGIT 960. Drug-containing tubes with GU values of ≥ 100 at the time when the drug-free control reach a value of 400 were considered resistant and those with values < 100 were recorded as susceptible. The MIC was defined as the lowest drug concentration that inhibits growth of more than 99% of the bacterial inoculum.
In vitro synergy evaluation
TAM16 was evaluated in two-drug combination studies for interactions with INH, RIF and EMB in vitro against Mtb mc27000 by combination index (CI) method (Chou, 2006). Briefly, TAM16 was combined at a constant ratio of 1:3; 1:0.5 and 1:30 with INH, RIF and EMB, respectively, based on the ratio of the MICs of the individual drugs to TAM16 to obtain equipotency combination ratios, and a 2-fold dilution series from the mixtures was prepared in DMSO. The dilution series of each drug combination as well as the serial dilutions of individual drugs in the combination were then tested in the same 96-well assay plate to obtain dose response data. Fractional inhibition was calculated by dividing the fluorescence data from treatments by the fluorescence from the DMSO only control wells. The CI and dose-reduction index (DRI) were calculated from the dose response data using CompuSyn software (http://www.combosyn.com) for determining the mode of interaction with CI < 1, CI = 1 and CI > 1 indicating synergistic, additive, and antagonistic interactions, respectively.
Kinetic solubility determination
The procedure for determining kinetic solubility of Pks13-TE inhibitors was derived and modified from methods described previously (Alelyunas et al., 2009). A 10 mM stock solution of TAM16 was prepared in 100% DMSO. 5 μL of the stock solution was added to 495 μL of sodium phosphate buffer (pH 7.4) taken in the wells of a 2 mL volume deep 96-well polypropylene plate (USA Scientific) to get a final concentration of TAM16 of 100 μM and containing 1% final DMSO concentration. A small stir bar was placed in the well and the plate was left on a stir plate with constant stirring for 24 hr at 25°C. At the end of the 24 hr the plate was centrifuged at 1,000 x g to precipitate any un-dissolved material. The supernatant solution was diluted 10-fold in sodium phosphate buffer (pH 7.4) and an aliquot was analyzed on the LC-MS as described under PK studies. TAM16 dissolved in methanol at 10 μM concentration was injected into the LC-MS and used as a single point calibration to estimate the concentration of TAM16 in the aqueous solution.
Plasma protein binding
Protein binding of TAM16 in mice plasma was determined using a Rapid Equilibrium Dialysis (RED) kit (ThermoFisher Scientific) with LC-MS analysis. TAM16 (10 μg/mL) and mice plasma with 1% DMSO were added to one side of the single-use RED plate dialysis chamber having an 8 kD MW cutoff membrane. PBS was added to the other side of the membrane. The plate was sealed and left on a shaker at 250 rpm in a 37°C incubator. After 5 hr of dialysis a 50 μL aliquot was collected from both sides of the membrane. 50 μL PBS was added to the plasma sample and 50 μL of plasma was added to the PBS sample in order to minimize matrix effect. Then 200 μL acetonitrile containing 1 μg/ml warfarin (internal standard) was added to precipitate the protein. The sample was mixed well and centrifuged. The clear supernatant was separated from the precipitate and 200 μL water containing 0.1% formic acid was added to the supernatant. 10 μL of the sample was used for LC-MS analysis as described above. The experiment was performed in triplicate.
Calculation of plasma protein binding
Where,
Pl = Ratio of mass intensities of TAM16 and Warfarin (Internal Standard) determined on the plasma side of the membrane.
Bu = Ratio of mass intensities of TAM16 and Warfarin (Internal Standard) determined on the buffer (PBS) side of the membrane.
Microsomal stability assay
20 mg/mL stock solution of mouse microsome S9 fractions (Life Technologies) were diluted to a 0.55 mg/mL working stock in 50 mM potassium phosphate buffer (pH 7.4). 0.5 μM final concentration of the compound was used. Verapamil was used as a control drug molecule to validate the assay. The reaction was started by adding 1 mM (final concentration) of freshly prepared NADPH solution. Two negative controls were used; microsomes inactivated by heating at 55°C for 10 min in a water bath, and phosphate buffer in the place of NADPH. Aliquots were removed from this reaction mix at time intervals of 0, 3, 6, 9, 15, 30, 45 and 60 min. The aliquots were added to vials containing 37% acetonitrile, water and an internal standard (0.1 μg/mL warfarin). The mixture was centrifuged to remove precipitated protein and the clear supernatant solution was evaporated to dryness. The dry sample vials were reconstituted with 50 μL methanol and used for LC-MS analysis as described in PK studies section.
Calculation of Intrinsic Clearance
Ratio of the mass intensities of TAM16 and the internal standard (Warfarin) were then plotted to fit an exponential decay curve and the rate constant (k) was derived from the curve. Intrinsic clearance was then calculated using the following equation:
Where,
CLi = Intrinsic Clearance.
k = rate constant.
V = incubation volume.mg-1 protein added = 1 × 0.5−1 = 2 ml.mg-1 protein.
Microsomal protein yield is a standard used for all species = 52.5 mg protein.g-1 liver.
Physicochemical properties calculations
Physicochemical properties (clogP, logD, TPSA and pKa) were calculated using ChemAxon chemistry engine as implemented on the Collaborative Drug Discovery web portal (www.collaborativedrug.com) and are summarized in Table 3.
Glutathione (GSH) and methoxylamine (MA) trapping
Metabolic stability of TAM16 was assessed following incubations with human liver microsomes (HLMs) (20 mg/mL, supplied by Xenotech as a pooled batch of 50 donors). The incubations were performed in a medium containing 0.05 M phosphate buffer (pH 7.4) with TAM16 at a final concentration of 10 μM in the presence of 0.5 mg/mL microsomal protein, 5 mM GSH/MA and a NADPH regenerating system (including 2.3 mM Glucose-6-Phosphate, 1.8 mM NADP, and 0.5 U/mL glucose 6-phosphate dehydrogenase in 2% (w/v) sodium bicarbonate). Positive controls included Clozapine and 17α-ethynylestradiol (EE) for GSH trapping and Amodiaquine for MA trapping at a final concentration of 5 μM. A ‘no compound control’ replacing TAM16/positive controls with DMSO was also included. Samples prepared in 96-plates were incubated on a shaking water bath/incubator at 37°C for 60-90 min and after incubation, reactions were stopped by adding acetonitrile (analytical grade). The incubation mixtures were then centrifuged at 2800 rpm for 10 min at 4°C. A 150 μL aliquot of supernatant was added to 150 μL water before analysis by UPLC-MS/MS (Waters Acquity UPLC with DAD and Waters Xevo Q-TOF). Chromatography was performed on a Waters BEH C18 (50 × 2.1mm 1.7μm) column at a flow of 0.5 mL/min with an injection volume of 5 μL. Detection of metabolites was performed by analysis with Metabolynx XS software (Waters) and by manual data searching. Analysis of positive controls was used to ensure that the assay was functional. Metabolic stability of TAM16 in mouse plasma was assessed in the same manner as that described for HLMs.
Assessment of glucuronide metabolites
Incubations were performed with addition of 5 μL TAM16 or positive control propranolol (from a stock solution of 0.5 M) to 450 μL of HLMs (1.11 mg/mL) in 0.05 M phosphate buffer (pH 7.4) containing 45 μL of cofactor uridine di-phosphoglucuronic acid (UDPGA) or UDPGA/NADPH added to wells of a 96 deep-well (2 mL) incubation plate. The samples were incubated at 37°C and 50 μL aliquots from incubations were removed at 0, 60 and 120 min after addition of the test compounds for analysis by UPLC-MS/MS. Chromatography was performed on a Waters BEH C18 (50 × 2.1mm 1.7μm) column heated at 40°C with a flow of 0.5 mL/min and an injection volume of 2 μL. Processing of samples was performed either by MetaboLynx software (Waters), setting each compound at t = 0 min to Control, or by manual processing of the data. Initially, the positive control (propranolol) was analyzed to ensure that glucuronide conjugates were formed.
Safety profiling
Cytochromes P450 (CYP) inhibition assays
CYP inhibition potential of TAM16 toward the major human isoforms CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 was determined using individually expressed CYP enzymes and known fluorescent probe substrates. Human EasyCYP bactosomes (1 nmol/mL, 10 mg/mL) derived from E.coli were supplied by Cypex. Incubation mix (220 μL) containing expressed CYP enzyme and probe substrate in 0.05M potassium phosphate buffer (pH 7.4) was added to each well of 96-well plate and place onto a thermomixer kept at 37°C. To this incubation plate, 5 μL of the serial dilution of the compounds was added, mixed well and the plate was incubated for at least 5 min at 37°C. Miconazole was included as the positive control. After incubation, 25 μL of NADPH regenerating system solution was added to each well of the incubation plate and measurement of fluorescent product was started immediately using the BMG Pherastar spectrophotometer with appropriate parameters (Table S6). Each well was measured for production of fluorescent metabolite every min for 10 min. The data analysis was done using Xcel Fit to calculate the rate of fluorescence units per min as a % of the average rate of the solvent control wells. The % of solvent control values were then plotted against the concentration range and IC50 value were determined by nonlinear regression fitting of the following 4 parameter logistic equation,
hERG activity assessment
The potential of TAM16 to inhibit the human cardiac hERG/IKr potassium channel functional activity was measured in an inducible hERG T-RExTM-CHO Cell line (ThermoFisher) using thallium influx as a surrogate indicator of potassium ion channel activity (Schmalhofer et al., 2010) at AbbVie, USA. Thallium enhances the fluorescent signal of BTC-AM dye (ThermoFisher). Cells were loaded with the dye for 90 min in a low potassium buffer, dye removed and compound added to the cells in a high potassium buffer in a 6 pt. dose response. After 30 min incubation with compound, channel activity was recorded upon addition of thallium buffer using a Tetra plate reader. The slope of the kinetic read was used to calculate channel activity.
Broad panel activity screening
Safety profiling of TAM16 for assessment of off-target interactions was performed at AbbVie, USA. The compound was evaluated across a panel of 21 liability targets (39 functional assays) which included cell based GPCRs and ion channels in both agonist and antagonist readout, and biochemical functional assays for nuclear hormone receptors and phosphodiesterases. The IC50 results were greater than 10 μM for all assays.
Ames mutagenicity assay
The two-strain Ames assay was a six-well modification of the standard Ames Petri plate incorporation test, reducing the volumes of reagents for the smaller size well. Salmonella strains TA98 and TA100 were exposed up to 1000 μg/well under conditions with and without Aroclor 1254-induced rat liver S9.
Chemical synthesis
Materials and general methods
TAM1 and its structural analogs TAM2-6 were purchased from a commercial vendor (ChemBridge Corporation, San Diego, CA). For the synthesis of analogs TAM7-24, all reagents and solvents used were reagent, analytical or HPLC grade. Anhydrous acetonitrile, diethyl ether, dichloromethane, benzene, toluene and N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), tetrahydrofuran (THF) and all other solvents and reagents were purchased from commercial suppliers (Sigma-Aldrich, EMD,AK Scientific, Alfa Aesar, Acros Organics, Combiblocks, Oakwoods, etc.). Thin-layer chromatography (TLC) was performed on E. Merck silica gel 60-F-254 plates and spots were visualized with UV light. Flash column chromatography was performed using 230–400 mesh silica gels (VWR). 1H NMR) and 13C NMR spectra were recorded in DMSO-d6, CD3OD and CDCl3 solutions on a Bruker Avance spectrometer, operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR. In every case trimethylsilane (TMS) was used as an internal standard. Chemical shifts are reported in parts per million (ppm, δ units) relative to TMS solvent signal, and coupling constant (J) values are reported in Hertz (Hz). Liquid chromatograph mass spectra (LCMS) were obtained using SHIMADZU2010 EV using methanol solvent. Analytical reversed-phase high performance liquid chromatography (HPLC) was performed on a Waters Separation Module 2695 system equipped with an auto sampler and a Waters 996 photodiode array detector. Purity of the final compounds was determined using chromatographic systems; column, Princeton SPHER- 100, C18 (particle size = 5 μm, pore size = 10 nm, dimensions = 50 mm x 4.6 mm); mobile phase A, water; mobile phase B, acetonitrile with 0.1% formic acid and 0.1% ammonium formate. Using a flow rate of 1.0 mL/min, gradient elution was performed from 20% B to 80% B over 10 min. In every case, 10 μL of a 1 mM solution was injected.
Synthesis of TAM8 and TAM11

Scheme 1: Reagents and conditions: (a) BnBr, K2CO3, CH3CN, reflux, 8 h; (b) MgBr2, benzene/ether, reflux, 6 h; (c) TBSCl, imidazole, DMF, 0°C-rt, 2 hr (for 3a) and MeI, K2CO3, reflux, 8 hr (for 3b); (d) phenylmagnesium bromide or cyclohexylmagnesium bromide, THF, 2 h; (e) H2/Pd-C, rt, 10 h; (f) FeCl3, (t-BuO)2, DCE, 100°C, 2 h; (g) TBAF, THF, 0°C, 1 hr (for 9a) and BBr3, CH2Cl2, - 78°C-rt, 3 hr (for 9b).
Following the literature procedures (Barrero et al., 2006, Hayashi et al., 2008, Panda et al., 2007), the requisite phenyl and cyclohexyl substituted methanols 5a–b were synthesized by the reaction between corresponding Grignard reagents and carbaldehydes 4a–b, which in turn were easily generated from the phenol derivative 5-(benzyloxy)-2-hydroxybenzaldehyde 3. Intermediate 3 was readily synthesized from commercially available 1 in two steps. Removal of the hydroxyl group and debenzylation of 5a–b were accomplished with hydrogen gas under Pd-catalysis to afford the desired intermediates 6a-b (Scheme 1).
2,5-bis(benzyloxy)benzaldehyde (2): To a solution of 2,5-dihydroxybenzaldehyde 1 (2.00 g, 14.48 mmol) in acetonitrile (40 mL), benzyl bromide (4.30 mL, 36.20 mmol) and K2CO3 (5.00 g, 36.20 mmol) were added. The mixture was stirred at reflux for 8 hr. After completion of the reaction, mixture was cooled to room temperature, water was added (50 mL), and extracted with EtOAc. The extract was washed with saturated solution of NH4Cl and brine, then dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by a silica gel column chromatography with hexane and EtOAc (9:1) to give 2 (3.90 g, yield 85%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 10.55 (s, 1H), 7.49 - 7.36 (m, 11 H), 7.23 - 7.20 (m, 1 H), 7.03 (d, J = 9.08 Hz, 1 H), 5.17 (s, 2 H), 5.08 (s, 2 H); 13C NMR (100 MHz, CDCl3) δ 189.37, 155.99, 153.09, 136.68, 136.30, 128.73 (2C), 128.61 (2C), 128.27, 128.08, 127.57 (2C), 127.37 (2C), 125.66, 124.14, 115.15, 111.85, 71.32, 70.67; MS (LCMS): m/z 317.30 (M-H)+.
5-(benzyloxy)-2-hydroxybenzaldehyde (3): To a suspension of 2,5-bis(benzyloxy)benzaldehyde 2 (3.50 g, 10.99 mmol) in a mixture of benzene and diethyl ether (7:1, 50 mL) was added magnesium bromide (2.43 g, 13.19 mmol) and stirred under refluxed for 6 hr. After cooling to room temperature, 1 N HCl was added to the resulting mixture, and extracted with EtOAc. The organic layer was washed with solution of saturated Na2CO3 and brine, and dried (Na2SO4). The solvent was evaporated in vacuo and the residue was purified by a silica gel column chromatography with hexane/EtOAc (9:1) to give the desired product 3 (2.06 g, yield 82%) as a yellow powder. 1H NMR (400 MHz, CDCl3) δ 10.69 (s, 1H), 10.69 (s, 1 H), 7.47 - 7.34 (m, 5 H), 7.26 - 7.22 (m, 1 H), 7.10 (d, J = 3.08 Hz, 1 H), 6.96 (d, J = 9.04 Hz, 1 H), 5.08 (s, 2 H), 5.08 (s, 2 H); 13C NMR (100 MHz, CDCl3) δ 196.06, 156.28, 151.84, 136.59, 128.68 (2C), 128.18, 127.47 (2C), 126.09, 120.13, 118.75, 116.96, 71.03; MS (LCMS): m/z 227.30 (M-H)+.
5-(benzyloxy)-2-((tert-butyldimethylsilyl)oxy)benzaldehyde (4a): To a solution of 3 (2.0 g, 8.76 mmol) in 20 mL of dichloromethane under nitrogen was added imidazole (1.0 g, 14.69 mmol) and TBSCl (1.98 g, 13.14 mmol). The resulting mixture was stirred at room temperature for 2 hr, and then extracted with EtOAc and washed with water, 1 N HCl, saturated NaHCO3 and brine. The crude product was purified by silica gel chromatography (hexane/EtOAc, 5:1) to afford 2.34 g (78% yield) of 4a as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 10.45 (s, 1 H), 7.52 - 7.32 (m, 6 H), 7.16 (dd, J = 8.90, 3.30 Hz, 1 H), 6.86 (d, J = 8.80 Hz, 1 H), 5.07 (s, 2 H), 1.05 (s, 9 H), 0.28 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 189.78, 153.47, 153.24, 136.69, 128.59 (2C), 128.05, 127.57 (2C), 127.20, 124.54, 121.60, 110.99, 70.61, 25.70 (3C), 18.34, 4.34; MS (LCMS): m/z 342.87 (M)+.
5-(benzyloxy)-2-methoxybenzaldehyde (4b): To a solution of 3 (1.0 g, 4.38 mmol) in 12 mL of acetone was added MeI (0.33 mL, 5.26 mmol) and K2CO3 (0.91 g, 6.57 mmol). Then the mixture was stirred under reflux for 8 hr. After cooling to room temperature, water was added and extracted with EtOAc, and washed with 1 N HCl, brine, dried (Na2SO4) and concentrated. The crude product was purified by silica gel chromatography (hexane/EtOAc, 6:1) to afford 0.86 g (81% yield) of 4a as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 10.47 (s, 1 H), 7.46 - 7.34 (m, 6 H), 7.24 - 7.21 (m, 1 H), 6.96 (d, J = 9.08 Hz, 1 H), 5.08 (s, 2 H), 3.91 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 189.42, 156.85, 152.78, 136.70, 128.58 (2C), 128.05, 127.54 (2C), 125.06, 124.18, 113.37, 112.00, 70.51, 56.15; MS (LCMS): m/z 242.88 (M)+.
(5-(benzyloxy)-2-((tert-butyldimethylsilyl)oxy)phenyl)(phenyl)methanol (5a): To a solution of 5-(benzyloxy)-2-((tert-butyldimethylsilyl)oxy)benzaldehyde 4a (2.0 g, 5.84 mmol) in dry THF (30 mL) under nitrogen was added phenyl magnesium bromide 7.00 mL (1 M) at room temperature and stirred for 2 hr. The reaction mixture was quenched by addition of saturated NH4Cl (2 mL) and THF was removed in vacuo. The residue was extracted thrice with ethyl acetate, washed with brine and dried over Na2SO4. Organic extract was concentrated and purified by silica gel column chromatography with 10% ethyl acetate in hexane to furnish carbinol product 5a (1.76 g, 72%) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.45 −7.29 (m, 10 H), 6.89 (d, J = 2.50 Hz, 1 H), 6.83 - 6.75 (m, 2 H), 6.08 (s, 1 H), 4.98 (s, 2 H), 0.96 (s, 9 H), 0.22 (d, J = 2.90 Hz, 6 H); 13C NMR (100 MHz, CDCl3) δ 153.09, 147.07, 142.89, 137.17, 134.66, 128.50 (2C), 128.23 (2C), 127.85, 127.55, 127.31, 126.30, 118.93, 115.01, 114.53, 71.74, 70.59, 25.77 (3C), 18.19, 1.01, −4.13; MS (LCMS): m/z 319.50 (M-H)+.
(5-(benzyloxy)-2-methoxyphenyl)(cyclohexyl)methanol (5b): The title compound was obtained from 4b and cyclohexylmagnesium bromide following the procedure for compound 5a in 66% yield after flash-chromatography (1:10 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.52 - 7.30 (m, 5 H), 6.95 (d, J = 2.79 Hz, 1 H), 6.90 - 6.77 (m, 2 H), 5.06 (s, 2 H), 4.55 (t, J = 6.90 Hz, 1 H), 3.82 (s, 3 H), 2.57 (d, J = 6.31 Hz, 1 H), 2.13-1.97 (m, 1 H), 1.87 - 1.58 - (m, 4 H), 1.48 - 1.08 (m, 5 H); MS (LCMS): m/z 309.05 (M-OH), 227.10 (M-C7H7)+.
(2-benzyl-4-(benzyloxy)phenoxy)(tert-butyl)dimethylsilane (6a): The compound 5a (1.60 g, 3.80 mmol) was hydrogenated over 10% Pd/C (0.20 g) and after usual work-up and purification furnished 6a (0.87 g, 73%) as white semi-solid. 1H NMR (400 MHz, CDCl3) δ 7.35 - 7.27 (m, 2 H), 7.26 - 7.18 (m, 3 H), 6.73 (d, J = 8.70 Hz, 1 H), 6.59 (dd, J = 8.70, 3.10 Hz, 1 H), 6.48 (d, J = 3.10 Hz, 1 H), 4.51 (bs, 1 H), 3.96 (s, 2 H), 1.01 (s, 9 H), 0.24 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 149.45, 147.34, 140.61, 132.71, 129.06 (2C), 128.35 (2C), 125.94, 119.12, 117.44, 113.48, 36.01, 25.83 (3C), 18.25, −4.04 (2C); MS (LCMS): m/z 315.25 (M)+.
3-(cyclohexylmethyl)-4-methoxyphenol (6b): The title compound was obtained from 5b following the procedure for compound 6a in 67% yield after flash-chromatography (1:10 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 6.78 - 6.71 (m, 1 H), 6.69 - 6.61 (m, 2 H), 5.12 (bs, 1 H), 3.79 (s, 3 H), 2.47 (d, J = 7.00 Hz, 2 H), 1.78 - 1.49 (m, 6 H), 1.32 - 1.11 (m, 6 H), 1.06 - 0.89 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 152.08, 148.89, 131.46, 118.10, 112.76, 111.90, 56.21, 38.27, 37.90, 33.29 (2C), 26.64, 26.37 (2C); MS (LCMS): m/z 220.73 (M)+.
Ethyl 4-benzyl-5-((tert-butyldimethylsilyl)oxy)-2-phenylbenzofuran-3-carboxylate (8a) (Guo et al., 2009): To a mixture of ethyl benzoylacetate 7 (0.14 mL, 0.83 mmol), phenol derivative 6a (0.80 g, 2.54 mmol), and FeCl3·6H2O (0.035 g, 0.13 mmol) 1,2-dichloroethane (3.0 mL) was added under nitrogen at room temperature. Then di-tert-butyl peroxide (0.34 mL, 1.86 mmol) was added dropwise to the reaction mixture and was stirred for 2 hr at 100°C. After completion of the reaction, mixture was cooled to room temperature and was quenched with saturated NaHCO3 and extracted with 25 mL of ethyl acetate. The organic phase was washed with 10 mL of saturated NaHCO3 and 10 mL of water. The extract was dried over Na2SO4 and concentrated. The crude mixture was filtered through a small silica gel column and used as such in the next step.
Ethyl 4-benzyl-5-hydroxy-2-phenylbenzofuran-3-carboxylate (9a) (TAM8): To a solution of crude mixture of 8a (0.80 g) in dry THF (25 mL) at 0°C was added tetra-n-butylammonium fluoride (TBAF) 2.0 mL (1M in THF). After being stirred at 0°C for 1 hr, the reaction mixture was diluted with ethyl acetate, washed with water and brine. The combined organic extract was dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification of the crude reaction mixture by silica gel column chromatography with ethyl acetate and hexane (1:10) as an eluent provided the desired product 9a as a colorless viscous liquid (10% overall yield in two steps) with its regioisomer as major product. 1H NMR (400 MHz, CDCl3) δ 8.05 - 7.93 (m, 2 H), 7.56 - 7.44 (m, 4 H), 7.40 - 7.19 (m, 6 H), 4.98 (bs, 1 H), 4.42 (q, J = 7.00 Hz, 2 H), 4.14 (s, 2 H), 1.41 (t, J = 7.00 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 164.07, 161.07, 150.89, 149.06, 139.79, 130.08, 129.81, 129.42 (3C), 128.80 (3C), 128.66 (2C), 127.98 (2C), 126.41 (2C), 112.53, 107.76, 60.61, 36.76, 14.26; MS (LCMS): m/z 373.10 (M)+.
Ethyl 6-benzyl-5-hydroxy-2-phenylbenzofuran-3-carboxylate (9’a): 1H NMR (400 MHz, CDCl3) δ 7.85 - 7.82 (m, 2 H), 7.62 (s, 1 H), 7.54 - 7.49 (m, 3 H), 7.37 - 7.22 (m, 6 H), 4.90 (bs, 1 H), 4.51 (q, J = 7.16 Hz, 2 H), 4.43 (s, 1 H), 4.17 (s, 1 H), 1.29 (t, J = 7.16 Hz, 3 H); MS (LCMS): m/z 373.09 (M)+.
Ethyl 4-(cyclohexylmethyl)-5-methoxy-2-phenylbenzofuran-3-carboxylate (8b). The title compound was obtained from 6b and 7 following procedure for compound 8a in 17% yield after flash-chromatography (1:10 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 8.01 (dd, J = 7.30, 2.20 Hz, 2 H), 7.55 - 7.43 (m, 4 H), 7.32 - 7.22 (m, 1 H), 4.42 (q, J = 7.00 Hz, 2 H), 3.93 (s, 3 H), 2.63 (d, J = 6.9 Hz, 2 H), 1.78 −1.60 (m, 6 H), 1.42 (t, J = 6.99 Hz, 3 H), 1.35 - 1.14 (m, 3 H), 1.09 - 0.95 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 164.18, 160.17, 155.23, 148.55, 130.06, 129.87 (2C), 129.38 (2C), 128.96, 127.95 (2C), 120.77, 112.59, 102.72, 60.43, 55.81, 38.55, 38.37, 33.29 (2C), 26.64, 26.37 (2C), 14.21; MS (LCMS): m/z 393.08 (M)+.
Ethyl 6-(cyclohexylmethyl)-5-methoxy-2-phenylbenzofuran-3-carboxylate (8’b): 1H NMR (400 MHz, CDCl3) δ 7.88 - 7.86 (m, 2 H), 7.69 (s, 1 H), 7.56 - 7.49 (m, 2 H), 7.37 −7.33 (m, 2 H), 4.51 (q, J = 7.12 Hz, 2 H), 2.96 (d, J = 6.96 Hz, 1 H), 2.67 (d, J = 6.88 Hz, 1 H), 1.80 - 1.65 (m, 6 H), 1.35 – 0.93 (m, 8 H); MS (LCMS): m/z 393.08 (M)+.
Ethyl 4-(cyclohexylmethyl)-5-hydroxy-2-phenylbenzofuran-3-carboxylate (9b) (TAM11): To a solution of compound 8b (0.05 g, 0.13 mmol) in dichloromethane (10 mL) stirred under N2 gas, was added boron tribromide (0.04 mL, 0.39 mmol) at −78°C and warm to room temperature and stirred for 3 hr. After completion of the reaction, reaction was quenched by addition of water and extracted with ethyl acetate, and solvent was evaporated under reduced pressure. Crude reaction mixture was purified by silica gel column chromatography with ethyl acetate/hexane (1:10) as an eluent to provide the desired product 9b 0.029 g, 61% yield as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 8.07 - 7.94 (m, 2 H), 7.57 - 7.42 (m, 4 H), 7.29 −7.25 (m, 1 H), 5.32 (bs, 1 H), 4.41 (q, J = 7.20 Hz, 2 H), 2.64 (d, J = 7.00 Hz, 2 H), 1.82 - 1.59 (m, 6 H), 1.41 (t, J = 7.20 Hz, 3 H), 1.29 - 1.16 (m, 3 H), 1.11 - 0.97 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 164.18, 160.78, 151.01, 148.92, 130.00, 129.93 (2C), 129.42, 127.96 (2C), 126.45, 125.81, 112.68, 108.67, 107.20, 60.57, 38.55, 33.29 (2C), 29.88, 26.54, 26.31 (2C), 14.26; MS (LCMS): m/z 379.09 (M)+.
Ethyl 6-(cyclohexylmethyl)-5-hydroxy-2-phenylbenzofuran-3-carboxylate (9’b): 1H NMR (400 MHz, CDCl3) δ 7.89 - 7.87 (m, 2 H), 7.58 - 7.48 (m, 3 H), 7.36 (s, 1 H), 7.31 (s, 1 H), 5.14 (bs, 1 H), 4.53 (q, J = 7.16 Hz, 2 H), 2.95 (d, J = 6.96 Hz, 1 H), 2.67 (d, J = 7.00 Hz, 1 H), 1.78 - 1.67 (m, 6 H), 1.36 (t, J = 7.16 Hz, 3 H), 1.36 - 1.11 (m, 3 H), 0.92 - 0.86 (m, 2 H); MS (LCMS): m/z 379.08 (M)+.
Synthesis of TAM7 and TAM9-16 (Scheme 2)

Scheme 2: Reagents and conditions: (a) Cu(OTf)2, toluene, reflux, 12 h; (b) formaldehyde, piperidine, reflux, 8 h; (c) BBr3, CH2Cl2, −78 to 0°C, 3 h; (d) NaOH, EtOH, H2O, reflux, 4 h; (e) i) (COCl)2, CH2Cl2, DMF (cat.), 0°C - rt, 3 hr, ii) methylamine, THF; (f) PPh3, DEAD, MeOH, overnight; (g) Tf2O, pyridine, rt, 1 h; (h) Pd(OAc)2, PPh3, formic acid, Et3N, 90°C, 8 hr.
Ethyl 5-hydroxy-2-phenylbenzofuran-3-carboxylate (12a) (Mothe et al., 2010) (TAM7): To a suspension of ethyl 3-oxo-3-phenylpropanoate 10a (2.40 mL, 13.88 mmol) and Cu(OTf)2 (5 mol %, 0.17 g, 0.46 mmol) in dry toluene (10 mL) under a nitrogen atmosphere was added drop wise a solution of p-benzoquinone 11 (1.0 g, 9.25 mmol) dissolved in toluene (8 mL). The reaction mixture was stirred at reflux for 12 hr. After completion of reaction, the reaction mixture was quenched with 10 mL of saturated NH4Cl solution and extracted with ethyl acetate (3x10 mL). The combined organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. Crude reaction mixture was purified by flash silica gel column chromatography (1:10 ethyl acetate/hexane) to afford the compound 13a, 1.07 g, 41% yield as a pale brown amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 9.45 (s, 1 H), 7.94 (dd, J = 6.68, 3.01 Hz, 2 H), 7.61 - 7.46 (m, 4 H), 7.39 (d, J = 2.49 Hz, 1 H), 6.87 (dd, J = 8.80, 2.49 Hz, 1 H), 4.32 (q, J = 7.12 Hz, 2 H), 1.32 (t, J = 7.12 Hz, 3 H),; 13C NMR (100 MHz, DMSO-d6) δ 163.55, 160.71, 154.91, 147.94, 130.76, 129.67, 129.64 (2C), 128.60 (2C), 127.83, 114.87, 112.16, 108.83, 107.03, 60.79, 14.44; MS (LCMS): m/z 282.93 (M)+.
Ethyl 5-hydroxy-2-(4-methoxyphenyl)benzofuran-3-carboxylate (12b): The title compound was obtained from 10b and p-benzoquinone 11 following procedure for compound 12a in 43% yield after flash-chromatography (1:10 EtOAc/hexane) as a pale gray amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 9.39 (s, 1 H), 7.94 (d, J = 8.84 Hz, 2 H), 7.45 (d, J = 8.80 Hz, 1 H), 7.35 (d, J = 2.32 Hz, 1 H), 7.07 (d, J = 8.84 Hz, 2 H), 6.85 - 6.80 (m, 1 H), 4.32 (q, J = 7.08 Hz, 2 H), 3.85 (s, 3 H), 1.33 (t, J = 7.08 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 163.75, 161.33, 160.98, 154.81, 147.61, 131.30 (2C), 128.04, 121.94, 114.37, 114.11 (2C), 111.93, 107.58, 107.05, 60.67, 55.82 14.51; MS (LCMS): m/z 313.00 (M)+.
Ethyl 5-hydroxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (13a) (Matsumoto et al., 2005) (TAM9): To a solution of 12a (1.0 g, 3.54 mmol) in EtOH (6 mL) was added formalin (37%, 1.2 eq.) and piperidine (0.35 mL, 3.54 mmol). The reaction mixture was stirred for approximately 8 hr at 80°C. The solution was then cooled to room temperature and diluted with water, and extracted with dichloromethane. The organic layer was dried over Na2SO4 and concentrated in vacuo. Crude residue was purified by silica gel column chromatography (40% ethyl acetate in n-hexane) and provided the desired product 13a (0.97 g, 72% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.77 (dd, J = 7.70, 1.69 Hz, 2 H), 7.53 - 7.41 (m, 3 H), 7.33 (d, J = 8.80 Hz, 1 H), 6.88 (d, J = 8.80 Hz, 1 H), 4.38 (q, 7.20 Hz, 2 H), 3.99 (s, 2 H), 2.63 - 2.45 (m, 4 H), 1.78 - 1.43 (m, 6 H), 1.30 (t, 7.20 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 166.35, 156.70, 155.50, 148.16, 130.03, 129.49, 128.33 (2C), 127.87 (2C), 125.49, 115.21, 112.24, 110.67, 110.09, 61.41, 57.98, 53.93 (2C), 25.85 (2C), 23.98, 13.98; MS (LCMS): m/z 380.15 (M)+.
5-hydroxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylic acid (15a) (TAM10): To a solution of ester derivative 13a (0.80 g, 2.10 mmol) in EtOH (10 mL) a solution of NaOH (0.34 g, 8.43 mmol) in water (5 mL) was added and then the mixture was refluxed for 4 hr. After completion of the reaction, reaction mixture was allowed to cool to room temperature and poured into ice cold water. Neutralization with dil. HCl solution resulted in precipitation of desired product as white solid. Solid so obtained was filtered, washed with water and dried to obtain the desired product 15a (0.43 g, 58% yield) as an amorphous white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.83-7.76 (m, 2 H), 7.37 - 7.16 (m, 4 H), 6.79 (d, J = 9.00 Hz, 1 H), 4.01 (s, 2 H), 2.87 - 2.71 (m, 4 H), 1.58 - 1.39 (m, 6 H); 13C NMR (100 MHz, DMSO-d6) δ 168.31, 153.50, 152.37, 147.64, 130.92, 129.05, 128.70 (2C), 127.97, 127.53 (2C), 119.28, 113.86, 113.27, 108.18, 51.34 (2C), 50.94, 23.81 (2C), 22.21; MS (LCMS): m/z 352.10 (M)+.
5-hydroxy-N-methyl-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (16a) (TAM12): To a solution of acid 15a (0.25 g, 0.71 mmol) in dichloromethane (8 mL), oxalyl chloride (0.08 mL, 0.93 mmol) and N,N-dimethylformamide (2 drops) were added at 0°C and stirred at room temperature for 3 hr. The reaction mixture was evaporated under reduced pressure. The residue was redissolved in THF (10 mL) and a solution of methylamine 0.54 mL (2 M in THF) was added and the mixture was stirred for 2 hr. After completion of the reaction, solvent was evaporated. Water was added and extracted with dichloromethane, dried (Na2SO4) and concentrated. Crude reaction mixture was purified by silica gel column chromatography (1% methanol in dichloromethane) to provide the desired product 16a (0.15 g, 57% yield) as a pale brown amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (s, 1 H), 7.76 (dd, J = 7.85, 1.54 Hz, 2 H), 7.54 - 7.30 (m, 4 H), 6.89 (d, J = 8.80 Hz, 1 H), 3.96 (s, 2 H), 2.84 - 2.80 (m, 7 H), 1.78 - 1.59 (m, 4 H), 1.40 - 1.17 (m, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 166.60, 159.43, 154.67, 154.07, 147.40, 128.74 (2C), 128.35, 120.20, 116.33, 113.73 (2C), 111.73, 111.67, 55.36 (2C), 53.05, 52.48, 26.83 (2C), 23.14, 21.76; MS (LCMS): m/z 364.89 (M)+.
Ethyl 5-hydroxy-2-(4-methoxyphenyl)-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (13b): The title compound was obtained from 12b following procedure for compound 13a in 67% yield after flash-chromatography (1:10 EtOAc/hexane) as an amorphous gray solid. 1H NMR (400 MHz, DMSO-d6) δ 7.68 (d, J = 8.80 Hz, 2 H), 7.39 (d, J = 8.80 Hz, 1 H), 7.09 (d, J = 8.80 Hz, 2 H), 6.82 (d, J = 8.80 Hz, 1 H), 4.32 (q, J = 7.00 Hz, 2 H), 3.84 (s, 3 H), 3.77 (s, 2 H), 1.55-1.34 (m, 4 H), 1.26 (t, J = 7.00 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 165.74, 160.89, 155.68, 154.20, 147.62, 129.33 (2C), 126.01, 121.91, 114.72 (2C), 114.65, 114.14, 110.75, 109.94, 61.60, 55.83 (2C), 55.45, 53.74, 25.87 (2C), 24.26, 14.30; MS (LCMS): m/z 409.87 (M)+.
Ethyl 5-hydroxy-2-(4-hydroxyphenyl)-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (14b) (TAM13): The title compound was obtained from 13b following procedure for compound 9a in 62% yield after flash-chromatography (4:10 EtOAc/hexane) as a gray amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 7.57 (d, J = 8.80 Hz, 2 H), 7.36 (d, J = 8.80 Hz, 1 H), 7.33 (d, J = 8.80 Hz, 1 H), 6.94 - 6.85 (m, 2 H), 6.80 (d, J = 8.80 Hz, 1 H), 4.31 (q, 7.20 Hz, 2 H), 3.78 (s, 2 H), 2.46 - 2.28 (m, 4 H) 1.54 - 1.36 (m, 6 H), 1.23 - 1.26 (t, 7.20 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ, 165.86, 159.48, 156.31, 154.23, 147.50, 129.46 (2C), 126.13, 120.38, 116.13 (2C), 114.39, 113.83, 110.71, 109.31, 61.52, 55.47, 53.70 (2C), 25.80 (2C), 24.17, 14.26; MS (LCMS): m/z 396.15 (M)+.
5-hydroxy-2-(4-methoxyphenyl)-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylic acid (15b): The title compound was obtained from 13b as described for 15a, and used as such in the next step.
5-hydroxy-2-(4-methoxyphenyl)-N-methyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (16b): The title compound was obtained from 15b as described for 16a in 60% yield after flash-chromatography (1% methanol in dichloromethane) as a pale brown amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 7.68 (d, J = 9.00 Hz, 2 H), 7.30 - 7.21 (m, 1 H), 6.97 (d, J = 8.8 Hz, 2 H), 6.88 (d, J = 8.8 Hz, 1 H), 6.37 (bs, 1 H), 3.95 (s, 2 H), 3.88 (s, 3 H), 2.98 (d, J = 4.80 Hz, 2 H), 2.81 - 2.53 (m, 4 H), 1.77 - 1.43 (m, 6 H); 13C NMR (100 MHz, DMSO-d6) δ 167.38, 160.53, 154.84, 153.75, 147.88, 128.41 (2C), 126.25, 122.05, 114.74, 114.28 (2C), 111.67, 111.07, 55.82, 55.34, 53.48 (2C), 26.96, 25.29 (2C), 23.48; MS (LCMS): m/z 395.03 (M)+.
5-hydroxy-2-(4-hydroxyphenyl)-N-methyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (17b) (TAM16): The title compound was obtained from 16b as described for 9b in 67% yield after flash-chromatography (2% methanol in dichloromethane) to provide the desired product 17b as a pale brown amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1 H), 8.80 (d, J = 4.69 Hz, 1 H), 7.72 - 7.44 (m, 3 H), 7.02 - 6.94 (m, 1 H), 6.93 (d, J = 8.66 Hz, 2 H), 4.30 (s, 2 H), 3.51 - 3.33 (m, 4 H), 2.81 (d, J = 4.70 Hz, 3 H), 1.79 - 1.73 (m, 4 H), 1.59 - 1.50 (m, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 166.60, 159.43, 154.67, 154.07, 147.40, 128.74 (2C), 128.35, 120.20, 116.33, 113.73 (2C), 111.73, 111.67, 55.36 (2C), 53.05, 52.48, 26.83 (2C), 23.14, 21.76; MS (LCMS): m/z 380.99 (M)+.
Ethyl 5-methoxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (18a) (TAM14): The title compound was obtained from 13a and methanol following the literature procedure(Newlander et al., 1997). To a solution of 13a (0.10 g, 0.26 mmol) in dry THF (5 mL), methanol (0.013 mL, 0.316 mmol), Ph3P (0.106 g, 0.39 mmol), and DEAD (0.07 mL, 0.39 mmol) were added. The reaction mixture was stirred under a nitrogen atmosphere for overnight. After completion of the reaction, mixture was concentrated and extracted with CH2Cl2 (2 × 15 mL). Organic extract was dried (Na2SO4), concentrated and purified by flash silica gel column chromatography (30% ethyl acetate–hexane) to afford the compound 18a (0.05 g, 48% yield) as a pale yellow liquid. 1H NMR (400 MHz, CDCl3) δ 7.90 - 7.76 (m, 2 H), 7.53 - 7.34 (m, 4 H), 6.99 (d, J = 8.95 Hz, 1 H), 4.36 (q, J = 7.20 Hz, 2 H), 3.90 (s, 3 H), 3.83 (s, 2 H), 2.29 (m, 4 H), 1.54 - 1.35 (m, 6 H), 1.29 (t, J = 7.20 Hz, 3 H). MS (LCMS): m/z 395.15 (M)+.
Ethyl 2-phenyl-4-(piperidin-1-ylmethyl)-5-(((trifluoromethyl)sulfonyl)oxy)benzofuran-3-carboxylate (19a): Compound 13a (0.10 g, 0.26 mmol) was dissolved in dry dichloromethane (3 mL) under nitrogen at room temperature followed by the addition of dry 2,6-lutidine (0.05 mL, 0.42 mmol). Trifluoromethanesulfonic anhydride (0.05 mL, 0.32 mmol) in dry dichloromethane (1 mL) was then added dropwise. The reaction mixture was stirred at room temperature for 1 hr. Water was added to the reaction mixture and extracted with CH2Cl2 (3 X 5 mL). The combined organic phase was washed with diluted HCl, water, brine and dried over Na2SO4, and concentrated. The crude reaction mixture was passed through a small of silica gel column and used as such in next step.
Ethyl 2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (20a) (TAM15): The title compound was obtained from 19a following the literature procedure (Peterson et al., 1987) in 23% yield after flash-chromatography (1:6 EtOAc/hexane). In brief, to a solution of triflate 19a (0.10 g, 0.19 mmol) in 3 mL of DMF, triphenylphosphine (2.0 mg, 0.008 mmol), palladium acetate (1.0 mg, 0.0044 mmol), triethylamine (0.08 mL, 0.58 mmol), and formic acid (0.015 mL, 0.39 mmol) were added. Then the mixture was stirred at 90 O C for 8 hr. Reaction mixture was diluted with water, extracted with ethyl acetate, dried over Na2SO4, and concentrated. The crude residue was chromatographed on silica gel with ethyl acetate-hexanes (1:10) to give 0.016 g (23%) of 20a as a colorless viscous liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 6.46 Hz, 2 H), 7.48 - 7.67 (m, 4 H), 7.34 (t, J = 7.78 Hz, 1 H), 7.21 (d, J = 7.34 Hz, 1 H), 4.32 (q, J = 7.12 Hz, 2 H), 3.65 (s, 2 H), 2.17 - 2.24 (m, 4 H), 1.28 - 1.39 (m, 6 H), 1.24 (t, J = 7.12 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 165.02, 154.91, 154.14, 133.45, 130.39, 129.33, 129.22 (2C), 127.93 (2C), 125.60, 125.47, 125.12, 111.83, 110.95, 61.51, 61.48, 54.09 (2C), 25.86 (2C), 24.59, 14.18; MS (LCMS): m/z 363.93 (M)+.
Synthesis of TAM19 and TAM20 (Scheme 3)

Scheme 3: Reagents and conditions: (a) FeCl3, (t-BuO)2, 100°C, 4 h; (b) NBS, AIBN, CCl4, 50°C, 5 h; (c) piperidine, NaI, K2CO3, acetone, 60°C, 6 h; (d) BBr3, CH2Cl2, −78 to 0°C, 3 hr, (e) NaOH, EtOH, H2O, 90°C, 6 h; (f) i) (COCl)2, CH2Cl2, DMF (cat.), 0°C - rt, 3 hr, ii) NH2Me in THF.
Ethyl 6-methoxy-4-methyl-2-phenylbenzofuran-3-carboxylate (23a): The title compound was obtained from 3-methoxy-5-methylphenol 21 and ethyl benzoylacetate 22a following procedure for compound 8a in 35% yield after flash-chromatography (1:6 EtOAc/hexane) as colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.87 – 7.75 (m, 2 H), 7.52 - 7.38 (m, 3 H), 6.92 (d, J = 1.90 Hz, 1 H), 6.75 (d, J = 1.30 Hz, 1 H), 4.42 (q, J = 7.10 Hz, 2 H), 3.87 (s, 3 H), 2.56 (s, 3 H), 1.36 (t, J = 7.10 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 165.98, 158.42, 155.05, 154.74, 132.43, 130.04, 129.15, 128.38 (2C), 127.48 (2C), 119.35, 114.36, 110.61, 93.14, 61.37, 55.61, 19.90, 13.98; MS (LCMS): m/z 310.94 (M)+.
Ethyl 4-(bromomethyl)-6-methoxy-2-phenylbenzofuran-3-carboxylate (24a): To a solution of ethyl 6-methoxy-4-methyl-2-phenylbenzofuran-3-carboxylate 23a (1.0 g, 3.22 mmol) in tetrachloromethane (20 mL), N-bromosuccinimide (0.69 g, 3.87 mmol), and a catalytic amount of azo(bis)isobutyronitrile (AIBN) (0.053 g, 0.32 mmol) were added and refluxed for 5 hr. The solvent was removed in vacuo, and crude residue was chromatographed on silica gel with ethyl acetate-hexanes (2:10) to give of 24a as a yellow viscous liquid. But this compound was unstable and was used as such in the next step.
Ethyl 6-methoxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (25a): To a solution of ethyl 4-(bromomethyl)-6-methoxy-2-phenylbenzofuran-3-carboxylate 15 (0.60 g, 1.54 mmol) in acetone (10 mL), NaI (0.23 g, 1.54 mmol), piperidine (0.18 g, 1.84 mmol) and K2CO3 (0.32 g, 2.31 mmol) were added. Then the resulting mixture was refluxed for about 6 hr and then, after cooling, was filtered and the solvent was evaporated. Crude reaction mixture was purified by silica gel column chromatography and provided the desired product 16 (0.39 g, 65% yield). 1H NMR (400 MHz, CDCl3) δ 7.86 - 7.58 (m, 3 H), 7.54 - 7.38 (m, 3 H), 7.10 (d, J = 1.61 Hz, 1 H), 4.66 (s, 2 H), 4.30 (q, J = 7.09 Hz, 2 H), 3.97 (s, 3 H), 3.83 - 2.37 (m, 4 H), 2.20 - 1.51 (m, 6 H), 1.18 (t, J = 7.12 Hz, 3 H); MS (LCMS): m/z 394.03 (M)+.
Ethyl 6-hydroxy-2-phenyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (26a) (TAM20): The title compound was obtained from 25a and boron tribromide as described for 9b in 64% yield after flash-chromatography (1% methanol in dichloromethane)) as an amorphous brown solid. 1H NMR (400 MHz, CD3OD) δ 7.74 - 7.66 (m, 2 H), 7.57 - 7.46 (m, 3 H), 7.13 (d, J = 2.10 Hz, 1 H), 7.01 (d, J = 2.10 Hz, 1 H), 4.63 (s, 2 H), 4.31 (q, 7.10 Hz, 2 H), 3.52 - 2.25 (m, 4 H), 2.01 - 1.65 (m, 6 H), 1.15 (t, 7.10 Hz, 3 H); 13C NMR (100 MHz, CD3OD) δ 166.06, 160.47, 156.19, 156.14, 130.13, 129.83, 129.05 (2C), 127.91, 127.81 (2C), 123.07, 118.65, 109.54, 99.28, 61.34, 59.04, 52.82 (2C), 22.73 (2C), 21.48, 12.54; MS (LCMS): m/z 380.00 (M)+.
Ethyl 6-methoxy-2-(4-methoxyphenyl)-4-methylbenzofuran-3-carboxylate (23b): The title compound was obtained from 3-methoxy-5-methylphenol 21 and ethyl 3-(4-methoxyphenyl)-3-oxopropanoate 22b following procedure for compound 8a in 44% yield after flash-chromatography (1:6 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.80 Hz, 2 H), 6.99 (d, J = 8.95 Hz, 2 H), 6.90 (d, J = 1.91 Hz, 1 H), 6.73 (d, J = 1.32 Hz, 1 H), 4.41 (q, J = 7.19 Hz, 2 H), 3.88 (s, 3 H), 3.86 (s, 3 H), 2.55 (s, 3 H), 1.37 (t, J = 7.19 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 166.03, 160.45, 158.14, 155.32, 154.82, 132.22, 129.18 (2C), 122.65, 119.44, 114.20, 113.85 (2C), 109.33, 93.15, 61.24, 55.60, 55.32, 20.07, 14.06; MS (LCMS): m/z 340.94 (M)+.
Ethyl 4-(bromomethyl)-6-methoxy-2-(4-methoxyphenyl)benzofuran-3-carboxylate (24b): The title compound was obtained from 23b and N-bromosuccinimide following procedure for compound 24a and was used as such in the next step.
Ethyl 6-methoxy-2-(4-methoxyphenyl)-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (25b): The title compound was obtained from 24b and piperidine following procedure for compound 25a in 63% yield after flash-chromatography (1:1 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.88 Hz, 2 H), 7.02 - 6.87 (m, 3 H), 6.83 (d, J = 2.08 Hz, 1 H), 4.37 (q, J = 7.16 Hz, 2 H), 3.88 (s, 3 H), 3.86 (s, 3 H), 3.68 (s, 2 H), 2.34 - 2.26 (m, 4 H), 1.51 - 1.41 (m, 6 H), 1.34 (t, J = 7.16 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 165.82, 160.35, 157.80, 155.08, 133.96, 129.25, 129.18, 122.70, 118.99, 113.90, 113.76 (2C), 113.63, 110.19, 94.07, 61.78, 61.04, 55.63, 55.31 (2C), 54.28, 25.94 (2C), 24.61, 14.97; MS (LCMS): m/z 423.97 (M)+.
Ethyl 2-(6-hydroxy-2-(4-hydroxyphenyl)-4-(piperidin-1-ylmethyl)benzofuran-3-yl)-2-oxoacetate (26b): The title compound was obtained from 25b with boron tribromide as described for 9b. Crude reaction mixture was passed through a bed of silica gel and used as such in the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.90 (bs, 1 H), 7.62 - 7.47 (m, 2 H), 7.20 - 7.16 (m, 1 H), 7.04 −6.98 (m, 1 H), 6.92 (d, J = 8.66 Hz, 2 H), 4.65 (d, J = 4.11 Hz, 2 H), 4.26 (q, J = 7.14 Hz, 2 H), 3.46 - 3.36 (m, 2 H), 3.10 - 2.94 (m, 2 H), 1.96 - 1.59 (m, 5 H), 1.51 - 1.36 (m, 1 H), 1.15 (t, J = 7.14 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 165.56, 159.88, 159.74, 156.02, 155.36, 131.00 (2C), 123.46, 120.51, 118.91, 115.80, 115.64 (2C), 108.52, 99.75, 61.57, 55.36, 54.07, 52.66, 22.74 (2C), 21.79, 14.05; MS (LCMS): m/z 423.97 (M)+.
6-methoxy-2-(4-methoxyphenyl)-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylic acid (27a): The title compound was obtained from 26a with aqueous NaOH as described for 15a. Crude reaction mixture was passed through a bed of silica gel and used as such in the next step.
2-(6-hydroxy-2-(4-hydroxyphenyl)-4-(piperidin-1-ylmethyl)benzofuran-3-yl)-2-oxoacetic acid (27b): The title compound was obtained from 26b with aqueous NaOH as described for 15a. Crude reaction mixture was passed through a bed of silica gel and used as such in the next step.
6-methoxy-2-(4-methoxyphenyl)-N-methyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (28a): The title compound was obtained from 27a following procedure for compound 16a in 68% yield after flash-chromatography (2% methanol in dichloromethane) as a brown viscous liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.94 (s, 1 H), 7.71 - 7.50 (m, 2 H), 7.46 −7.43 (m, 3 H), 7.18 - 7.14 (m, 2 H), 4.32 (s, 2 H), 2.88 - 2.83 (m, 4 H), 2.82 (d, J = 4.60 Hz, 3 H), 1.79 - 1.69 (m, 5 H), 1.41 - 1.36 (m, 1 H); 13C NMR (100 MHz, DMSO-d6) δ 166.20, 156.74, 155.07, 152.36, 129.65, 129.53 (2C), 129.43, 126.51 (2C), 123.10, 119.61, 112.71, 115.62, 99.53, 56.06, 52.55 (2C), 26.84 (2C), 22.86, 21.90; MS (LCMS): m/z 365.03(M)+.
6-hydroxy-2-(4-hydroxyphenyl)-N-methyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (28b) (TAM19): The title compound was obtained from 27b as described for 9a in 60% yield after flash-chromatography (2% methanol in dichloromethane) to provide the desired product 29b as a pale yellow amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 8.36 (d, J = 4.21 Hz, 1 H), 7.60 - 7.49 (m, 2 H), 6.91 - 6.81 (m, 3 H), 6.75 (d, J = 1.61 Hz, 1 H), 3.48 (s, 2 H), 2.77 (d, J = 4.55 Hz, 3 H), 2.30 - 2.21 (m, 4 H), 1.55 - 1.28 (m, 6 H); 13C NMR (100 MHz, DMSO-d6) δ 166.53, 159.17, 156.21, 154.67, 152.44, 128.22 (2C), 122.49, 120.29, 119.91, 117.31, 116.32 (2C), 111.30, 99.51, 56.09, 52.42 (2C), 49.01, 26.80 (2C), 22.85, 21.94; MS (LCMS): m/z 380.99 (M)+.
Synthesis of TAM17 and TAM18 (Scheme 4)

Scheme 4: Reagents and conditions: (a) N2CHCO2Et, HBF4-OEt2, rt, 2 h; (b) LDA, I2, −78°C, 1 h; (c) arylboronic acid, Pd(PPh3)4, Na2CO3, 100°C, 10 h; (d) BBr3, −78°C-rt, 4h; (e) piperidine, formaldehyde, 80°C, 3 h; (f) NaOH, EtOH-H2O, reflux, 4 h; (g) i) (COCl)2, cat. DMF, CH2Cl2, 2 hr, ii) NH2Me in THF.
Ethyl 5-methoxybenzofuran-3-carboxylate (30) (He et al., 2014): HBF4·Et2O (0.09 mL, 0.66 mmol) was added to a solution of compound 29 (1.0 g, 6.57 mmol) in CH2Cl2 (4 mL), and then a solution of ethyl diazoacetate (0.90 g, 7.89 mmol) in CH2Cl2 (4 mL) was added dropwise. Once gas evolution ceased, the reaction mixture was stirred for 2 hr, then concentrated to half, and conc. H2SO4 (0.7 mL was added to the mixture. After 30 min, the acidic mixture was neutralized with Na2CO3 (aq.). After completion of the reaction, water was added to crude mixture and extracted with CH2Cl2, and the solvent was evaporated. Crude reaction mixture was purified by silica gel column chromatography (10% ethyl acetate–hexane) and provided the desired product 30 in 1.05 g, 73% yield as a pale yellow viscous liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.23 (s, 1 H), 7.55 (d, J = 2.60 Hz, 1 H), 7.42 (d, J = 8.90 Hz, 1 H), 6.97 (dd, J = 9.00, 2.60 Hz, 1 H), 4.43 (q, J = 7.10 Hz, 2 H), 3.90 (s, 3 H), 1.44 (t, J = 7.10 Hz, 3 H); MS (LCMS): m/z 220.83 (M)+.
Ethyl 2-iodo-5-methoxybenzofuran-3-carboxylate (31): To a mixture of compound 30 (1 g, 4.54 mmol) and iodine (1.44 g, 11.35 mmol) in 12 mL dry THF under N2 gas was dropwisely added LDA solution (15.90 mL, 1 M in THF) at −78°C and stirred at this temperature for 1 hr. After completion of the reaction (by LCMS) the reaction was quenched with saturated NH4Cl solution and concentrated. The residue was diluted with water and extracted by EtOAc. The organic extracts was washed with brine, and dried over NaSO4. The solvent was removed and the residue was purified by silica gel column chromatography (10% EtOAc in hexane) and afforded compound 31 (1.25 g, 80% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.50 (d, J = 2.64 Hz, 1 H), 7.39 (d, J = 9.04 Hz, 1 H), 6.90 (dd, J = 9.04, 2.65 Hz, 1 H), 4.47 (q, J = 7.16 Hz, 2 H), 3.88 (s, 3 H), 1.37 (t, J = 7.16 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 162.68, 156.93, 153.61, 126.50, 119.21, 114.05, 111.49, 108.17, 103.80, 60.61, 55.85, 14.27; MS (LCMS): m/z 346.31 (M)+.
Ethyl 2-(6-hydroxypyridin-3-yl)-5-methoxybenzofuran-3-carboxylate (32a): A mixture of compound 31 (0.20 g, 0.58 mmol), (6-hydroxypyridin-3-yl)boronic acid (0.096 g, 0.69 mmol) and Pd(PPh3)4 (0.033 g, 0.03 mmol) in 1 mL aq. Na2CO3 (0.12 g, 1.15 mmol) and dioxane (7 mL) under nitrogen was stirred at 100°C in a two necked round bottom flask for 10 hr. After cooling to room temperature, the reaction mixture was concentrated. The residue was dissolved in CH2Cl2 and washed with water and brine. The combined organic extracts were dried over anhydrous Na2SO4. The solvent was removed and the residue was purified by silica gel column chromatography (1:3 EtOAc/hexane) to give compound 32a (0.13 g, 74%) as a colorless solid. 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 2.50 Hz, 1 H), 7.99 (dd, J = 9.70, 2.50 Hz, 1 H), 7.56 (d, J = 8.90 Hz, 1 H), 7.44 (d, J = 2.50 Hz, 1 H), 6.98 (dd, J = 8.90, 2.60 Hz, 1 H), 6.47 (d, J = 9.70 Hz, 1 H), 4.35 (q, J = 7.20 Hz, 2 H), 3.83 (s, 3 H), 1.37 (t, J = 7.20 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 163.68, 162.05, 158.67, 156.97, 148.05, 140.54, 139.33, 127.74, 119.73, 113.87, 112.22, 107.89, 107.29, 105.05, 60.96, 56.02, 14.46; MS (LCMS): m/z 313.80 (M)+.
Ethyl 5-hydroxy-2-(6-hydroxypyridin-3-yl)benzofuran-3-carboxylate (33a): The title compound was obtained from 32a and boron tribromide as described for 14a in 65% yield after flash-chromatography (1:1 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 2.30 Hz, 1 H), 7.96 (dd, J = 9.70, 2.50 Hz, 1 H), 7.42 (d, J = 8.80 Hz, 1 H), 7.34 (d, J = 2.30 Hz, 1 H), 6.81 (dd, J = 8.80, 2.50 Hz, 1 H), 6.43 (d, J = 9.50 Hz, 1 H), 4.33 (q, J = 7.10 Hz, 2 H), 1.36 (t, J = 7.10 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 163.85, 162.89, 158.70, 154.92, 147.27, 140.35, 140.02, 127.83, 119.19, 114.19, 111.79, 108.02, 107.09, 106.88, 60.78, 14.53; MS (LCMS): m/z 299.76 (M)+.
Ethyl 5-hydroxy-2-(6-hydroxypyridin-3-yl)-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (34a) (TAM17): The title compound was obtained from 33a and piperidine as described for 13a in 74% yield after flash-chromatography (2% methanol in dichloromethane) as a colorless viscous liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.38 - 7.32 (m, 1 H), 8.19 (bs, 1 H), 8.01 - 7.90 (m, 1 H), 7.74 (dd, J = 9.6, 2.6 Hz, 1 H), 7.45 - 7.34 (m, 1 H), 6.83 (d, J = 8.8 Hz, 1 H), 6.47 (d, J = 9.5 Hz, 1 H), 4.36 - 4.25 (m, 2 H), 3.86 (s, 2 H), 2.48 - 2.33 (m, 4 H), 1.64 - 1.35 (m, 6 H), 1.28 (t, J = 7.20 Hz, 3 H). MS (LCMS): m/z 396.96 (M)+.
Ethyl 2-(3-fluoro-4-hydroxyphenyl)-5-methoxybenzofuran-3-carboxylate (32b): The title compound was obtained from 31 and (3-fluoro-4-hydroxyphenyl)boronic acid as described for 32a and after usual work up, crude reaction mixture was passed through a silica gel column and used as such in the next step.
Ethyl 2-(3-fluoro-4-hydroxyphenyl)-5-hydroxybenzofuran-3-carboxylate (33b): The title compound was obtained from 32b and borontribromide as described for 14a in 76% yield after flash-chromatography (1:1 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.87 (dd, J = 12.76, 1.91 Hz, 1 H), 7.67 (dd, J = 8.58, 1.25 Hz, 1 H), 7.44 (d, J = 8.80 Hz, 1 H), 7.36 (d, J = 2.35 Hz, 1 H), 7.08 (t, J = 8.80 Hz, 1 H), 6.83 (dd, J = 8.80, 2.49 Hz, 1 H), 4.33 (q, J = 7.04 Hz, 2 H), 1.34 (t, J = 7.12 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 163.69, 159.83, 154.83, 151.88, 149.49, 147.54, 127.96, 126.47, 120.77, 117.83, 117.51, 114.55, 111.94, 107.80, 107.11, 60.78, 14.45; MS (LCMS): m/z 316.64 (M)+.
Ethyl 2-(3-fluoro-4-hydroxyphenyl)-5-hydroxy-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylate (34b): The title compound was obtained from 33a and piperidine as described for 13a in 74% yield after flash-chromatography (1% methanol in dichloromethane) as an amorphous pale yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.52 (dd, J = 12.32, 2.10 Hz, 1 H), 7.44 - 7.32 (m, 2 H), 7.14 - 7.05 (m, 1 H), 6.82 (d, J = 8.80 Hz, 1 H), 4.32 (q, J = 7.04 Hz, 2 H), 3.76 (s, 2 H), 2.46 - 2.25 (m, 4 H), 1.33 - 1.28 (m, 6 H), 1.26 (t, J = 7.04 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 165.56, 154.78, 154.27, 152.34, 149.95, 147.48, 125.92, 124.70, 120.70, 118.47, 115.70, 114.82, 114.08, 110.77, 110.15, 61.64, 55.49, 53.73 (2C), 25.81 (2C), 24.20, 14.22; MS (LCMS): m/z 413.93 (M)+.
2-(3-fluoro-4-hydroxyphenyl)-5-hydroxy-4-(piperidin-1-ylmethyl)benzofuran-3-carboxylic acid (35b): The title compound was obtained from 34b with aqueous NaOH as described for 15a. Crude reaction mixture was passed through a bed of silica gel and used as such in the next step.
2-(3-fluoro-4-hydroxyphenyl)-5-hydroxy-N-methyl-4-(piperidin-1-ylmethyl)benzofuran-3-carboxamide (37b) (TAM18): The title compound was obtained from 35b as described for 14a in 74% yield after flash-chromatography (2% methanol in dichloromethane) as an amorphous pale brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.64 (d, J = 4.7 Hz, 1 H), 7.51 - 7.32 (m, 3 H), 7.09 (t, J = 8.8 Hz, 1 H), 6.75 (d, J = 8.8 Hz, 1 H), 3.76 (s, 2 H), 2.81 (d, J = 4.5 Hz, 3 H), 3.50 - 2.28 (m, 4 H), 1.66 - 1.34 (m, 6 H); 13C NMR (100 MHz, DMSO-d6) δ 166.06, 154.59, 152.55, 150.79, 150.15, 147.21, 146.58, 146.46, 126.99, 123.08, 121.48, 118.76, 114.38, 113.52, 110.71, 55.91, 53.76 (2C), 26.68 (2C), 25.78, 23.95; MS (LCMS): m/z 398.82 (M)+.
Synthesis of TAM21-TAM24
Ethyl 5-hydroxy-4-((3-(hydroxymethyl)piperidin-1-yl)methyl)-2-phenylbenzofuran-3-carboxylate (TAM21): To the solution of 12a (0.54 g, 3.0 mmol) in EtOH (10 mL) was added formalin (37%, 1.2 eq.) and 3-piperidinemethanol (0.42 g, 3.75 mmol). The reaction mixture was stirred for approximately 8h at 80°C. The solution was then cooled to room temperature and diluted with water, and extracted with dichloromethane. The organic layer was dried over Na2SO4 and concentrated in vacuo. Crude residue was purified by silica gel column chromatography (40% ethyl acetate in n-hexane) and provided the desired product (1.11 g, 90% yield) as a white amorphous powder. 1H NMR (400 MHz, DMSO) δ 7.71 (dd, J = 7.9, 1.3 Hz, 2H), 7.58 – 7.46 (m, 3H), 7.42 (d, J = 8.8 Hz, 1H), 6.85 (d, J = 8.8 Hz, 1H), 4.32 (q, J = 7.1 Hz, 2H), 3.90 – 3.70 (m, 2H), 3.27 (dd, J = 10.6, 5.2 Hz, 1H), 3.21 – 3.08 (m, 1H), 2.88 (d, J = 9.8 Hz, 1H), 2.75 (d, J = 10.8 Hz, 1H), 1.94 (t, J = 10.6 Hz, 1H), 1.70 (t, J = 10.6 Hz, 1H), 1.61 (m, 3H), 1.36 (dd, J = 25.8, 13.6 Hz, 1H), 1.23 (t, J = 7.1 Hz, 3H), 0.91 (dd, J = 20.7, 11.2 Hz, 1H).13C NMR (101 MHz, DMSO) δ 165.04, 154.80, 153.73, 147.40, 129.73, 129.04, 128.70, 127.17, 125.31, 114.65, 113.93, 110.80, 110.42, 64.10, 61.22, 56.39, 54.74, 53.26, 38.71, 26.77, 24.48, 13.68; MS (LCMS): m/z 409.94 (M+).
Ethyl 5-hydroxy-4-((2-(hydroxymethyl)piperidin-1-yl)methyl)-2-phenylbenzofuran-3-carboxylate (TAM22): To the solution of 12a (0.28 g, 1.0 mmol) in EtOH (3 mL) was added formalin (37%, 1.2 eq.) and 2-piperidinemethanol (0.14 g, 1.25 mmol). The reaction mixture was stirred for approximately 8h at 80°C. The solution was then cooled to room temperature and diluted with water, and extracted with dichloromethane. The organic layer was dried over Na2SO4 and concentrated in vacuo. Crude residue was purified by silica gel column chromatography (40% ethyl acetate in n-hexane) and provided the desired product (0.35 g, 86% yield) as a yellowish amorphous powder. 1H NMR (400 MHz, DMSO) δ 7.70 (d, J = 7.1 Hz, 2H), 7.58 – 7.45 (m, 3H), 7.42 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 8.8 Hz, 1H), 4.46 (d, J = 14.6 Hz, 1H), 4.34 (q, J = 7.0 Hz, 2H), 3.77 (d, J = 14.6 Hz, 1H), 3.60 (d, J = 4.6 Hz, 2H), 2.78 (d,J = 11.7 Hz, 1H), 2.45 (d, J = 4.3 Hz, 1H), 2.30 – 2.12 (m, 1H), 1.59 – 1.44 (m, 2H), 1.46 (dd, J = 21.9, 13.6 Hz, 2H), 1.44 – 1.29 (m, 2H), 1.24 (t, J = 7.1 Hz, 3H).13C NMR (101 MHz, DMSO) δ 165.51, 163.23, 154.77, 154.55, 147.20, 129.80, 128.97, 128.78, 128.43, 127.02, 125.07, 114.98, 113.11, 110.54, 110.19, 61.95, 61.52, 61.19, 51.42, 50.49, 27.25, 24.11, 22.20, 13.66; m/z 409. 87 (M+).
Ethyl 5-hydroxy-4-((3-(hydroxymethyl)piperidin-1-yl)methyl)-2-(4-hydroxyphenyl)benzofuran-3-carboxylate (TAM23): To the solution of 12b (0.31 g, 1.0 mmol) in EtOH (3 mL) was added formalin (37%, 1.2 eq.) and 3-piperidinemethanol (0.14 g, 1.25 mmol). The reaction mixture was stirred for approximately 8h at 80°C. The solution was then cooled to room temperature and diluted with water, and extracted with dichloromethane. The organic layer was dried over Na2SO4 and concentrated in vacuo. Crude residue was purified by silica gel column chromatography (40% ethyl acetate in n-hexane) and provided the desired product (0.28 g, 66% yield). 1H NMR (400 MHz, DMSO) δ 7.54 (d, J = 8.6 Hz, 2H), 7.35 (d, J = 8.8 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 6.78 (d, J = 8.8 Hz, 1H), 4.30 (d, J = 7.1 Hz, 1H), 3.94 – 3.63 (m, 2H), 3.25 (dd, J = 10.4, 4.9 Hz, 1H), 3.22 – 3.06 (m, 1H), 2.87 (d, J = 11.5 Hz, 1H), 2.74 (d, J = 11.1 Hz, 1H), 1.93 (t, J = 11.0 Hz, 1H), 1.83 – 1.48 (m, 4H), 1.36 (dd, J = 24.3, 9.8 Hz, 1H), 1.24 (t, J = 7.1 Hz, 3H), 0.90 (d, J = 9.7 Hz, 1H).13C NMR (101 MHz, DMSO) δ 165.85, 164.56, 159.56, 156.19, 154.20, 147.49, 130.05, 129.42, 126.12, 120.30, 116.05, 114.39, 114.08, 110.65, 109.32, 64.62, 61.56, 56.90, 55.45, 53.77, 39.24, 27.29, 25.00, 14.29; MS (LCMS): m/z 425.97 (M+).
5-Hydroxy-4-((3-(hydroxymethyl)piperidin-1-yl)methyl)-N-methyl-2-phenylbenzofuran-3-carboxamide (TAM24): To the solution of ester derivative TAM21 (0.82 g, 2.0 mmol) in EtOH (10 mL), a solution of NaOH (0.34 g, 8.43 mmol) in water (5 mL) was added and the resulting mixture was refluxed for 4 hr. After the completion of the reaction, reaction mixture was allowed to cool to room temperature and poured into ice cold water. Neutralization with dil. HCl solution resulted in precipitation of white acid. Acid so obtained was filtered, washed with water and dried and directly used for next step.To the solution of acid (0.38 g, 1.0 mmol) in dichloromethane (10 mL) was added oxalyl chloride (0.11 mL, 1.41 mmol) and N,N-dimethylformaide (2 drops) was added at 0°C and stirred at room temperature for 3h. The reaction mixture was evaporated under reduced pressure. The residue was re-dissolved in THF (10 mL) and a solution of methylamine (0.75 mL, 2.0 M in THF) was added and the mixture was stirred for 2h. After the completion of reaction, solvent was evaporated. Water was added and extracted with dichloromethane. The organic layer was dried over Na2SO4 and concentrated in vacuo. Crude residue was purified by silica gel column chromatography (1%–3% methanol in dichloromethane) to provide the desired product (0.28 g, 71% yield) as a pale yellow amorphous solid. 1H NMR (400 MHz, DMSO) δ 8.65 (d, J = 4.6 Hz, 1H), 7.77 – 7.72 (m, 2H), 7.50 (t, J = 7.5 Hz, 2H), 7.41 (dd, J = 11.8, 5.8 Hz, 2H), 6.78 (d, J = 8.8 Hz, 1H), 3.90 – 3.75 (m, 2H), 3.30 (dd, J = 10.6, 5.2 Hz, 1H), 3.19 (dd, J = 10.6, 7.1 Hz, 1H), 3.00 (d, J = 10.1 Hz, 1H), 2.94 – 2.83 (m, 1H), 2.79 s, 3H), 2.13 – 2.00 (m, 1H), 1.84 (t, J = 10.7 Hz, 1H), 1.67 (dd, J = 16.9, 6.8 Hz, 3H), 1.50 (dd, J = 16.1, 8.1 Hz, 1H), 0.98 (dd, J = 21.8, 11.7 Hz, 1H).13C NMR (101 MHz, DMSO) δ 166.06, 163.88, 154.67, 151.51, 147.51, 129.94, 129.57, 129.42, 126.93, 126.26, 114.83, 114.71, 112.69, 111.02, 64.42, 56.78, 55.82, 53.74, 39.11, 26.88, 26.71, 24.77; MS (LCMS): m/z 494.97 (M+).
Purity of TAM1 analogs as determined by HPLC using Water/Acetonitrile (0.1% Formic and 0.1% Ammonium Formate Acid).
| Compound | Rt (min) | Purity (%) |
|---|---|---|
| 9a/TAM8 | 4.83 | 97.5 |
| 9b/TAM11 | 5.47 | 97.3 |
| 13a/TAM9 | 4.18 | 97.7 |
| 14b/TAM13 | 3.87 | 96.4 |
| 15a/TAM10 | 2.93 | 97.5 |
| 16a/TAM12 | 3.48 | 97.6 |
| 17b/TAM16 | 2.98 | 98.6 |
| 18a/TAM14 | 4.64 | 96.8 |
| 20a/TAM15 | 4.71 | 97.8 |
| 26a/TAM20 | 3.83 | 99.1 |
| 34a/TAM17 | 2.76 | 96.2 |
| 37b/TAM18 | 2.84 | 98.3 |
| TAM21 | 3.26 | 97.5 |
| TAM22 | 3.37 | 96.8 |
| TAM23 | 2.76 | 97.8 |
| TAM24 | 2.68 | 98.9 |
Quantification and Statistical Analysis
All potency determinations and dose response curves were produced using Prism 5.0 (GraphPad Software). Animal efficacy data were analyzed using Prism 4.0 (GraphPad) and SigmaPlot 11.0 (Jandel Corporation) software. Number of samples and statistical tests used are provided in the STAR Methods.
Data and Software Availability
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession codes PDB: 5V3W (Apo Pks13-TE), PDB: 5V3X (Pks13-TE:TAM1), PDB: 5V3Y (Pks13-TE:TAM16), PDB: 5V3Z (Pks13-TE(D1607N)), PDB: 5V40 (Pks13-TE:TAM6), PDB: 5V41 (Pks13-TE:TAM5), and PDB: 5V42 (Pks13-TE:TAM3).
Author Contributions
A.A. designed and performed biochemical, whole-cell, and X-ray crystallography experiments; analyzed and interpreted data; and participated in writing the manuscript. M.K.P. designed and optimized synthetic strategies, selected commercial analogs, and contributed to manuscript writing; M.K.P. and J.C.S. designed inhibitors; N.S. performed physicochemical and PK studies; D.W. and S.G. developed animal use protocol and assisted in PK studies; E.C.B., P.K., F.A.S., and P.D.v.H. determined the MIC on drug-resistant strains; N.K.D., L.W., C.H., G.T.R., P.C.K., and A.J.L. conducted mouse efficacy studies and interpreted data; C.W., D.M., and D.F. contributed to SAR data analysis, analog design, and synthetic strategies; A.A., S.W., and E.J.R. isolated resistance mutants; O.E., S.N., J.R., and P.S. performed DMPK studies; T.S. performed THP-1 infection experiments; R.v.d.M. performed whole-genome sequencing data analysis of clinical strains from South Africa; R.S. assisted with biochemical assays and MIC determination; R.C.D. synthesized analogs; T.R.I. performed whole-genome sequencing data analysis and contributed to the writing; J.C.S. designed and supervised the study, analyzed and interpreted data, and co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Acknowledgments
This work was supported by grants from the Bill and Melinda Gates Foundation (OPP1024055), NIAID-NIH (TB Structural Genomics grant P01A1095208), Welch foundation (A-0015), and Structure-guided Drug Discovery Coalition (OPP1032548) to J.C.S.; the NIH/NIAID IDIQ Contract Task Order HHSN272201000009I/HHSN27200001 (Task A04) for the efficacy testing to A.J.L. The authors thank Midwest Center for Structural Genomics for providing LIC vectors; the staff at beamlines 19-ID and 23-ID of the Advanced Photon Source, Argonne National Laboratory for assistance during the X-ray data collection; scientists from AbbVie who provided in vitro safety data: M. Srikumaran, M.D. Kurnick, and G.M. Freiberg (hERG and enzyme/receptor assays) and J. Nicolette and A. Kondratiuk (Ames assay); K.D. Read from DDU for support and discussions. We also acknowledge the following for technical assistance: K. Loesch, W. Dong (HDF assays); H.L. Huang (MIC assays); A. Srivastava (genome sequencing of resistant mutants); L.M. Guthrie (PK studies); F. Zhou and M.K. Gupta (TAM16 purification); WuXi Apptec (TAM16 synthesis and purification); and S. Courtright (manuscript editing). E.C.B and P.K thank Rico Hömke for expert technical assistance and the University of Zurich for continuous support. R.v.d.M., F.A.S., and P.D.v.H. thank the South African MRC and NRF for funding assistance. The authors declare no competing financial interests. J.C.S, A.A., and M.K.P. are listed as inventors on a patent relating to this work filed by Texas A&M University.
Published: June 29, 2017
Footnotes
Supplemental Information includes five figures and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.06.025.
An audio PaperClip is available at http://dx.doi.org/10.1016/j.cell.2017.06.025#mmc3.
Supplemental Information
This excel file contains all of the drug-resistance conferring mutations found from whole genome sequencing in each of the clinical strains tested in this study.
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alelyunas Y.W., Liu R., Pelosi-Kilby L., Shen C. Application of a Dried-DMSO rapid throughput 24-h equilibrium solubility in advancing discovery candidates. Eur. J. Pharm. Sci. 2009;37:172–182. doi: 10.1016/j.ejps.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A., Dubnau E., Quemard A., Balasubramanian V., Um K.S., Wilson T., Collins D., de Lisle G., Jacobs W.R., Jr. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- Barrero A.F., del Moral J.F.Q., Herrador M.M., Arteaga P., Cortés M., Benites J., Rosellón A. Mild and rapid method for the generation of o-quinone methide intermediates. Synthesis of puupehedione analogues. Tetrahedron. 2006;62:6012–6017. [Google Scholar]
- Barry C.E., 3rd, Lee R.E., Mdluli K., Sampson A.E., Schroeder B.G., Slayden R.A., Yuan Y. Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid Res. 1998;37:143–179. doi: 10.1016/s0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- Bellizzi J.J., 3rd, Widom J., Kemp C., Lu J.Y., Das A.K., Hofmann S.L., Clardy J. The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proc. Natl. Acad. Sci. USA. 2000;97:4573–4578. doi: 10.1073/pnas.080508097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein J., Lott W.A., Steinberg B.A., Yale H.L. Chemotherapy of experimental tuberculosis. V. Isonicotinic acid hydrazide (nydrazid) and related compounds. Am. Rev. Tuberc. 1952;65:357–364. doi: 10.1164/art.1952.65.4.357. [DOI] [PubMed] [Google Scholar]
- Bhatt A., Molle V., Besra G.S., Jacobs W.R., Jr., Kremer L. The Mycobacterium tuberculosis FAS-II condensing enzymes: their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 2007;64:1442–1454. doi: 10.1111/j.1365-2958.2007.05761.x. [DOI] [PubMed] [Google Scholar]
- Bhusal Y., Shiohira C.M., Yamane N. Determination of in vitro synergy when three antimicrobial agents are combined against Mycobacterium tuberculosis. Int. J. Antimicrob. Agents. 2005;26:292–297. doi: 10.1016/j.ijantimicag.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Bosne-David S., Barros V., Verde S.C., Portugal C., David H.L. Intrinsic resistance of Mycobacterium tuberculosis to clarithromycin is effectively reversed by subinhibitory concentrations of cell wall inhibitors. J. Antimicrob. Chemother. 2000;46:391–395. doi: 10.1093/jac/46.3.391. [DOI] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra T., Gokhale R.S. Polyketide versatility in the biosynthesis of complex mycobacterial cell wall lipids. Methods Enzymol. 2009;459:259–294. doi: 10.1016/S0076-6879(09)04612-6. [DOI] [PubMed] [Google Scholar]
- Chou T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Chovancova E., Pavelka A., Benes P., Strnad O., Brezovsky J., Kozlikova B., Gora A., Sustr V., Klvana M., Medek P. CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012;8:e1002708. doi: 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E., 3rd Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Donnelly M.I., Zhou M., Millard C.S., Clancy S., Stols L., Eschenfeldt W.H., Collart F.R., Joachimiak A. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr. Purif. 2006;47:446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta N.K., Pinn M.L., Karakousis P.C. Reduced emergence of isoniazid resistance with concurrent use of thioridazine against acute murine tuberculosis. Antimicrob. Agents Chemother. 2014;58:4048–4053. doi: 10.1128/AAC.02981-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Franzblau S.G., Witzig R.S., McLaughlin J.C., Torres P., Madico G., Hernandez A., Degnan M.T., Cook M.B., Quenzer V.K., Ferguson R.M., Gilman R.H. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat J.F., Madej T., Bryant S.H. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Glickman M.S., Cox J.S., Jacobs W.R., Jr. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol. Cell. 2000;5:717–727. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- Guardia A., Gulten G., Fernandez R., Gómez J., Wang F., Convery M., Blanco D., Martínez M., Pérez-Herrán E., Alonso M. N-Benzyl-4-((heteroaryl)methyl)benzamides: a new class of direct NADH-dependent 2-trans enoyl-acyl carrier protein reductase (InhA) inhibitors with antitubercular activity. ChemMedChem. 2016;11:687–701. doi: 10.1002/cmdc.201600020. [DOI] [PubMed] [Google Scholar]
- Guo X., Yu R., Li H., Li Z. Iron-catalyzed tandem oxidative coupling and annulation: an efficient approach to construct polysubstituted benzofurans. J. Am. Chem. Soc. 2009;131:17387–17393. doi: 10.1021/ja907568j. [DOI] [PubMed] [Google Scholar]
- Hayashi K., Yamazoe A., Ishibashi Y., Kusaka N., Oono Y., Nozaki H. Active core structure of terfestatin A, a new specific inhibitor of auxin signaling. Bioorg. Med. Chem. 2008;16:5331–5344. doi: 10.1016/j.bmc.2008.02.085. [DOI] [PubMed] [Google Scholar]
- He S., Li P., Dai X., McComas C.C., Du C., Wang P., Lai Z., Liu H., Yin J., Bulger P.G. Facile functionalization at the C2 position of a highly substituted benzofuran. Tetrahedron Lett. 2014;55:2212–2216. [Google Scholar]
- Heym B., Alzari P.M., Honoré N., Cole S.T. Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol. Microbiol. 1995;15:235–245. doi: 10.1111/j.1365-2958.1995.tb02238.x. [DOI] [PubMed] [Google Scholar]
- Ioerger T.R., O’Malley T., Liao R., Guinn K.M., Hickey M.J., Mohaideen N., Murphy K.C., Boshoff H.I.M., Mizrahi V., Rubin E.J. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS ONE. 2013;8:e75245. doi: 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins H.E., Zignol M., Cohen T. Quantifying the burden and trends of isoniazid resistant tuberculosis, 1994-2009. PLoS ONE. 2011;6:e22927. doi: 10.1371/journal.pone.0022927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson T., Jurva U., Grönberg G., Weidolf L., Masimirembwa C. Novel metabolites of amodiaquine formed by CYP1A1 and CYP1B1: structure elucidation using electrochemistry, mass spectrometry, and NMR. Drug Metab. Dispos. 2009;37:571–579. doi: 10.1124/dmd.108.025171. [DOI] [PubMed] [Google Scholar]
- Kamerbeek J., Schouls L., Kolk A., van Agterveld M., van Soolingen D., Kuijper S., Bunschoten A., Molhuizen H., Shaw R., Goyal M., van Embden J. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 1997;35:907–914. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M.H., Biermann K., Tandberg S., Hsu T., Jacobs W.R., Jr. Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. 2007;6 doi: 10.1002/9780471729259.mc10a02s6. A:10A.2:10A.2.1–10A.2.21. [DOI] [PubMed] [Google Scholar]
- Lechartier B., Hartkoorn R.C., Cole S.T. In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012;56:5790–5793. doi: 10.1128/AAC.01476-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechartier B., Rybniker J., Zumla A., Cole S.T. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med. 2014;6:158–168. doi: 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U.H., S. Rao S.P., Kondreddi R.R., Noble C.G., Camacho L.R., Tan B.H., Ng S.H., Ng P.S., Ma N.L., Lakshminarayana S.B. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci. Transl. Med. 2015;7:269ra3. doi: 10.1126/scitranslmed.3010597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto J., Ishizu M., Kawano R.-i., Hesaka D., Shiragami T., Hayashi Y., Yamashita T., Yasuda M. Generation of quinone methide from aminomethyl (hydroxy) arenes precursors in aqueous solution. Tetrahedron. 2005;61:5735–5740. [Google Scholar]
- Matthews B.W. Solvent content of protein crystals. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- Monti S.A., Castillo G.D. Reverse Mannich reaction of some 5-hydroxyindoles. J. Org. Chem. 1970;35:3764–3767. [Google Scholar]
- Moriarty N.W., Grosse-Kunstleve R.W., Adams P.D. Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothe S.R., Susanti D., Chan P.W.H. Efficient synthesis of 3-acyl-5-hydroxybenzofurans via copper (II) triflate-catalyzed cycloaddition of unactivated 1, 4-benzoquinones with 1, 3-dicarbonyl compounds. Tetrahedron Lett. 2010;51:2136–2140. [Google Scholar]
- Nardini M., Dijkstra B.W. Alpha/beta hydrolase fold enzymes: the family keeps growing. Curr. Opin. Struct. Biol. 1999;9:732–737. doi: 10.1016/s0959-440x(99)00037-8. [DOI] [PubMed] [Google Scholar]
- Newlander K.A., Chenera B., Veber D.F., Yim N.C., Moore M.L. Simple silyl linker for the solid phase organic synthesis of aryl-containing molecules. J. Org. Chem. 1997;62:6726–6732. [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pan P., Tonge P.J. Targeting InhA, the FASII enoyl-ACP reductase: SAR studies on novel inhibitor scaffolds. Curr. Top. Med. Chem. 2012;12:672–693. doi: 10.2174/156802612799984535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda G., Parai M.K., Das S.K., Shagufta, Sinha M., Chaturvedi V., Srivastava A.K., Manju Y.S., Gaikwad A.N., Sinha S. Effect of substituents on diarylmethanes for antitubercular activity. Eur. J. Med. Chem. 2007;42:410–419. doi: 10.1016/j.ejmech.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Peterson G.A., Kunng F.-A., McCallum J.S., Wulffe W.D. Palladium catalyzed reduction of aryl triflates-utilization in thesynthesis of angelicin, olivin and chromomycinone from phenolsproduced in the benzannulation reaction of chromium carbenecomplexes. Tetrahedron Lett. 1987;28:1381–1384. [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Portevin D., De Sousa-D’Auria C., Houssin C., Grimaldi C., Chami M., Daffé M., Guilhot C. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. USA. 2004;101:314–319. doi: 10.1073/pnas.0305439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri L.E. Biosynthesis of mycobacterial lipids by polyketide synthases and beyond. Crit. Rev. Biochem. Mol. Biol. 2014;49:179–211. doi: 10.3109/10409238.2014.896859. [DOI] [PubMed] [Google Scholar]
- Richardson R.D., Smith J.W. Novel antagonists of the thioesterase domain of human fatty acid synthase. Mol. Cancer Ther. 2007;6:2120–2126. doi: 10.1158/1535-7163.MCT-07-0187. [DOI] [PubMed] [Google Scholar]
- Rozwarski D.A., Grant G.A., Barton D.H., Jacobs W.R., Jr., Sacchettini J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- Sambandamurthy V.K., Derrick S.C., Hsu T., Chen B., Larsen M.H., Jalapathy K.V., Chen M., Kim J., Porcelli S.A., Chan J. Mycobacterium tuberculosis DeltaRD1 DeltapanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine. 2006;24:6309–6320. doi: 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- Schmalhofer W.A., Swensen A.M., Thomas B.S., Felix J.P., Haedo R.J., Solly K., Kiss L., Kaczorowski G.J., Garcia M.L. A pharmacologically validated, high-capacity, functional thallium flux assay for the human Ether-à-go-go related gene potassium channel. Assay Drug Dev. Technol. 2010;8:714–726. doi: 10.1089/adt.2010.0351. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Springer B., Lucke K., Calligaris-Maibach R., Ritter C., Böttger E.C. Quantitative drug susceptibility testing of Mycobacterium tuberculosis by use of MGIT 960 and EpiCenter instrumentation. J. Clin. Microbiol. 2009;47:1773–1780. doi: 10.1128/JCM.02501-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes R.W., Doxsee D. The receptor-mediated uptake, survival, replication, and drug sensitivity of Mycobacterium tuberculosis within the macrophage-like cell line THP-1: a comparison with human monocyte-derived macrophages. Cell. Immunol. 1999;197:1–9. doi: 10.1006/cimm.1999.1554. [DOI] [PubMed] [Google Scholar]
- Streicher E.M., Victor T.C., van der Spuy G., Sola C., Rastogi N., van Helden P.D., Warren R.M. Spoligotype signatures in the Mycobacterium tuberculosis complex. J. Clin. Microbiol. 2007;45:237–240. doi: 10.1128/JCM.01429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K., Wang C., Besra G.S. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C., Klei H., Adams P.D., Moriarty N.W., Cohn J.D. Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallogr. D Biol. Crystallogr. 2006;62:915–922. doi: 10.1107/S0907444906017161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres J.N., Paul L.V., Rodwell T.C., Victor T.C., Amallraja A.M., Elghraoui A., Goodmanson A.P., Ramirez-Busby S.M., Chawla A., Zadorozhny V. Novel katG mutations causing isoniazid resistance in clinical M. tuberculosis isolates. Emerg. Microbes Infect. 2015;4:e42. doi: 10.1038/emi.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Embden J.D., Cave M.D., Crawford J.T., Dale J.W., Eisenach K.D., Gicquel B., Hermans P., Martin C., McAdam R., Shinnick T.M. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J. Clin. Microbiol. 1993;31:406–409. doi: 10.1128/jcm.31.2.406-409.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilcheze C., Jacobs W.R., Jr. Resistance to isoniazid and ethionamide in Mycobacterium tuberculosis: genes, mutations, and causalities. Microbiol. Spectr. 2014;2 doi: 10.1128/microbiolspec.MGM2-0014-2013. MGM2-0014-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C., Morbidoni H.R., Weisbrod T.R., Iwamoto H., Kuo M., Sacchettini J.C., Jacobs W.R., Jr. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 2000;182:4059–4067. doi: 10.1128/jb.182.14.4059-4067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R., Kumar P., Parashar V., Vilchèze C., Veyron-Churlet R., Freundlich J.S., Barnes S.W., Walker J.R., Szymonifka M.J., Marchiano E. Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat. Chem. Biol. 2013;9:499–506. doi: 10.1038/nchembio.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . WHO; 2014. Global Tuberculosis Report 2014. [Google Scholar]
- Zhang W., Chakravarty B., Zheng F., Gu Z., Wu H., Mao J., Wakil S.J., Quiocho F.A. Crystal structure of FAS thioesterase domain with polyunsaturated fatty acyl adduct and inhibition by dihomo-gamma-linolenic acid. Proc. Natl. Acad. Sci. USA. 2011;108:15757–15762. doi: 10.1073/pnas.1112334108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Heym B., Allen B., Young D., Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- Zumla A., Raviglione M., Hafner R., von Reyn C.F. Tuberculosis. N. Engl. J. Med. 2013;368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- Zuniga E.S., Early J., Parish T. The future for early-stage tuberculosis drug discovery. Future Microbiol. 2015;10:217–229. doi: 10.2217/fmb.14.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This excel file contains all of the drug-resistance conferring mutations found from whole genome sequencing in each of the clinical strains tested in this study.