

Abstract
Background: Peroxisome biogenesis disorders (PBDs) may have a variable clinical expression, ranging from severe, lethal to mild phenotypes with progressive evolution. PBDs are autosomal recessive disorders caused by mutations in PEX genes, which encode proteins called peroxins, involved in the assembly of the peroxisome.
Patient Description: We herein report a patient who is currently 9 years old and who is compound heterozygous for two novel mutations in the PEX3 gene.
Results: Mild biochemical abnormalities of the peroxisomal parameters suggested a Zellweger spectrum defect in the patient. Sequence analysis of the PEX3 gene identified two novel heterozygous, pathogenic mutations.
Conclusion: Mutations in PEX3 usually result in a severe, early lethal phenotype. We report a patient compound heterozygous for two novel mutations in the PEX3 gene, who is less affected than previously reported patients with a defect in the PEX3 gene. Our findings indicate that PEX3 defects may cause a disease spectrum similar as previously observed for other PEX gene defects.
Keywords: Peroxisomal disorders, PEX3, Zellweger spectrum disorders
Introduction
Peroxisomal disorders can be subdivided into peroxisome biogenesis disorders (PBDs; MIM 601539) and the single peroxisomal enzyme deficiencies (Wanders 1999; Wanders and Waterham 2004). PBDs include the Zellweger spectrum disorders (ZSDs), which are Zellweger Syndrome (ZS), Neonatal Adrenoleukodystrophy (NALD) and Infantile Refsum Disease (IRD), and Rhizomelic Chondrodysplasia Punctata (RCDP) type 1 (Wanders 1999; Wanders and Waterham 2004; Muntau et al. 2000a, b; Shimozawa et al. 2005). Patients with a ZSD may present with a continuum of symptoms of varying severity; the ZS presentation represents the most severe clinical phenotype. ZSDs can be caused by mutations in any of at least 13 different PEX genes, which encode proteins called peroxins that are involved in peroxisomal protein import and peroxisome assembly (Ebberink et al. 2012).
The PEX1, PEX6 and PEX12 genes are the most affected genes in patients with a ZSD (Ebberink et al. 2012). In contrast, PEX3 defects are very rare. Patients with a defect in PEX3 are usually severely affected, thus resulting in early lethality.
We herein report a patient, who is currently 9 years old and who is compound heterozygous for two novel mutations in the PEX3 gene. The patient is less affected than previously reported patients with a defect in the PEX3 gene.
Patient Description
This 9-year-old boy is the first child of non-consanguineous healthy parents. The pregnancy was uneventful and he was born at term by caesarean delivery due to podalic version. He presented with neonatal jaundice that resolved spontaneously. He had a high forehead, posteriorly rotated, low set ears and inverted nipples. At the age of 8 months, he presented with psychomotor retardation, axial and peripheral muscular hypotonia and nephrocalcinosis. Evaluation at the age of 18 months revealed progressive spastic paraparesis, neurogenic bladder and nystagmus. At the evaluation at 4 years of age, he showed severe spastic paraparesis, brisk reflexes and axial hypotonia. He had neither cutaneous abdominal reflex nor tickling. He managed to remain seated but needed motor assistance using a wheelchair for ambulation. He also needed constant care and assistance. The ophthalmologic exam showed bilateral cataracts at the age of 4 years, which were surgically corrected at the age of 5. At 5 years of age, he had a non-febrile focal clonic seizure on left arm and left hemiface lasting less than 5 min. He repeated a similar episode with 24 h associated to an abnormal EEG so he was treated with clobazam. He had an isolated fever record 24 h later the event. He was on clobazam until 9 years of age and did not repeat any other seizure (Fig. 1).
Fig. 1.
Patient at 8 months and at 4 years of age
Central nervous system (CNS) magnetic resonance imaging (MRI), CNS spectroscopy and CNS MRI angiography performed at 26 months and at 3 and 4 years of age were normal. However, increased gadolinium reinforcement at the medullary cone on spine MRI was described on the studies performed at 3 and 4 years of age. Somatosensory evoked potential showed spinal cord compromise at 19 months and at 3 years of age. Elbow and knee radiographies performed at 2 years of age were normal. Visual evoked potentials, electroretinography and auditory evoked potentials performed at 4 years of age were also normal.
He had a normal 46 XY karyotype. Screening for HTLV1 by PCR on CSF was negative. Laboratory and basic metabolic investigations performed (creatine kinase, aldolase, lactic acid, ammonia levels, uric acid, pristanic and phytanic acids, urinary organic and bile acids, and transferrin isoelectric focusing) were all normal. However, erythrocyte plasmalogen levels were C16 DMA/C16:0 0.025 (reference values 0.045–0.082) and C18DMA/C18:0 0.065 (reference values 0.125–0.265); and the ratios for the very long chain fatty acids (VLCFAs) were elevated in plasma at the age of 3 years: C24:0/C22:0 1.30 (reference values 0.68–0.98) and C26:0/C22:0 0.0383 (reference values 0.0064–0.0216).
Peroxisomal parameters were subsequently studied in cultured skin fibroblasts. VLCFA levels were also elevated in fibroblasts: C22:0 4.63 (μmol/g) (reference values 3.84–10.20), C24:0 8.91 (μmol/g) (reference values 7.76–17.66) and C26:0 0.47 (μmol/g) (reference values 0.18–0.38). Ratio C24:0/C22:0 1.93 (reference values 1.55–2.30) and ratio C26:0/C22:0 0.10 (reference values 0.03–0.07).
Immunofluorescence microscopy analysis with antibodies against catalase, a peroxisomal matrix protein, revealed an abnormal peroxisomal mosaic pattern with cells with import-competent peroxisomes and cells without import-competent peroxisomes (Fig. 2). Moreover, the patient’s cells with import-competent peroxisomes showed lower numbers and increased size of peroxisomes. Immunoblot analysis of the peroxisomal proteins acyl-CoA oxidase 1 (ACOX1) and 3-ketoacyl-CoA thiolase revealed normal processing of these proteins within the peroxisome. The activity of the peroxisomal enzyme dihydroxyacetone phosphate acyltransferase (DHAPAT), the first enzyme of the plasmalogen biosynthesis pathway, was low normal compared to the reference values. Despite the relative mild biochemical abnormalities of the peroxisomal parameters, these results strongly suggested a Zellweger spectrum defect in the patient. To determine which PEX gene would be defective in the patient, we sequenced all exons and flanking intron sequences of the PEX1, PEX2, PEX5, PEX10, PEX12, PEX13, PEX14, PEX16, PEX19 and PEX26 genes, but did not identify potential pathogenic mutations. However, sequence analysis of the PEX3 gene (reference sequence MN_003630.2) identified two heterozygous, pathogenic mutations in the PEX3 gene: c.898C>T (p.Arg300*) inherited from the mother and c.991G>A (p.Gly331Arg) inherited from the father. Both mutations have not been reported previously. The c.898C>T mutation interrupts the reading frame by a premature STOP codon, which theoretically leads to a truncated PEX3 protein. Alternatively, the PEX3 mRNA might be targeted for nonsense mediated decay. The c.991G>A mutation changes the glycine at position 331 into an arginine. This glycine is highly conserved among PEX3 proteins from different species ranging from fruit fly to man, and the amino acid change is predicted as deleterious by SIFT, disease causing by MutationTaster and probably damaging by PolyPhen-2 prediction software (Fig. 3).
Fig. 2.
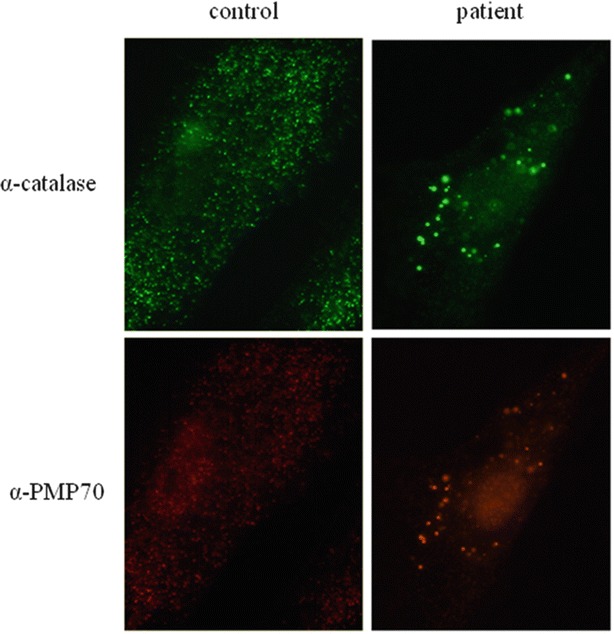
Immunofluorescence microscopy analysis for catalase, a peroxisomal matrix protein, and PMP70, a peroxisomal membrane protein (PMP) in cultured fibroblasts from the patient
Fig. 3.

Sequence analysis of PEX3. Pathogenic mutations
Discussion
We herein report the clinical, neuroradiological, biochemical and molecular characterization of a male patient affected with a ZSD due to two novel heterozygous mutations in the PEX3 gene. The relatively mild clinical and biochemical phenotype of our patient expands the clinical spectrum for PEX3 patients and indicates that dependent on the severity of the disease-causing mutations PEX3 defects result in a disease spectrum similar as previously observed for other PEX gene defects.
Patients with PBDs can be classified into 13 genetic complementation groups reflecting defects in 13 different PEX genes. These complementation groups can be subdivided into two groups according to the dysfunction. Defects in PEX3, PEX16 and PEX19 result in a complete defect in peroxisomal assembly and cell lines often do not show peroxisomal membrane structures (ghosts) (Ebberink et al. 2011). In cell lines with defects in the other PEX genes, peroxisomal membrane structures are always present and sometimes partial matrix protein import can be observed (Wanders 1999; Shimozawa et al. 2005; Muntau et al. 2000a, b; Ghaedi et al. 2000; Matsui et al. 2013).
PEX3 is an integral membrane protein. It is supposed to play a role in the insertion of membrane proteins into the peroxisomal membrane. Since PEX3 interacts with PEX16 and PEX19 in yeast and human cells, it has been proposed that a defective PEX3 affects this interaction and leads to degradation or mislocalization of the peroxisomal membrane proteins (PMPs) (Muntau et al. 2000a, b; Matsui et al. 2013).
The commonly observed clinical presentation of patients with mutations in PEX3 includes the severe ZS phenotype (Muntau et al. 2000a, b; Ghaedi et al. 2000). Clinical findings in patients presenting with ZS include facial dysmorphism, neonatal jaundice, hepatomegaly, liver dysfunctions, calcification of joints, renal cortical cysts, cataracts, hearing impairment, retinal degeneration, seizures, polymicrogyria and developmental delay (Wanders 1999). ZSD atypical phenotypes presenting with a relatively mild clinical phenotype with slow progression and mild elevation of biochemical markers have recently been described (Regal et al. 2010; Sevin et al. 2011; Thoms and Gärtner 2012; Zeharia et al. 2007).
In addition, Matsui et al. recently reported another patient with a milder phenotype due to a PEX3 mutation (Matsui et al. 2013). Similarly, the patient described here presented a mosaic pattern of catalase positive particles and peroxisomal membrane structures. It has been suggested that the severity of the phenotype could be related to the number of residual peroxisomes in tissues and the preservation of peroxisomal function. The mosaic pattern found in our patient’s fibroblasts and the relative mild biochemical aberrations are in agreement with this hypothesis.
Pathological findings on brain MRI in ZSD include abnormalities in neuronal migration and differentiation, such as cortical dysplasia and neuronal heterotopias, particularly perisylvian polymicrogyria or incomplete opercularization. Germinolytic cysts along the frontal horns of the lateral ventricles and cerebellar cortical dysplasia are also described. White matter abnormalities consist of dysmyelination rather than demyelination, involving cerebral and cerebellar hemispheres (Wanders 1999). Cerebellar atrophy without any other changes has also been described in phenotypes presenting as autosomic recessive cerebellar ataxias. Late-onset white matter disease is a distinct phenotype in ZSD, and the physiopathological explanation for this phenotype is still unresolved. Patients can present with abrupt clinical deterioration preceded by normal development or moderate delay in the first months of life. Clinical findings include hypotonia, retinopathy, sensory deafness, seizures and cerebral demyelination in centrum semiovale, sparing U fibres and even central cerebellar white matter. When conventional MRI techniques fail to detect white matter abnormalities, then diffusion-weighted and diffusion tensor imaging may be useful. Although our patient presented a normal CNS on MRI follow-up, progressive changes cannot be definitely ruled out.
Conclusions
Several patients with ZSD presenting with atypical phenotypes have been recently reported. These patients showed milder phenotypes, slow progression of symptoms, spastic paraparesis, ataxia, normal or mild elevation of biochemical markers and normal images on CNS MRI. Mutations in PEX3, PEX16 and PEX19 genes usually result in a severe, early lethal phenotype and in cells entirely lacking remnant peroxisomal membranes. However, our patient still has peroxisomal remnants and displays a much milder phenotype than that previously observed in patients with PEX3 mutations. Our findings indicate that PEX3 defects may cause a disease spectrum similar as previously observed for other PEX gene defects.
Declaration of Conflicting Interests
Delfina Marchione works for Genzyme Sanofi Company.
The authors Clarisa Maxit, Inés Denzler, Guillermo Agosta, Janet Koster, Ronald Wanders, Sacha Ferdinandusse and Hans Waterham declare no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Author Contributions
CM, ID, MD and AG: conception, manuscript, acquisition of clinical data, preparation and revision of intellectual content, and final approval of the manuscript. KJ, WRJA, FS and WHR: acquisition of biochemical and genetic data, review and critique, and final approval of the manuscript.
Ethical Approval
This work was performed in agreement with the ethical rules of Hospital Italiano de Buenos Aires. No ethical approval was necessary for the writing of this case report.
The authors received an informed consent form from the patient’s parents.
This article does not contain any studies with human or animal subjects performed by any of the authors.
Contributor Information
I. Denzler, Email: ines.denzler@hospitalitaliano.org.ar
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Ebberink MS, Mooijer P, Gootjes J, et al. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat. 2011;32:59–69. doi: 10.1002/humu.21388. [DOI] [PubMed] [Google Scholar]
- Ebberink MS, Koster J, Visser G, et al. A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11β gene. J Med Genet. 2012;49:307–313. doi: 10.1136/jmedgenet-2012-100778. [DOI] [PubMed] [Google Scholar]
- Ghaedi K, Honsho M, Shimozawa N, et al. PEX3 is the causal gene responsible for peroxisome membrane assembly-defective Zellweger syndrome of complementation group G. Am J Hum Genet. 2000;67:976–981. doi: 10.1086/303086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui S, Funahashi M, Honda A, Shimozawa N. Newly identified milder phenotype of peroxisome biogenesis disorder caused by mutated PEX3 gene. Brain Dev. 2013;35:842–848. doi: 10.1016/j.braindev.2012.10.017. [DOI] [PubMed] [Google Scholar]
- Muntau AC, Mayerhofer PU, Paton BC, et al. Defective peroxisome membrane synthesis due to mutations in human PEX3 causes Zellweger syndrome, complementation group G. Am J Hum Genet. 2000;67:967–975. doi: 10.1086/303071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntau AC, Holzinger A, Mayerhofer P, et al. The human PEX3 gene encoding a peroxisomal assembly protein: genomic organization, positional mapping, and mutation analysis in candidate phenotypes. Biochem Biophys Res Commun. 2000;268:704–710. doi: 10.1006/bbrc.2000.2193. [DOI] [PubMed] [Google Scholar]
- Regal L, Ebberink M, Goemans N, et al. Mutations in PEX10 are a cause of autosomal recessive ataxia. Ann Neurol. 2010;68:259–263. doi: 10.1002/ana.22035. [DOI] [PubMed] [Google Scholar]
- Sevin C, Ferdinandusse S, Waterham HR, et al. Autosomal recessive cerebellar ataxia caused by mutations in the PEX2 gene. Orphanet J Rare Dis. 2011;10:6–8. doi: 10.1186/1750-1172-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozawa N, Nagase T, Takemoto Y, et al. Molecular and neurologic findings of peroxisome biogenesis disorders. J Child Neurol. 2005;20:326–329. doi: 10.1177/08830738050200041001. [DOI] [PubMed] [Google Scholar]
- Thoms S, Gärtner J. First PEX11b patient extends spectrum of peroxisomal biogenesis disorder phenotypes. J Med Genet. 2012;49:314–316. doi: 10.1136/jmedgenet-2012-100899. [DOI] [PubMed] [Google Scholar]
- Wanders RJ. Peroxisomal disorders: clinical, biochemical and molecular aspects. Neurochem Res. 1999;24:565–580. doi: 10.1023/A:1022592014988. [DOI] [PubMed] [Google Scholar]
- Wanders R, Waterham H. Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin Genet. 2004;67:107–133. doi: 10.1111/j.1399-0004.2004.00329.x. [DOI] [PubMed] [Google Scholar]
- Zeharia A, Ebberink M, Wanders R, et al. A novel PEX12 mutation identified as the cause of a peroxisomal biogenesis disorder with mild clinical phenotype, mild biochemical abnormalities in fibroblasts and a mosaic catalase immunofluorescence pattern, even at 40°C. J Hum Genet. 2007;52:599–606. doi: 10.1007/s10038-007-0157-y. [DOI] [PubMed] [Google Scholar]