

Abstract
Key points
Afterhyperpolarizations (AHPs) generated by repetitive action potentials in supraoptic magnocellular neurons regulate repetitive firing and spike frequency adaptation but relatively little is known about PIP2’s control of these AHPs.
We examined how changes in PIP2 levels affected AHPs, somatic [Ca2+]i, and whole cell Ca2+ currents.
Manipulations of PIP2 levels affected both medium and slow AHP currents in oxytocin (OT) neurons of the supraoptic nucleus.
Manipulations of PIP2 levels did not modulate AHPs by influencing Ca2+ release from IP3‐triggered Ca2+ stores, suggesting more direct modulation of channels by PIP2.
PIP2 depletion reduced spike‐evoked Ca2+ entry and voltage‐gated Ca2+ currents.
PIP2 appears to influence AHPs in OT neurons by reducing Ca2+ influx during spiking.
Abstract
Oxytocin (OT)‐ and vasopressin (VP)‐secreting magnocellular neurons of the supraoptic nucleus (SON) display calcium‐dependent afterhyperpolarizations (AHPs) following a train of action potentials that are critical to shaping the firing patterns of these cells. Previous work demonstrated that the lipid phosphatidylinositol 4,5‐bisphosphate (PIP2) enabled the slow AHP component (sAHP) in cortical pyramidal neurons. We investigated whether this phenomenon occurred in OT and VP neurons of the SON. Using whole cell recordings in coronal hypothalamic slices from adult female rats, we demonstrated that inhibition of PIP2 synthesis with wortmannin robustly blocked both the medium and slow AHP currents (I mAHP and I sAHP) of OT, but not VP neurons with high affinity. We further tested this by introducing a water‐soluble PIP2 analogue (diC8‐PIP2) into neurons, which in OT neurons not only prevented wortmannin's inhibitory effect, but slowed rundown of the I mAHP and I sAHP. Inhibition of phospholipase C (PLC) with U73122 did not inhibit either I mAHP or I sAHP in OT neurons, consistent with wortmannin's effects not being due to reducing diacylglycerol (DAG) or IP3 availability, i.e. PIP2 modulation of AHPs is not likely to involve downstream Ca2+ release from inositol 1,4,5‐trisphosphate (IP3)‐triggered Ca2+‐store release, or channel modulation via DAG and protein kinase C (PKC). We found that wortmannin reduced [Ca2+]i increase induced by spike trains in OT neurons, but had no effect on AHPs evoked by uncaging intracellular Ca2+. Finally, wortmannin selectively reduced whole cell Ca2+ currents in OT neurons while leaving VP neurons unaffected. The results indicate that PIP2 modulates both the I mAHP and I sAHP in OT neurons, most likely by controlling Ca2+ entry through voltage‐gated Ca2+ channels opened during spike trains.
Keywords: after‐hyperpolarization, after‐hyperpolarization current, caged calcium, calcium, oxytocin, PIP2
Key points
Afterhyperpolarizations (AHPs) generated by repetitive action potentials in supraoptic magnocellular neurons regulate repetitive firing and spike frequency adaptation but relatively little is known about PIP2’s control of these AHPs.
We examined how changes in PIP2 levels affected AHPs, somatic [Ca2+]i, and whole cell Ca2+ currents.
Manipulations of PIP2 levels affected both medium and slow AHP currents in oxytocin (OT) neurons of the supraoptic nucleus.
Manipulations of PIP2 levels did not modulate AHPs by influencing Ca2+ release from IP3‐triggered Ca2+ stores, suggesting more direct modulation of channels by PIP2.
PIP2 depletion reduced spike‐evoked Ca2+ entry and voltage‐gated Ca2+ currents.
PIP2 appears to influence AHPs in OT neurons by reducing Ca2+ influx during spiking.
Abbreviations
- AHP
afterhyperpolarization
- DAG
diacylglycerol
- IAHP
afterhyperpolarization current
- MNC
magnocellular neurosecretory cell
- OT
oxytocin
- PKC
protein kinase C
- PLC
phospholipase C
- PVN
paraventricular nucleus
- SON
supraoptic nucleus
- VP
vasopressin
Introduction
Oxytocin (OT)‐ and vasopressin (VP)‐secreting magnocellular neurosecretory cells (MNCs) play a crucial role in many physiological functions including lactation, parturition (OT cells) and cardiovascular regulation (VP cells) (Armstrong et al. 2010). The release of both hormones is optimized by a burst‐firing pattern of action potentials, albeit of different form in the two cell types (Dutton & Dyball, 1979; Bicknell & Leng, 1981; Cazalis et al. 1985). Sustained stimulation of these cells (most notably VP) results in significant reduction of hormone release over time, whereas periods of quiescence reverse this secretory fatigue (Bicknell, 1988). Furthermore, the burst‐firing is synchronized among all OT neurons in the supraoptic nucleus (SON) and paraventricular nucleus (PVN) (Belin et al. 1984) during lactation, whereas VP neurons burst asynchronously (Sabatier & Leng, 2007). Many cell types, including MNCs, express calcium‐activated potassium currents that (i) produce afterhyperpolarizations (AHPs) following a single action potential or train of action potentials and (ii) shape firing patterns. In MNCS, the AHP typically comprises three components with varying time courses mediated by different channels: fast (fAHP), medium (mAHP), and slow (sAHP). The fast AHP current (I fAHP) is contributed by KCa1.1 (BK) channels as well as voltage‐gated K+ channels, and lasts <15 ms (Dopico et al. 1999; Hlubek & Cobbett, 2000; Roper et al. 2003). The medium AHP current (I mAHP) lasts ∼500 ms, is underlain by KCa2.3 (SK3) channels (Stocker & Pedarzani, 2000; Tacconi et al. 2001), and is blocked by apamin (Bourque & Brown, 1987). The channel underlying the slow AHP current (I sAHP) in MNCS (and most neuron types) is unknown, but is Ca2+ dependent and lasts on the order of seconds (Ghamari‐Langroudi & Bourque, 2004; Andrade et al. 2012). Pinpointing an exact mechanism underlying the sAHP has been difficult because the I sAHP characteristics differ among neuronal cell types (Andrade et al. 2012). However, there are some common features, including activation by rises in [Ca2+]i, modulation by neurotransmitters, and voltage independence. This gap in our understanding is important because the I sAHP controls spike frequency adaptation and is affected by major intracellular signals, including Ca2+, in many cell types of several brain regions. With regard to MNCs, the I mAHP and I sAHP are of particular interest due to their significant influence on burst firing patterns. Further, the AHPs in OT neurons get larger during late pregnancy and lactation (Stern & Armstrong, 1996; Teruyama & Armstrong, 2002), coinciding with an increase in pulsatile OT release from the neurohypophysis during labour and lactation (Wakerly & Lincoln, 1971; Higuchi et al. 1986) and the adoption of synchronized bursting firing patterns (Poulain & Wakerley, 1982).
While we do not know how Ca2+ activates the unknown AHP channels, a study in neocortical pyramidal cells demonstrated that phosphatidylinositol 4,5‐bispohsphate (PIP2) facilitates the I sAHP but not the SK‐mediated I mAHP (Villalobos et al. 2011). This is intriguing because PIP2 is known to influence the properties of a host of channel types and is activated by Gq‐coupled receptors (Hille, 1994; Suh & Hille, 2002, 2008; Suh et al. 2010; Kruse & Hille, 2013), including receptors that are activated by neurotransmitters known to attenuate the I sAHP, (e.g. acetylcholine via muscarinic receptors). These features, combined with the slow current kinetics and strong Ca2+ dependence, support the hypothesis that a biochemical signalling cascade contributes to modulation of the I sAHP, which may influence more than one channel type (Andrade et al. 2012).
Methods
Ethical approval
These studies were performed on virgin adult Sprague‐Dawley female rats (Harlan Laboratories, Indianapolis, IN, USA) weighing between 150–220 g. The UTHSC IACUC review board approved all experiments and the experiments conform to the principles of UK regulations as described in (Drummond, 2009). Animals were on an ad libitum diet. For use in experiments, rats were deeply anesthetized with either sodium pentobarbital (100 mg kg−1) or ketamine–xylasine (10% xylasine; 100 mg kg−1) and perfused through the heart with artificial cerebrospinal fluid (aCSF) with NaCl replaced by 210 mm sucrose. The rats were decapitated via guillotine. The brains were then removed and subsequently sliced for use in whole cell patch clamp electrophysiology. The work described in this report complies with ethical standards and protocols under which The Journal of Physiology operates as described in Grundy (2015). AHPs in OT neurons undergo significant plastic changes during the female reproductive cycle (Teruyama & Armstrong, 2002, 2005). Because of this, we limit our study to females because these changes offer insights into the MNC‐specific mechanisms of AHP generation.
Slice preparation
Coronal brain slices 250 μm thick were cut in ice‐cold aCSF with 210 mm sucrose replacing NaCl, using a Leica VT1000S vibratome. After cutting, the brain slices were transferred to an aCSF‐filled holding chamber and warmed for 15–20 min at 32°C. aCSF was continuously bubbled with 95% O2–5% CO2, and contained (in mm): 20 d‐glucose, 0.45 ascorbic acid, 2.5 KCl, 1 MgSO4, 1.25 NaH2PO4.H2O, 26 NaHCO3, 125 NaCl, 2 CaCl2. Slices were then transferred to aCSF at room temperature, where they remained for at least 40 min prior to recording.
Electrophysiology
Slices were placed in the well of a Plexiglass chamber attached to a modified stage on an Olympus BX51WI upright microscope and perfused with aCSF containing 5 mm CsCl to block the slow depolarizing after‐potential (sDAP) (Ghamari‐Langroudi & Bourque, 1998; Teruyama & Armstrong, 2005, 2007). The aCSF was bubbled constantly with 95% O2–5% CO2, warmed to 32°C ± 1°C, and flowed at ∼2 ml min−1. Whole cell voltage clamp recordings were obtained using an Axon Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). Traces were digitized using an Axon 1440A Digitizer at 10 kHz and filtered at 2 kHz on a Dell desktop computer running Clampex 9 software (Molecular Devices).
Recording pipettes (4–8 MΩ) were pulled from borosilicate glass with an outer diameter of 1.5 mm using a P‐1000 flaming/brown horizontal micropipette puller (Sutter Instruments, Sovato, CA, USA). The pipette internal solution for analysing AHP tail currents consisted of (in mm): 135 KMeSO4, 8 NaCl, 10 Hepes, 2 Mg‐ATP, 0.3 Na‐GTP, 0.1 leupeptin, 6 phosphocreatine, 0.2 EGTA with pH 7.2–7.4 and 285–295 mosmol (kg H2O)−1. 0.1% biocytin (Sigma‐Aldrich, USA) was added to an aliquot on the day of the experiment for visualization during immunochemical identification of cell type. The liquid junction potential for the KMeSO4 internal was ∼−10 mV, and was not corrected. For certain experiments, 30 μm diC8‐PIP2 (Echelon Biosciences, Salt Lake City, UT, USA) reconstituted in H2O was added to the internal solution.
I AHP tail currents were evoked using a voltage clamp protocol of a 17 spike, 5 ms pulse train at 20 Hz from a holding potential of −60 mV to +20 mV; the I AHP was clamped at −60 mV immediately following the train. This was done to mimic a train of action potentials, and at 20 Hz, 17 spikes produced an I AHP amplitude near maximum in pilot experiments (data not shown). In MNCs I AHP amplitude correlates with spike count (Ghamari‐Langroudi & Bourque, 2004). Baseline I AHP recordings were taken in the presence of 10 μm 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX), 40 μm 2R)‐amino‐5‐phosphonovaleric acid (AP5), and 100 μm picrotoxin to block fast synaptic currents. Experimental reagents include 0.001–10 μm wortmannin (Sigma‐Aldrich), 100 nm apamin (Sigma‐Aldrich), 10 μm 2‐(4‐morpholinyl)‐8‐phenyl‐4H‐1‐benzopyran‐4‐one (LY294,002) (Sigma‐Aldrich) to block PI3K, 10 μm 1‐[6‐[[(17β)‐3‐methoxyestra‐1,3,5(10)‐trien‐17‐yl]amino]hexyl]‐1H‐pyrrole‐2,5‐dione (U73122) (Sigma‐Aldrich) to block PLC, and 300 μm (cells were incubated before the experiment in 1 mm) myo‐inositol (Sigma‐Aldrich) (Villalobos et al. 2011) to supplement the substrate for PIP2 production. Cells whose series resistance exceeded 20 MΩ and/or changed by more than 20% during the recording were discarded. I AHPs were averaged over 2 or more runs.
For measuring whole cell Ca2+ currents, the external bath solution contained (in mm): 80 NaCl, 50 TEA‐Cl, 5 CsCl, 1 MgCl2, 10 Hepes, 10 glucose, 0.0005 tetrodotoxin (TTX), 0.1 picrotoxin, 4 CaCl2. The pipette internal solution contained: 180 N‐methyl‐d‐glucamine, 4 MgCl2, 40 Hepes, 10 EGTA, 12 phosphocreatine, 2 Mg‐ATP, 0.4 Na‐GTP. The liquid junction potential for this internal solution is ∼−5 mV and was not corrected. Cells were held at −70 mV. For I–V curves, cells were hyperpolarized to −90 mV for 200 ms followed by 10 mV 1000 ms steps up to +10 mV. Cd2+ at 400 μm was bath‐applied at the end of each trial to confirm the Ca2+ current. I–V curves were derived from the steady‐state measurement of these steps. Currents were leak subtracted by scaling the current in response to a +10 mV step from baseline.
Immunochemistry
Slices were fixed in 4% paraformaldehyde and 0.2% picric acid in phosphate buffered saline (PBS) and stored at 4°C post‐experimentally. Biocytin‐labelled neurons were processed for double labelling with either anti‐OT‐ or VP‐neurophysins. The anti‐VP‐neurophysin is a rabbit polyclonal antibody provided by Alan Robinson (UCLA, Emeritus), and was used at 1:20,000. The anti‐OT‐neurophysin antibody (PS36) is a mouse monoclonal antibody provided by Harold Gainer (National Institutes of Health, Emeritus) and was used at 1:500. All antibodies and labelling reagents were dissolved in PBS + 0.5% Triton X‐100 (PBST). After 36–72 h of incubation at 4°C, the slices were washed with PBST and incubated in a cocktail of secondary antibodies including Alexa Fluor 488 goat anti‐rabbit immunoglobulin G (IgG) and Alexa Fluor 594 goat anti‐mouse IgG (1:200) along with Avidin‐AMCA (1:200) for reaction with the biocytin. Double staining of biocytin and one antibody complemented with the negative staining of the other identified neurons as OT or VP (Fig. 1).
Figure 1. An example of a positively identified OT cell using immunolabelling.
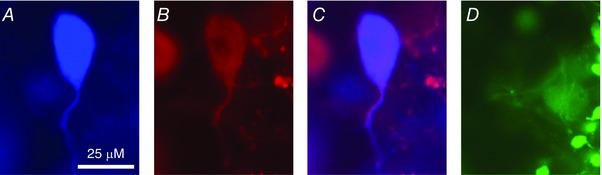
A, a single cell loaded with biocytin during recording, and subsequently labelled with Avid‐AMCA. The tissue was then labelled for OT‐ and VP‐neurophysins (NP) by double immunofluorescence using Alexa Fluor 594‐ and Alexa Fluor 488‐conjugated antibodies, respectively. This biocytin‐labelled cell was immunoreactive to the OT‐NP (B and C) but not to VP‐NP (D).
Calcium imaging and uncaging
Electrophysiology and Ca2+ imaging were performed simultaneously on a single computer using a custom windows‐based program (CCD32; written by Dr J. Callaway, UTHSC, Memphis, TN, USA, based on software developed by Lasser‐Ross et al. (1991). Voltage clamp recordings were acquired using an Axopatch 700A (Molecular Devices, Sunnyvale, CA, USA). Electrodes were pulled using borosilicate electrodes (4–8 MΩ). The pipette internal solution for analysing AHP tail currents was as previously described, except with 0.2 mm EGTA replaced with 0.1 mm fura‐2. Images were obtained with a CCD Imago Sensicam camera, using a Polychrome V monochromater (TILL Photonics, Planegg, Germany) to control excitation wavelength and intensity. Calcium fluorescence was obtained by exciting fura‐2 at 380 nm using a USHIO UXL‐150MO 150 W xenon arc lamp. We measured fluorescence changes at an emission wavelength of 520 ± 40 nm. Photobleaching was corrected by subtracting a Ca2+ signal from a control sweep at equal length, in which the cell was not stimulated and held at a hyperpolarized holding potential (−70 mV) to minimize Ca2+ entry. We subtracted background autofluorescence by using a reference area near the cell. Measurements were made from the soma, excluding the nucleus.
To uncage Ca2+, we used 2 mm DM‐nitrophen as the Ca2+ caging compound loaded into the internal solution with 40% calcium occupancy (0.8 mm CaCl2). In the internal solution, 0.2 mm EGTA was replaced with 0.05 mm fluo‐4 for Ca2+ imaging. Fluo‐4 was excited at 488 nm and measured at 520 ± 40 nm. Additionally, Mg‐ATP was replaced by Na‐ATP to avoid loading the caging compound with Mg2+. We used a xenon flashlamp (Rapp Optoelectronic, JML‐C2) for photolysis and discharged 72.6 J in ∼1 ms with UV light. Upon photolysis, the K d of DM‐nitrophen for Ca2+ increases from 5 nm to 3 mm, rapidly releasing Ca2+ into the cell (Sah & Clements, 1999).
All Ca2+ imaging data are reported as percentage relative change in fura‐2 fluorescence (%ΔF/F). These data were analysed either with the custom acquisition program described above, or with Igor Pro 7.0 (Wavemetrics Inc., Portland, OR, USA).
Dissociated cells and PIP2 distribution
Adult female rats (150–220 g) (n = 5) were anaesthetized with ketamine–xylasine (dose) and perfused through the heart with cold sucrose solution as described above for slices. We adapted the protocol of Shah et al. (2014) for assessing PIP2 labelling in dissociated SON neurons. Tissue blocks from the SON area were excised from 200 μm sections taken with a vibratome, and incubated in oxygenated Pipes (in mm: 110 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 20 Pipes and 25 glucose, pH 7.1, containing trypsin (0.6 mg ml−1; Sigma Aldrich) for 90 min at 34°C. The tissue was transferred to oxygenated Pipes without trypsin for 30 min at room temperature, then gently triturated with three different sizes of fire‐polished pasture pipettes. The cells were plated into two wells from a 6‐well plate (Cat. P06‐1.5H‐N, In Vitro Scientific, Sunnyvale, CA, USA) and allowed to settle at room temperature for 30 min. One well was then treated with 100 nm wortmannin in Pipes for 30 min at room temperature, while the second well was untreated. The media in each well was then replaced with fixative (4% paraformaldehyde with 0.2% picric acid in 0.01 m PBS) and incubated overnight at 4°C. The fixative was rinsed 3× with PBS and the cells then treated with 10% non‐fat dry milk in PBS for 1 h at room temperature to block non‐specific antibody interactions. The cells were incubated overnight at 4°C for double labelling with an antibody raised in rabbit against OT–NP (oxytocin–neurophysin, 1:5000) + mouse anti‐PIP2 antibody (1:1000, Cat. ADI‐915‐062‐100, Enzo Life Sciences), rinsed 3× in PBS then incubated in a cocktail of secondary antibodies for 1 h at room temperature. The secondary antibodies used were goat‐anti‐rabbit (Alexa Fluor 568 nm, Invitrogen, Carlsbad, CA, USA) and goat‐anti‐mouse (Alexa Flour 488 nm, Invitrogen) conjugated IgGs at 1:200 dilutions with PBS. Cells were rinsed 3× in PBS then stained with DAPI (300 nm, Cat. D9542, Sigma Aldrich) in PBS for 5 min. Cells were rinsed 3× in PBS and left immersed in PBS for microscopic analysis.
Images were acquired with a Zeiss 710 confocal inverted microscope (Carl Zeiss Microscopy, Thornwood, NY, USA) using a 63× oil‐immersion objective (n.a. 1.4). OT‐positive neurons were located and 1 μm optical sections were collected from the top to the bottom of each cell sampled. The laser power (18%) and the pin‐hole (2.5 AU) were constant for all the images taken and well below saturation. Only cells that were positive for both OT–NP and PIP2 antibodies as well as exhibiting clear DAPI nuclear staining were considered for the study. For each group, from each animal, at least 31 images were taken. Thus a total of more than 150 cells per group (control and wortmannin treated) were analysed for this study. Data were analysed blind. One investigator made the dissociated cell preparation, drug treatment and antibody staining. A second investigator acquired the images blind to the two groups, then coded the digital images for measurements by the first investigator. Thereafter the second investigator decoded the cells, and the first investigator made the appropriate statistical analysis.
Images were analysed with Image J (NIH, Bethesda, MD, USA). Each Z‐stack had between 15 and 25 optical sections, thus a PIP2 image for analysis was selected from each cell based on the maximum size of the nucleus through the stack. For measuring the average membrane (or near membrane) intensity of PIP2 staining for each cell, the line tool with freehand line option was used to draw a line (5 pixels) around the edge of the cell and the mean pixel intensity value was recorded. To measure the average intensity of the cytoplasm, an area excluding the nucleus and the perimeter of the cell was selected using the polygon tool (1 pixel), and the mean intensity was recorded. Such mean intensities recorded were then subtracted from the mean background intensity for each cell. To measure mean background intensity from each cell, a uniform dark spot was selected using the polygon tool (1 pixel), and the mean intensity was recorded. The data are presented as cytoplasm: membrane ratios. These ratios were then averaged for each group in each animal, and analysed with a Wilcoxon ranked‐sum non‐parametric test, using JMP Pro 12.
Statistics and analysis
In MNCs the I fAHP is a fast, transient event lasting <15 ms (Dopico et al. 1999), and was not evaluated herein. The I mAHP decay tau is ∼500 ms in MNCs (Teruyama & Armstrong, 2005), whereas the I sAHP has a decay tau of 1–2 s in MNCs (Ghamari‐Langroudi & Bourque, 2004). With this consideration, we operationally defined the I mAHP and I sAHP as the amplitude of the AHP tail current (I AHP) at 100 ms and 1000 ms after the stimulus, respectively. While measurement of the I mAHP at 100 ms is likely to contain a small contribution from the I sAHP, it is dominated by the I mAHP due to the slower onset kinetics of the I sAHP (Teruyama & Armstrong, 2005). Furthermore, we demonstrate in the Results section that the SK channel blocker apamin (100 nm) inhibits the I mAHP by 70.5 ± 5.5% (data not shown). In some experiments, the I sAHP was selectively evaluated after isolation in the presence of 100 nm apamin.
All electrophysiological traces were analysed in ClampFit 10.2 (Molecular Devices) or Igor Pro. Because the I mAHP and I sAHP are relatively small currents, special considerations were taken when measuring them in voltage clamp. During analysis, the currents were further filtered using a Gaussian lowpass filter at 1 kHz. Measurements of amplitude were averaged over a 30 ms segment of current (30 points). This was done to marginalize the contribution of electrical noise to individual points. All statistics were performed in SPSS. Unless stated otherwise, data were compared using a Student's paired samples t test. For the experiment comparing controls to apamin and apamin + wortmannin (Fig. 5), we ran a repeated measures ANOVA with a Bonferroni post hoc test for pairwise comparisons. All reported values are represented as means ± SEM.
Figure 5. Wortmannin inhibits the apamin‐treated I AHP in OT neurons while having no effect in VP neurons.
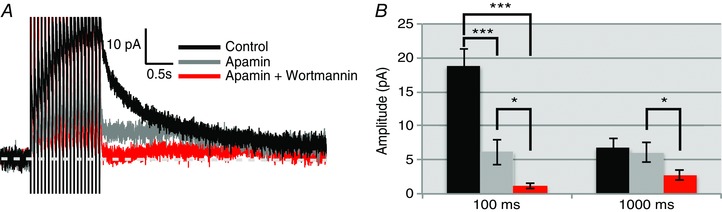
Apamin (100 nm) was applied to MNCs to block the I mAHP in both cell types, followed by wortmannin (1 μm) to observe effects on the isolated I sAHP. A, voltage clamp trace of I AHP from an OT neuron. This contains a baseline recording (black trace), apamin treated trace to block the I mAHP (grey), and a trace with apamin and wortmannin (red). Wortmannin inhibited the apamin‐isolated I sAHP. B, summary data of OT (n = 6) neurons. There was a significant main effect (repeated measures ANOVA, F (2,12) = 45.97, *** P < 0.001). Apamin inhibits the Im AHP at 100 ms (*** P < 0.001). Measurements of the I sAHP (1000 ms) indicate that apamin has little effect at this time, while subsequent wortmannin application significantly reduces the isolated I sAHP (* P < 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
Results
Depleting PIP2 from cells inhibits the I mAHP and I sAHP of OT but not VP neurons
Previous work demonstrated that PIP2 levels are critical for generation of I sAHP, but not I mAHP, in cortical pyramidal neurons (Villalobos et al. 2011). To test this in SON neurons, we bath‐applied wortmannin and measured the macroscopic AHP tail current (I AHP) at two time points (see Methods section: Data and analysis). Wortmannin is a pharmacological agent known to inhibit PIP2 levels by blocking the rate‐limiting enzyme of PIP2 production, PI4Kα (Nakanishi et al. 1995) (Fig. 2). Application of wortmannin (10 μm) caused a significant decrease in both I mAHP and I sAHP of OT supraoptic neurons (n = 10, P < 0.001; 100 ms: control 22.12 ± 1.9 pA vs. wortmannin 2.67 ± 0.3 pA; 1000 ms: control 6.48 ± 0.9 pA vs. wortmannin 1.01 ± 0.4 pA; Fig. 3). In contrast to OT cells, VP cells demonstrated no significant change in I mAHP and I sAHP in response to wortmannin (n = 9, P > 0.05; 100 ms: control 19.67 ± 4.2 pA vs. wortmannin 23.72 ± 5.1 pA; 1000 ms: control 5.44 ± 1 pA vs. wortmannin 6.12 ± 1.1 pA; Fig. 3). To characterize wortmannin's effect on AHPs further, we generated a dose–response curve measuring percentage of I mAHP inhibition as a function of wortmannin concentration. We used the I mAHP time point because current amplitudes were much larger, allowing clear measurements at all doses. Wortmannin's effects on OT neurons were dose dependent, characterized by a sigmoidal dose–response relationship with an IC50 = 58 nm and a Hill coefficient of 1.6 (Fig. 3 D). This indicates that wortmannin exhibits specific effects at nanomolar concentrations, with non‐cooperative binding. While the effect of wortmannin was similarly dose dependent on the I sAHP, with stronger inhibition at increased wortmannin concentrations, dose–response plots could not be reliably fitted with a Hill equation due to the small amplitude of the IsAHP (≤20 pA; data not shown).
Figure 2. Diagram of the relevant PIP2 pathways.
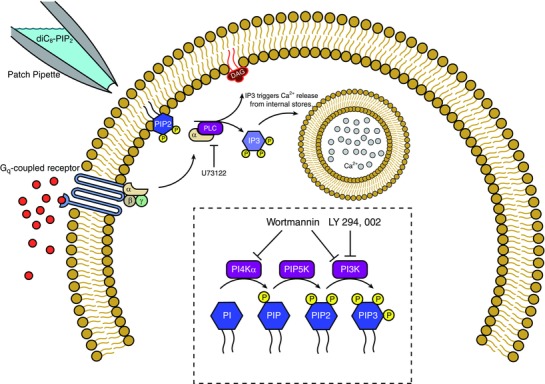
Upon stimulation of Gq‐coupled receptors, the α component of the G‐protein activates phospholipase‐C (PLC), which cleaves membrane‐bound PIP2 into diacylglycerol (DAG) and inositol trisphosphate (IP3). DAG activates protein kinase C while IP3 triggers release of Ca2+ ions from internal Ca2+ stores. U73122 blocks the activity of PLC. PIP2 has been demonstrated to modulate several ion channels in previous studies, such as Kv7 channels (Suh & Hille, 2007) and SK channels (Zhang et al. 2014). Inset, the dashed box displays the pathway for production of different PIP molecules (blue hexagons), and the enzymes that phosphorylate each of them (purple boxes). Wortmannin blocks synthesis of PIP2 and PIP3 production while LY294,002 only blocks PIP3 production. Both work by blocking the synthetic enzymes.
Figure 3. Wortmannin (10 μm), a PIP2 and PIP3 inhibitor, blocks the I mAHP and I sAHP of OT neurons but not VP neurons.
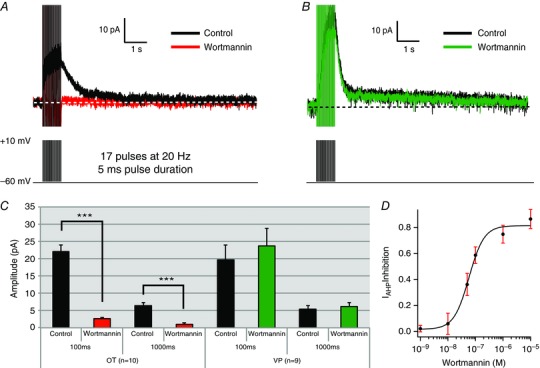
A and B, voltage clamp recording of a supraoptic magnocellular OT (A) and VP (B) cell before and after wortmannin application. Wortmannin application inhibits both the I mAHP and I sAHP in OT cells while having no effect in VP cells. C, summary data of the wortmannin effect on OT and VP cells: I AHP amplitude was measured at 100 ms (I mAHP) and 1000 ms (I sAHP) after the end of the pulse. OT cells demonstrate statistically significant inhibition of both the I mAHP and I sAHP (n = 10, *** P < .001). In contrast, VP cells demonstrate no significant change in I mAHP or I sAHP amplitude (n = 9, P > 0.05). D, dose–response curve for wortmannin in OT neurons. We plotted peak I mAHP inhibition as a function of wortmannin concentration. Wortmannin exerted robust inhibition at nanomolar concentrations (IC50 = 58 nm). The Hill Coefficient of 1.6 ± 0.02 indicates relatively non‐cooperative binding. A minimum of 5 cells was collected for each concentration of wortmannin. [Color figure can be viewed at wileyonlinelibrary.com]
Although wortmannin is an inhibitor of PIP2 synthesis, it also depletes PIP3 by inhibiting phosphoinositide 3‐kinase (PI3K) (Meyers & Cantley, 1997; Vanhaesebroeck et al. 2001). Furthermore, PI3K is the higher affinity target in comparison to PI4Kα (Meyers & Cantley, 1997; Vanhaesebroeck et al. 2001). We thus bath‐applied the specific PI3K inhibitor LY294,002 (10 μm: Vlahos et al. 1994; Lee et al. 2007; Wang et al. 2016) to both OT and VP neurons to test for PIP3 involvement. This control has been used previously in studies of PIP2‐affected AHPs in cortex (Villalobos et al. 2011). As in pyramidal neurons, LY294,002 had no significant effect on the I mAHP or I sAHP of either cell type (OT: n = 7, P > 0.05; control 46.1 ± 6.4 pA vs. wortmannin 44.18 ± 7.4 pA; VP: n = 4, P > 0.05; control 36.48 ± 10.4 pA vs. wortmannin 38.58 ± 10.5 pA; Fig. 4), consistent with wortmannin reducing the AHPs by its action on PI4Kα.
Figure 4. LY294,002 has no effect on the I AHP .
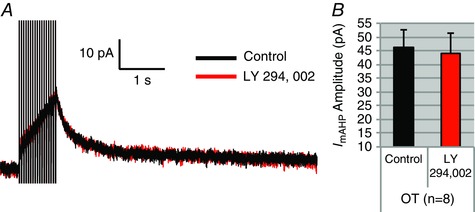
Because wortmannin also blocks PIP3, we tested the selective PIP3 blocker LY294,002 on the I AHP. A, an example of LY294,002's effect on an OT neuron. B, summary data of I mAHP amplitude in OT neurons. There was no significant difference (n = 8, P > 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
To ensure that wortmannin's effects reflected a response specific to AHPs and not a change in general cell health, 10 MNCs (5 OT and 5 VP) were assessed in current clamp recordings, where we found no significant differences in spike amplitude (OT: P > 0.05; 61.5 ± 2.8 mV control vs. 59.2 ± 3.2 mV wortmannin; VP: P > 0.05; 65.6 ± 4.0 mV control vs. 65.0 ± 3.8 mV wortmannin), spike half‐width (OT: P > 0.05; 2.3 ± 0.1 ms control vs. 2.5 ± 0.1 ms wortmannin; VP: P > 0.05; 2.5 ± 0.1 ms control vs. 2.4 ± 0.1 ms wortmannin), resting membrane potential (OT: P > 0.05; −60.0 ± 0.6 mV control vs. −60.2 ± 0.7 mV wortmannin; VP: P > 0.05; −58.9 ± 2.6 mV control vs. −60.6 ± 2.2 mV wortmannin), or input resistance (OT: P > 0.05; 467.37 ± 57.5 MΩ control vs. 444.49 ± 55.3 MΩ wortmannin; VP: P > 0.05; 545.75 ± 110.8 MΩ control vs. 528 ± 107.3 MΩ wortmannin) in cells before and after wortmannin application.
Wortmannin inhibits the I sAHP in the presence of apamin in OT neurons
To isolate wortmannin's effects on the sAHP, we tested its effects in the presence of apamin, which blocks SK channels and thus the I mAHP in MNCs (Bourque & Brown, 1987; Erickson et al. 1993; Armstrong et al. 1994; Kirkpatrick & Bourque, 1996; Teruyama & Armstrong, 2005). Since we bath‐applied apamin (100 nm) first, followed by the addition of wortmannin (1 μm), we first ran a repeated measures ANOVA that revealed a significant main effect (P < 0.001). Post hoc analysis found that in OT neurons, apamin resulted in robust (∼70%) inhibition of the I mAHP (measured at 100 ms: n = 7, P < 0.001; control 18.76 ± 2.5 pA vs. apamin 6.14 ± 1.8 pA; Fig. 5) while having no effect on the I sAHP (measured at 1 s: n = 7, P > 0.05; control 6.70 ± 1.5 pA vs. apamin 6.10 ± 1.4 pA; Fig. 5). Application of wortmannin inhibited the I sAHP in the presence of apamin (measured at 1 s: n = 7, P < 0.05; apamin 6.10 ± 1.4 pA vs. wortmannin + apamin 2.81 ± 0.8 pA; Fig. 5). We also observed further inhibition of the I AHP at 100 ms (n = 7, P < 0.05; apamin 6.14 ± 1.8 pA vs. wortmannin + apamin 1.2 ± 0.4 pA; Fig. 5). For comparison, wortmannin had no effect on apamin‐treated VP neurons (measured at 1 s: n = 6, P > 0.05; apamin 6.79 ± 1.6 pA vs. wortmannin + apamin 6.23 ± 1.9 pA). These data confirm that wortmannin acts on the isolated I sAHP in addition to I mAHP in OT cells.
Wortmannin reduced PIP2 immunoreactivity in OT neurons
To determine whether the application of wortmannin at a similar dose to our electrophysiological experiments was associated with changes in the cellular distribution of PIP2 in OT neurons, we calculated the ratio of the staining intensity of PIP2 immunoreactivity of the cytoplasm vs. membrane in dissociated cell preparations and compared control neurons with those exposed to 100 nm wortmannin (n = 5 animals in each group). As shown in Fig. 6, this ratio was significantly increased by wortmannin, suggesting depletion of membrane PIP2. This type of redistribution is similar to that reported for PIP2 in response to muscarinic modulation in sympathetic neurons (Suh & Hille, 2002; Delmas & Brown, 2005).
Figure 6. Effect of wortmannin on PIP2 expression in OT neurons.
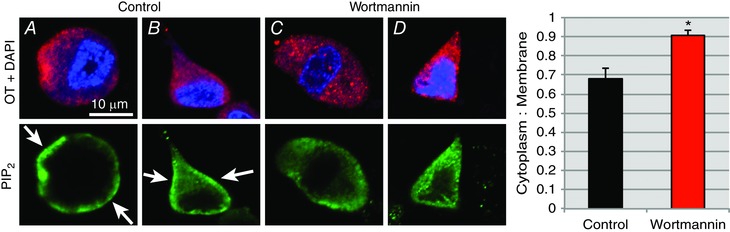
Double immunofluorescence confocal microscopy revealed that PIP2 (green) in OT neurons is expressed more densely in the cell membrane in the control group (A and B; lower panels). Upon wortmannin (100 nm) treatment for 30 min, OT neurons show a decreased PIP2 expression in the cell membranes (C and D; lower panels) relative to the cytosol. The upper panels show the expression of OT‐NP (red) expression and DAPI (blue) staining for nucleus. The histogram on the right side shows the mean cytoplasm: membrane ratios between the two groups, and indicates a significant difference (control vs. wortmannin; * P ≤ 0.016). n = 5 animals per group.
diC8‐PIP2 in the pipette prevents I mAHP and I sAHP inhibition by wortmannin
We have shown that application of wortmannin blocks both the medium and slow I AHPs in OT neurons. We interpret this as wortmannin blocking PIP2 synthesis and thus reducing total PIP2 levels. In contrast, wortmannin has no significant effect on these I AHPs in VP neurons. We next tested whether directly increasing the amount of available PIP2 inside the cell affected AHPs and whether it prevented the inhibition by wortmannin. We supplied the water‐soluble PIP2 analogue diC8‐PIP2 (30 μm) into neurons through the patch pipette to provide a constant source of PIP2. Wortmannin had no significant effect on I mAHP and I sAHP in OT neurons when diC8‐PIP2 was dialysed into the cell (100 ms: n = 8, P > 0.05; diC8‐PIP2 17.62 ± 1.9 pA vs. diC8‐PIP2 + wortmannin 15.18 ± 2.4 pA; 1000 ms: n = 8, P > 0.05; diC8‐PIP2 4.13 ± 0.8 pA vs. diC8‐PIP2 + wortmannin 3.82 ± 1 pA; Fig. 7).
Figure 7. Dialysing OT neurons with diC8‐PIP2 prevents inhibition of the I AHP by wortmannin.
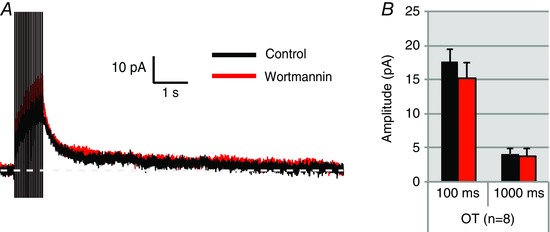
A, I AHP of an OT neuron dialysed with diC8‐PIP2. diC8‐PIP2 prevents inhibition of the I AHP by wortmannin. B, summary data for 8 OT cells tested as in A. Measurements of the I AHP 100 ms (I mAHP) and 1000 ms (I sAHP) after the pulse reveal that there are no significant changes in I AHP peak amplitude after wortmannin application in diC8‐PIP2 dialysed cells. (OT: n = 8, P > 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
Supplying the PIP2 precursor myo‐inositol also prevents inhibition by wortmannin
Another means of saturating the PIP2 supply is by adding the obligatory precursor myo‐inositol in the bath. Myo‐inositol is actively transported into the cell and increases PIP2 production (Fisher et al. 1992, 2002; Villalobos et al. 2011). We pre‐incubated slices in aCSF in 1 mm myo‐inositol for 1 h. For recording, slices were then transferred to a solution of 300 μm myo‐inositol in aCSF. Wortmannin (1 μm) had no effect on OT AHPs in the presence of myo‐inositol (100 ms: n = 7, P > 0.05; myo‐inositol 14.03 ± 1.6 pA vs. myo‐inositol + wortmannin 14.27 ± 1.5 pA; 1000 ms: n = 7, P > 0.05; myo‐inositol 4.11 ± 0.8 pA vs. myo‐inositol + wortmannin 4.49 ± 0.6 pA; Fig. 8), similar to the results with diC8‐PIP2 in the pipette (Fig. 7).
Figure 8. Wortmannin fails to inhibit OT neurons in aCSF containing myo‐inositol.
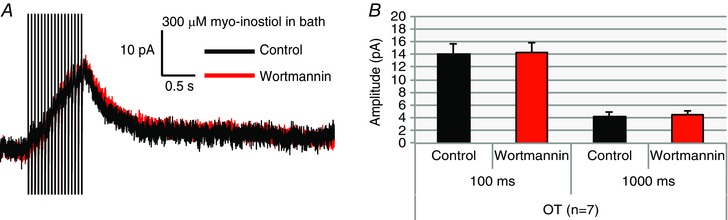
Slices were preincubated in aCSF containing 1 mm myo‐inositol, an important precursor molecule for PIP2 synthesis. When the slice was transported to the stage for patch clamp, the aCSF perfused onto the slice contained 300 μm myo‐inositol during recording. A, voltage clamp trace of an OT neuron before and after application of wortmannin. Wortmannin had no effect on the OT I AHP when the cells were bathed in 300 μm myo‐inositol. B, summary data for OT neurons. There were no significant differences in amplitude of current at 100 ms and 1000 ms after the pulse (n = 7, P > 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
diC8‐PIP2 slows rundown of the I mAHP in OT neurons
Rundown is a term used to describe the decreased amplitude of I AHPs over long recording sessions (Fig. 9 A). Since a previous study in neocortical pyramidal neurons showed that enhancement or inhibition of PIP2 slows or hastens the rundown of the sAHP, respectively (Villalobos et al. 2011), we supplied the cells with water‐soluble diC8‐PIP2 (30 μm), and then evaluated rundown of the peak I AHP in cells with or without diC8‐PIP2. OT cells dialysed with diC8‐PIP2 display slower rundown compared to controls over 30 min (Fig. 9 B).
Figure 9. Inclusion of water‐soluble PIP2 analogue diC8‐PIP2 (30 μm) in the recording pipette slows I AHP rundown in OT.
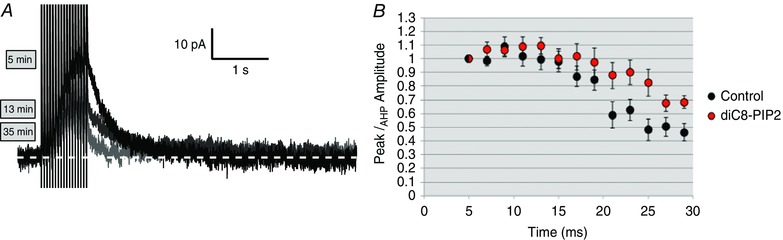
A, I AHP recordings in an MNC under normal conditions demonstrating rundown over a 35 min time period. Both OT and VP cells display I AHP rundown over long recording periods. B, OT cells dialysed with diC8‐PIP2 display slower rundown and ran down less compared to controls at 30 min (n = 9). [Color figure can be viewed at wileyonlinelibrary.com]
Changes in I mAHP and I sAHP via PIP2 are unlikely to be the result of PLC‐dependent phenomena
So far, we have demonstrated that a presumed reduction of PIP2 by wortmannin caused a pronounced inhibition of the I mAHP and I sAHP in OT neurons, suggesting that PIP2 is required for generation of both AHPs. While these results demonstrate the I mAHP and I sAHP require PIP2, they do not provide any evidence for a mechanism by which PIP2 can modulate I AHPs. For example, PIP2 could directly modulate the AHP channels. Alternatively, reduced PIP2 levels could potentially act indirectly through PLC‐induced inositol 1,4,5‐trisphosphate (IP3) or diacylglycerol (DAG) since PIP2 is a precursor for breakdown by PLC into DAG and IP3 (Suh & Hille, 2008). That is, blocking PIP2 production by inhibiting the rate‐limiting enzyme of production (PI4Kα) with wortmannin could cause a drop in PIP2, which could in turn would reduce PLC‐induced IP3 availability (IP3 binds to IP3 receptors on the endoplasmic reticulum and induces Ca2+ release into the cytoplasm from internal stores: Dickson et al. 2013).
To test this, we used the selective PLC inhibitor U73122 (10 μm) to directly block the conversion of PIP2 into DAG and IP3 (Bleasdale et al. 1990; Pérez et al. 2010). U73122 has been widely used in the nervous system, including the study of PLC‐dependent phenomena in SON (Sabatier et al. 1998; Li et al. 1999; Bonfardin et al. 2010). In OT neurons, U73122 had no effect on either the I mAHP or the I sAHP amplitude (100 ms: n = 8, P > 0.05; control 21.48 ± 2.4 pA vs. U73122 20.53 ± 3.1 pA; 1000 ms: n = 8, P > 0.05; control 3.45 ± 0.6 pA vs. U73122 3.42 ± 0.5 pA; Fig. 10). The decay time constant was also not significantly affected (n = 8, P > 0.05, control τ = 233.32 ± 62.9 ms vs. U73122 τ = 507.61 ± 185.5 ms; Fig. 10 B). These results show it is unlikely that PIP2 modulates the I AHP by indirectly affecting downstream targets of PLC. This would primarily include Ca2+ release from stores via IP3, or working through DAG and its product, protein kinase C (also known to modulate ion channels: Suh & Hille, 2008).
Figure 10. The PLC inhibitor U73122 (10 μm) failed to inhibit the I mAHP or I sAHP in OT neurons.
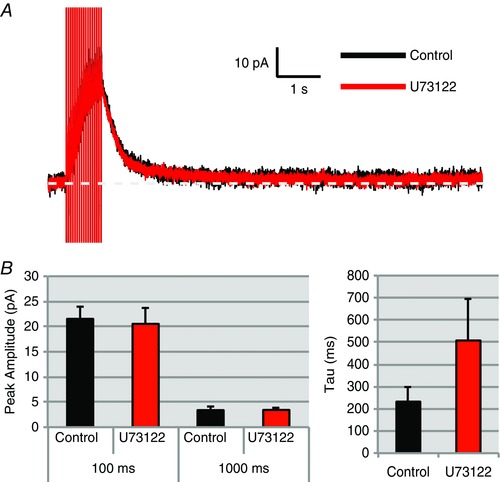
A, voltage clamp trace of an OT neuron before and after application of U73122. B, summary data for OT neurons: current amplitude at 100 ms (I mAHP), 1000 ms (I sAHP), and I AHP decay tau (n = 8, P > 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
Changes in the I mAHP and I sAHP in OT neurons may be due to changes in Ca2+ influx
Unlike neocortical pyramidal cells where only the I sAHP is affected by alterations in PIP2 levels (Villalobos et al. 2011), all of our results in OT neurons occur in parallel for the I mAHP and I sAHP. A parsimonious explanation for this parallel effect is that PIP2 exerts its effect upstream from the AHP K+ channels on Ca2+ entry or Ca2+ availability. We hypothesized that PIP2 may act on regulating Ca2+ entry or otherwise alter [Ca2+]i. To test this, we used Ca2+ imaging to determine whether wortmannin's inhibition of the AHPs in OT neurons affected [Ca2+]i. We used the high affinity Ca2+ indicator fura‐2 (100 nm), as in our previous publications (Roper et al. 2003, 2004; Abel et al. 2004; Teruyama & Armstrong, 2005). The relative change in fura‐2 fluorescence (%ΔF/F) is linearly proportional to bulk [Ca2+]i changes in the cytoplasm when %ΔF/F is less than ∼0.5 (Abel et al. 2004). Since the peaks of bulk [Ca2+]i were less than 0.5, we used the %ΔF/F value as an index for changes in [Ca2+]i. During the stimulus train, we observed a rapid rise in [Ca2+]i that decayed monoexponentially (Fig. 11 C and D). In OT neurons, we found that simultaneously with the reduction in I AHPs (100 ms: n = 9, P > 0.001; control 30.23 ± 4.5 pA vs. wortmannin 9.77 ± 2.5 pA; 1000 ms: P < 0.001; control 7.64 ± 0.5 pA vs. wortmannin 4.09 ± 0.5 pA), wortmannin (1 μm) also reduced the spike‐evoked increase in somatic [Ca2+]i in OT neurons (n = 9, P < 0.001; control 44.09 ± 2.3 %ΔF/F vs. wortmannin 30.77 ± 2.4 %ΔF/F; Fig. 11 A, C and E). Consistent with our previous experiments showing no inhibition of AHPs, wortmannin had no effect on VP neuron I mAHPs or I sAHPs (100 ms: n = 7, P > 0.05; control 23.01 ± 4.6 pA vs. wortmannin 21.18 ± 3.98 pA; 1000 ms: P > 0.05; control 5.34 ± 1.1 pA vs. wortmannin 4.41 ± 0.9 pA; Fig. 11), or on [Ca2+]i (control 47.40 ± 2.6 %ΔF/F vs. wortmannin 44.03 ± 2.9 %ΔF/F; ) (Fig. 11 B, D and E).
Figure 11. Wortmannin (1 μm) inhibits the I mAHP, I sAHP and somatic Ca2+ while having no effect on these measures in VP neurons.
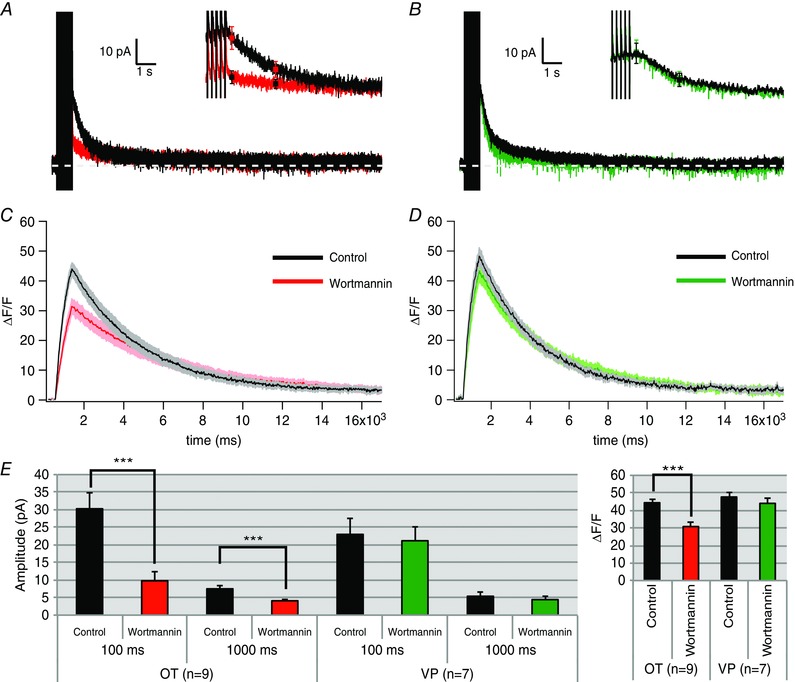
A and B, averaged voltage clamp traces of OT (A) and VP (B) cells after wortmannin application; inset is higher temporal resolution of same trace with the mean ± SEM values superimposed at 100 ms and 1000 ms after the pulse. C and D, average intracellular Ca2+ response of OT (C) and VP (D) cells to wortmannin expressed as the change in fluorescence divided by the total fluorescence (%ΔF/F; shaded area of the curve is SEM). E, summary data at 100 ms (I mAHP) and 1000 ms (I sAHP) amplitude (OT: n = 9, 100 ms and 1000 ms *** P < 0.001; VP: n = 7, 100 ms and 500 ms P > 0.05) and summary data of Ca2+ transient peak amplitude (OT: n = 9, *** P < 0.001; VP: n = 7, P > 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
PIP2 depletion has no effect on AHPs generated by uncaging Ca2+
The previous Ca2+ data suggest that PIP2 modulates AHPs by changing Ca2+ entry or Ca2+ availability to the K+ channels but does not rule out additional effects on the AHP channel itself. To test whether or not PIP2 modulated AHP channels, we rapidly increased somatic [Ca2+]i of the cell via photolytic release of Ca2+ from its caging compound, DM‐nitrophen (2 mm). Neurons were dialysed with DM‐nitrophen at 40% Ca2+ occupancy via the recording pipette and stimulated using a UV light flash from a xenon flash bulb. The result of this rapid Ca2+ release is an AHP generated without stimulating a train of action potentials in the cell, and thus an AHP independent of Ca2+ entry through voltage‐gated Ca2+ ion channels (Sah & Clements, 1999). Using fluo‐4 (50 μm) as the dye indicator for Ca2+, we elicited AHPs with spiking and uncaging in every cell tested. We opted to use current clamp for these experiments for two reasons. (1) Signal fidelity was much better in current clamp. It was sometimes difficult to see an uncaged AHP in voltage clamp, while it was always clearly present in current clamp. (2) While still present in current clamp, rundown of both the currents and the Ca2+ transient was slower than the rundown we observed in voltage clamp.
For AHPs generated from current injections, we evoked action potentials with suprathreshold current using 20 10 ms pulses at 20 Hz, from a resting potential of ∼−55 mV (controlled by DC current injection). For AHPs generated from uncaging Ca2+, we administered a UV light flash of ∼1 ms duration at 72.6 J at the same membrane potential. To control for rundown, we measured AHPs at two time points for each of two groups: one control group and one group that received wortmannin application between the first and second measurements. Consistent with previous results in voltage clamp, both mAHPs and sAHPs generated by spiking were significantly inhibited by wortmannin application (100 ms: n = 7, P < 0.001; control 10.8 ± 0.6 mV vs. wortmannin 6.7 ± 1.2 mV; 1000 ms: n = 7, P = 0.018; control 8.3 ± 1.4 mV vs. wortmannin 4.6 ± 0.8 mV; Fig. 12 A). The corresponding Ca2+ peak transients using fluo‐4 were also inhibited significantly (n = 7, P < 0.05; control 45.2 ± 6.9 %ΔF/F vs. wortmannin 23.7 ± 4.1 %ΔF/F; Fig. 12 B). However, when AHPs were generated by uncaging Ca2+ in these same cells, we found no significant differences between groups in either the peak or 1000 ms uncaged AHP amplitude (peak: P > 0.05; control (n = 7) 10.0 ± 1.2 mV vs. wortmannin (n = 8) 13.9 ± 3.0 mV; 1000 ms: P > 0.05; control 7.9 ± 3.6 mV vs. wortmannin 10.5 ± 1.8 mV). Additionally, we found no change in the corresponding peak Ca2+ transient (n = 7, P > 0.05; control 128.6 ± 13.2 %ΔF/F vs. wortmannin 23.7 ± 4.1 %ΔF/F; Fig. 12 C and D). This demonstrates that not only is PIP2 likely to modulate AHPs via spike‐induced Ca2+ entry, but also that PIP2 is not likely to gate the AHP channels themselves.
Figure 12. Wortmannin has no effect on AHPs or somatic Ca2+ when AHPs are generated via uncaging Ca2+ in OT neurons.
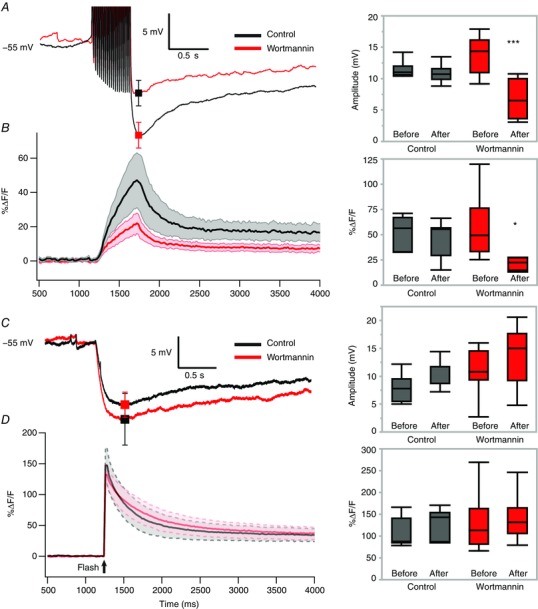
Measurements of AHPs were taken at two time points to control for the rundown present in this experiment. In one group, wortmannin was administered between the two measurements. All example traces are group averages of the second measurement from the control and wortmannin groups. A, left, averaged current clamp traces of AHPs generated by trains of current injections with a superimposed mean ± SEM at 100 ms. A, right, box plot summary data of this effect (** P = 0.008). B, left, corresponding Ca2+ signal average for traces shown in A. Shaded area represents error. B, right, box plot summary data for Ca2+ transients generated by spike‐generated AHPs (left) (* P < 0.05). C, left, averaged current clamp traces of AHPs generated by uncaging Ca2+ inside the cell with a superimposed mean ± SEM at the peak (P > 0.05). C, right, box plot summary data for uncaging‐generated AHPs. D, left, corresponding Ca2+ signal average for traces shown in C. Shaded area represents error. D, right, box plot summary data for Ca2+ transients generated by an uncaging AHP (P > 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
PIP2 depletion inhibits whole cell Ca2+ currents in OT neurons
The results of our Ca2+ imaging and uncaging data suggest that PIP2 affects the AHP by modulating Ca2+ entry through voltage‐gated Ca2+ channels. To further test this, we isolated whole cell Ca2+ currents in slices using 0.0005 mm TTX, 50 mm TEA, and 5 mm CsCl, measured steady‐state current responses to voltage steps, and plotted I–V curves before and after wortmannin application. Space clamp is a prevalent issue when measuring these currents in slice recordings of dendritic neurons, so cells exhibiting marked space clamp errors were excluded from analysis (criteria included delayed responses to voltage steps and escaping tail currents). Peak current occurred at a step to −10 mV, a result consistent with previous studies done on whole cell Ca2+ currents in these neurons (Fisher & Bourque, 1995; Foehring & Armstrong, 1996; Teruyama & Armstrong, 2005). Application of 400 μm Cd2+ after each trial resulted in a near complete block of current. When stepping positively, we started to observe inward current at the −40 mV step. At −10 mV where steady‐state Ca2+ currents are largest, we observed a statistically significant inhibition by wortmannin (n = 7, P = 0.018; control −459.0 ± 41.1 pA vs. wortmannin −260.8 ± 23.9 pA; Fig. 13) in OT neurons. By comparison, VP neurons displayed no change in Ca2+ currents after application of wortmannin (n = 5, P > 0.05; control ‐482.5 ± 30.9 pA vs. wortmannin −493.8 ± 28.7 pA; Fig. 13). This result demonstrates PIP2 modulates Ca2+ entry through voltage‐gated Ca2+ channels.
Figure 13. Wortmannin inhibits whole cell Ca2+ currents in OT neurons while having no effect in VP neurons.
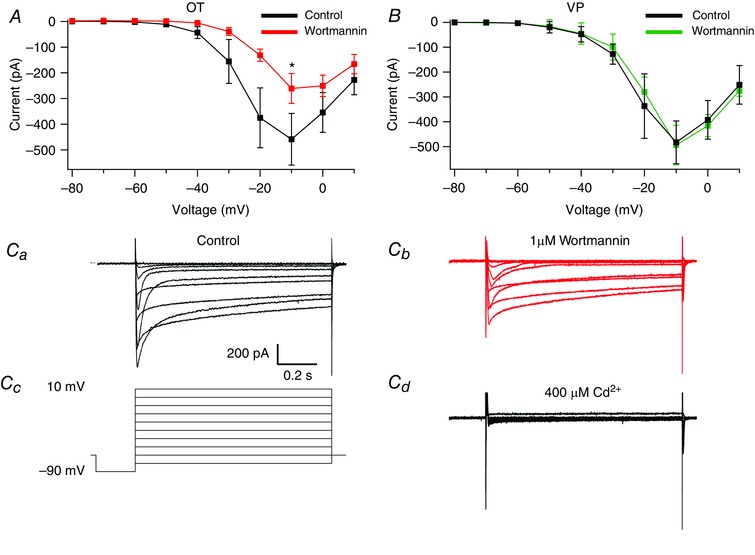
Ca2+ currents were isolated and subsequently measured before and after wortmannin (1 μm). I–V curves were plotted from steady‐state measurements of these currents. A, I–V curve of Ca2+ currents before and after wortmannin in OT neurons. Comparison at the highest amplitude steady‐state current (−10 mV) revealed a significant difference (* P < 0.05). B, I–V curve of Ca2+ currents before and after wortmannin in VP neurons. C, example traces from a single OT neuron of isolated whole cell Ca2+ currents generated by voltage steps from −90 mV to +10 mV under control conditions (Ca) and after 1 μm wortmannin application (Cb). The voltage protocol (Cc) and 400 μm Cd2+ (Cd) are shown for the same cell. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
The AHP and its underlying currents are important regulators of intrinsic neuronal excitability. In SON neurons, they control the length and frequency of bursts of action potentials during phasic firing (Kirkpatrick & Bourque, 1996; Ghamari‐Langroudi & Bourque, 2004). Despite the extensive work on the I sAHP, the gating mechanism via Ca2+ is currently unknown. Furthermore, the mechanism appears to vary greatly depending on the neuronal cell type. In neocortical pyramidal cells, the unknown sAHP channels were found to track cytoplasmic [Ca2+]i (Abel et al. 2004) and to be sensitive to PIP2 manipulations (Villalobos et al. 2011). PIP2 depletion by wortmannin reduced the I sAHP, increasing PIP2 levels by adding myo‐inositol dramatically slowed I sAHP rundown, and increasing PIP2 by overexpression of PIP5K facilitated calcium activation of the I sAHP (Villalobos et al. 2011). Together, these data suggest that in pyramidal neurons, PIP2 may affect Ca2+ sensitivity of the I sAHP channel. Several K+ channel types are known to require PIP2 for activation, including KCNQ (Kv7) and KATP, among others (Hilgemann & Ball, 1996; Delmas & Brown, 2005 (p.7); Suh & Hille, 2008; Zaydman et al. 2013 (p.7); Eckey et al. 2014). In addition, the rundown during recordings of Kv7 and KATP currents is a result of PIP2 depletion (Hughes et al. 2007; Logothetis et al. 2007). Ca2+ channels also play a critical role in AHPs, as different channel species are coupled to the AHP depending on the cell type (Andrade et al. 2012). Many of these voltage‐gated channels interact with PIP2, and have been shown to have smaller currents when PIP2 is depleted from the cells (Suh et al. 2010; Cruz et al. 2016). Because PIP2 availability so greatly affected I sAHP generation in neocortical pyramidal cells, we tested the extent to which it could regulate the phenotypically similar I AHPs in SON.
Both OT and VP cells share core AHP features, including robust calcium dependence, voltage independence, modulation by neurotransmitters, and a relationship between amplitude and spike count (Alger & Nicoll, 1980; Lorenzon & Foehring, 1993; Ghamari‐Langroudi & Bourque, 2004; Teruyama & Armstrong, 2005; Armstrong et al. 2010; Andrade et al. 2012). Suggestions for the potassium channel underlying the sAHP channel across the nervous system include KCNQ, KATP, and IK channels. KCNQ channels have been an exceptional focus of study in this regard, considering the exhaustive documentation of their modulation by PIP2 (Suh & Hille, 2002, 2007; Loussouarn et al. 2003; Li et al. 2005; Winks et al. 2005; Liu et al. 2008; Kim et al. 2016b). While KCNQ channels may be promising candidates for at least a component of the sAHP in some cells due to their modulation by PIP2 (Loussouarn et al. 2003; Kim et al. 2016b), it is not clear whether a similar mechanism will hold for our results. For example, Kim et al. (2016b) revealed a complicated dynamic in CA1 pyramidal cells between the I sAHP, the voltage dependence of KCNQ3 (Kv7.3) channels, and hippocalcin, a Ca2+ sensor protein critical for sAHP generation in CA1 neurons (Tzingounis et al. 2007). However, hippocalcin has not been found in the SON (Paterlini et al. 2000), and although KCNQ channels are expressed in the SON (Zhang et al. 2009b), and muscarinic suppression of sAHPs in SON has been reported, the sAHP does not show any voltage dependence (Ghamari‐Langroudi & Bourque, 2004), some of which would be expected if underlain by KCNQ channels. The latter authors did rule out BK, SK, and IK channels, however, based on the insensitivity of the sAHP to toxins targeting these toxins (Ghamari‐Langroudi & Bourque, 2004).
In SON neurons, we demonstrate an observable mechanistic difference between two similar cell types in the same nucleus. Depletion of PIP2 inhibits both the medium and slow components of the AHP in OT neurons while having no discernable effect on VP neurons. This is a novel and perhaps surprising result. Previous studies described cell type differences in firing patterns and plasticity during the reproductive cycle, but this is the first description of an OT–VP difference in an AHP mechanism (Bourque et al. 1985; Armstrong, 1995; Teruyama & Armstrong, 2002, 2005). Perhaps the differences are related to the fact that AHP and spike frequency undergo massive plastic changes in OT neurons during pregnancy and lactation, while VP neurons remain mostly unaffected in this regard. Thus PIP2 activity could be a key regulator of AHP changes during the reproductive cycle.
Differences between our results in SON and previous results in neocortical neurons are twofold. First, PIP2 depletion blocked both the I mAHP and I sAHP in OT neurons (with no effect in VP neurons), whereas it only affected the I sAHP in neocortical neurons (Villalobos et al. 2011). This is demonstrated by wortmannin block at 100 ms (mAHP) and 1000 ms (sAHP) after the stimulus (Fig. 3) and inhibition of isolated I sAHP currents in the presence of apamin (Fig. 5). Second, [Ca2+]i transients are reduced after PIP2 depletion in OT neurons (Fig. 11) whereas neocortical [Ca2+]i transients were unaffected by PIP2 depletion. These differences reflect a novel distinction between AHP mechanisms of different cell types. The results in cortex are likely to reflect a change in Ca2+ sensitivity of the AHP while the results in OT neurons appear to reflect a change in Ca2+ entry through voltage‐gated Ca2+ channels.
Wortmannin‐induced inhibition of I AHPs results from a restriction of PIP2 availability
Wortmannin consistently and effectively blocked the I mAHP and I sAHP in OT neurons in a dose‐dependent manner (Figs 3 and 11). Because wortmannin has multiple targets and drastic effects on OT AHPs, careful dissection of its effects was carried out in this study. One potential confound is non‐specific effects of wortmannin, since this drug inhibits more than PI4Kα. In fact, wortmannin is an inhibitor of PI3K activity as well as the PI3K–Akt signal transduction pathway (Nakanishi et al. 1995; Brunn et al. 1996). PI3K is actually the higher affinity target of wortmannin (IC50 = 2–4 nm), as opposed to its effect on PI4Kα (IC50 ≈ 50 nm) (Nakanishi et al. 1995). The previously reported IC50 for wortmannin's effect on PI4Kα is consistent with our dose–response curve generated by the proportion of peak I AHP inhibition (IC50 = 58 nm), which was well fitted by a single Langmuir isotherm with a Hill coefficient of 1.6. Consistent with the IC50 matching PI4Kα inhibition, the more specific PI3K inhibitor LY294,002 failed to affect the I mAHP and I sAHP in OT neurons (Fig. 4). Elevating PIP2 levels by supplementing the internal solution with diC8‐PIP2 (Fig. 7), or by exposing the cells to myo‐inositol (Fig. 8), prevented I mAHP and I sAHP inhibition by wortmannin. We also demonstrated that wortmanin increases the cytoplasm:membrane ratio of PIP2 in dissociated OT neurons (Fig. 6). This result has also been reported with oxotremorine‐M in MNCs with immunochemistry (Shah et al. 2014), and is consistent with a study in which wortmannin prevented the recovery of M‐currents after PIP2 depletion via oxotremorine‐M, confirming its inhibitory activity on PIP2 synthesis (Suh & Hille, 2002). Together, these results demonstrate that wortmannin blocks the I AHP by inhibiting PIP2 production and that PIP2 availability is critical for generation of the I mAHP and I sAHP in OT neurons. The robust inhibition of both medium and slow components suggests PIP2 affects a common mechanism between the two. The most recognizable connection is the Ca2+ dependence of the I mAHP and I sAHP.
PIP2 depletion alters Ca2+entry through voltage‐gated Ca2+ channels in OT neurons
We hypothesized that PIP2 affected either Ca2+ entry or Ca2+ availability to the AHP channels. We first considered that depleting PIP2 was effectively limiting Ca2+ release from IP3‐gated Ca2+ stores, shown to be a source of AHP‐related Ca2+ in dopamine neurons (Morikawa et al. 2000). To the extent that U73122 is specific to PLC inhibition, this application had no effect on the I mAHP or I sAHP of OT neurons (Fig. 10), consistent with the hypothesis that downstream targets of PLC activity are not involved. In contrast, both Ca2+ transients and whole cell Ca2+ currents were suppressed by wortmannin in OT, but not VP neurons, suggesting that PIP2 is interacting with voltage‐gated Ca2+ channels in these cells. This conclusion was further strengthened by a lack of wortmannin's effect on uncaging‐invoked AHPs (Fig. 12). Thus, PIP2 depletion simultaneously inhibits AHPs, Ca2+ transients, and Ca2+ currents in OT neurons. Though this result contrasts with observations in cortical neurons, the data do not necessarily rule out Ca2+ sensitivity as contributing to the effect on the I sAHP.
Although we observed clear inhibition of HVA currents with wortmannin, several voltage‐activated Ca2+ channels are candidates for the PIP2 modulation. It is presently unknown which specific Ca2+ channel types couple to the medium and slow AHP in OT and VP neurons in the SON. MNCs express high‐voltage‐activated (HVA) L‐, N‐, P/Q‐type, and R type channels, as demonstrated with mRNA (Glasgow et al. 1999), immunochemistry (Joux et al. 2001) and pharmacological block of whole cell currents in voltage clamp (Fisher & Bourque, 1995; Foehring & Armstrong, 1996), although these studies did not differentiate between OT and VP neurons. Importantly, HVAs have been demonstrated to interact with PIP2 (Suh et al. 2010; Kim et al. 2016a; Cruz et al. 2016). Transient, low‐threshold‐activated (LVA) type Ca2+ channels may also contribute to AHPs, and these have been reported in MNCs by some (Erickson et al. 1993; Fisher & Bourque, 1995; Israel et al. 2008), although not by all investigators (Foehring & Armstrong, 1996; Luther & Tasker, 2000; Joux et al. 2001; Luther et al. 2002). Nevertheless, LVAs clearly contribute to sAHPs in thalamic paraventricular neurons (Zhang et al. 2009a). Channel coupling may also be different for the mAHP and sAHP. Previous work describes mAHPs being activated by restricted microdomains of membrane Ca2+, while the sAHP has a tighter, cooperative sigmoidal relationship with bulk somatic [Ca2+]i (Wilson & Callaway, 2000; Abel et al. 2004). Therefore, the sAHP inhibition caused by PIP2 depletion could be caused by an inhibition of multiple Ca2+ channel species providing diffusible Ca2+. Interestingly, N‐type channels contribute to peptide release in OT and VP neurons, while P/Q channels contribute only in VP neurons in this regard (Wang & Fisher, 2014). This trait might provide a clue about cell‐type differences for PIP2 modulation of HVA channels.
It is important to explicitly mention that our results do not assume the same mechanisms for both the I mAHP and I sAHP, just that PIP2 is a regulator of both, and both are dependent on modulation of Ca2+ entry. For example, previous work demonstrated that PIP2 is a necessary cofactor for SK channel activation, binding to the CaM–SK interface on these channels to shift Ca+ sensitivity (Zhang et al. 2014). Though this work was done on SK2 channels, the SK3 channel complex also contains CaM, and its modulation in this manner remains a possibility for gating the I mAHP in SON.
Additional information
Competing interests
No authors report a competing interest.
Author contributions
The slice preparation, immunochemical identification of neurons, and calcium imaging/uncaging were performed by M.K.K. in W.E.A.’s laboratory. PIP2 immunochemistry was performed by G.K.C. Calcium imaging/uncaging assistance, software, and data interpretation were provided by J.C.C. All authors contributed to conception and design of experiments. The manuscript was drafted by M.K.K. and edited by all authors. All authors approved the final version of the manuscript, all persons listed as authors qualify for authorship, and all those who qualify for authorship are included.
Funding
This work was supported by NIH grants R01HD072056 (W.E.A.) and R01NS044163 (R.C.F.). M. Kirchner and L. Wang were supported in part by Neuroscience Centre of Excellence, UTHSC.
References
- Abel HJ, Lee JCF, Callaway JC & Foehring RC (2004). Relationships between intracellular calcium and afterhyperpolarizations in neocortical pyramidal neurons. J Neurophysiol 91, 324–335. [DOI] [PubMed] [Google Scholar]
- Alger BE & Nicoll RA (1980). Epileptiform burst afterhyperpolarization: calcium‐dependent potassium potential in hippocampal CA1 pyramidal cells. Science 210, 1122–1124. [DOI] [PubMed] [Google Scholar]
- Andrade R, Foehring RC & Tzingounis AV (2012). The calcium‐activated slow AHP: cutting through the Gordian knot. Front Cell Neurosci 6, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong WE (1995). Morphological and electrophysiological classification of hypothalamic supraoptic neurons. Prog Neurobiol 47, 291–339. [PubMed] [Google Scholar]
- Armstrong WE, Smith BN & Tian M (1994). Electrophysiological characteristics of immunochemically identified rat oxytocin and vasopressin neurones in vitro . J Physiol 475, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong WE, Wang L., Li C & Teruyama R (2010). Performance, properties and plasticity of identified oxytocin and vasopressin neurones in vitro . J Neuroendocrinol 22, 330–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin V, Moos F & Richard P (1984). Synchronization of oxytocin cells in the hypothalamic paraventricular and supraoptic nuclei in suckled rats: direct proof with paired extracellular recordings. Exp Brain Res 57, 201–203. [DOI] [PubMed] [Google Scholar]
- Bicknell RJ (1988). Optimizing release from peptide hormone secretory nerve terminals. J Exp Biol 139, 51–65. [DOI] [PubMed] [Google Scholar]
- Bicknell RJ & Leng G (1981). Relative efficiency of neural firing patterns for vasopressin release in vitro . Neuroendocrinology 33, 295–299. [DOI] [PubMed] [Google Scholar]
- Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ & Bunting S (1990). Selective inhibition of receptor‐coupled phospholipase C‐dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther 255, 756–768. [PubMed] [Google Scholar]
- Bonfardin VDJ, Fossat P, Theodosis DT & Oliet SHR (2010). Glia‐dependent switch of kainate receptor presynaptic action. J Neurosci 30, 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque CW & Brown DA (1987). Apamin and d‐tubocurarine block the after‐hyperpolarization of rat supraoptic neurosecretory neurons. Neurosci Lett 82, 185–190. [DOI] [PubMed] [Google Scholar]
- Bourque CW, Randle JC & Renaud LP (1985). Calcium‐dependent potassium conductance in rat supraoptic nucleus neurosecretory neurons. J Neurophysiol 54, 1375–1382. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC & Abraham RT (1996). Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3‐kinase inhibitors, wortmannin and LY294002. EMBO J 15, 5256–5267. [PMC free article] [PubMed] [Google Scholar]
- Cazalis M, Dayanithi G & Nordmann JJ (1985). The role of patterned burst and interburst interval on the excitation‐coupling mechanism in the isolated rat neural lobe. J Physiol 369, 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz L de la, Puente EI, Reyes‐Vaca A, Arenas I, Garduño J, Bravo‐Martínez J & Garcia DE (2016). PIP2 in pancreatic β‐cells regulates voltage‐gated calcium channels by a voltage‐independent pathway. Am J Physiol Cell Physiol 311, C630–C640. [DOI] [PubMed] [Google Scholar]
- Delmas P & Brown DA (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6, 850–862. [DOI] [PubMed] [Google Scholar]
- Dickson EJ, Falkenburger BH & Hille B (2013). Quantitative properties and receptor reserve of the IP3 and calcium branch of Gq‐coupled receptor signaling. J Gen Physiol 141, 521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopico AM, Widmer H, Wang G, Lemos JR & Treistman SN (1999). Rat supraoptic magnocellular neurones show distinct large conductance, Ca2+‐activated K+ channel subtypes in cell bodies versus nerve endings. J Physiol 519, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB (2009). Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol 587, 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton A & Dyball REJ (1979). Phasic firing enhances vasopressin release from the rat neurohypophysis. J Physiol 290, 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckey K, Wrobel E, Strutz‐Seebohm N, Pott L, Schmitt N & Seebohm G (2014). Novel Kv7.1‐phosphatidylinositol 4,5‐bisphosphate interaction sites uncovered by charge neutralization scanning. J Biol Chem 289, 22749–22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson KR, Ronnekleiv OK & Kelly MJ (1993). Role of a T‐type calcium current in supporting a depolarizing potential, damped oscillations, and phasic firing in vasopressinergic guinea pig supraoptic neurons. Neuroendocrinology 57, 789–800. [DOI] [PubMed] [Google Scholar]
- Fisher SK, Heacock AM & Agranoff BW (1992). Inositol lipids and signal transduction in the nervous system: an update. J Neurochem 58, 18–38. [DOI] [PubMed] [Google Scholar]
- Fisher SK, Novak JE & Agranoff BW (2002). Inositol and higher inositol phosphates in neural tissues: homeostasis, metabolism and functional significance. J Neurochem 82, 736–754. [DOI] [PubMed] [Google Scholar]
- Fisher TE & Bourque CW (1995). Voltage‐gated calcium currents in the magnocellular neurosecretory cells of the rat supraoptic nucleus. J Physiol 486, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehring RC & Armstrong WE (1996). Pharmacological dissection of high‐voltage‐activated Ca2+ current types in acutely dissociated rat supraoptic magnocellular neurons. J Neurophysiol 76, 977–983. [DOI] [PubMed] [Google Scholar]
- Ghamari‐Langroudi M & Bourque CW (1998). Caesium blocks depolarizing after‐potentials and phasic firing in rat supraoptic neurones. J Physiol 510, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari‐Langroudi M & Bourque C (2004). Muscarinic receptor modulation of slow afterhyperpolarization and phasic firing in rat supraoptic nucleus neurons. J Neurosci 24, 7718–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasgow E, Kusano K, Chin H, Mezey E, Young WS & Gainer H (1999). Single cell reverse transcription‐polymerase chain reaction analysis of rat supraoptic magnocellular neurons: neuropeptide phenotypes and high voltage‐gated calcium channel subtypes. Endocrinology 140, 5391–5401. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi T, Uchide K, Honda K & Negoro H (1986). Oxytocin release during parturition in the pelvic‐neurectomized rat. J Endocrinol 109, 149–154. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW & Ball R (1996). Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2 . Science 273, 956–959. [DOI] [PubMed] [Google Scholar]
- Hille B (1994). Modulation of ion‐channel function by G‐protein‐coupled receptors. Trends Neurosci 17, 531–536. [DOI] [PubMed] [Google Scholar]
- Hlubek MD & Cobbett P (2000). Differential effects of K+ channel blockers on frequency‐dependent action potential broadening in supraoptic neurons. Brain Res Bull 53, 203–209. [DOI] [PubMed] [Google Scholar]
- Hughes S, Marsh SJ, Tinker A & Brown DA (2007). PIP2‐dependent inhibition of M‐type (Kv7.2/7.3) potassium channels: direct on‐line assessment of PIP2 depletion by Gq‐coupled receptors in single living neurons. Pflügers Archiv 455, 115–124. [DOI] [PubMed] [Google Scholar]
- Israel J‐M, Poulain DA & Oliet SHR (2008). Oxytocin‐induced postinhibitory rebound firing facilitates bursting activity in oxytocin neurons. J Neurosci 28, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joux N, Chevaleyre V, Alonso G, Boissin‐Agasse L, Moos FC, Desarménien MG & Hussy N (2001). High voltage‐activated Ca2+ currents in rat supraoptic neurones: biophysical properties and expression of the various channel alpha1 subunits. J Neuroendocrinol 13, 638–649. [DOI] [PubMed] [Google Scholar]
- Kim D‐I, Kweon H‐J, Park Y, Jang D‐J & Suh B‐C (2016a). Ca2+ controls gating of voltage‐gated calcium channels by releasing the β2e subunit from the plasma membrane. Sci Signal 9, ra67‐ra67. [DOI] [PubMed] [Google Scholar]
- Kim KS, Duignan KM, Hawryluk JM, Soh H & Tzingounis AV (2016b). The voltage activation of cortical KCNQ channels depends on global PIP2 levels. Biophys J 110, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick K & Bourque CW (1996). Activity dependence and functional role of the apamin‐sensitive K+ current in rat supraoptic neurones in vitro . J Physiol 494, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse M & Hille B (2013). The phosphoinositide sensitivity of the KV channel family. Channels 7, 530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser‐Ross N, Miyakawa H, Lev‐Ram V, Young SR & Ross WN (1991). High time resolution fluorescence imaging with a CCD camera. J Neurosci Methods 36, 253–261. [DOI] [PubMed] [Google Scholar]
- Lee J, Jung K, Kim YS & Park D (2007). Diosgenin inhibits melanogenesis through the activation of phosphatidylinositol‐3‐kinase pathway (PI3K) signaling. Life Sci 81, 249–254. [DOI] [PubMed] [Google Scholar]
- Li Y, Gamper N, Hilgemann DW & Shapiro MS (2005). Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5‐bisphosphate. J Neurosci 25, 9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Miyata S & Hatton GI (1999). Inositol 1,4,5‐trisphosphate‐sensitive Ca2+ stores in rat supraoptic neurons: involvement in histamine‐induced enhancement of depolarizing afterpotentials. Neuroscience 93, 667–674. [DOI] [PubMed] [Google Scholar]
- Liu B, Liang H, Liu L & Zhang H (2008). Phosphatidylinositol 4,5‐bisphosphate hydrolysis mediates histamine‐induced KCNQ/M current inhibition. Am J Physiol Cell Physiol 295, C81–C91. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Jin T, Lupyan D & Rosenhouse‐Dantsker A (2007). Phosphoinositide‐mediated gating of inwardly rectifying K+ . Pflügers Archiv 455, 83–95. [DOI] [PubMed] [Google Scholar]
- Lorenzon NM & Foehring RC (1993). The ontogeny of repetitive firing and its modulation by norepinephrine in rat neocortical neurons. Brain Res Dev Brain Res 73, 213–223. [DOI] [PubMed] [Google Scholar]
- Loussouarn G, Park K‐H, Bellocq C, Baró I, Charpentier F & Escande D (2003). Phosphatidylinositol‐4,5‐bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage‐gated potassium channels: a functional homology between voltage‐gated and inward rectifier K+ channels. EMBO J 22, 5412–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA, Daftary SS, Boudaba C, Gould GC, Halmos KC & Tasker JG (2002). Neurosecretory and non‐neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J Neuroendocrinol 14, 929–932. [DOI] [PubMed] [Google Scholar]
- Luther JA & Tasker JG (2000). Voltage‐gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J Physiol 523, 193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers R & Cantley LC (1997). Cloning and characterization of a wortmannin‐sensitive human phosphatidylinositol 4‐kinase. J Biol Chem 272, 4384–4390. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K & Williams JT (2000). Inositol 1,4,5‐triphosphate‐evoked responses in midbrain dopamine neurons. J Neurosci 20, RC103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S, Catt KJ & Balla T (1995). A wortmannin‐sensitive phosphatidylinositol 4‐kinase that regulates hormone‐sensitive pools of inositolphospholipids. Proc Natl Acad Sci USA 92, 5317–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterlini M, Revilla V, Grant AL & Wisden W (2000). Expression of the neuronal calcium sensor protein family in the rat brain. Neuroscience 99, 205–216. [DOI] [PubMed] [Google Scholar]
- Pérez C, Vega R & Soto E (2010). Phospholipase C‐mediated inhibition of the M‐potassium current by muscarinic‐receptor activation in the vestibular primary‐afferent neurons of the rat. Neurosci Lett 468, 238–242. [DOI] [PubMed] [Google Scholar]
- Poulain DA & Wakerley JB (1982). Electrophysiology of hypothalamic magnocellular neurones secreting oxytocin and vasopressin. Neuroscience 7, 773–808. [DOI] [PubMed] [Google Scholar]
- Roper P, Callaway J & Armstrong W (2004). Burst initiation and termination in phasic vasopressin cells of the rat supraoptic nucleus: a combined mathematical, electrical, and calcium fluorescence study. J Neurosci 24, 4818–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper P, Callaway J, Shevchenko T, Teruyama R & Armstrong W (2003). AHP's, HAP's and DAP's: how potassium currents regulate the excitability of rat supraoptic neurones. J Comput Neurosci 15, 367–389. [DOI] [PubMed] [Google Scholar]
- Sabatier N & Leng G (2007). Bistability with hysteresis in the activity of vasopressin cells. J Neuroendocrinol 19, 95–101. [DOI] [PubMed] [Google Scholar]
- Sabatier N, Richard P & Dayanithi G (1998). Activation of multiple intracellular transduction signals by vasopressin in vasopressin‐sensitive neurones of the rat supraoptic nucleus. J Physiol 513, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P & Clements JD (1999). Photolytic manipulation of [Ca2+]i reveals slow kinetics of potassium channels underlying the afterhyperpolarization in hipppocampal pyramidal neurons. J Neurosci 19, 3657–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah L, Bansal V, Rye PL, Mumtaz N, Taherian A & Fisher TE (2014). Osmotic activation of phospholipase C triggers structural adaptation in osmosensitive rat supraoptic neurons. J Physiol 592, 4165–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JE & Armstrong WE (1996). Changes in the electrical properties of supraoptic nucleus oxytocin and vasopressin neurons during lactation. J Neurosci 16, 4861–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M & Pedarzani P (2000). Differential distribution of three Ca2+‐activated K+ channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci 15, 476–493. [DOI] [PubMed] [Google Scholar]
- Suh B‐C & Hille B (2002). Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5‐bisphosphate synthesis. Neuron 35, 507–520. [DOI] [PubMed] [Google Scholar]
- Suh B‐C & Hille B (2007). Electrostatic interaction of internal Mg2+ with membrane PIP2 seen with KCNQ K+ channels. J Gen Physiol 130, 241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B‐C & Hille B (2008). PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 37, 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B‐C, Leal K & Hille B (2010). Modulation of high‐voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5‐bisphosphate. Neuron 67, 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacconi S, Carletti R, Bunnemann B, Plumpton C, Merlo Pich E & Terstappen GC (2001). Distribution of the messenger RNA for the small conductance calcium‐activated potassium channel SK3 in the adult rat brain and correlation with immunoreactivity. Neuroscience 102, 209–215. [DOI] [PubMed] [Google Scholar]
- Teruyama R & Armstrong WE (2002). Changes in the active membrane properties of rat supraoptic neurones during pregnancy and lactation. J Neuroendocrinol 14, 933–944. [DOI] [PubMed] [Google Scholar]
- Teruyama R & Armstrong WE (2005). Enhancement of calcium‐dependent afterpotentials in oxytocin neurons of the rat supraoptic nucleus during lactation. J Physiol 566, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruyama R & Armstrong WE (2007). Calcium‐dependent fast depolarizing afterpotentials in vasopressin neurons in the rat supraoptic nucleus. J Neurophysiol 98, 2612–2621. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Kobayashi M, Takamatsu K & Nicoll RA (2007). The diffusible calcium sensor, hippocalcin, gates the calcium activation of the slow afterhyperpolarization in hippocampal pyramidal neurons. Neuron 53, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck, B , Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ & Waterfield MD (2001). Synthesis and function of 3‐phosphorylated inositol lipids. Annu Rev Biochem 70, 535–602. [DOI] [PubMed] [Google Scholar]
- Villalobos C, Foehring RC, Lee JC & Andrade R (2011). Essential role for phosphatidylinositol 4,5‐bisphosphate in the expression, regulation, and gating of the slow afterhyperpolarization current in the cerebral cortex. J Neurosci 31, 18303–18312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY & Brown RF (1994). A specific inhibitor of phosphatidylinositol 3‐kinase, 2‐(4‐morpholinyl)‐8‐phenyl‐4H‐1‐benzopyran‐4‐one (LY294002). J Biol Chem 269, 5241–5248. [PubMed] [Google Scholar]
- Wakerly JB & Lincoln DW (1971). Milk ejection in the rat: recordings of intramammary pressure during suckling. J Endocrinol 51, 13–14. [PubMed] [Google Scholar]
- Wang D & Fisher TE (2014). Expression of CaV 2.2 and splice variants of CaV 2.1 in oxytocin‐ and vasopressin‐releasing supraoptic neurones. J Neuroendocrinol 26, 100–110. [DOI] [PubMed] [Google Scholar]
- Wang Q, Xia X, Deng X, Li N, Wu D, Zhang L, Yang C, Tao F & Zhou J (2016). Lambda‐cyhalothrin disrupts the up‐regulation effect of 17β‐estradiol on post‐synaptic density 95 protein expression via estrogen receptor α‐dependent Akt pathway. J Environ Sci China 41, 252–260. [DOI] [PubMed] [Google Scholar]
- Wilson CJ & Callaway JC (2000). Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol 83, 3084–3100. [DOI] [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA & Marsh SJ (2005). Relationship between membrane phosphatidylinositol‐4,5‐bisphosphate and receptor‐mediated inhibition of native neuronal M channels. J Neurosci 25, 3400–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, Shi J & Cui J (2013). Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci USA 110, 13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Renaud LP & Kolaj M (2009a). Properties of a T‐Type Ca2+channel‐activated slow afterhyperpolarization in thalamic paraventricular nucleus and other thalamic midline neurons. J Neurophysiol 101, 2741–2750. [DOI] [PubMed] [Google Scholar]
- Zhang M, Meng X‐Y, Cui M, Pascal JM, Logothetis DE & Zhang J‐F (2014). Selective phosphorylation modulates the PIP2 sensitivity of the CaM–SK channel complex. Nat Chem Biol 10, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang D, Liu X‐H, Kosala WR, Rajapaksha JS & Fisher TE (2009b). An osmosensitive voltage‐gated K+ current in rat supraoptic neurons. Eur J Neurosci 29, 2335–2346. [DOI] [PubMed] [Google Scholar]