

Abstract
The incidence of succinic semialdehyde dehydrogenase (SSADH) deficiency, an autosomal recessive inherited disorder of GABA degradation, is unknown. Upon a recent diagnosis of a new family of affected fraternal twins from the Punjabi ethnic group of India, case ascertainment from the literature and our database was done to determine the number of confirmed cases along with their geographic distribution. The probands presented with global developmental delay, infantile onset epilepsy, and a persistent neurodevelopmental disorder upon diagnosis at 10 years of age with intellectual disability, expressive aphasia, and behavioral problems most prominent for hyperactivity. Gamma-hydroxybutyric aciduria and homozygous ALDH5A1 c.608C>T; p.Pro203Leu mutations were confirmed. Identification of all available individual cases with clinical details available including geographic or ethnic origin revealed 182 patients from 40 countries, with the largest number of patients reported from the USA (24%), Turkey (10%), China (7%), Saudi Arabia (6%), and Germany (5%). This study provides an accounting of all published cases of confirmed SSADH deficiency and provides data useful in planning further studies of this rare inborn error of metabolism.
Introduction
Succinic semialdehyde dehydrogenase (SSADH) deficiency (OMIM 271980, 610045) is a rare autosomal recessive disorder of GABA degradation initially described in 1981 (Jakobs et al. 1981). GABA-transaminase (GABA-T) converts GABA to succinic semialdehyde, which is metabolized to succinic acid by SSADH. In SSADH deficiency, there is an excessive buildup of GABA and γ-hydroxybutyrate (GHB). High levels of GABA and GHB have been implicated in the neurological manifestations of SSADH deficiency, and recent evidence suggests multiple metabolic perturbations may be associated with the pathophysiology including markers of oxidative stress, dysregulation of autophagy including the mTOR pathway, and accumulation of the semialdehyde intermediate.
SSADH deficiency does not typically feature intermittent deterioration or neurodevelopmental regression and may be relegated to a nonprogressive encephalopathy or atypical neurobehavioral syndrome with either a missed diagnosis or identification late into adulthood (Lapalme-Remis et al. 2015). We report affected twins from the second family reported from India and the first since the early report of three affected siblings from 1997 that added 23 new cases to the literature and described the phenotype (Gibson et al. 1997). We subsequently reviewed the literature to identify all reported cases since the initial description of urinary gamma-hydroxybutyric aciduria (Jakobs et al. 1981) and added patients from our database with a confirmed diagnosis.
Methods
Clinical and genetic confirmation of SSADH deficiency of a Punjabi family from Northern India came to our attention as the second affected family reported from India and the first in 20 years. We reviewed the literature and our database for confirmed cases of SSADH deficiency and report on the geographic origin of reported patients. Collection and reporting of subjects from our database was approved by the Boston Children’s Hospital Institutional Review Board.
Case Reports and Results
Ten-year-old dizygotic twin boys born to a North Indian couple of the Punjabi ethnic group presented for evaluation of developmental disability and a history of seizures. There was no known history of consanguinity or family history of neurodevelopmental disorders.
Twin A presented with cognitive impairment, profound deficit of expressive language, and incoordination. Behavior was notable for hyperactivity and, at times, aggressiveness. Developmental assessment indicated moderate intellectual deficiency with WISC-III Full Scale IQ measurement of 43, gross motor skill level approximating age 4 years, and fine motor skills at 14 months. Activities of daily living were affected such that the patient could not dress independently or drink from a cup without spillage. The proband had onset of convulsive seizures at 3 months of age. Delayed developmental with hypotonia was noted then. The past medical history disclosed no gestational or perinatal problems, but acquisition of developmental milestones was notable for head control at 12 months, independent sitting 30 months, independent standing at 36 months, and walking at 42 months. On neurological examination, the sparse vocabulary used was very inarticulate and there was decreased attention. Motor examination revealed hypotonia and chorea, and deep tendon reflexes were hypoactive.
Twin B similarly presented with cognitive impairment and hyperactive behavior. Developmental assessment showed moderate intellectual disability with a WISC-III Full Scale IQ measurement of 38 and impairment of motor coordination along with hyperactive behavior. This patient had infantile onset of convulsive seizures at 3 months of age, treated with phenobarbital and without a recurrence since the age of 4 years. Phenobarbital was tapered off after two seizure-free years. Levetiracetam was initiated recently when the breakthrough seizure occurred in the twin brother.
There had been no gestational or perinatal complications. The developmental history similarly showed that hypotonia was noted in infancy at the time of seizure onset and there was delayed acquisition of milestones. Neurological examination showed minimal expressive and disarticulate language, hypotonia, and hyporeflexia.
Brain MRI was obtained in Twin A and showed symmetrical hyperintensities of the globus pallidi on T2-weighted and FLAIR sequences (Fig. 1). Urine was sent on both twins for organic acid analysis, and gas chromatography-mass spectrometry (GC-MS) revealed significantly increased levels of 4-hydroxybutyric acid and 4,5-dihydroxyhexanoic acid. These findings led to ALDH5A1 sequencing which revealed homozygous c.608C>T; pPro203Leu mutations, previously described as disease causing, in both subjects. The parents were confirmed as heterozygous as were two siblings.
Fig. 1.
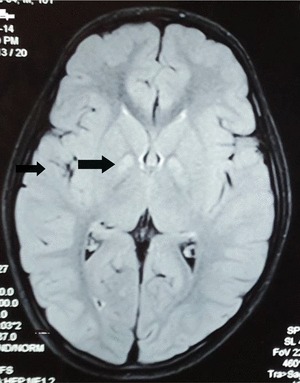
Magnetic resonance imaging of the brain. Axial MRI FLAIR sequence with hyperintensities of globus pallidi (arrowhead)
Case ascertainment dating back to the initial report of gamma-hydroxybutyric aciduria identified 91 unique patients with clinical details provided (Aoshima et al. 2002; Bekri et al. 2004; Brown et al. 1987; Dayan et al. 2006; Deng et al. 2011; Divry et al. 1983; Escalera et al. 2010; Gogou et al. 2016; Haan et al. 1985; Ishiguro et al. 2001; Jakobs et al. 1981; Jiang et al. 2013; Kratz 2009; Kwok et al. 2012; Lemes et al. 2006; Li et al. 2015; Lin et al. 2015; Liu et al. 2016; Neu et al. 2002; Niemi et al. 2014; O’Rourke et al. 2010; Onkenhout et al. 1989; Pearl et al. 2003; Peters et al. 1999; Puttmann et al. 2013; Racaru et al. 2010; Rashed et al. 1994; Rating et al. 1984; Saronwala et al. 2008; Spilioti et al. 2013; Tay et al. 2015; Wang et al. 2016; Yamakawa et al. 2012; Zhao et al. 2003; Ziyeh et al. 2002). We furthermore identified 91 additional subjects with confirmed SSADH deficiency in our database published in aggregate (Parviz et al. 2014). Overall, we were able to identify 182 unique cases of SSADH deficiency. The ethnicity of the patients is shown in Table 1; when only geographic residence was provided, this was used instead. There were a total of 40 ethnicities or countries represented in the patient population (Fig. 2). The countries with the greatest number of patients reported were the USA (24%), Turkey (10%), China (7%), Saudi Arabia (6%), and Germany (5%). Together, these five countries accounted for approximately half of all reported patients.
Table 1.
Ethnicity/geographic distribution of reported SSADH deficiency cases
| Country | Total |
|---|---|
| United States | 43 |
| Turkey | 18 |
| China | 13 |
| Saudi | 10 |
| Germany | 9 |
| Australia | 7 |
| Netherlands | 7 |
| UK | 7 |
| Pakistan | 6 |
| Greece | 5 |
| Spain | 5 |
| Iran | 4 |
| Japan | 4 |
| India | 3 |
| Ireland | 3 |
| Israel | 3 |
| Italy | 3 |
| Afghanistan | 2 |
| Argentina | 2 |
| Bulgaria | 2 |
| Canada | 2 |
| France | 2 |
| Korea | 2 |
| Lebanon | 2 |
| Lifu | 2 |
| Taiwan | 2 |
| Albania | 1 |
| Algeria | 1 |
| Belgium | 1 |
| Denmark | 1 |
| Inuit | 1 |
| Luxemburg | 1 |
| Malaysia | 1 |
| Sicily | 1 |
| Sweden | 1 |
| Syria | 1 |
| Tunisia | 1 |
| UAE | 1 |
| Uruguay | 1 |
| Yemen | 1 |
| Total | 182 |
Fig. 2.
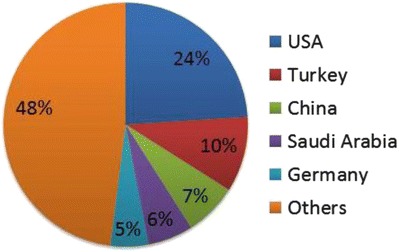
Countries reporting cases of SSADH deficiency (N = 40)
Discussion
The median age at diagnosis of SSADH deficiency is 2 years, but underdiagnosis is suspected. Nearly 80% of reported patients are diagnosed by the age of 5 years although 10% of patients are diagnosed after the first decade (Lapalme-Remis et al. 2015). An adult was recently diagnosed at age 62 during a terminal illness marked by repeated status epilepticus, although his family described a typical childhood course yet without a specific diagnosis or suspicion of a metabolic disorder (Lapalme-Remis et al. 2015). The phenotype typically is neurodevelopmental impairment with intellectual deficiency, marked expressive language impairment, and epilepsy. Typical neurological features include hypotonia, ataxia, and hyporeflexia with some patients manifesting chorea. The neuropsychiatric profile includes often disabling obsessive compulsive disorder and anxiety in addition to inattention, hyperactive behavior, and sleep disturbances (Gibson et al. 2003; Pearl et al. 2003).
This is the first report of a family from India since the relatively large series of patients that added 23 new cases to the literature in 1997 (Gibson et al. 1997). The previously reported family had three affected children, the oldest diagnosed posthumously based on a consistent phenotype with death at 13 years. The siblings in that family were diagnosed at ages 9 and 3 years, with laboratory detection using urine organic acids and confirmation based on enzyme activity determination.
The twins in the present report had a delay in diagnosis to 10 years of age and otherwise presented with delay of acquired infantile milestones, early hypotonia, intellectual deficiency, and marked impairment of expressive language. The infantile onset of epilepsy with remission in early childhood is less typical but does occur. Epilepsy often appears during later childhood or adolescence and is more prevalent in the adult than pediatric cohort (Lapalme-Remis et al. 2015). The findings on examination of hypotonia, chorea, and hyporeflexia are typical of the disorder. Globus pallidus hyperintensities on T2-weighted MRI and gamma-hydroxybutyric aciduria along with elevated 4,5-dihydroxyhexanoic acid are diagnostic markers of the disorder (Pearl et al. 2009). Epileptiform discharges on EEG tend to be generalized but focal spikes have been described (Pearl et al. 2009). More than 35 ALDH5A1 mutations including missense, nonsense, and splice mutations have been reported. The c.608C>T; p.Pro203Leu mutation has been previously reported and affects an amino acid highly conserved across all known mammalian species and was undetected in approximately 1,300 control alleles (Akaboshi et al. 2003).
We were able to identify 182 subjects with SSADH deficiency based on the literature and our database, from 40 countries with five countries reporting half of all patients. The nonspecific phenotype of the disorder, usually following the pattern of a nonprogressive encephalopathy that may have superimposed epilepsy, mandates a high index of clinical suspicion for a metabolic disorder, and we suspect these are the countries with centers more likely to pursue metabolic diagnostic studies in this clinical scenario. Underdiagnosis, late diagnosis, and disproportional geographic representation are common as with other neurometabolic disorders.
Footnotes
Summary Statement: This study provides the worldwide geographic distribution of SSADH deficiency from all identified published cases and subjects in the investigators’ database.
Contributor Information
Phillip L. Pearl, Email: Phillip.Pearl@childrens.harvard.edu
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Akaboshi S, Hogema BM, Novelletto A, et al. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum Mutat. 2003;22:442–450. doi: 10.1002/humu.10288. [DOI] [PubMed] [Google Scholar]
- Aoshima T, Kajita M, Sekido Y, et al. Mutation analysis in a patient with succinic semialdehyde dehydrogenase deficiency: a compound heterozygote with 103-121del and 1460T > A of the ALDH5A1 gene. Hum Hered. 2002;53:42–44. doi: 10.1159/000048603. [DOI] [PubMed] [Google Scholar]
- Bekri S, Fossoud C, Plaza G, et al. The molecular basis of succinic semialdehyde dehydrogenase deficiency in one family. Mol Genet Metab. 2004;81:347–351. doi: 10.1016/j.ymgme.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Brown GK, Cromby CH, Manning NJ, Pollitt RJ. Urinary organic acids in succinic semialdehyde dehydrogenase deficiency: evidence of alpha-oxidation of 4-hydroxybutyric acid, interaction of succinic semialdehyde with pyruvate dehydrogenase and possible secondary inhibition of mitochondrial beta-oxidation. J Inherit Metab Dis. 1987;10:367–375. doi: 10.1007/BF01799979. [DOI] [PubMed] [Google Scholar]
- Dayan C, Ülker M, Akgün YH, Günaydin S, Atay T, Arpaci B. Succinic semialdehyde dehydrogenase deficiency: three sibling in a family (case report) J Neurol Sci (Turkish) 2006;23:129–134. [Google Scholar]
- Deng XL, Yin F, Xiang QL, Liu CT, Peng J. Succinic semialdehyde dehydrogenase deficiency. Zhongguo Dang Dai Er Ke Za Zhi. 2011;13:740–742. [PubMed] [Google Scholar]
- Divry P, Baltassat P, Rolland MO, et al. A new patient with 4-hydroxybutyric aciduria, a possible defect of 4-aminobutyrate metabolism. Clin Chim Acta. 1983;129:303–309. doi: 10.1016/0009-8981(83)90033-5. [DOI] [PubMed] [Google Scholar]
- Escalera GI, Ferrer I, Marina LC, et al. Succinic semialdehyde dehydrogenase deficiency: decrease in 4-OH-butyric acid levels with low doses of vigabatrin. An Pediatr (Barc) 2010;72:128–132. doi: 10.1016/j.anpedi.2009.09.018. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Christensen E, Jakobs C, et al. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria): case reports of 23 new patients. Pediatrics. 1997;99:567–574. doi: 10.1542/peds.99.4.567. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Gupta M, Pearl PL, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma-hydroxybutyric aciduria) Biol Psychiatry. 2003;54:763–768. doi: 10.1016/S0006-3223(03)00113-6. [DOI] [PubMed] [Google Scholar]
- Gogou M, Spilioti M, Tramma D, Papadopoulou-Alataki E, Evangeliou A. Succinic semialdehyde dehydrogenase deficiency presenting as autism spectrum disorder. Indian J Pediatr. 2016;83:1036–1037. doi: 10.1007/s12098-015-2003-0. [DOI] [PubMed] [Google Scholar]
- Haan EA, Brown GK, Mitchell D, Danks DM. Succinic semialdehyde dehydrogenase deficiency--a further case. J Inherit Metab Dis. 1985;8:99. doi: 10.1007/BF01819287. [DOI] [PubMed] [Google Scholar]
- Ishiguro Y, Kajita M, Aoshima T, Watanabe K, Kimura M, Yamaguchi S. The first case of 4-hydroxybutyric aciduria in Japan. Brain Dev. 2001;23:128–130. doi: 10.1016/S0387-7604(01)00181-4. [DOI] [PubMed] [Google Scholar]
- Jakobs C, Bojasch M, Monch E, Rating D, Siemes H, Hanefeld F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin Chim Acta. 1981;111:169–178. doi: 10.1016/0009-8981(81)90184-4. [DOI] [PubMed] [Google Scholar]
- Jiang SZ, Shu JB, Zhang YQ, Fan WX, Meng YT, Song L. Analysis of ALDH5A1 gene mutation in a Chinese Han family with succinic semialdehyde dehydrogenase deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2013;30:389–393. doi: 10.3760/cma.j.issn.1003-9406.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Kratz SV. Sensory integration intervention: historical concepts, treatment strategies and clinical experiences in three patients with succinic semialdehyde dehydrogenase (SSADH) deficiency. J Inherit Metab Dis. 2009;32:353–360. doi: 10.1007/s10545-009-1149-1. [DOI] [PubMed] [Google Scholar]
- Kwok JS, Yuen CL, Law LK, Tang NL, Cherk SW, Yuen YP. A novel ALDH5A1 mutation in a patient with succinic semialdehyde dehydrogenase deficiency. Pathology. 2012;44:280–282. doi: 10.1097/PAT.0b013e32835140c2. [DOI] [PubMed] [Google Scholar]
- Lapalme-Remis S, Lewis EC, De Meulemeester C, et al. Natural history of succinic semialdehyde dehydrogenase deficiency through adulthood. Neurology. 2015;85:861–865. doi: 10.1212/WNL.0000000000001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemes A, Blasi P, Gonzales G, et al. Succinic semialdehyde dehydrogenase (SSADH) deficiency: molecular analysis in a South American family. J Inherit Metab Dis. 2006;29:587. doi: 10.1007/s10545-006-0277-0. [DOI] [PubMed] [Google Scholar]
- Li X, Ding Y, Liu Y, et al. Succinic semialdehyde dehydrogenase deficiency of four Chinese patients and prenatal diagnosis for three fetuses. Gene. 2015;574:41–47. doi: 10.1016/j.gene.2015.07.078. [DOI] [PubMed] [Google Scholar]
- Lin CY, Weng WC, Lee WT. A novel mutation of ALDH5A1 gene associated with succinic semialdehyde dehydrogenase deficiency. J Child Neurol. 2015;30:486–489. doi: 10.1177/0883073814544365. [DOI] [PubMed] [Google Scholar]
- Liu N, Kong XD, Kan QC, et al. Mutation analysis and prenatal diagnosis in a Chinese family with succinic semialdehyde dehydrogenase and a systematic review of the literature of reported ALDH5A1 mutations. J Perinat Med. 2016;44:441–451. doi: 10.1515/jpm-2014-0164. [DOI] [PubMed] [Google Scholar]
- Neu P, Seyfert S, Brockmoller J, Dettling M, Marx P. Neuroleptic malignant syndrome in a patient with succinic semialdehyde dehydrogenase deficiency. Pharmacopsychiatry. 2002;35:26–28. doi: 10.1055/s-2002-19830. [DOI] [PubMed] [Google Scholar]
- Niemi AK, Brown C, Moore T, Enns GM, Cowan TM. Evidence of redox imbalance in a patient with succinic semialdehyde dehydrogenase deficiency. Mol Genet Metab Rep. 2014;1:129–132. doi: 10.1016/j.ymgmr.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke DJ, Ryan S, King MD. Head bobbing due to succinic semialdehyde dehydrogenase deficiency. Neurology. 2010;74:2025. doi: 10.1212/WNL.0b013e3181e398cf. [DOI] [PubMed] [Google Scholar]
- Onkenhout W, Maaswinkel-Mooij PD, Poorthuis BJ. 4-Hydroxybutyric aciduria: further clinical heterogeneity in a new case. Eur J Pediatr. 1989;149:194–196. doi: 10.1007/BF01958280. [DOI] [PubMed] [Google Scholar]
- Parviz M, Vogel K, Gibson KM, Pearl PL. Disorders of GABA metabolism: SSADH and GABA-transaminase deficiencies. J Pediatr Epilepsy. 2014;3:217–227. doi: 10.3233/PEP-14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl PL, Novotny EJ, Acosta MT, Jakobs C, Gibson KM. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann Neurol. 2003;54(Suppl 6):S73–S80. doi: 10.1002/ana.10629. [DOI] [PubMed] [Google Scholar]
- Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis. 2009;32:343–352. doi: 10.1007/s10545-009-1034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters H, Cleary M, Boneh A. Succinic semialdehyde dehydrogenase deficiency in siblings: clinical heterogeneity and response to early treatment. J Inherit Metab Dis. 1999;22:198–199. doi: 10.1023/A:1005439111908. [DOI] [PubMed] [Google Scholar]
- Puttmann L, Stehr H, Garshasbi M, et al. A novel ALDH5A1 mutation is associated with succinic semialdehyde dehydrogenase deficiency and severe intellectual disability in an Iranian family. Am J Med Genet A. 2013;161A:1915–1922. doi: 10.1002/ajmg.a.36030. [DOI] [PubMed] [Google Scholar]
- Racaru VM, Pinard JM, Cheliout-Heraut F. Sleep disorders in succinic semialdehyde dehydrogenase deficiency: a family report. Eur J Paediatr Neurol. 2010;14:282–287. doi: 10.1016/j.ejpn.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Rashed M, Ozand PT, al Aqeel A, Gascon GG. Experience of king faisal specialist hospital and research center with Saudi organic acid disorders. Brain Dev. 1994;16(Suppl):1–6. doi: 10.1016/0387-7604(94)90090-6. [DOI] [PubMed] [Google Scholar]
- Rating D, Hanefeld F, Siemes H, et al. 4-Hydroxybutyric aciduria: a new inborn error of metabolism. I. Clinical review. J Inherit Metab Dis. 1984;7(Suppl 1):90–92. doi: 10.1007/BF03047381. [DOI] [PubMed] [Google Scholar]
- Saronwala A, Tournay A, Gargus JJ (2008) Taurine treatment of succinate semialdehyde dehydrogenase (SSADH) deficiency reverses MRI-documented globus lesion and clinical syndrome [abstract]. Proc Am Coll Med Genet 103
- Spilioti M, Evangeliou AE, Tramma D, et al. Evidence for treatable inborn errors of metabolism in a cohort of 187 Greek patients with autism spectrum disorder (ASD) Front Hum Neurosci. 2013;7:858. doi: 10.3389/fnhum.2013.00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay CG, Ariffin H, Yap S, Rahmat K, Sthaneshwar P, Ong LC. Succinic semialdehyde dehydrogenase deficiency in a Chinese boy: a novel ALDH5A1 mutation with severe phenotype. J Child Neurol. 2015;30:927–931. doi: 10.1177/0883073814540523. [DOI] [PubMed] [Google Scholar]
- Wang KY, Barker PB, Lin DD (2016) A case of acute onset succinic semialdehyde dehydrogenase deficiency: neuroimaging findings and literature review. Child’s Nerv Syst 32:1305–1309 [DOI] [PubMed]
- Yamakawa Y, Nakazawa T, Ishida A, et al. A boy with a severe phenotype of succinic semialdehyde dehydrogenase deficiency. Brain Dev. 2012;34:107–112. doi: 10.1016/j.braindev.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Zhao XP, Liu GS, Song YZ. A case of succinic semialdehyde dehydrogenase deficiency. Zhonghua Er Ke Za Zhi. 2003;41:719. [PubMed] [Google Scholar]
- Ziyeh S, Berlis A, Korinthenberg R, Spreer J, Schumacher M. Selective involvement of the globus pallidus and dentate nucleus in succinic semialdehyde dehydrogenase deficiency. Pediatr Radiol. 2002;32:598–600. doi: 10.1007/s00247-002-0717-4. [DOI] [PubMed] [Google Scholar]