

Abstract
Keratan sulfate (KS) is commonly elevated in urine samples from patients with mucopolysaccharidosis type IVA (MPS IVA) and is considered pathognomonic for the condition. Recently, a new method has been described by Martell et al. to detect and measure urinary KS utilizing LC-MS/MS. As a part of the validation of this method in our laboratory, we studied the sensitivity and specificity of elevated urine KS levels using 25 samples from 15 MPS IVA patients, and 138 samples from 102 patients with other lysosomal storage disorders, including MPS I (n = 9), MPS II (n = 13), MPS III (n = 23), MPS VI (n = 7), beta-galactosidase deficiency (n = 7), mucolipidosis (ML) type II, II/III and III (n = 51), alpha-mannosidosis (n = 11), fucosidosis (n = 4), sialidosis (n = 5), Pompe disease (n = 3), aspartylglucosaminuria (n = 4), and galactosialidosis (n = 1). As expected, urine KS values were significantly higher (fivefold average increase) than age-matched controls in all MPS IVA patients. Urine KS levels were also significantly elevated (threefold to fourfold increase) in patients with GM-1 gangliosidosis, MPS IVB, ML II and ML II/III, and fucosidosis. Urine KS was also elevated to a smaller degree (1.1-fold to 1.7-fold average increase) in patients with MPS I, MPS II, and ML III. These findings suggest that while the UPLC-MS/MS urine KS method is 100% sensitive for the detection of patients with MPS IVA, elevated urine KS is not specific for this condition. Therefore, caution is advised when interpreting urinary keratan sulfate results.
Electronic supplementary material: The online version of this chapter (doi:10.1007/8904_2016_1) contains supplementary material, which is available to authorized users.
Introduction
Keratan sulfate (KS) is a complex glycosaminoglycan composed of repeating disaccharides of d-galactose and N-acetyl-d-glucosamine (Zhang et al. 2005). Keratan sulfate proteoglycans have been found in the cornea, cartilage, and bones. Degradation of KS occurs in the lysosomes by a stepwise series of enzymatic reactions (Wraith 1995). Deficiency of one of the enzymes involved in this process results in storage of partially degraded KS causing cellular and organ dysfunction (Wraith 1995).
Morquio syndrome (MPS IVA; OMIM # 253000) is an autosomal recessive lysosomal storage disorder (LSD) resulting from an inability to degrade KS and chondroitin-6-sulfate. Patients typically present with short stature, kyphosis, genu valgum, enlarged organs such as liver or spleen, ocular abnormalities, and the radiological finding of dysostosis multiplex. Neurologic function is typically not affected. The age of onset is often in early childhood but milder variants may present in adolescence (Mendelsohn et al. 2013). Recently, enzyme replacement therapy with recombinant human N-acetyl-galactosamine-6-sulfatase (rhGALNS, elosulfase alfa) for the treatment of MPS IVA (Lyseng-Williamson 2014) has been approved by the Food and Drug Administration. Elevation of urinary KS is associated with MPS IVA and measurement of total urine glycosaminoglycans (GAGs) or qualitative GAG analysis to detect KS are commonly used clinical screening tests (Wood et al. 2013). However, a high false negative rate has been reported for these assays and false negative results are more common in mildly affected patients (Whitley et al. 1989; Piraud et al. 1993; Gray et al. 2007).
Oguma et al. (2001) followed by Martell et al. (2011) reported LC-MS/MS assays that measure two specific KS disaccharides after enzymatic digestion of the GAG polymer with keratanase II. These disaccharides, Gal-1-4GlcNAc(6S) (1S) and Gal(6S)-1-4GlcNAc(6S) (2S), were found to be increased in all of the MPS IVA patients suggesting this assay would be highly sensitive for the detection of patients with this condition. Additionally, using an ELISA assay, Tomatsu et al. (2005) showed that plasma KS was increased in patients with mucolipidosis type II (I cell disease; OMIM#252500) or III (OMIM#252600) as well as in patients with MPS I (OMIM#607014), II (OMIM#309900), III (OMIM#A:252900,B:252920,C:252930), VI (OMIM#253200), and VII (OMIM#253220). Elevation of urine KS has also been previously observed in patients with fucosidosis (OMIM#230000) (Greiling et al. 1978).
Here, we expand on the previous studies by analyzing 25 additional urine samples from Morquio A patients. Urinary KS was also measured in 138 urine samples from 102 patients with various LSDs: MPS I (n = 9), MPS II (n = 13), MPS III (n = 23), MPS VI (n = 7), beta-galactosidase deficiency (OMIM#230650 and 253010) (n = 7), mucolipidosis (ML) type II, II/III (n = 12), and III (n = 39), alpha-mannosidosis (OMIM#248500) (n = 11), fucosidosis (n = 4), sialidosis (OMIM#256550) (n = 5), Pompe disease (OMIM#232300) (n = 3), aspartylglucosaminuria (OMIM#208400) (n = 4), and galactosialidosis (OMIM#256540) (n = 1), as well as 105 controls. The clinical sensitivity and specificity of urine KS for the detection of MPS IVA were examined, and showed that urine KS is a more sensitive diagnostic marker than total urine GAGs for MPS IVA. Also, evaluation of urine KS levels in younger (<3 years) unaffected individuals is described to better delineate age-dependent reference ranges for this biomarker compared to previous studies. Finally, clinical laboratories should be cautioned that patients with other LSDs may show elevations in urinary KS. Therefore, while urine KS is a sensitive biomarker for the detection of MPS IVA patients, its accumulation is not specific to this condition.
Materials and Methods
Patient Samples
Samples were submitted to the Greenwood Genetic Center biochemical diagnostic laboratory for diagnostic evaluation or as part of the Longitudinal Studies of the Glycoproteinoses (NCT01891422). Analyses were performed on randomly collected urine samples from patients with mucopolysaccharidosis (MPS) I (n = 9), II (n = 13), III (n = 23), IVA (n = 18), and VI (n = 7), as well as from patients with other LSDs: beta-galactosidase deficiency (n = 7), mucolipidosis II (n = 4), II/III (n = 8), and III (n = 39), alpha-mannosidosis (n = 11), fucosidosis (n = 4), sialidosis (n = 3), Pompe disease (n = 3), aspartylglucosaminuria (n = 4), and galactosialidosis (n = 1), in whom the diagnosis was confirmed by enzyme and/or mutation analysis. Control urine samples were collected from healthy volunteers (n = 25; age: 12 months – 63 years) and patients with other, unrelated metabolic disorders (n = 80; age: 20 days to 36 years), and used to establish age-dependent, normal reference ranges. All the samples were stored at −20°C prior to use.
Urine Keratan Sulfate Measurements
Urine keratan sulfate disaccharides Galβ1-4GlcNAc(6S) (predominant species) and Gal(6S)β1-4GlcNAc(6S) were produced by keratanase II enzymatic digestion of keratan sulfate (KS) as described previously (Oguma et al. 2001; Martell et al. 2011). The internal standards (heavy-isotope-labeled Galβ1-4GlcNAc(6S) 13C6, and Gal(6S)β1-4GlcNAc(6S) 13C6), the reference standards (Galβ1-4GlcNAc(6S) and Gal(6S)β1-4GlcNAc(6S)), KS (Bovine cornea, Na salt), and keratanase II (Bacillus sp.) were all obtained from GlycoSyn, Lower Hutt, New Zealand. Urine keratan sulfate was detected via stable isotope dilution using ultra performance liquid chromatography (UPLC), utilizing a Thermo Hypercarb 5 μm 50 × 2.1 mm column, and tandem mass spectrometry (MS/MS), utilizing an Acquity/Xevo TQD instrument (Waters Corporation, Milford, MA). Transitions (Galβ1-4GlcNAc(6S): 462.05 → 97.01, 13C6-Galβ1-4GlcNAc(6S): 467.78 → 97.01, Gal(6S)β1-4GlcNAc(6S): 563.87 → 462.69) 13C6-Gal(6S)β1-4GlcNAc(6S): 569.75 → 468.81) were monitored by multiple reaction monitoring in negative ion mode. The monosulfated and the disulfated disaccharides of KS were calculated by means of separate standard curves, and the final KS concentration was reported as the sum of both. The standard curves for both KS disaccharides included concentrations of 0.103, 0.313, 0.625, 1.25, 2.5, 5.0, 10.0, and 20.0 μg/ml. The average inter-day precision in five samples analyzed was 9.1% and 8% for monosulfated KS and disulfated KS, respectively. The average intra-day precision in five samples analyzed was 4.9% and 5.9% for monosulfated KS and disuflated KS, respectively. The lower limit of quantification (LLOQ) is 0.75 ug/ml. The average percent deviation at the LLOQ is −10.87% (for monosulfated KS) and −4.74% (for disulfated KS), and the intra-day variability at the LLOQ is 9.08% (for monosulfated KS) and 14.91% (for disulfated KS).
Urine Total Glycosaminoglycans Analysis
GAGs were measured in 25 samples from 15 MPS IVA patients using the Blyscan kit (1,9-dimethyl-methylene blue staining (DMB)) (Blyscan Sulfated Glycosaminoglycan Assay Biocolor Ltd. Northern Ireland, UK) and comparing absorbance at 656 nm to that of a standard curve. Age-related reference ranges were developed previously in the laboratory.
Statistical Analysis
KS values in urine samples from patients with MPS IVA or other LSDs were normalized to the upper limit of the appropriate age-specific normal reference range. An unpaired t test was used to compare normalized KS levels in the samples from patients with MPS IVA with those from control samples, and from patients with other LSDs. Values were considered statistically significant at p < 0.05.
Results
Establishing Normal Reference Ranges
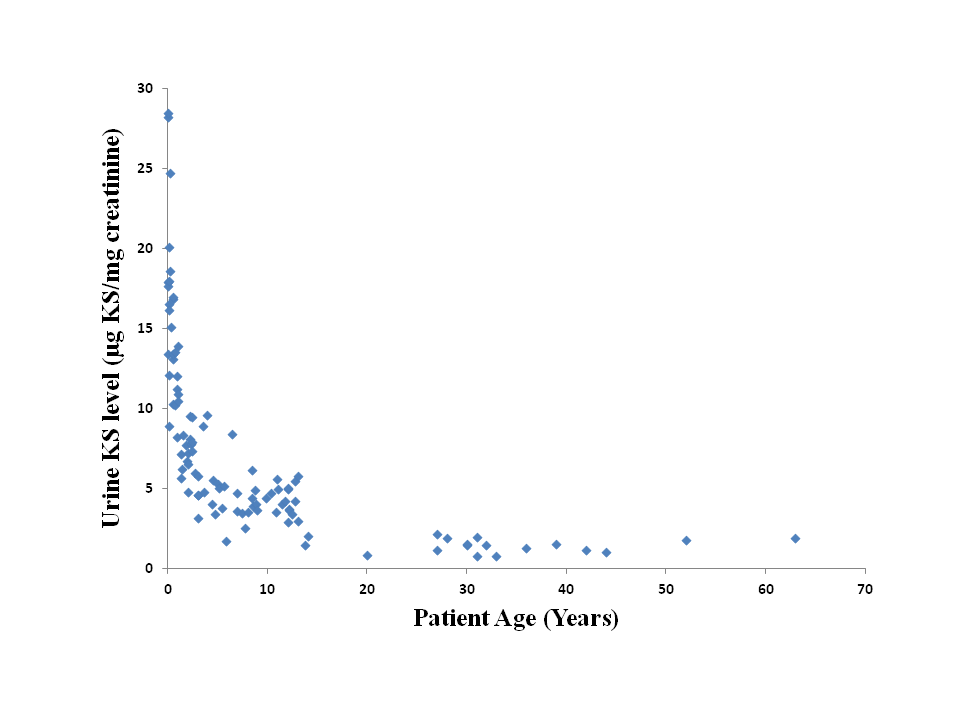
In order to establish age-dependent urine KS normal reference ranges, urine KS was measured in 105 unaffected controls (20 days – 63 years of age). It was observed that urine KS levels decrease with age (Supplementary Fig. 1), as has been previously observed by Martell et al. (2011). This phenomenon was also observed for GAGs (de Jong et al. 1989; Whitley et al. 1989; Piraud et al. 1993; Gray et al. 2007; Wood et al. 2012). Therefore, it was important to establish age-dependent reference ranges to accurately interpret KS levels in patient samples. Decrease in urine KS was especially evident in patients under 3 years of age. Therefore, this patient population was divided further into three age groups: 0–4 months of age (n = 14), 5–18 months of age (n = 16), and 19–36 months of age (n = 14). Urine KS values observed in these age groups were 8.9–28.5; 5.6–17.0; and 4.8–9.6 μg urine KS/mg creatinine, respectively. Further age-dependent normal ranges included 3–5 years of age (1.7–9.6 μg urine KS/mg creatinine), 6–10 years of age (2.5–6.2 μg urine KS/mg creatinine), 11–14 years of age (1.5–5.8 μg urine KS/mg creatinine), and >14 years of age (0.8–2.1 μg urine KS/mg creatinine). The cut-off values (or the upper limit of normal) referred to in this manuscript represent the highest values observed in each of the age-groups, excluding any outliers. In order to compare individual patient results, urine keratan sulfate values were normalized to the upper limit of the appropriate age-specific normal reference range.
Urine KS in MPS IVA Patients
Urine KS levels were measured in 25 samples from 15 Morquio syndrome type A patients (Fig. 1). As expected, all of the MPS IVA patient samples had elevated urine keratan sulfate levels that ranged from a 1.22–8.27 fold increase (average 4.51-fold increase), when normalized to the upper limit of the age-appropriate normal range.
Fig. 1.
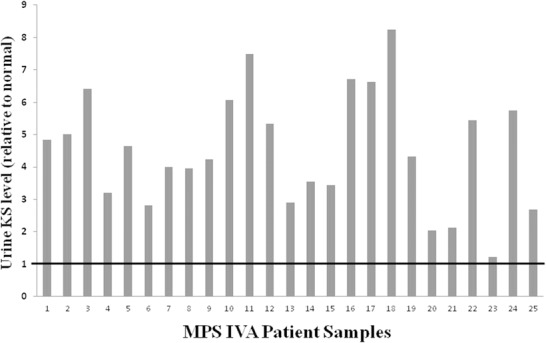
Urine keratan sulfate is elevated in all MPS IVA patients. Urine keratan sulfate level in 25 MPS IVA patient samples is shown relative to the upper limit of the appropriate age-specific reference range (indicated by the bold black line) such that the Y axis value represents the fold elevation of urine KS in each patient
The sensitivity of urine KS analysis versus standard total urine GAG analysis was compared for the detection of patients with MPS IVA (Table 1). Whereas urine KS was elevated in all 25 MPS IVA samples (100% sensitivity), total urine GAGs were elevated in only 20 of the 25 MPS IVA samples (83% sensitivity), when compared to the appropriate age-specific reference range. The degree to which urine KS levels were elevated (average 4.51-fold) was also significantly higher than that for total urine GAGs (average 1.63-fold; P < 10−8). This demonstrates that the quantification of urine KS is a more sensitive biomarker than total urine GAGs for the diagnosis of MPS IVA.
Table 1.
Urine keratan sulfate (KS) and glycosaminoglycan (GAG) measurements from 25 MPS IVA patients
| Age (Yr) | Total urine GAGs (mg/mmol crnn) | Urine KS (μg/mg crnn) |
|---|---|---|
| 2 | 27.21 (<24) | 46.51 (<9.6) |
| 2 | 30.39 (<24) | 48.14 (<9.6) |
| 2 | 28.32 (<24) | 61.51 (<9.6) |
| 3 | 33.80 (<16) | 30.68 (<9.6) |
| 3 | 46.02 (<16) | 44.62 (<9.6) |
| 4 | 16.70 (<16) | 25.83 (<9.6) |
| 5 | 19.75 (<16) | 27.13 (<9.6) |
| 5 | 26.45 (<16) | 38.36 (<9.6) |
| 5 | 13.68 (<16) | 38.03 (<9.6) |
| 6 | 22.02 (<12) | 26.32 (<6.2) |
| 6 | 30.89 (<12) | 37.66 (<6.2) |
| 6 | 10.71 (<12) | 46.48 (<6.2) |
| 6 | 20.08 (<12) | 33.07 (<6.2) |
| 6 | 25.35 (<12) | 18.00 (<6.2) |
| 6 | 25.33 (<12) | 21.98 (<6.2) |
| 7 | 11.01 (<12) | 21.31 (<6.2) |
| 7 | 21.51 (<12) | 41.67 (<6.2) |
| 7 | 24.42 (<12) | 41.13 (<6.2) |
| 7 | 25.89 (<12) | 51.04 (<6.2) |
| 9 | 26.55 (<12) | 26.79 (<6.2) |
| 10 | 12.44 (<12) | 12.64 (<6.2) |
| 10 | 9.57 (<12) | 13.14 (<6.2) |
| 10 | 20.92 (<12) | 33.79 (<6.2) |
| 14 | 7.57 (<6.5) | 7.09 (<5.8) |
| >15 | 12.12 (<6.5) | 12.34 (<2.2) |
Values in parentheses are the appropriate age-specific upper limit of the reference range and patient values within the normal range are in bold font. Total GAGs were measured by DMB incorporation and keratan sulfate by LC-MS/MS. All 25 samples had elevated urine KS but only 21/25 (83%) had elevated total GAGs
Urine KS in Patients with Other Lysosomal Storage Disorders
In order to assess the clinical specificity of elevated urine KS levels and the clinical utility of urine KS as a screening biomarker for MPS IVA, urine KS was measured in samples from patients diagnosed with LSDs other than MPS IVA, including selected mucopolysaccharidoses, mucolipidosis types II, II/III, III and other glycoproteinoses, Pompe disease, and beta-galactosidase deficiency.
Selected Mucopolysaccharidoses
Urinary KS was measured in samples from patients with MPS I (n = 9), MPS II (n = 13), MPS III (n = 23), and MPS VI (n = 7) (Fig. 2). Urine KS levels in patients with MPS I and MPS II were on average 1.1 and 1.7 fold elevated, respectively, relative to the appropriate age-specific normal range. Specifically 3/9 MPS I and 9/13 MPS II patient samples had elevated urine KS. These elevations, though mild, were still statistically significant (MPS I, P < 0.05, and MPS II, P < 0.005); however these elevations were significantly lower than in patients with MPS IVA (P < 10−7). On average MPS III and MPS VI patients were found to have normal average urine KS levels; however, 3/23 MPS III and 1/7 MPS VI patient samples had elevated urine KS levels (1.11-fold to 2.33-fold, and 1.9-fold, respectively).
Fig. 2.
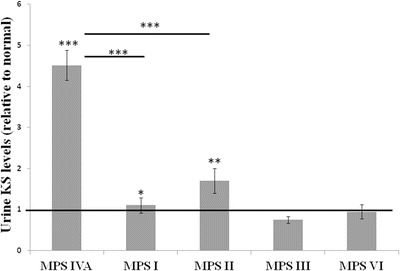
Mild elevations of urine KS observed in other MPS disorders. Average urine keratan sulfate level for patients with five different MPS disorders is shown relative to the upper limit of the appropriate age-specific reference range (indicated by the bold black line). The Y axis represents the fold elevation of urine KS in each patient population. Patients with MPS I and MPS II on average had higher urine KS levels than controls, but significantly lower levels than MPS IVA patients. Error bars represent standard error of the means. MPS I-VI = mucopolysaccharidosis I-VI. * = P < 0.05; ** = P < 0.01; *** = P < 0.001. Statistical analysis performed by T-test
GNPTAB: Associated Mucolipidoses
Based on the previous report by Tomatsu et al. (2005) that KS could also be elevated in patients with mucolipidoses (ML), as well as the fact that our laboratory found total urinary GAGs to be elevated in a subset of ML II, II/III, and III patients (data not published), urine KS levels were evaluated in a large cohort of samples from patients with ML II (n = 4), II/III (n = 8), and III (n = 39). We observed that patients with a severe skeletal phenotype, specifically individuals with ML II and ML II/III, had significantly higher fold elevations of urine KS compared to unaffected individuals (P < 10−5) (Fig. 3). Moreover, in these patients, urine KS levels were as elevated (approximately fourfold) as those observed in patients with MPS IVA (P = 0.13). Patients with ML III, who present with a milder skeletal phenotype as compared to ML II and ML II/III patients, also had significantly elevated urine KS levels, as compared to unaffected patients (P < 10−5); however, 16/39 ML III patients had normal urine KS. Urine KS level in ML III patient samples was significantly lower (on average 1.3-fold elevated) than urine KS levels observed in ML II and ML II/III (P < 10−5), or MPS IVA patients (P < 10−7) (Fig. 3).
Fig. 3.
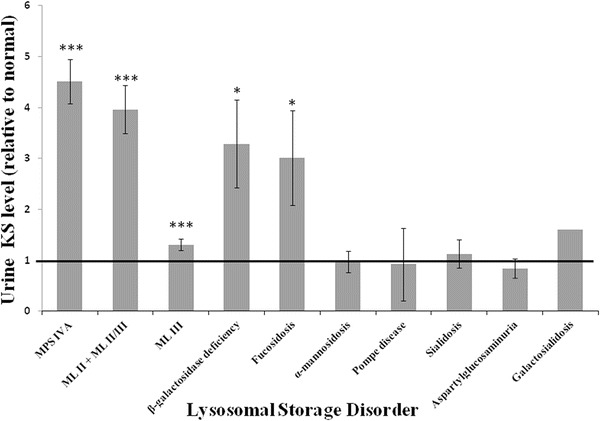
Urine KS in patients with various lysosomal storage disorders. The Y axis represents the average fold elevation relative to the upper limit of the age-specific reference range of urine KS (indicated by the bold black line) in each patient population. Patients with ML, fucosidosis, and beta-galactosidase deficiency have significantly higher urine KS levels than controls. No significant difference was observed between MPSIVA and ML II + II/III, fucosidosis, and beta-galactosidase deficiency patient samples. Error bars represent standard error of the means. * = P < 0.05; *** = P < 0.001; ### = P < 0.001. Statistical analysis performed by T-test
Other Lysosomal Storage Disorders
Urine KS levels were also measured in a cohort of patients with various glycoproteinoses, including alpha-mannosidosis (n = 10), fucosidosis (n = 2; four samples), sialidosis (n = 4; five samples), aspartylglucosaminuria (n = 3; four samples), and galactosialidosis (n = 1), as well as in patients with beta-galactosidase deficiency (n = 6; seven samples) and Pompe disease (n = 3). Urine KS levels varied depending on the disorder, with the highest urine KS levels present in samples from patients with fucosidosis and beta-galactosidase deficiency (Fig. 3). These patients’ urine KS levels were significantly elevated (approximately threefold), as compared to age-appropriate normal controls (fucosidosis P = 0.04; beta-galactosidase deficiency P = 0.01), and there was no significant difference between urine KS levels in these patients compared to those in patients with MPS IVA (P = 0.10 and P = 0.11, respectively). One sample from a patient with galactosialidosis demonstrated 1.6-fold elevated urine KS level compared to the age-appropriate reference range. Average urine KS levels in patients with alpha-mannosidosis, Pompe disease, sialidosis, and aspartylglucosaminuria were essentially normal.
Discussion
Elevated keratan sulfate is a well-established biochemical feature of MPS IVA, in which N-acetyl-galactosamine-6-sulfatase deficiency results in impaired catabolism of this glycosaminoglycan (Glossl and Kresse 1978; Yutaka et al. 1982). In the present study, an ultraperformance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) method was utilized to examine the clinical sensitivity and specificity of urine KS analysis for patients with not only MPS IVA, but also for patients with other LSDs. We also, for the first time, delineated urine KS ranges for unaffected individuals under the age of 3 years.
Previous reports have documented the age-dependent decrease in keratan sulfate levels (de Jong et al. 1989; Whitley et al. 1989; Piraud et al. 1993; Gray et al. 2007; Martell et al. 2011; Wood et al. 2012). However, these studies had a limited number of samples for patients under the age of 3 years. Our data demonstrate that patients within this age group (n = 43) display the most dramatic age-dependent decrease in keratan sulfate (Supplementary Fig. 1). Therefore, three distinct normal ranges within this cohort of patients were created: 8.9–28.5 μg/mg creatinine (0–4 months), 5.6–17 μg/mg creatinine (5–18 months), and 4.8–9.6 μg/mg creatinine (19–36 months). Having proper age-specific normal ranges is imperative for the accurate interpretation of urine keratan sulfate results, especially in young children.
Urine KS levels were elevated in all MPS IVA patient samples analyzed (n = 25) (Fig. 1). However, GAGs were elevated in only 83% of the same MPS IVA patient samples (Table 1). False negative results have been well documented for patients with MPS IVA using either the DMB incorporation assay for the quantitative measurement of total urine GAGs or the qualitative separation of individual GAG species by electrophoresis or thin layer chromatography (Whitley et al. 1989; Piraud et al. 1993; Gray et al. 2007). Quantitative measurement of urine KS via UPLC-MS/MS offers increased sensitivity for the detection of patients with MPS IVA compared to the assays commonly used by most laboratories today.
To evaluate the specificity of the urine KS assay, samples obtained from patients with other LSDs were analyzed. Urine KS levels were at least mildly elevated in patients with several other LSDs: MPS I, MPS II, fucosidosis, beta-galactosidase deficiency, and mucolipidosis II, II/III, and III (Figs. 2 and 3). Beta-galactosidase hydrolyzes terminal beta-linked galactose residues from GM1 gangliosides, glycoproteins, and from keratan sulfate (Okada and O’Brien 1968; O’Brien et al. 1976). A subset of patients with beta-galactosidase deficiency are classified as having Morquio syndrome type B (O’Brien et al. 1976); therefore, it is not surprising that patients with this enzyme deficiency have elevated urine keratan sulfate levels.
Elevated urine KS levels have previously been reported by Tomatsu et al. in patients with Mucolipidosis II or III (Tomatsu et al. 2005). Patients with these conditions have a deficiency of N-acetylglucosaminyl-1-phosphotransferase, which prevents the import of most lysosomal hydrolases into the lysosome. This leads to the accumulation of various GAG species in different tissues (Leroy et al. 1972; Thomas et al. 1973; Reitman et al. 1981), and likely explains the accumulation of urine keratan sulfate observed in these patients. However, the Tomatsu et al. study only included 11 samples, and the type of ML, or clinical severity, was not indicated. We analyzed urine KS in 51 samples from ML patients with a well-defined clinical phenotype and a confirmed diagnosis of either ML II, ML II/III, or ML III (Leroy et al. 2014; Lyseng-Williamson 2014). Urine KS was elevated in all samples from patients with ML II or ML II/III and the average urine KS level in these patients (fourfold increase relative to age-matched controls) was approximately the same as that in MPS IVA patients (4.7-fold increase). Alternatively, although on average patients with ML III have elevated urine KS levels compared to controls (p < 10−5), 16/39 of these patient had normal urine KS levels. Furthermore, the urine KS levels in ML III patients were significantly lower than those in ML II and ML II/III patients (p < 10−5). Therefore, urine KS appears to correlate with the severity of skeletal involvement, as patients with MPS IVA and ML II or ML II/III have the highest urine KS levels of all patients analyzed in this study. A correlation between urine KS level and clinical severity has already been reported in MPS IVA patients (Tomatsu et al. 2004).
The explanation for elevated urine KS levels in patients with MPS I, MPS II, and fucosidosis is less clear. The enzymes deficient in these disorders are not directly involved in keratan sulfate catabolism. Tomatsu et al. (2005) have reported that keratan sulfate is elevated in both plasma and urine samples from patients with all mucopolysaccharidosis disorders (types I, II, III, IV, VI, and VII) compared to age-matched controls. It was speculated that elevated heparan sulfate directly inhibits GALNS enzyme activity, resulting in a secondary elevation of keratan sulfate (Rowan et al. 2013; Tomatsu et al. 2014). This could explain the mild elevation of urine KS that was observed in MPS I and MPS II patients (Fig. 2). However, urine KS was only slightly elevated in 3/23 samples from MPS III patients, in whom heparan sulfate is the primary biomarker. Therefore, other explanations for the mild increase of urine KS in these patients should be explored. Greiling et al. demonstrated that patients with fucosidosis have elevated urine KS levels (Greiling et al. 1978). They speculated that the impaired removal of alpha-fucose residues from keratan sulfate due to alpha-fucosidase deficiency could lead to KS accumulation in these patients. However, further studies are needed to follow up on these findings.
Our study demonstrates that the quantitative measurement of urine KS via UPLC-MS/MS is a highly sensitive test for the detection and diagnosis of patients with MPS IVA. However, elevated urine KS levels are not specific to patients with this condition. Patients with other LSDs such as beta-galactosidase deficiency (including MPS IVB), Mucolipidosis II, II/III, or III, MPS I, MPS II, and fucosidosis may also excrete higher levels of urine KS than age-matched controls, in some cases to the same degree as patients with MPS IVA. Therefore, caution is advised when interpreting elevated urine KS results. The results should be interpreted within the context of the patient’s clinical features, and an abnormal urine KS result should always be followed by enzyme analysis to make a definitive diagnosis.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
{kind=link}
Urine keratan sulfate levels in unaffected individuals is age-dependent. Urine KS was measured in 105 unaffected controls (20 days – 63 years of age) (PNG 10 kb)
Acknowledgements
We would like to thank Yana Bierezovskaya, Nicole Miller (BioMarin), David Millington, and Hayogue Zhang (Duke University) for assistance in the development of this method. Drs. Roger Stevenson and Charles Schwartz provided thoughtful reviews of early versions of this manuscript.
This work was partially supported by the Greenwood Genetic Center Foundation and South Carolina Department of Disabilities and Special Needs. It was also supported in part by the Lysosomal Disease Network. The Lysosomal Disease Network (U54NS065768) is a part of the National Institutes of Health (NIH) Rare Diseases Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS), the National Institute of Neurological Disorders and Stroke (NINDS), and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors received funding from BioMarin Pharmaceuticals to support development of the urinary KS assay.
Synopsis
Urine keratan sulfate elevations are not specific to MPS IVA and may be found in other LSDs.
Compliance with Ethics Guidelines
Conflict of Interest
Tim Wood, Laura Pollard, and Katarzyna Ellsworth received support from BioMarin Pharmaceuticals to develop/validate the urine KS assay. Additionally they received speaker honoraria from BioMarin Pharmaceuticals. Sara Cathey declares no conflicts of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included as part of the Longitudinal Studies of the Glycoproteinoses (NCT01891422). The remaining samples were sent to the clinical biochemical laboratory at the Greenwood Genetic Center for clinical testing. These samples were de-identified prior to use.
Animal Rights
This article does not contain any studies with animal subjects performed by the any of the authors.
Details of the Contributions of Individual Authors
Katarzyna Ellsworth performed the laboratory work for this study as well as the review of data. She was involved in the writing of the manuscript and preparation of figures.
Laura Pollard was involved in data analysis, manuscript editing, and the preparation of figures.
Tim Wood was involved in data analysis, manuscript editing, and the preparation of figures.
Sara Cathey was involved in data analysis, manuscript editing, and the preparation of figures. Dr. Cathey also provided clinical information about specific patients.
Contributor Information
Tim Wood, Email: tim@ggc.org.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- de Jong JG, Wevers RA, Laarakkers C, Poorthuis BJ. Dimethylmethylene blue-based spectrophotometry of glycosaminoglycans in untreated urine: a rapid screening procedure for mucopolysaccharidoses. Clin Chem. 1989;35:1472–1477. [PubMed] [Google Scholar]
- Glossl J, Kresse H. A sensitive procedure for the diagnosis of N-acetyl-galactosamine-6-sulfate sulfatase deficiency in classical Morquio’s disease. Clin Chim Acta. 1978;88:111–119. doi: 10.1016/0009-8981(78)90157-2. [DOI] [PubMed] [Google Scholar]
- Gray G, Claridge P, Jenkinson L, Green A. Quantitation of urinary glycosaminoglycans using dimethylene blue as a screening technique for the diagnosis of mucopolysaccharidoses: an evaluation. Ann Clin Biochem. 2007;44:360–363. doi: 10.1258/000456307780945688. [DOI] [PubMed] [Google Scholar]
- Greiling H, Stuhlsatz HW, Cantz M, Gehler J. Increased urinary excretion of keratan sulfate in fucosidosis. J Clin Chem Clin Biochem. 1978;16:329–334. doi: 10.1515/cclm.1978.16.6.329. [DOI] [PubMed] [Google Scholar]
- Leroy JG, Ho MW, MacBrinn MC, Zielke K, Jacob J, O’Brien JS. I-cell disease: biochemical studies. Pediatr Res. 1972;6:752–757. doi: 10.1203/00006450-197210000-00002. [DOI] [PubMed] [Google Scholar]
- Leroy JG, Sillence D, Wood T, et al. A novel intermediate mucolipidosis II/IIIalphabeta caused by GNPTAB mutation in the cytosolic N-terminal domain. Eur J Hum Genet. 2014;22:594–601. doi: 10.1038/ejhg.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyseng-Williamson KA. Elosulfase Alfa: a review of its use in patients with mucopolysaccharidosis type IVA (Morquio A syndrome) BioDrugs. 2014;28:465–475. doi: 10.1007/s40259-014-0108-z. [DOI] [PubMed] [Google Scholar]
- Martell LA, Cunico RL, Ohh J, Fulkerson W, Furneaux R, Foehr ED. Validation of an LC-MS/MS assay for detecting relevant disaccharides from keratan sulfate as a biomarker for Morquio A syndrome. Bioanalysis. 2011;3:1855–1866. doi: 10.4155/bio.11.172. [DOI] [PubMed] [Google Scholar]
- Mendelsohn NJ, Wood T, Olson RA, et al. Spondyloepiphyseal dysplasias and bilateral Legg-Calve-Perthes disease: diagnostic considerations for mucopolysaccharidoses. JIMD Rep. 2013;11:125–132. doi: 10.1007/8904_2013_231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JS, Gugler E, Giedion A, et al. Spondyloepiphyseal dysplasia, corneal clouding, normal intelligence and acid beta-galactosidase deficiency. Clin Genet. 1976;9:495–504. doi: 10.1111/j.1399-0004.1976.tb01603.x. [DOI] [PubMed] [Google Scholar]
- Oguma T, Toyoda H, Toida T, Imanari T. Analytical method for keratan sulfates by high-performance liquid chromatography/turbo-ionspray tandem mass spectrometry. Anal Biochem. 2001;290:68–73. doi: 10.1006/abio.2000.4940. [DOI] [PubMed] [Google Scholar]
- Okada S, O’Brien JS. Generalized gangliosidosis: beta-galactosidase deficiency. Science. 1968;160:1002–1004. doi: 10.1126/science.160.3831.1002. [DOI] [PubMed] [Google Scholar]
- Piraud M, Boyer S, Mathieu M, Maire I. Diagnosis of mucopolysaccharidoses in a clinically selected population by urinary glycosaminoglycan analysis: a study of 2,000 urine samples. Clin Chim Acta. 1993;221:171–181. doi: 10.1016/0009-8981(93)90031-X. [DOI] [PubMed] [Google Scholar]
- Reitman ML, Varki A, Kornfeld S. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5′-diphosphate-N-acetylglucosamine: glycoprotein N-acetylglucosaminylphosphotransferase activity. J Clin Invest. 1981;67:1574–1579. doi: 10.1172/JCI110189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan DJ, Tomatsu S, Grubb JH, Montano AM, Sly WS. Assessment of bone dysplasia by micro-CT and glycosaminoglycan levels in mouse models for mucopolysaccharidosis type I, IIIA, IVA, and VII. J Inherit Metab Dis. 2013;36:235–246. doi: 10.1007/s10545-012-9522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GH, Taylor HA, Reynolds LW, Miller CS. Mucolipidosis 3 (Pseudo-Hurler polydystrophy): multiple lysosomal enzyme abnormalities in serum and cultured fibroblast cells. Pediatr Res. 1973;7:751–756. doi: 10.1203/00006450-197309000-00004. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Okamura K, Maeda H, et al. Keratan sulphate levels in mucopolysaccharidoses and mucolipidoses. J Inherit Metab Dis. 2005;28:187–202. doi: 10.1007/s10545-005-5673-3. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Okamura K, Taketani T, et al. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr Res. 2004;55:592–597. doi: 10.1203/01.PDR.0000113767.60140.E9. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Shimada T, Mason RW, et al. Establishment of glycosaminoglycan assays for mucopolysaccharidoses. Metabolites. 2014;4:655–679. doi: 10.3390/metabo4030655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley CB, Draper KA, Dutton CM, Brown PA, Severson SL, France LA. Diagnostic test for mucopolysaccharidosis. II. Rapid quantification of glycosaminoglycan in urine samples collected on a paper matrix. Clin Chem. 1989;35:2074–2081. [PubMed] [Google Scholar]
- Wood T, Bodamer OA, Burin MG, et al. Expert recommendations for the laboratory diagnosis of MPS VI. Mol Genet Metab. 2012;106:73–82. doi: 10.1016/j.ymgme.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Wood TC, Harvey K, Beck M, et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36:293–307. doi: 10.1007/s10545-013-9587-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraith JE. The mucopolysaccharidoses: a clinical review and guide to management. Arch Dis Child. 1995;72:263–267. doi: 10.1136/adc.72.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutaka T, Okada S, Kato T, Inui K, Yabuuhi H. Galactose 6-sulfate sulfatase activity in Morquio syndrome. Clin Chim Acta. 1982;122:169–180. doi: 10.1016/0009-8981(82)90276-5. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kariya Y, Conrad AH, Tasheva ES, Conrad GW. Analysis of keratan sulfate oligosaccharides by electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77:902–910. doi: 10.1021/ac040074j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Urine keratan sulfate levels in unaffected individuals is age-dependent. Urine KS was measured in 105 unaffected controls (20 days – 63 years of age) (PNG 10 kb)