

Abstract
The sodium/iodide symporter (NIS) mediates active iodide (I−) accumulation in the thyroid, the first step in thyroid hormone (TH) biosynthesis. Mutations in the SLC5A5 gene encoding NIS that result in a non-functional protein lead to congenital hypothyroidism due to I− transport defect (ITD). ITD is a rare autosomal disorder that, if not treated promptly in infancy, can cause mental retardation, as the TH decrease results in improper development of the nervous system. However, in some patients, hypothyroidism has been ameliorated by unusually large amounts of dietary I−. Here we report the first NIS knockout (KO) mouse model, obtained by targeting exons 6 and 7 of the Slc5a5 gene. In NIS KO mice, in the thyroid, stomach, and salivary gland, NIS is absent, and hence there is no active accumulation of the NIS substrate pertechnetate (99mTcO4 −). NIS KO mice showed undetectable serum T4 and very low serum T3 levels when fed a diet supplying the minimum I− requirement for rodents. These hypothyroid mice displayed oxidative stress in the thyroid, but not in the brown adipose tissue or liver. Feeding the mice a high-I− diet partially rescued TH biosynthesis, demonstrating that, at high I− concentrations, I− enters the thyroid through routes other than NIS.
Introduction
Iodine is an essential constituent of the thyroid hormones (THs), which are phenolic rings joined by an ether link and iodinated at either three positions (3,5,3′-tri-iodo-l-thyronine, or T3) or four positions (3,5,3′,5′-tetra-iodo-l-thyronine, or T4). The THs are required for the proper development of the central nervous system1, skeleton2, and lungs3 in the fetus and the newborn and for intermediary metabolism at all ages4. Therefore, both hyper and hypothyroidism are diseased states with serious systemic ramifications. Iodide (I−) is an extremely scarce nutrient in the environment and is supplied only in the diet. The Na+/I− symporter (NIS), a member of solute carrier family 5 (SLC5), is the plasma membrane protein that actively transports I− into the thyroid follicular cells, using as its driving force the Na+ gradient generated by the Na+/K+ ATPase. NIS transports I− electrogenically with a 2 Na+ : 1 I− stoichiometry1. Since our group cloned the cDNA coding for NIS5, we have reported in vivo and in vitro that NIS is regulated transcriptionally and post-transcriptionally by thyroid stimulating hormone (TSH)6–9. We have identified residues that coordinate NIS substrates10–12, and elucidated the basis for the efficient transport of I− by NIS at the physiological (submicromolar) concentrations of I− in the plasma13. Besides the thyroid gland, NIS is functionally expressed in other tissues including lactating breast14, breast cancer9, 14, salivary glands14, and stomach1, 9, 14.
NIS-mediated I− transport is the key first step in TH biosynthesis, which occurs partly intracellularly and partly in the colloid, an extracellular compartment. I− reaches the colloid through an I− efflux process that has not been fully elucidated. The genetic characterization of patients affected by Pendred syndrome, a disorder characterized by goiter and a partial I− organification defect15, led to the identification of pendrin (SLC26A4) as a protein that may mediate I− efflux into the colloid. However, pendrin knockout (KO) mice do not display hypothyroidism16, indicating that other mechanisms are also involved in thyroid apical I− efflux. The Na+/monocarboxylate transporter (SMCT) (Slc5a8) is expressed at the apical membrane of thyroid follicular cells17. SMCT was initially proposed to mediate I− efflux into the colloid, because of its high sequence similarity to NIS (~70%)18. However, we demonstrated that SMCT does not transport I− 17 and Slc5a8 KO mice are not hypothyroid19. The calcium-activated chloride channel anoctamin1 (Ano1) is expressed at the apical membrane of thyroid follicular cells20. HEK 293 T and PCCl3 cells co-transfected with plasmids encoding NIS and Ano1 show increased I− efflux in the presence of ionomycin, a Ca+ ionophore, suggesting that Ano1 may play a role in I− efflux20. Ano1 KO mice die within their first month of life and displayed severe tracheomalacia, with gaps in the cartilage rings all along the trachea21. Independently of how I− crosses the apical surface, once it reaches the colloid, it is oxidized to iodine and covalently incorporated into thyroglobulin (TG) by dual oxidases (DUOX) 1 and 2 and thyroperoxidase (TPO)1. TH biosynthesis and release is stimulated by TSH, which is produced by the pituitary gland. TSH binds to its receptor, which is located at the basolateral membrane of the thyroid follicular cells, and stimulates the endocytosis and proteolytic cleavage of iodinated TG, resulting in the release of T3 and T4 into the bloodstream22. Iodine in uncoupled monotyrosine (MIT) and diiodotyrosine (DIT) is reutilized by DEHAL, an iodotyrosine deiodinase that catalyzes the dehalogenation of mono- and diiodotyrosine, allowing I− to be recycled for further TH biosynthesis.
The total or partial impairment of NIS function due to mutations in the SLC5A5 gene (which encodes NIS) has long been known to cause congenital I− transport defect (ITD), which, if left untreated, leads to stunted growth and cognitive deficits1. ITD follows an autosomal recessive inheritance pattern and is diagnosed by reduced or absent thyroid I− uptake and a low saliva-to-plasma I− ratio (normal value is >30). ITD is clinically characterized by hypothyroidism, goiter, and mental impairment of varying degrees. However, there are reports of a few ITD patients whose I− intake was unusually high (~100 times higher than the 150 µg/day recommended by the World Health Organization) and whose hypothyroidism was less severe23, 24. This suggests that, in the absence of a functional NIS, high dietary I− supply may result in some entry of I− into the follicular cells through non-NIS paths, leading to at least modest TH biosynthesis.
We have generated a NIS KO mouse model and used it to test the hypothesis that high concentrations of I− in the plasma, resulting from high dietary I−, make it possible for I− to enter the thyroid follicular cells in the absence of NIS. Existing animal models of hypothyroidism rely on the use of propylthiouracil (PTU) or methimazole (MMI)25–27; these are well-known antithyroid agents that inhibit I− organification, but also have side effects28–30. Thus, we took advantage of our NIS KO model to analyze, under conditions free of pharmacological manipulation, some endocrine and metabolic alterations resulting from the hypothyroid state of these mice.
Results
NIS KO mice on a standard chow diet (CD) produce low levels of T4 but have unaltered levels of T3
To generate a constitutive NIS KO mouse model, we obtained embryonic stem cells from the knockout mouse project (KOMP) repository. Gene targeting was used to modify the Slc5a5 gene (Fig. 1a), which encodes NIS. By homologous recombination, a cassette containing a polyA signal was inserted after exon 5, while exons 6 and 7 were flanked by loxP sites (Fig. 1a). The resulting allele was Slc5a5 TM1a in KOMP nomenclature. A fully verified clone was injected into blastocysts to generate chimeric mice, which transmitted the Slc5a5 TM1a allele to their offspring. These chimeric mice were crossed with mice expressing flippase ubiquitously to remove the LacZ, neo, and polyA signal cassettes. An additional crossing with mice expressing Cre ubiquitously was necessary to remove NIS exons 6 and 7 (Fig. 1a). The expected PCR products were amplified from genomic DNA extracted from wild-type (WT) and NIS KO mice (Fig. 1b,c). Exons 6 and 7 encode transmembrane segments (TMSs) 7 and 8 (Fig. 1d), which are crucial for NIS function12. Moreover, if exon 8 were to splice with exon 5, Cre recombination would generate a frame shift in exon 8, which would produce a stop codon and hence a truncated NIS protein. Mice heterozygous for the disrupted NIS allele were crossed, and the resulting litters displayed Mendelian ratios.
Figure 1.
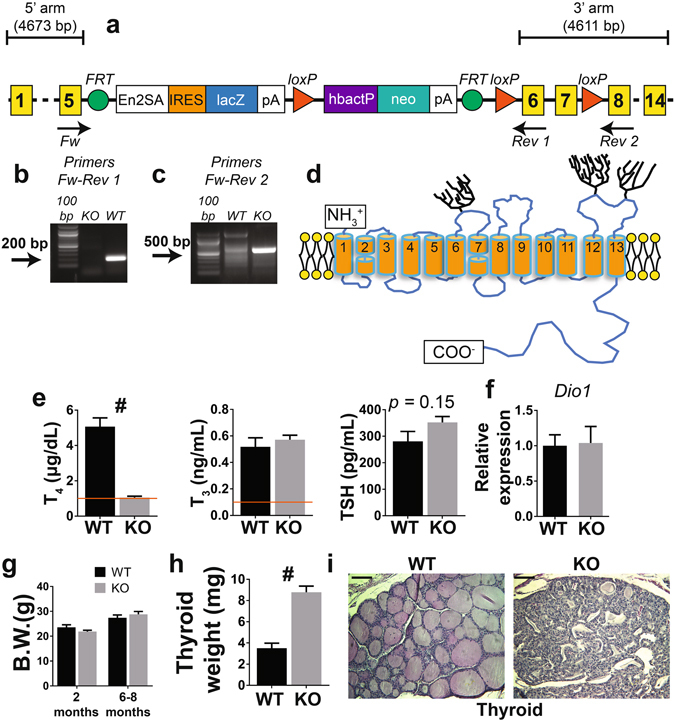
Schematic representation of the targeted region of the Slc5a5 gene, which codes for NIS, and metabolic characterization of WT and NIS KO mice fed a CD. (a) The targeted Slc5a5 allele. Yellow squares mark the exons. (b) The PCR product obtained using the primers Fw and Rev 1. NIS KO mice, which lack exons 6 and 7, do not show any PCR product, while WT mice show the expected 200 bp amplicon. (c) The PCR product obtained using the primers Fw and Rev 2. After FRT and Cre recombination, a 700-bp PCR product is obtained in NIS KO mice, due to the excision of exons 6 and 7. (d) NIS secondary structure model showing the 13 transmembrane segments (TMSs) and carbohydrates (trees). The targeted exons (6 and 7) code for TMSs 7 and 8. (e) Serum TH and TSH levels in 6–8-month-old male WT and NIS KO mice fed a CD. The horizontal line indicates the detection limit of the T4 and T3 ELISA kits (n = 4–5). (f) Liver Dio1 mRNA expression levels (n = 5). (g) Body weight of male WT and NIS KO mice 2 and 6–8 months old fed a CD (n = 5). (h) Thyroid weight of 6–8-month-old male WT and NIS KO mice fed a CD (n = 5). (i) Hematoxylin and eosin staining of thyroid sections obtained from the dissected thyroids in h. Scale bar = 50 µm. En2SA = engrailed 2 gene splice acceptor, IRES = internal ribosome entry site, pA = polyadenylation, hbactP = human beta actin promoter, neo = neomycin resistance. #Indicates p < 0.01.
We then determined serum T4, T3, and TSH levels in NIS KO mice fed a standard CD and compared the results to data from age- and sex-matched WT mice with the same genetic background (Fig. 1e). Surprisingly, NIS KO mice produced T4 and T3. T4 levels were lower (1.05 ± 0.07 µg/dL) than in WT mice (5.05 ± 0.6 µg/dL), while T3 levels (0.51 ± 0.06 ng/mL) were similar to those in WT mice (0.56 ± 0.02 ng/ml). TSH values were higher (albeit non-significantly) in NIS KO mice (352 ± 22 pg/mL) than in WT animals (280 ± 37 pg/mL) (Fig. 1e). The unaltered T3 levels in the serum of NIS KO mice fed a CD may be explained by the observation that D131 was expressed at the same levels in the livers of NIS KO mice and WT mice (Fig. 1f). There were no differences in body weight between NIS KO and WT mice either at 2 or at 6–8 months of age (Fig. 1g). The thyroids of NIS KO mice were larger (8.8 ± 0.6 mg) than those of WT mice (3.5 ± 0.5 mg) (Fig. 1h), a finding consistent with the low T4 and high TSH levels in NIS KO mice. The characteristic follicular structure was lost in the thyroids of 6–8-month-old NIS KO mice, as shown by hematoxylin and eosin-stained sections of these glands (Fig. 1i).
That the NIS KO mice did produce some T4 could be explained in one of two ways: 1) NIS KO mice were not successfully generated, so that some residual NIS was present to mediate I− uptake, or 2) if NIS KO mice were successfully generated, I− entered the thyroid follicular cells by routes other than NIS.
TH biosynthesis still occurs in NIS KO mice, but not owing to residual NIS activity
To determine whether NIS was indeed deleted in our NIS KO mouse model, we investigated NIS expression in NIS KO and WT mice by immunohistochemistry (IHC) and western blot (WB) in the thyroid, salivary glands, and stomach. As expected, in WT mice, IHC showed typical NIS staining at the basolateral plasma membrane of the thyroid follicular cells, mucin-secreting cells in the stomach, and salivary ductal cells (Fig. 2a). In stark contrast, NIS KO mice did not display any specific staining in these three tissues, suggesting that the protein was not expressed there (Fig. 2a). These findings were confirmed by the WB results: there was no immunoreactivity in tissues from NIS KO mice, whereas the typical polypeptide bands corresponding to NIS were evident in the tissues from WT mice (Fig. 2b and Supplementary Figure 1). The electrophoretic mobility of NIS is different in the thyroid, stomach, and salivary glands because, as we have reported previously32, NIS is glycosylated to a different extent in each of these three tissues (Fig. 2b).
Figure 2.
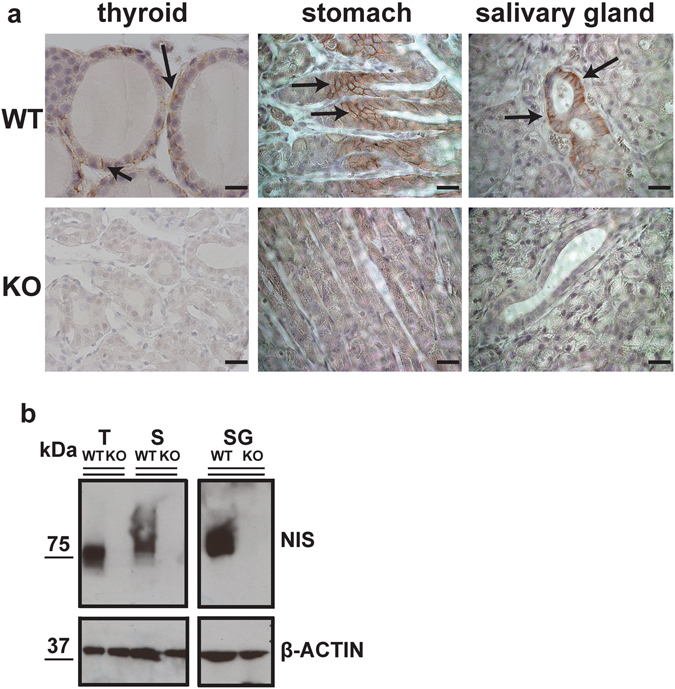
NIS protein expression is abolished in NIS KO mice. (a) Sections of thyroid, stomach, and salivary glands obtained from WT or NIS KO mice fed a CD were incubated with anti-NIS Ab and IHC performed as described in Materials and Methods. Arrows indicate the typical basolateral NIS staining observed in organs from WT mice. NIS KO mice did not show any specific staining. Scale bar = 10 µm. (b) WB analysis of NIS expression in organs from WT and NIS KO mice. Images were cropped from the full images presented in Supplementary Figure 1.
To investigate the unlikely possibility that the NIS KO mice produced a truncated functional protein undetectable by WB or IHC, because our NIS antibody recognizes the last 16 residues of the carboxy terminus of NIS6 (Fig. 1d), we carried out hybrid SPECT (single-photon emission computed tomography)/CT imaging of WT and NIS KO mice fed a CD using the NIS substrate pertechnetate (99mTcO4 −)33. 99mTcO4 −, a NIS substrate that is widely used in clinical medicine, has several advantages over I− isotopes: it is inexpensive, has a short half-life (~6 h), and is not covalently incorporated into TG in the thyroid. As expected, WT mice accumulated 99mTcO4 − in the regions corresponding to the thyroid and salivary glands, and in the stomach (Fig. 3a,b). In NIS KO mice, 99mTcO4 − was detected only in the bladder, as it is eliminated in the urine (Fig. 3a,b).
Figure 3.
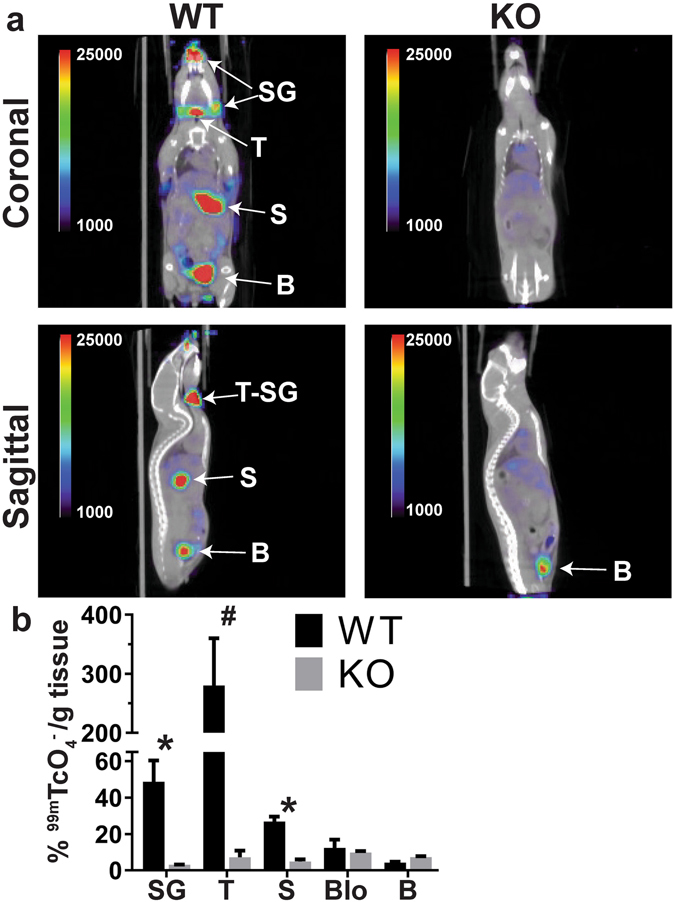
Lack of active accumulation of a NIS substrate in NIS KO mice. (a) SPECT/CT analysis of 3-month-old female WT and NIS KO mice fed a CD (coronal and sagittal views). WT mice accumulate 99mTcO4 − in the thyroid (T), salivary glands (SG), and stomach (S). NIS KO mice accumulate 99mTcO4 − only in the bladder (B). Images are displayed as % injected dose/cc. (b) Quantitation of 99mTcO4 − in the corresponding dissected organs 3 hours after injection of the isotope (n = 2–3) with gamma well counting. Activity is expressed as % injected dose/gram of tissue. T = thyroid, SG = salivary gland, S = stomach, Blo = blood, B = bladder. *Indicates p < 0.05, # p < 0.01.
In conclusion, NIS KO mice do not accumulate 99mTcO4 − in the tissues where WT mice display NIS-mediated active transport of 99mTcO4 − because NIS KO mice do not express NIS.
NIS KO mice on a minimal iodide diet (MID) have very low T4, low T3, and high TSH
The minimum required supply of I− for rodents is 0.15 µg per gram of food34. The standard CD provides 6 µg of I− per gram of food (i.e., 40 times the minimum required supply). We hypothesized that such a great excess of I− in the diet would sufficiently increase the plasma I− concentration to make it possible for I− to enter the follicular cells via non-specific mechanisms.
To test this hypothesis, we measured T4, T3, and TSH levels in NIS KO mice fed a MID, which supplies 0.15 µg of I− per gram of food (the minimum recommended amount)34. Indeed, these animals had extremely low serum T4 levels and low serum T3 levels (0.16 ± 0.02 ng/mL) (Fig. 4a,b). In accordance with the reduced serum T3 levels, NIS KO mice showed a strong downregulation (~75%) of D1 expression in their liver, compared to WT mice fed the same MID (Fig. 4f). TSH levels were increased in NIS KO mice compared to WT mice fed a MID (Fig. 4c). In conclusion, some THs are biosynthesized in mice totally devoid of NIS as long as their I− supply is sufficiently great to allow I− to enter the thyroid, most likely via non-specific routes.
Figure 4.
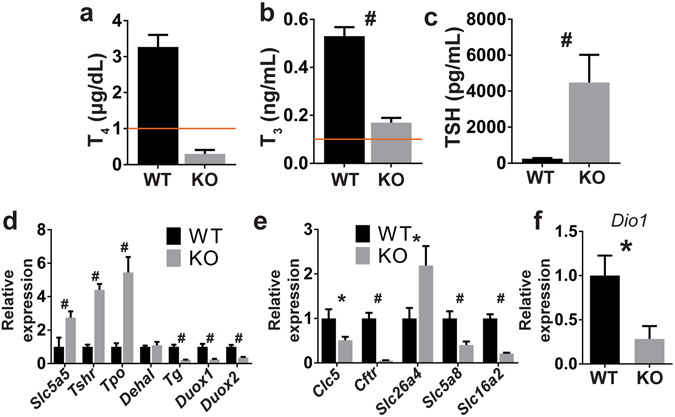
Reduced I− intake impairs TH biosynthesis in NIS KO mice. (a,b) Serum T4 and T3 levels of male WT and NIS KO mice fed a MID. The horizontal line indicates the detection limit of the T4 and T3 ELISA kits. (c) Serum TSH levels of male WT and NIS KO mice fed a MID (n = 5). (d) Thyroid mRNA expression levels of genes involved in TH biosynthesis (n = 7–8). (e) Thyroid mRNA expression levels of apical thyroid proteins. Each group contained 4–5 males and 3 females fed a MID. No sex differences were observed. (f) Liver Dio1 mRNA expression levels in males (n = 7–8). Slc5a5 encodes NIS, Tshr = thyroid stimulating hormone receptor, Tpo = thyroid peroxidase, Dehal = iodotyrosine deiodinase, Tg = thyroglobulin, Duox 1 and 2 = Dual oxidase 1 and 2, Clc5 = chloride voltage-gated channel 5, Cftr = cystic fibrosis transmembrane conductance regulator, Slc26a4 encodes pendrin, Slc5a8 encodes SMCT, Slc16a2 = solute carrier family 16 member 2, and Dio1 = iodothyronine deiodinase 1. *Indicates p < 0.05, # p < 0.01.
TSH-stimulated expression of genes involved in TH biosynthesis is upregulated in NIS KO mice on a MID
I− organification takes place immediately after the anion exits the thyroid follicular cells apically to enter the colloid22. I− organification is more efficient under hypothyroid than euthyroid conditions, because high levels of TSH stimulate the overexpression of certain genes involved in TH biosynthesis22. NIS KO mice on a MID showed increased expression of the Tshr, Tpo, and the truncated Slc5a5 mRNA (Fig. 4d). However, Tg and Duox 1 and 2, which are not upregulated by TSH35, 36, were all strongly downregulated in these mice, as were the apical anion channels Clc5 and Cftr, the apically expressed Na+-dependent monocarboxylate transporter (SMCT, encoded by Slc5a8), and the basolaterally expressed MCT8 [encoded by solute carrier family 16 member 2 (Slc16a2)], whereas the expression of iodotyrosine deiodinase (Dehal) did not change in NIS KO mice. Interestingly, Slc26a4, which encodes pendrin, was upregulated in NIS KO mice (Fig. 4e), which is consistent with the proposal that pendrin is one of the conduits that facilitate I− transport into the colloid.
NIS KO mice on a MID exhibit distinct metabolic adaptations in the thyroid, brown adipose tissue (BAT), and liver
Reactive oxygen species (ROS) are tightly regulated at multiple levels for homeostasis. In addition, the generation of ROS is affected in a complex fashion by pharmacological intervention. For these reasons, it is often difficult to draw conclusions from studies of ROS. For example, THs have generally been claimed to increase metabolic rates and ROS production37, 38; therefore, lower ROS levels are expected in hypothyroidism. However, the antithyroid drug MMI, used to generate hypothyroid mouse models, actually increases oxidative stress39. Therefore, our NIS KO mice on a MID represent a unique drug-free model for studying the effect of hypothyroidism on ROS production. NIS KO mice on a MID showed reduced expression of Duox genes in the thyroid (Fig. 4d) but, in agreement with previous findings, increased concentrations of H2O2 and free radicals (FRs) [ROS and reactive nitrogen species (RNS)]40, 41 (Fig. 5a,b). Interestingly, the superoxide dismutases (SODs) Sod1 and 2 (which produce H2O2); catalase (Cat, which disproportionates H2O2 to H2O and O2); and Nfe2l2, Gpx1, Txn1, Gstp1, and Gsta1 (genes involved in ROS disposal) were all strongly downregulated in the thyroids of these mice (Fig. 5c). Taken together, these results suggest that TSH stimulation increases oxidative stress in the thyroid by downregulating ROS disposal, because the balance between production and elimination is skewed in the direction of higher ROS levels in the NIS KO mice.
Figure 5.
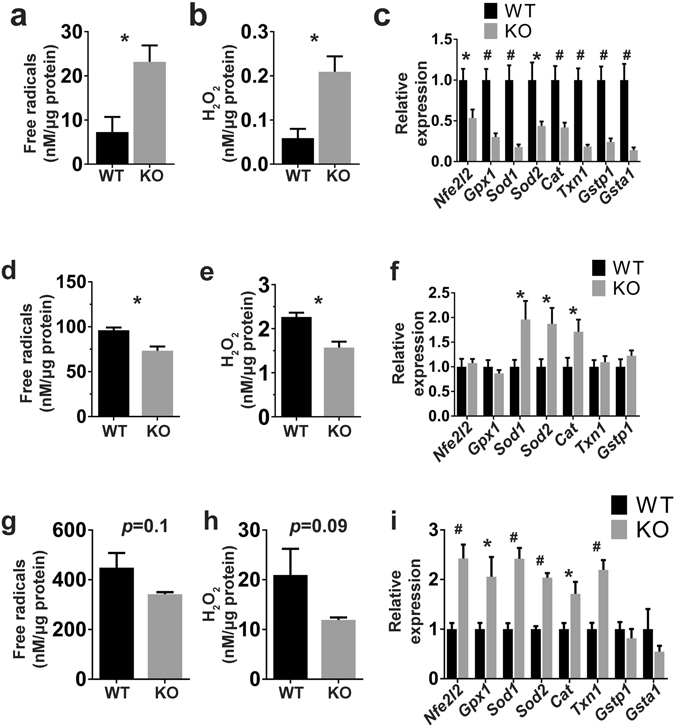
Hypothyroidism produces oxidative stress in the thyroid and a less oxidative intracellular environment in BAT and liver. (a,b) Quantitation of FR and H2O2 levels in thyroid extracts from WT and NIS KO mice. (c) Expression of genes involved in FR metabolism in the thyroid (n = 7–8). (d,e) Quantitation of FR and H2O2 levels in BAT extracts. (f) Expression of genes involved in FR metabolism in BAT. (g,h) Quantitation of FR and H2O2 levels in liver extracts. (i) Expression of genes involved in free radical metabolism in the liver. Each group contained 4–5 males and 2–3 females fed a MID. Nfe2l2 = nuclear factor, erythroid 2 like 2, Gpx1 = glutathione peroxidase 1, Sod 1 and 2 = superoxide dismutase 1 and 2, Cat = Catalase, Txn1 = thioredoxin 1, Gstp1 = glutathione S-transferase pi 1, and Gsta1 = glutathione S-transferase alpha 1. *Indicates p < 0.05, # p < 0.01.
THs stimulate mitochondrial activity4. The increased mitochondrial activity leads to higher ROS production in target tissues such as BAT and liver4, 38, 42. To study the effect of hypothyroidism on ROS generation in BAT and liver, we measured H2O2 and FR levels in these tissues. BAT showed a significant reduction in both H2O2 and FR levels (Fig. 5d,e) as well as upregulation of Sod1, Sod2, and Cat (Fig. 5f). Upregulation of these enzymes may favor the breakdown of H2O2 into H2O and O2, thereby reducing H2O2 levels. Liver tissue, on the other hand, showed a non-significant reduction in H2O2 and FR levels (Fig. 5g,h), but consistent upregulation of several genes involved in H2O2 and FR metabolism (Fig. 5i). These results suggest that hypothyroidism causes lower FR levels not only through reduced mitochondrial activity but also via upregulation of genes involved in H2O2 and FR metabolism.
Discussion
Mutations in the SLC5A5 gene, which encodes NIS, lead to congenital hypothyroidism (CH) due to an I− transport defect (ITD). ITD is a rare autosomal recessive condition diagnosed by reduced or absent thyroidal I− uptake and a low saliva-to-plasma I− ratio (<30). To date, 15 ITD-causing NIS mutations have been reported1. Invaluable mechanistic and structural information has been obtained by characterizing the amino acid positions bearing mutations in ITD patients and elucidating the molecular requirements of NIS at those positions. For example, the first ITD-causing mutation T354P led us to identify the Na2 binding site of NIS10, 11, 43.
Interestingly, the same NIS mutation was discovered in the three children of a Japanese couple, as was the mutation V59E44. The patients inherited these two mutations (V59E and T354P) from their healthy mother and father, respectively. The T354P and V59E NIS mutant proteins, when expressed in COS7 cells, were both trafficked to the cell surface, but totally inactive. Surprisingly, the three siblings displayed different degrees of mental retardation. The oldest was nursed for longer than the second oldest, and evinced a less severe cognitive deficit. The youngest was not nursed, and displayed a more severe cognitive deficit than either of her siblings. It was discovered that the mother was addicted to laminaria, an alga extremely rich in I−. As NIS is expressed in the lactating breast14, where it mediates the transport of I− to the milk, our hypothesis was that breastfeeding the oldest and second-oldest children supplied them with a high-I− diet, enabling them to produce higher levels of THs than the youngest child and thereby lessening their cognitive deficit. In the studies reported here, we tested this hypothesis by generating a NIS KO mouse model (Fig. 1), which recapitulates the conditions of ITD, and which provides evidence that even in the absence of NIS expression (Fig. 2), extremely high dietary I− can partially restore TH biosynthesis (Fig. 1e). Consistent with these findings, studies of ITD patients that preceded our cloning of NIS5 revealed that some of these patients’ symptoms were ameliorated when they were given 14 (or even 100) mg potassium iodide daily23, 24, which is ~90 (or ~640) times the daily amount recommended for adults by the Food and Nutrition Board45 and the World Health Organization46. We recapitulated this condition by feeding NIS KO mice a CD supplying 40 times the recommended minimum amount of I− for rodents34.
Here, we formally demonstrated that NIS is the only protein that actively accumulates I− in the thyroid, stomach, and salivary glands (Fig. 3a). We also showed that I− can enter the thyroid through non-specific mechanisms, most likely basolaterally expressed Cl− channels or cotransporters. NIS, a specific active transporter that mediates I− accumulation in the thyroid, must have been crucial in evolution, as serum [Cl−]s are >106 times higher than serum [I−]s (~100 mM vs. ~300 nM)1. A higher dietary I− in turn increases serum [I−]s, which generates a concentration gradient that allows I− to diffuse into the thyroid follicular cells. A critical mechanism for maintaining this gradient is the upregulation of genes regulated by TSH (Fig. 4d): these genes code for proteins that increase the efficiency of I− organification, which makes the I− gradient favorable for the passive diffusion of I− into the thyroid. TSH has been reported to increase expression only of the protein pendrin, but not of Slc26a4, the gene encoding it, in vitro 47. However, we observed increased levels of Slc26a4 gene expression in our NIS KO mice (Fig. 4e), which, as shown in Fig. 4c, have higher TSH levels than WT mice.
The overexpression in NIS KO mice of TSH-regulated genes involved in TH biosynthesis is consistent with the notion that, in the absence of NIS, high dietary I− partially rescues TH biosynthesis. This is likely because an inwardly directed I− concentration gradient is generated, allowing I− to diffuse passively into the thyroid follicular cells through non-specific routes, followed by the immediate oxidation of I− to iodine and the incorporation of this iodine into TG by TPO. As previously proposed36, when there is only a small amount of iodine available, the marked downregulation of TG may be a mechanism for preventing it from being incorporated into tyrosine residues that will not end up being part of T4 (or T3) molecules.
Mitochondria are the main source of ROS in the cell48. The mitochondrial electron transport chain generates superoxide radicals (O2 −). SODs promptly convert O2 − into H2O2 and O2 48, 49. Although ROS are key second messengers in several signal transduction pathways50, 51, an excess of them can lead to oxidative stress characterized by increased cellular macromolecule damage, as often occurs in aging52. THs regulate mitochondrial activity, and severe hyperthyroidism is associated with cellular damage due to increased ROS production49. Reduced thyroid function, by contrast, is associated with longevity in both human and animal models53–55. It has been hypothesized that THs stimulate mitochondrial activity, which contributes to increased ROS production and spurs on the aging process53, 55. Here, we measured FR levels in BAT, liver, and thyroid under hypothyroid conditions. BAT is rich in mitochondria, important for thermoregulation, and one of the main targets of THs. Under hypothyroid conditions, we found that BAT displays significantly reduced FR and H2O2 levels (Fig. 5d,e). However, the FR–related genes were not downregulated, and Sod1, Sod2, and Cat were even upregulated (Fig. 5f). THs also regulate the expression of metabolic genes in the liver, which plays a central role in glucose and lipid metabolism31, 56, 57. In the liver, we found that FR and H2O2 levels were reduced (Fig. 5g,h), although not significantly, and several FR–related genes were upregulated (Fig. 5i). Our results indicate that hypothyroidism affects genes involved in FR metabolism.
The H2O2-rich environment in the thyroid is key for TH biosynthesis, as H2O2 is essential for I− oxidation. TSH and I− have antagonistic effects on H2O2 production in thyrocytes. TSH induces H2O2 production, whereas I− inhibits it58, 59. These opposing effects guarantee protection against oxidative stress in euthyroid conditions when TSH levels are within the physiological range and I− levels are adequate, but allow improved I− organification in hypothyroid conditions when TSH is increased and I− is scarce. It is not surprising that hypothyroid conditions increase metabolism in the thyroid. Thyrocytes are forced to proliferate by chronic TSH stimulation, an adaptive response that attempts to compensate for reduced TH levels. This may be the cause of the increased FR and H2O2 levels we observed (Fig. 5a,b). DUOX 1 and 2 are part of the TH biosynthetic pathway: they produce H2O2, which is crucial for I− organification. Interestingly, Duox genes are strongly downregulated in the thyroids of hypothyroid mice (Fig. 4d). This may be a mechanism to protect the thyroid from the higher H2O2 levels caused by the increased metabolic activity of the thyrocytes. Although the oxidative environment produced by hypothyroidism may be interpreted as an adaptive response aimed at making I− oxidation more efficient, it may be dangerous, as it may damage macromolecules, including lipids, proteins and DNA40, 60. This oxidative environment can, in the long term, cause DNA mutations that can lead to thyroid cancer61.
In summary, we report here, for the first time, the generation of a constitutive NIS KO mouse model, which is characterized by hypothyroidism, with reduced TH and increased TSH levels. We have demonstrated that I− can enter the thyroid via mechanisms other than NIS, and that, in hypothyroidism, peripheral tissues display a less oxidative intracellular environment, whereas the thyroid, stimulated by TSH, evinces an increased intracellular concentration of free radicals.
Materials and Methods
Animals
Mouse protocols were approved by the Yale University International Animal Care and Use Committee (IACUC). All experiments were performed in accordance with relevant guidelines and regulations. Five-to-seven-month-old male C57BL/6 J/N-A mice with mixed genetic backgrounds were used unless otherwise specified. Sex-, age-, and diet-matched WT mice with the same genetic background were used as controls. Mice were fed a chow diet (CD) (Harlan 2018, which provides 6 µg of I− per gram) up until they were switched to the minimal iodide diet (MID) (Harlan TD.150677, which provides 0.15 µg of I− per gram), two weeks prior to the metabolic studies. Both the CD and the MID provide ~20% of calories from protein, ~60% from carbohydrates, and ~20% from fat. NIS KO mice were generated by following the standard procedure reported before62.
Slc5a5-targeted embryonic stem (ES) cells harboring the C57BL/6N-Atm1Brd (http://www.informatics.jax.org/allele/key/814110) genetic background were injected into C57BL/6 J blastocysts to obtain chimeric mice. Chimeras were crossed with B6.Cg-Tg(ACTFLPe)9205Dym/J mice (https://www.jax.org/strain/005703) harboring the C57BL/6 J genetic background. Litters positive for the Slc5a5 targeted allele were crossed with FVB/N-Tg(ACTB-cre)2Mrt/J mice (https://www.jax.org/strain/003376), which had been backcrossed for over 20 generations with C57BL/6 J mice. NIS KO mice were crossed with C57BL/6 J WT mice to expand the colony.
Western blot
SDS-PAGE, electrotransfer to PVDF membranes, and immunoblotting were carried out as described6. Membranes were incubated with 4 nM of an affinity-purified polyclonal anti-rat NIS Ab (diluted 1:2000 in TBST 5% Non-Fat Omniblok milk Americanbio AB10109) directed against 16 residues of the cytosolic NIS carboxyl terminus6. Equal loading was assessed by stripping and reprobing the same blot with 0.5 µg/ml monoclonal anti-β-actin Ab (Cell Signaling 4970) diluted 1:1000 in TBST 5% Non-Fat Omniblok milk. Horseradish peroxidase (HRP)-linked secondary anti-mouse and anti-rabbit Abs were from Jackson ImmunoResearch (West Grove, PA, USA). Proteins were visualized using the enhanced chemiluminescence western blot detection system (Amersham Biosciences).
Immunohistochemistry
Organs were fixed overnight in 10% neutral buffered formalin prior to paraffin inclusion63.
5-μm sections were deparaffinated in xylene and rehydrated through graded alcohols. Then antigen retrieval was performed at 95 °C for 15 min with 10 mM citrate buffer solution. After cooling slides, endogenous peroxidase was quenched with 3% hydrogen peroxide. Slides were incubated with 5% goat serum. Slides were incubated overnight with an affinity purified anti-rat NIS Ab6 diluted 1:3000 in PBS 0.5% BSA. The remainder of the immunoperoxidase procedure was carried out according to the supplier’s instructions provided in the EnVision System-HRP (DAKO K4010). Slides were analyzed by light microscopy.
MicroSPECT/CT
At each imaging session animals were injected with 99mTcO4 − [0.962 ± 0.137 mCi (mean ± SEM)] via the tail vein for microSPECT/CT imaging using a dedicated high-resolution small-animal hybrid imaging system (GammaMedica, X-SPECT)33. One and a half hours following injection of 99mTcO4 −, ungated CT imaging was performed (50 kVp/800 μA; projections, 512) to identify the anatomic structures. Immediately following the CT scan, microSPECT images were acquired using the following acquisition parameters: 32 projections, 60 seconds per projection, and 140 keV photopeak ± 20% window. Imaging occurred under light isoflurane anesthesia (1.5–2.0% isoflurane/98.0–98.5% oxygen).
MicroSPECT/CT Image Reconstruction and Analyses
Reconstructed microSPECT images were reoriented according to the CT anatomic images and external point sources, fused, and exported in “Analyze” format (Analyze, Mayo Clinic) for further processing using Amide Medical Imaging Data Examiner (amide.sf.net)33. Images were corrected for injected dose and decay from injection time and displayed as % injected dose/cc.
99mTcO4− quantitation in tissues
Mice were euthanized with saturated KCl (1–2 mmol/kg) and their tissues rapidly extracted. Each tissue piece’s radioactivity was measured by gamma well counting (Cobra Packard). Raw counts were corrected for spill-up/spill-down, background, decay, and weight. Corrected counts were converted to mCi/g with the use of previously determined counter efficiency. Activity in each tissue segment was then calculated as percentage of injected dose (%ID) by correcting for decay to the time of radiotracer injection. The calculated %ID was computed by dividing corrected tissue counts (mCi/g) by the corrected injected dose (mCi) and expressing it as %ID per gram tissue (%ID/g)33.
Hormone, free radical, and H2O2 measurements
Hormone levels were determined using age-matched male mice. TH levels were quantitated with the Thyroxine (T4) ELISA (Mouse/Rat) Kit (Sigma SE120090 and Calbiotech T4044T)64, 65 and the Triiodothyronine (T3) ELISA (Mouse/Rat) Kit (Sigma SE120091 and Calbiotech T3043T)65, 66.
TSH levels were quantitated using the Milliplex map mouse pituitary magnetic bead panel (Millipore MPTMAG-49K-01), following the manufacturer’s instructions.
Free radicals and H2O2 were measured using the OxiSelectTM in Vitro ROS/RNS Assay kit (Green fluorescence; Cell Biolabs Inc STA-347)67–69. The proprietary non-fluorescent dichlorodihydrofluorescein DiOxyQ (DCFH-DiOxyQ) probe was primed to form the non-fluorescent DCFH-DiOxy and then converted to the non-fluorescent DCFH. DCFH can react with ROS and H2O2 to form fluorescent DCF. Tissues were homogenized in PBS and incubated with DCFH and catalytic agents. The fluorescence intensity was analyzed using a Tecan infinite M1000 with the excitation at 480 nm and emission at 530 nm. Data were normalized against a DCF or an H2O2 standard curve. All kits were used according to the manufacturer’s instructions.
Real Time PCR
RNA from frozen dissected tissues was extracted by using Trizol reagent (Ambion 15596026). cDNA was synthesized by using the iScript cDNA Synthesis Kit (Biorad 1708891). Five ng of cDNA were amplified by using Power SYBR® Green PCR Master Mix (Applied Biosystems 4368577), according to the manufacturer’s instructions. The Light Cycler 480 System (Roche Life Science) was used to carry out amplification. For each sample, the expression of the genes of interest was normalized to the expression of Rn18S measured under the same conditions. Specific primers for the analyzed genes were designed by using Primer-BLAST http://www.ncbi.nlm.nih.gov/tools/primer-blast/ if not otherwise specified. Primer sequences are given in Supplementary Table 1.
Statistical analysis
All the quantitative measurements represent the average ± SEM of 4–8 mice per group if not differently indicated. The differences between the groups were determined using a two-tailed Student’s T-test. All calculations were carried out in Microsoft Excel. *Indicates P < 0.05, # P < 0.01.
Electronic supplementary material
Acknowledgements
We thank Dr. Silvia Ravera for critical reading of the manuscript and valuable discussions. This study was supported by NIH Grant DK-41544 (to N.C.), NIH Grant HL-098069 (to A.J.S.), and a grant from the American Thyroid Association (to A.R.-N.). We also acknowledge the technical support of Xiangning Wang for SPECT imaging.
Author Contributions
N.C. designed research; A.J.S. oversaw imaging component of studies, G.F., R.R.K., A.R.N., N.B. performed research; and G.F., R.R.K., and N.C. wrote the paper. All authors read and approved the final version of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04326-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Portulano C, Paroder-Belenitsky M, Carrasco N. The Na+/I− symporter (NIS): mechanism and medical impact. Endocr Rev. 2014;35:106–149. doi: 10.1210/er.2012-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bassett JH, Williams GR. Role of Thyroid Hormones in Skeletal Development and Bone Maintenance. Endocr Rev. 2016;37:135–187. doi: 10.1210/er.2015-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bizzarro, M. J. & Gross, I. Effects of hormones on fetal lung development. Obstet Gynecol Clin North Am31, 949–961, xii (2004). [DOI] [PubMed]
- 4.Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94:355–382. doi: 10.1152/physrev.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature. 1996;379:458–460. doi: 10.1038/379458a0. [DOI] [PubMed] [Google Scholar]
- 6.Levy O, et al. Characterization of the thyroid Na+/I− symporter with an anti-COOH terminus antibody. Proc Natl Acad Sci USA. 1997;94:5568–5573. doi: 10.1073/pnas.94.11.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohno M, Zannini M, Levy O, Carrasco N, Di Lauro R. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependermt transcription. Mol Cell Biol. 1999;19:2051–2060. doi: 10.1128/MCB.19.3.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riedel C, Levy O, Carrasco N. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. Journal of Biological Chemistry. 2001;276:21458–21463. doi: 10.1074/jbc.M100561200. [DOI] [PubMed] [Google Scholar]
- 9.Wapnir IL, et al. The Na+/I− symporter mediates iodide uptake in breast cancer metastases and can be selectively down-regulated in the thyroid. Clin Cancer Res. 2004;10:4294–4302. doi: 10.1158/1078-0432.CCR-04-0074. [DOI] [PubMed] [Google Scholar]
- 10.De la Vieja A, Reed MD, Ginter CS, Carrasco N. Amino acid residues in transmembrane segment IX of the Na+/I− symporter play a role in its Na+ dependence and are critical for transport activity. J Biol Chem. 2007;282:25290–25298. doi: 10.1074/jbc.M700147200. [DOI] [PubMed] [Google Scholar]
- 11.Ferrandino G, et al. Na+ coordination at the Na2 site of the Na+/I− symporter. Proc Natl Acad Sci USA. 2016;113:E5379–5388. doi: 10.1073/pnas.1607231113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paroder-Belenitsky M, et al. Mechanism of anion selectivity and stoichiometry of the Na+/I− symporter (NIS) P Natl Acad Sci USA. 2011;108:17933–17938. doi: 10.1073/pnas.1108278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicola, J. P., Carrasco, N. & Amzel, L. M. Physiological sodium concentrations enhance the iodide affinity of the Na+/I− symporter. Nat Commun5 (2014). [DOI] [PMC free article] [PubMed]
- 14.Tazebay UH, et al. The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat Med. 2000;6:871–878. doi: 10.1038/78630. [DOI] [PubMed] [Google Scholar]
- 15.Everett LA, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 16.Calebiro D, et al. Absence of primary hypothyroidism and goiter in Slc26a4 (−/−) mice fed on a low iodine diet. J Endocrinol Invest. 2011;34:593–598. doi: 10.3275/7262. [DOI] [PubMed] [Google Scholar]
- 17.Paroder V, et al. Na+/monocarboxylate transport (SMCT) protein expression correlates with survival in colon cancer: Molecular characterization of SMCT. P Natl Acad Sci USA. 2006;103:7270–7275. doi: 10.1073/pnas.0602365103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez AM, et al. Identification and characterization of a putative human iodide transporter located at the apical membrane of thyrocytes. J Clin Endocr Metab. 2002;87:3500–3503. doi: 10.1210/jcem.87.7.8797. [DOI] [PubMed] [Google Scholar]
- 19.Frank H, et al. Lactaturia and loss of sodium-dependent lactate uptake in the colon of SLC5A8-deficient mice. Journal of Biological Chemistry. 2008;283:24729–24737. doi: 10.1074/jbc.M802681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Twyffels L, et al. Anoctamin-1/TMEM16A is the major apical iodide channel of the thyrocyte. Am J Physiol Cell Physiol. 2014;307:C1102–1112. doi: 10.1152/ajpcell.00126.2014. [DOI] [PubMed] [Google Scholar]
- 21.Rock JR, Futtner CR, Harfe BD. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev Biol. 2008;321:141–149. doi: 10.1016/j.ydbio.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Mondal, S., Raja, K., Schweizer, U. & Mugesh, G. Chemistry and Biology in the Biosynthesis and Action of Thyroid Hormones. Angew Chem Int Ed Engl (2016). [DOI] [PubMed]
- 23.Toyoshima K, Matsumoto Y, Nishida M, Yabuuchi H. Five cases of absence of iodide concentrating mechanism. Acta Endocrinol (Copenh) 1977;84:527–537. doi: 10.1530/acta.0.0840527. [DOI] [PubMed] [Google Scholar]
- 24.Wolff J. Congenital Goiter with Defective Iodide Transport. Endocrine Reviews. 1983;4:240–254. doi: 10.1210/edrv-4-3-240. [DOI] [PubMed] [Google Scholar]
- 25.Vallortigara J, Alfos S, Micheau J, Higueret P, Enderlin V. T3 administration in adult hypothyroid mice modulates expression of proteins involved in striatal synaptic plasticity and improves motor behavior. Neurobiol Dis. 2008;31:378–385. doi: 10.1016/j.nbd.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 26.Weiner J, et al. Thyroid hormone status defines brown adipose tissue activity and browning of white adipose tissues in mice. Sci Rep. 2016;6:38124. doi: 10.1038/srep38124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarkar, D. & Singh, S. K. Effect of neonatal hypothyroidism on prepubertal mouse testis in relation to thyroid hormone receptor alpha 1 (THRalpha1). Gen Comp Endocrinol (2016). [DOI] [PubMed]
- 28.Fumarola A, Di Fiore A, Dainelli M, Grani G, Calvanese A. Medical Treatment of Hyperthyroidism: State of the Art. Exp Clin Endocr Diab. 2010;118:678–684. doi: 10.1055/s-0030-1253420. [DOI] [PubMed] [Google Scholar]
- 29.Andersen SL, Laurberg P. Managing hyperthyroidism in pregnancy: current perspectives. Int J Womens Health. 2016;8:497–504. doi: 10.2147/IJWH.S100987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laurberg P, Andersen SL. Antithyroid Drug Use in Pregnancy and Birth Defects: Why Some Studies Find Clear Associations, and Some Studies Report None. Thyroid. 2015;25:1185–1190. doi: 10.1089/thy.2015.0182. [DOI] [PubMed] [Google Scholar]
- 31.Zavacki AM, et al. Type 1 iodothyronine deiodinase is a sensitive marker of peripheral thyroid status in the mouse. Endocrinology. 2005;146:1568–1575. doi: 10.1210/en.2004-1392. [DOI] [PubMed] [Google Scholar]
- 32.Levy O, et al. N-linked glycosylation of the thyroid Na+/I− symporter (NIS). Implications for its secondary structure model. J Biol Chem. 1998;273:22657–22663. doi: 10.1074/jbc.273.35.22657. [DOI] [PubMed] [Google Scholar]
- 33.Su HL, et al. Noninvasive targeted imaging of matrix metalloproteinase activation in a murine model of postinfarction remodeling. Circulation. 2005;112:3157–3167. doi: 10.1161/CIRCULATIONAHA.105.583021. [DOI] [PubMed] [Google Scholar]
- 34.National Research Council (US). Subcommittee on Laboratory Animal Nutrition. Nutrient requirements of laboratory animals, (National Academy of Sciences, Washington, D.C., 1995).
- 35.Milenkovic M, et al. Duox expression and related H2O2 measurement in mouse thyroid: onset in embryonic development and regulation by TSH in adult. Journal of Endocrinology. 2007;192:615–626. doi: 10.1677/JOE-06-0003. [DOI] [PubMed] [Google Scholar]
- 36.Vassart G, et al. Structure, Expression and Regulation of the Thyroglobulin Gene. Molecular and Cellular Endocrinology. 1985;40:89–97. doi: 10.1016/0303-7207(85)90162-5. [DOI] [PubMed] [Google Scholar]
- 37.Fernandez V, et al. Thyroid hormone-induced oxidative stress in rodents and humans: A comparative view and relation to redox regulation of gene expression. Comp Biochem Phys C. 2006;142:231–239. doi: 10.1016/j.cbpc.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Sinha RA, et al. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy. 2015;11:1341–1357. doi: 10.1080/15548627.2015.1061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heidari R, Babaei H, Eghbal M. Mechanisms of methimazole cytotoxicity in isolated rat hepatocytes. Drug Chem Toxicol. 2013;36:403–411. doi: 10.3109/01480545.2012.749272. [DOI] [PubMed] [Google Scholar]
- 40.Maier J, et al. Iodine deficiency activates antioxidant genes and causes DNA damage in the thyroid gland of rats and mice. Bba-Mol Cell Res. 2007;1773:990–999. doi: 10.1016/j.bbamcr.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 41.Poncin S, et al. Oxidative stress in the thyroid gland: From harmlessness to hazard depending on the iodine content. Endocrinology. 2008;149:424–433. doi: 10.1210/en.2007-0951. [DOI] [PubMed] [Google Scholar]
- 42.Lee JY, et al. Triiodothyronine induces UCP-1 expression and mitochondrial biogenesis in human adipocytes. Am J Physiol-Cell Ph. 2012;302:C463–C472. doi: 10.1152/ajpcell.00010.2011. [DOI] [PubMed] [Google Scholar]
- 43.Levy O, Ginter CS, De la Vieja A, Levy D, Carrasco N. Identification of a structural requirement for thyroid Na+/I− symporter (NIS) function from analysis of a mutation that causes human congenital hypothyroidism. FEBS Lett. 1998;429:36–40. doi: 10.1016/S0014-5793(98)00522-5. [DOI] [PubMed] [Google Scholar]
- 44.Reed-Tsur MD, D la Vieja A, Ginter CS, Carrasco N. Molecular characterization of V59E NIS, a Na+/I− symporter mutant that causes congenital I- transport defect. Endocrinology. 2008;149:3077–3084. doi: 10.1210/en.2008-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prete A, Paragliola RM, Corsello SM. Iodine Supplementation: Usage “with a Grain of Salt”. Int J Endocrinol. 2015;2015:312305. doi: 10.1155/2015/312305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zimmermann MB, Galetti V. Iodine intake as a risk factor for thyroid cancer: a comprehensive review of animal and human studies. Thyroid Res. 2015;8:8. doi: 10.1186/s13044-015-0020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pesce L, et al. TSH Regulates Pendrin Membrane Abundance and Enhances Iodide Efflux in Thyroid Cells. Endocrinology. 2012;153:512–521. doi: 10.1210/en.2011-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21:569–576. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugawara S. Reactive Oxygen Species and Thyroid Diseases. Systems Biology of Free Radicals and Antioxidants. 2014;1:3521–3538. doi: 10.1007/978-3-642-30018-9_150. [DOI] [Google Scholar]
- 50.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 51.Woo CH, et al. Tumor necrosis factor-alpha generates reactive oxygen species via a cytosolic phospholipase A2-linked cascade. J Biol Chem. 2000;275:32357–32362. doi: 10.1074/jbc.M005638200. [DOI] [PubMed] [Google Scholar]
- 52.Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med. 2013;60:1–4. doi: 10.1016/j.freeradbiomed.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 53.Gesing A, Lewinski A, Karbownik-Lewinska M. The thyroid gland and the process of aging; what is new? Thyroid Res. 2012;5:16. doi: 10.1186/1756-6614-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buffenstein R, Pinto M. Endocrine function in naturally long-living small mammals. Mol Cell Endocrinol. 2009;299:101–111. doi: 10.1016/j.mce.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Atzmon G, Barzilai N, Hollowell JG, Surks MI, Gabriely I. Extreme longevity is associated with increased serum thyrotropin. J Clin Endocrinol Metab. 2009;94:1251–1254. doi: 10.1210/jc.2008-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Visser WE, et al. Tissue-Specific Suppression of Thyroid Hormone Signaling in Various Mouse Models of Aging. PLoS One. 2016;11:e0149941. doi: 10.1371/journal.pone.0149941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dozin B, Magnuson MA, Nikodem VM. Thyroid-Hormone Regulation of Malic Enzyme-Synthesis - Dual Tissue-Specific Control. Journal of Biological Chemistry. 1986;261:290–292. [PubMed] [Google Scholar]
- 58.Corvilain B, Laurent E, Lecomte M, Vansande J, Dumont JE. Role of the Cyclic Adenosine-3′,5′-Monophosphate and the Phosphatidylinositol Ca2+ Cascades in Mediating the Effects of Thyrotropin and Iodide on Hormone Synthesis and Secretion in Human Thyroid Slices. J Clin Endocr Metab. 1994;79:152–159. doi: 10.1210/jcem.79.1.8027219. [DOI] [PubMed] [Google Scholar]
- 59.Corvilain B, Vansande J, Dumont JE. Inhibition by Iodide of Iodide Binding to Proteins - the Wolff-Chaikoff Effect Is Caused by Inhibition of H2o2 Generation. Biochem Bioph Res Co. 1988;154:1287–1292. doi: 10.1016/0006-291X(88)90279-3. [DOI] [PubMed] [Google Scholar]
- 60.Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res-Fund Mol M. 2011;711:193–201. doi: 10.1016/j.mrfmmm.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 61.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer How are they linked? Free Radical Bio Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Behringer, R. Manipulating the mouse embryo: a laboratory manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 2014).
- 63.Bianco AC, et al. American Thyroid Association Guide to investigating thyroid hormone economy and action in rodent and cell models. Thyroid. 2014;24:88–168. doi: 10.1089/thy.2013.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watson LA, et al. Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J Clin Invest. 2013;123:2049–2063. doi: 10.1172/JCI65634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piatkowska E, et al. The Impact of Carrot Enriched in Iodine through Soil Fertilization on Iodine Concentration and Selected Biochemical Parameters in Wistar Rats. PLoS One. 2016;11:e0152680. doi: 10.1371/journal.pone.0152680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Viana-Huete V, et al. Essential Role of IGFIR in the Onset of Male Brown Fat Thermogenic Function: Regulation of Glucose Homeostasis by Differential Organ-Specific Insulin Sensitivity. Endocrinology. 2016;157:1495–1511. doi: 10.1210/en.2015-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang YC, et al. Deficiency of NPGPx, an oxidative stress sensor, leads to obesity in mice and human. EMBO Mol Med. 2013;5:1165–1179. doi: 10.1002/emmm.201302679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soni KK, et al. Dose-dependent effects of cisplatin on the severity of testicular injury in Sprague Dawley rats: reactive oxygen species and endoplasmic reticulum stress. Drug Des Devel Ther. 2016;10:3959–3968. doi: 10.2147/DDDT.S120014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yin Q, et al. Antioxidant Defenses in the Brains of Bats during Hibernation. PLoS One. 2016;11:e0152135. doi: 10.1371/journal.pone.0152135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.