

Abstract
Key points
Patients with post‐traumatic stress disorder (PTSD) are at a significantly higher risk of developing hypertension and cardiovascular disease. The mechanisms underlying this increased risk are not known.
Studies have suggested that PTSD patients have an overactive sympathetic nervous system (SNS) that could contribute to cardiovascular risk; however, sympathetic function has not previously been rigorously evaluated in PTSD patients.
Using direct measurements of sympathetic nerve activity and pharmacological manipulation of blood pressure, we show that veterans with PTSD have augmented SNS and haemodynamic reactivity during both combat‐related and non‐combat related mental stress, impaired sympathetic and cardiovagal baroreflex sensitivity, and increased inflammation.
Identifying the mechanisms contributing to increased cardiovascular (CV) risk in PTSD will pave the way for developing interventions to improve sympathetic function and reduce CV risk in these patients.
Abstract
Post‐traumatic stress disorder (PTSD) is associated with increased cardiovascular (CV) risk. We tested the hypothesis that PTSD patients have augmented sympathetic nervous system (SNS) and haemodynamic reactivity during mental stress, as well as impaired arterial baroreflex sensitivity (BRS). Fourteen otherwise healthy Veterans with combat‐related PTSD were compared with 14 matched Controls without PTSD. Muscle sympathetic nerve activity (MSNA), continuous blood pressure (BP) and electrocardiography were measured at baseline, as well as during two types of mental stress: combat‐related mental stress using virtual reality combat exposure (VRCE) and non‐combat related stress using mental arithmetic (MA). A cold pressor test (CPT) was administered for comparison. BRS was tested using pharmacological manipulation of BP via the Modified Oxford technique at rest and during VRCE. Blood samples were analysed for inflammatory biomarkers. Baseline characteristics, MSNA and haemodynamics were similar between the groups. In PTSD vs. Controls, MSNA (+8.2 ± 1.0 vs. +1.2 ± 1.3 bursts min–1, P < 0.001) and heart rate responses (+3.2 ± 1.1 vs. −2.3 ± 1.0 beats min–1, P = 0.003) were significantly augmented during VRCE. Similarly, in PTSD vs. Controls, MSNA (+21.0 ± 2.6 vs. +6.7 ± 1.5 bursts min–1, P < 0.001) and diastolic BP responses (+6.3 ± 1.0 vs. +3.5 ± 1.0 mmHg, P = 0.011) were significantly augmented during MA but not during CPT (P = not significant). In the PTSD group, sympathetic BRS (–1.2 ± 0.2 vs. –2.0 ± 0.3 burst incidence mmHg−1, P = 0.026) and cardiovagal BRS (9.5 ± 1.4 vs. 23.6 ± 4.3 ms mmHg−1, P = 0.008) were significantly blunted at rest. PTSD patients had significantly higher highly sensitive‐C‐reactive protein levels compared to Controls (2.1 ± 0.4 vs. 1.0 ± 0.3 mg L−1, P = 0.047). Augmented SNS and haemodynamic responses to mental stress, blunted BRS and inflammation may contribute to an increased CV risk in PTSD.
Keywords: arterial baroreflex, blood pressure, inflammation, stress, sympathetic activity
Key points
Patients with post‐traumatic stress disorder (PTSD) are at a significantly higher risk of developing hypertension and cardiovascular disease. The mechanisms underlying this increased risk are not known.
Studies have suggested that PTSD patients have an overactive sympathetic nervous system (SNS) that could contribute to cardiovascular risk; however, sympathetic function has not previously been rigorously evaluated in PTSD patients.
Using direct measurements of sympathetic nerve activity and pharmacological manipulation of blood pressure, we show that veterans with PTSD have augmented SNS and haemodynamic reactivity during both combat‐related and non‐combat related mental stress, impaired sympathetic and cardiovagal baroreflex sensitivity, and increased inflammation.
Identifying the mechanisms contributing to increased cardiovascular (CV) risk in PTSD will pave the way for developing interventions to improve sympathetic function and reduce CV risk in these patients.
Abbreviations
- AU
arbitrary unit
- BP
blood pressure
- BRS
baroreflex sensitivity
- CAPS
Clinician Administered PTSD Scale
- CPT
cold pressor test
- CV
cardiovascular
- DBP
diastolic blood pressure
- hsCRP
highly sensitive C‐reactive protein
- IL
interleukin
- MA
mental arithmetic
- MAP
mean arterial pressure
- MSNA
muscle sympathetic nerve activity
- OEF
Operation Enduring Freedom
- OIF
Operation Iraqi Freedom
- OND
Operation New Dawn
- PCL‐M
PTSD Checklist – Military Version
- PTSD
post‐traumatic stress disorder
- SBP
systolic blood pressure
- SNA
sympathetic nerve activity
- SNS
sympathetic nervous system
- VRCE
virtual reality combat exposure
Introduction
Post‐traumatic stress disorder (PTSD) is a highly prevalent mental health condition amongst both military Veterans and the general population in those who have been exposed to psychologically traumatic events. This disorder is associated with a significantly increased risk for the development of cardiovascular (CV) disease and hypertension (Kang et al. 2006; Bedi & Arora, 2007; Kubzansky et al. 2007; Boscarino, 2008; Cohen et al. 2009; Kubzansky et al. 2009; Edmondson et al. 2013; Vaccarino et al. 2013; Beristianos et al. 2016). Multiple large epidemiological studies have demonstrated an independent link between PTSD and the risk of CV disease and hypertension (Kang et al. 2006; Bedi & Arora, 2007; Kubzansky et al. 2007; Boscarino, 2008; Cohen et al. 2009; Kubzansky et al. 2009; Edmondson et al. 2013; Vaccarino et al. 2013; Beristianos et al. 2016); however, the physiological mechanisms underlying this association have not been determined. Prior studies have demonstrated a decrease in low frequency heart rate (HR) variability (Cohen et al. 1998; Chang et al. 2013; Shah et al. 2013) and increased urinary noradrenaline levels (Wingenfeld et al. 2015) in PTSD patients, suggestive of heightened sympathetic nervous system (SNS) activity, which could be a potential mechanism contributing to an increased risk of CV disease and hypertension. Despite these findings, direct assessments of sympathetic nerve activity (SNA) have never previously been measured in this population or used to assess SNS and haemodynamic reactivity to mental stress, nor have they been used to investigate aberrancies in SNS regulation such as blunted arterial baroreflex sensitivity that could contribute to chronic SNS overactivity and subsequently to an increased CV risk.
The present study aimed to investigate haemodynamic and SNS reactivity and regulation at rest and during mental stress in young PTSD patients, prior to the development of hypertension and CV disease. The study population included young, otherwise healthy military Veterans with and without PTSD from the wars in Iraq and Afghanistan (Operation Enduring Freedom, Operation Iraqi Freedom and Operation New Dawn; OEF/OIF/OND, respectively). We tested the hypothesis that Veterans with PTSD have significantly greater SNS and haemodynamic responses during mental stress associated with their PTSD symptoms (i.e. combat‐related mental stress). We further hypothesized that these augmented sympathetic and haemodynamic responses extend to other forms of mental stress (i.e. non‐combat‐related mental stress) and are mediated by blunted arterial baroreflex sensitivity (BRS) and increased inflammation. Characterizing SNS reactivity and regulation in PTSD could uncover key physiological alterations in autonomic function in PTSD patients, which could, in turn, expedite the development of therapies to mitigate SNS overactivation and thereby improve long‐term CV risk in these patients.
Methods
Ethical approval
This study conformed to the standards set by the Declaration of Helsinki and was approved by the Emory University Institutional Review Board (IRB #54697) and the Atlanta VA Medical Centre Research and Development Committee. All participants provided signed informed consent for study participation via forms approved by the aforementioned regulatory committees.
Study population
The study population consisted of 28 participants: 14 patients with PTSD and 14 age‐matched Controls without PTSD. All study participants (both PTSD and Controls) were physically healthy OEF/OIF/OND Veterans, without hypertension or other medical comorbidities. Participants were recruited from mental health clinics serving OEF/OIF/OND Veterans at the Atlanta VA Medical Center and surrounding clinics. Exclusion criteria for all participants included hypertension, smoking, diabetes, heart or vascular disease, illicit drug use, excessive alcohol use (> 2 drinks per day), hyperlipidaemia, autonomic dysfunction, medications known to affect SNS (clonidine, β‐blockers, angiotensin‐converting enzyme inhibitors), treatment with monoamine oxidase inhibitors and any serious systemic disease. Participants with a prior documented diagnosis of PTSD from medical records were recruited to the PTSD group and those without a prior diagnosis were recruited to the Control group. The diagnosis of PTSD was confirmed in PTSD patients and excluded in Controls using the Clinician Administered PTSD Scale for DSM (CAPS)‐IV (Blake et al. 1995) or the PTSD Checklist – Military Version (PCL‐M) (Blanchard et al. 1996). PTSD patients required a CAPS severity score of ≥ 45 to confirm the presence of PTSD (Weathers et al. 2001).
Measurements and procedures
Blood pressure
Beat‐to‐beat arterial BP was measured continuously and non‐invasively using digital pulse photoplethysmography (CNAP; CNSystems, Graz, Austria). Absolute values of BP were calibrated with upper arm BP readings at the start and every 15 min throughout the study. This device has been validated to reflect accurate absolute and beat‐to‐beat fluctuations in BP when compared to invasive intra‐arterial catheter readings (Jeleazcov et al. 2010; Ilies et al. 2012).
Muscle sympathetic nerve activity
Multiunit postganglionic sympathetic nerve activity directed to muscle (MSNA) was recorded from the peroneal nerve using microneurography, as described previously (Vallbo et al. 1979; Wallin & Fagius, 1988). A tungsten microelectrode (tip diameter 5–15 μm; Bioengineering, University of Iowa, Iowa City, IA, USA) was inserted into the nerve and a reference microelectrode was inserted in close proximity. The signals were amplified (total gain 50 000–100 000), filtered (700–2000 Hz), rectified and integrated (time constant 0.1 s) to obtain a mean voltage display of sympathetic nerve activity (Nerve Traffic Analyzer, Model 662C‐4; Bioengineering, University of Iowa). The neurogram was recorded using LabChart 7 (PowerLab 16sp; ADInstruments, Sydney, Australia) along with continuous ECG recordings using a bioamp. All MSNA recordings met previously established criteria (Delius et al. 1972a, b; Mano et al. 2006) and were analysed by a single investigator who was blinded to the participant's status as a Control or patient. MSNA was expressed as burst frequency (bursts per minute) and total activity (arbitrary units per minute).
Virtual reality combat exposure (VRCE)
Participants wore head‐mounted goggles with peripheral display and stereo earphones for auditory input (SamSung Gear VR, Ridgefield Park, NJ, USA). These goggles displayed computer‐generated first‐person battle scenarios from Iraq (Virtual Iraq for OIF/OND) and Afghanistan (Virtual Afghanistan for OEF) (Rizzo et al. 2010, 2011; Rothbaum et al. 2010, 2014; McLay et al. 2011; Roy et al. 2013) for a duration of 3 min, followed by an additional 4 min during concomitant arterial baroreflex testing. These VRCE clips have been shown to increase reactivity of psychophysiological measures, such as acoustic startle, fear‐enhanced startle and galvanic skin response (Roy et al. 2013), are associated with elevations in startle reactivity and salivary cortisol levels (Rothbaum et al. 2014; Norrholm et al. 2016) and have been reported by patients as immersive and anxiogenic.
Mental stress via mental arithmetic (MA)
Participants were asked to serially subtract a one or two digit number from a three or four digit number and were urged to do so as quickly and accurately as possible, for a duration of 3 min (Carter et al. 2005; Carter & Ray, 2009). An investigator used flash cards with a three or four digit number and were urged by two additional study team members in white coats to answer ‘faster’ and ‘get it right’. This test has been shown to induce mental stress and increase MSNA (by ∼5–15 bursts min–1) and BP (by ∼10–16 mmHg), as well as HR (by∼16–24 beats min–1), in both healthy humans and patient groups (Matsukawa et al. 1991b; Noll et al. 1996; Dishman et al. 2003; Kuniyoshi et al. 2003; Carter et al. 2013).
Arterial BRS testing using the Modified Oxford technique
The gold standard method for evaluation of arterial BRS is performed by measuring changes in MSNA and R‐R interval during arterial BP changes induced by nitroprusside and phenylephrine (Rudas et al. 1999). Sodium nitroprusside (100 μg in 10 mL of normal saline (NS)) was bolused through an antecubital i.v. catheter, followed 60 s later by a bolus of phenylephrine (150 μg in 10 mL of NS) during continuous MSNA, ECG and haemodynamic monitoring. Medications were room temperature at the time of administration. These medications induce a decrease of ∼15 mmHg, followed by an increase of ∼15 mmHg in arterial BP.
Cold pressor test (CPT)
CPT is a standard human physiology research technique with a powerful sympathoexcitatory and pressor effect that was performed by submerging the participant's hand up to the wrist in an ice‐water slurry (∼0–1°C) for 1 min (Wood et al. 1984; Victor et al. 1987; Fagius et al. 1989; Cui et al. 2002; Dishman et al. 2003; Park et al. 2012). SNS activation during CPT is not regulated by baroreflexes (Cui et al. 2002) and allows for discernment between baroreflex‐mediated vs. generalized or non‐baroreflex mediated increases in SNS activation.
Inflammatory biomarkers
Blood was drawn in all subjects at baseline for the following inflammatory biomarkers: highly sensitive C‐reactive protein (hsCRP), interleukin (IL)‐2 and IL‐6. hsCRP was determined by immunoturbidometric method on the Beckman AU460 (Beckman Diagnostics, LaBrea, CA, USA) using reagent kits from Sekisui Diagnostics (Lexington, MA, USA). IL‐2 and IL‐6 measurements were determined using enzyme‐linked immunosorbent assay kits from R&D Systems (Minneapolis, MN, USA).
CAPS‐IV and the PCL‐M
The diagnosis of PTSD was confirmed in PTSD patients and excluded in Controls using CAPS‐IV or PCL‐M. CAPS‐IV was administered and interpreted by a single trained investigator for all participants. A severity score of ≥ 45 on either survey was required to confirm the presence of PTSD (Weathers & Ruscio, 1999; Weathers et al. 2001).
Experimental protocol
Screening visits
All participants presented for one or two screening visits prior to experimental procedures. During the screening visit(s), BP was taken a total of three times (separated by 5 min) and averaged to ensure the absence of hypertension and obtain baseline values. CAPS‐IV or the PCL‐M was performed during the screening visit to confirm the diagnosis of PTSD in patients and to exclude the diagnosis of PTSD in Controls.
Experimental study visit
All participants were studied in the morning, after abstaining from food, caffeine and alcohol for at least 12 h, as well as smoking and exercise for at least 24 h. The study room was quiet, semidark and temperate (∼21° C). Participants were placed in a supine position on a comfortable stretcher. A 20 gauge i.v. catheter was placed into the antecubital vein of the arm and blood was obtained for inflammatory biomarkers. Finger cuffs were fitted and placed on the fingers of the dominant arm for continuous beat‐to‐beat arterial BP measurements, and an upper arm cuff was placed for intermittent automatic calibrations with the finger cuffs. ECG patch electrodes were placed for continuous ECG recordings and a belt‐type sensor was placed for monitoring continuous respiratory rates. The leg was positioned for microneurography, and the tungsten microelectrode was inserted and manipulated to obtain a satisfactory nerve recording. After 10 min of rest, baseline BP, HR and MSNA were recorded continuously for 15 min. After baseline measurements, BRS testing was performed by infusing i.v. boluses of 100 μg of nitroprusside, followed 60 s later by 150 μg of phenylephrine, as described above. After 30 min of rest to ensure sufficient washout of drugs and return to baseline conditions, participants underwent mental arithmetic for 3 min and VRCE for 3 min. The order of these manoeuvres was randomized and separated by 30 min of rest. In a subset of participants, VRCE was extended for an additional 4 min, during which time BRS testing was repeated by infusing nitroprusside and phenylephrine during concomitant VRCE. After 30 min of rest, the cold pressor test was performed for 1 min.
Data analysis
MSNA
MSNA, BP and ECG data from the study visit were exported from Labchart 7 into WinCPRS (Absolute Aliens, Turku, Finland) for analysis. The software automatically marks the R‐waves from the continuous ECG recording that was then reviewed and edited manually if needed. MSNA bursts were detected and marked by the software using the criteria: 3:1 burst to noise ratio within a 0.5 s search window, with an average latency of 1.2–1.4 s from the previous R‐wave. The MSNA neurograms were also inspected for accuracy of detection by a single investigator (JP) and edited if needed. For total activity measurements, burst amplitudes were normalized by assigning the largest burst amplitude during at least 5 min of baseline recordings to be equated to a measurement of 1000 arbitrary units (AU). MSNA was expressed as burst frequency (bursts min–1), burst incidence [bursts (100 heart beats)–1], and total activity (AU min–1), which is a sum of the amplitudes of normalized bursts min–1.
Assessment of BRS
Sympathetic BRS was defined as the slope of the linear regression between values of MSNA (burst incidence and total activity) on the y‐axis and the diastolic BP on the x‐axis derived during the 2 min of drug boluses and following 2 min of data. Linear regression was performed using the WinCPRS program that grouped diastolic BP into 3 mmHg bins and then averaged the MSNA burst incidence and normalized total activity values within each bin. Cardiovagal BRS was assessed as the slope of the linear regression between R‐R intervals measured from the continuous ECG recording and the systolic BP during drug infusions. Only regressions with a coefficient of determination (R 2) > 0.70 were included in the analysis. The operating point around which the arterial baroreflex operates was derived from the mean resting systolic and diastolic blood pressure, with the corresponding mean R‐R interval, MSNA burst incidence and normalized MSNA total activity during at least 5 min of baseline recordings.
Statistical analysis
Statistical analysis was performed using SAS, version 9.2 (SAS Institute Inc., Cary, NC, USA). Baseline characteristics and baroreflex slopes were compared between the two groups (PTSD vs. Controls) using two‐tailed two‐sample t tests for continuous variables and Fisher's exact test for categorical variables. Repeated measures ANOVA was performed using PROC GLM to determine differences between the groups with respect to change in MSNA, systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial pressure (MAP) and HR with time during each intervention: MA, VRCE and CPT. When the overall F test was significant, the contrast option for post hoc analysis was used to compare the difference from baseline to each time point between the groups. For correlation analyses, the t statistic for correlation coefficient was used to evaluate whether or not a linear relationship existed between hsCRP levels and MSNA responses, as well as cardiovagal and sympathetic baroreflex sensitivity. Results are expressed as the mean ± SEM. P < 0.05 was considered statistically significant.
Results
Baseline characteristics
There was no significant difference in age, sex, race, weight or body mass index between groups (Table 1). Groups were well‐matched for age with a mean ± SEM age of 34.4 ± 1.7 years in PTSD patients and 32.2 ± 1.5 years in Controls. The majority of participants in each group were African‐American and male. There were no significant differences in the presence of comorbid depression or use of serotonin reuptake inhibitors between the groups. Mean baseline blood pressure and HR were similar between the groups. There was no significant difference in resting MSNA quantified as burst frequency, burst incidence or total activity between the groups. As expected, the mean CAPS‐IV score was significantly higher amongst the PTSD patients compared to Controls without PTSD.
Table 1.
Baseline characteristics and measurements
| PTSD | Controls | ||
|---|---|---|---|
| (n = 14) | (n = 14) | P value | |
| Age | 34.4 ± 1.7 | 32.2 ± 1.5 | 0.331 |
| Sex (male/female) | 12/2 | 13/1 | 1.0 |
| Race (black/white) | 10/4 | 12/2 | 0.645 |
| Weight (kg) | 93.8 ± 6.2 | 88.1 ± 4.8 | 0.416 |
| BMI (kg m–2) | 30.1 ± 1.6 | 28.4 ± 1.2 | 0.393 |
| Depression (yes/no) | 2/12 | 2/12 | 1.0 |
| SSRI (yes/no) | 4/10 | 2/12 | 0.645 |
| SBP (mmHg) | 133 ± 3 | 128 ± 4 | 0.351 |
| DBP (mmHg) | 81 ± 2 | 77 ± 4 | 0.389 |
| MAP (mmHg) | 99 ± 3 | 95 ± 4 | 0.451 |
| HR (beats min–1) | 68 ± 4 | 62 ± 2 | 0.267 |
| MSNA (bursts min–1) | 19.8 ± 1.3 | 19.9 ± 2.5 | 0.952 |
| MSNA [bursts (100 heart beats)–1] | 32.2 ± 3.1 | 32.7 ± 4.2 | 0.929 |
| MSNA (AU min–1) | 9934 ± 1022 | 10950 ± 1648 | 0.605 |
| CAPS‐IV score | 81 ± 9 | 39 ± 3 | 0.001 |
Baseline measurements of body mass index (BMI), SBP, DBP, HR and MSNA calculated as burst frequency (bursts min–1) and burst incidence were compared between PTSD and Controls without PTSD. SSRI, serotonin reuptake inhibitor. CAPS‐IV was used to confirm PTSD diagnosis in subjects and exclude PTSD in Controls. Values are the mean ± SE. * P < 0.05 was considered statistically significant.
VRCE
These experiments aimed to determine whether the haemodynamic and MSNA responses during combat‐related mental stress (i.e. mental stress related to PTSD symptoms) were augmented amongst young Veterans with PTSD. During 3 min of VRCE, there was no statistically significant difference in the SBP, DBP and MAP, responses during VRCE between the groups (Fig. 1 A–C)(Table 2). However, there was a small but significantly greater increase in HR during VRCE amongst PTSD patients compared to Controls (ANOVA, F test, P = 0.003) (Fig. 1 D). There was also a significantly greater increase in MSNA quantified as change in burst frequency (ANOVA, F test, P < 0.001) and total activity (ANOVA, F test, P < 0.001) (Fig. 1 E and F) amongst PTSD patients compared to Controls.
Figure 1. Hemodynamic and sympathetic reactivity during virtual reality combat exposure.
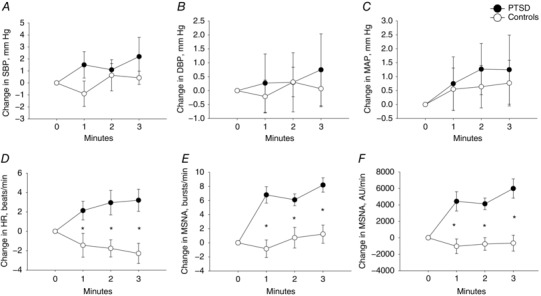
Change in SBP (A), DBP (B), mean arterial pressure (MAP) (c), heart rate (HR) (D), muscle sympathetic nerve activity (MSNA) quantified as burst frequency (bursts/min) (E) and total activity (AU/min) (F) during 3 min of virtual reality combat exposure in PTSD patients (filled circles) and Controls (open circles). * P < 0.05 between PTSD and Control groups.
Table 2.
Haemodynamic and sympathetic nerve responses during sympathoexcitation in PTSD and Controls
| VRCE | MA | CPT | ||||
|---|---|---|---|---|---|---|
| M0 | M3 | M0 | M3 | M0 | M1 | |
| SBP (mmHg) | ||||||
| PTSD | 129 ± 4 | 131 ± 4 | 127 ± 4 | 134 ± 4 | 123 ± 4 | 128 ± 6 |
| Controls | 127 ± 6 | 128 ± 6 | 129 ± 5 | 132 ± 5 | 120 ± 5 | 120 ± 6 |
| DBP (mmHg) | ||||||
| PTSD | 74 ± 2 | 75 ± 2 | 73 ± 2 | 79 ± 3 | 77 ± 3 | 83 ± 3 |
| Controls | 77 ± 4 | 77 ± 5 | 79 ± 4 | 82 ± 5 | 73 ± 4 | 78 ± 4 |
| MAP (mmHg) | ||||||
| PTSD | 93 ± 2 | 94 ± 2 | 93 ± 2 | 99 ± 3 | 94 ± 2 | 101 ± 3 |
| Controls | 95 ± 5 | 96 ± 5 | 97 ± 5 | 101 ± 5 | 91 ± 4 | 96 ± 5 |
| HR (beats min–1) | ||||||
| PTSD | 65 ± 3 | 68 ± 3* | 64 ± 4 | 71 ± 4 | 68 ± 4 | 75 ± 3 |
| Controls | 66 ± 5 | 64 ± 5 | 67 ± 4 | 70 ± 5 | 65 ± 4 | 73 ± 5 |
| MSNA (bursts min–1) | ||||||
| PTSD | 19.3 ± 1.6 | 27.6 ± 1.9* | 17.4 ± 1.4 | 38.0 ± 3.2* | 24.5 ± 1.7 | 31.8 ± 2.0 |
| Controls | 22.3 ± 5.0 | 23.5 ± 4.9 | 24.5 ± 3.9 | 30.5 ± 3.2 | 22.5 ± 3.9 | 34.2 ± 5.9 |
| MSNA (AU min–1) | ||||||
| PTSD | 12960 ± 1082 | 17873 ± 2448* | 8814 ± 1305 | 25629 ± 4206 | 15993 ± 1898 | 26814 ± 2636 |
| Controls | 11855 ± 2599 | 11207 ± 3611 | 13464 ± 3151 | 20015 ± 3082 | 13517 ± 2950 | 21478 ± 4445 |
SBP, DBP, MAP, HR and MSNA quantified as burst frequency (bursts min–1) and total activity (AU min–1) at rest (minute 0; M0) and during minute 3 (M3) of VRCE and MA, as well as during minute 1 (M1) of CPT in PTSD vs. Controls. Values are the mean ± SE. *Significant difference in response between PTSD and Control groups (P < 0.05).
MA
These experiments aimed to determine whether the haemodynamic and MSNA responses during non‐combat‐related mental stress (i.e. mental stress that was not related to PTSD symptoms) were augmented amongst young Veterans with PTSD. During 3 min of MA, there was no significant difference in the SBP response amongst PTSD patients compared to Controls (Fig. 2 A) (Table 2). However, there was a significantly greater increase in the DBP response amongst PTSD patients (ANOVA, F test, P = 0.011) (Fig. 2 B), although there was no statistically significant difference in the MAP and HR responses during MA between the groups (Fig. 2 C and D). There was a significantly greater increase in MSNA quantified as change in burst frequency (ANOVA, F test, P < 0.001) amongst PTSD patients compared to Controls (Fig. 2 E) and a trend towards a significant difference when quantified as change in MSNA total activity (ANOVA, F test, P = 0.056) (Fig. 2 F).
Figure 2. Hemodynamic and sympathetic reactivity during mental arithmetic.
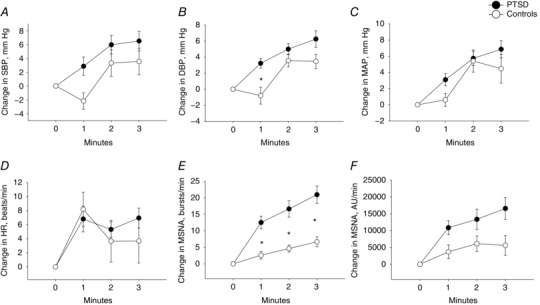
Change in SBP (A), DBP (B), MAP (C), HR (D), MSNA quantified as burst frequency (bursts min–1) (E) and total activity (AU min–1) (F) during 3 min of mental arithmetic in PTSD patients (filled circles) and Controls (open circles). * P < 0.05 between PTSD and Control groups.
CPT
These experiments aimed to determine whether there was a difference in the haemodynamic and MSNA responses between PTSD patients and Controls during a non‐mental stress type of sympathoexcitatory stimulus that is not mediated by arterial baroreflexes (Cui et al. 2002) for comparison. During 1 min of CPT, there was no significant difference in the MSNA response quantified as the change in burst frequency or change in total activity between the PTSD and Control groups (Fig. 3 A and B) (Table 2). There was also no significant difference in BP and HR responses during CPT between the groups (Fig. 3 C and D).
Figure 3. Hemodynamic and sympathetic reactivity during the cold pressor test.
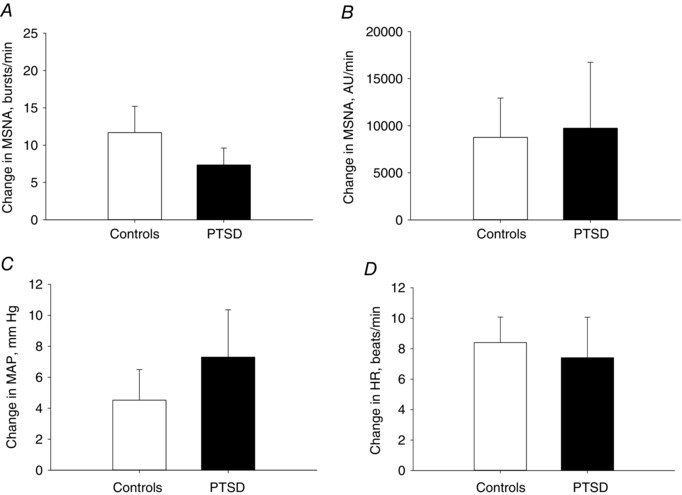
Change in MSNA quantified as burst frequency (bursts min–1) (A) and total activity (AU min–1) (B), MAP (C) and HR (D) during 1 min of CPT in PTSD patients vs. Controls.
Arterial BRS
Sympathetic BRS was quantified as the slope of the linear relationship between MSNA burst incidence and diastolic BP during pharmacological manipulation of BP. This standard quantification method controls for potential differences in HR responses during baroreflex testing. At baseline, the mean slope of the individual linear regressions between MSNA burst incidence and diastolic BP was significantly less steep in PTSD patients vs. Controls, demonstrating decreased sympathetic BRS in PTSD patients (P = 0.026) (Fig. 4 A and B). Similarly, under baseline conditions, the mean slope of the individual linear regressions between RRI and systolic BP was significantly less steep in PTSD patients vs. Controls, demonstrating decreased cardiovagal BRS in the PTSD group (P = 0.008) (Fig. 4 C and D).
Figure 4. Sympathetic and cardiovagal baroreflex sensitivity at rest.
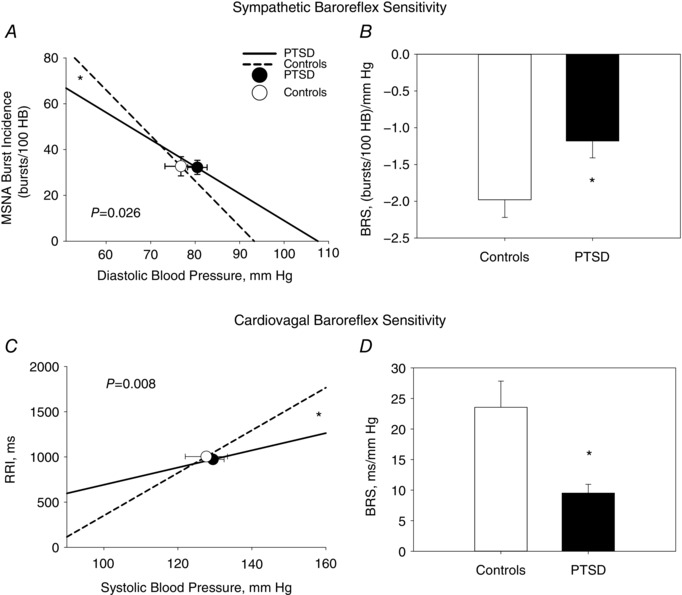
The linear relationships between diastolic blood pressure and MSNA burst incidence (A) and between SBP and R‐R interval (RRI) (C) are shown at rest and were derived from the mean slope values from individual linear regressions performed during baroreflex testing in PTSD (continuous line) vs. Controls (dashed line). The mean slope of the individual linear regressions between DBP and MSNA represents mean sympathetic BRS and was significantly less steep in PTSD [−1.2 ± 0.2 bursts (100 heart beats)–1 mmHg–1; R 2 = 0.77 ± 0.02] vs. Controls [−2.0 ± 0.3 bursts (100 heart beats)–1 mmHg–1; R 2 = 0.85 ± 0.02; P = 0.026] (A and B). The mean slope of the individual linear regressions between systolic blood pressure and RRI represents mean cardiovagal BRS and was significantly less steep in PTSD (9.5 ± 1.4 ms mmHg–1; R 2 = 0.74 ± 0.02) vs. Controls (23.6 ± 4.3 ms mmHg−1; R 2 = 0.75 ± 0.02; P = 0.008) (C and D). The operating point around which the baroreflex operates is derived from baseline measures and signified by the closed circles (PTSD) and open circles (Controls). * P < 0.05 between PTSD and Control groups.
Arterial baroreflex sensitivity was repeated in a subset of PTSD (N = 9) and Controls (N = 11) during concomitant VRCE. Several of the participants in each group did not have repeat baroreflex testing as a result of side effects during IV nitroprusside administration, including headache, palpitations and hypotension. There was no difference in baseline BRS in the subset of Controls and PTSD subjects that had repeat baroreflex testing. During VRCE, there was no difference in the mean slope of the individual linear regressions between MSNA burst incidence and diastolic BP between PTSD patients and Controls, demonstrating no significant difference in sympathetic BRS during VRCE between the groups (P = 0.135) (Fig. 5 A and B). However, the mean slope of the individual linear regressions between the RRI and systolic BP was significantly less steep in PTSD patients vs. Controls, demonstrating decreased cardiovagal BRS in PTSD patients during VRCE (P = 0.014) (Fig. 5 C and D).
Figure 5. Sympathetic and cardiovagal baroreflex sensitivity during virtual reality combat exposure.
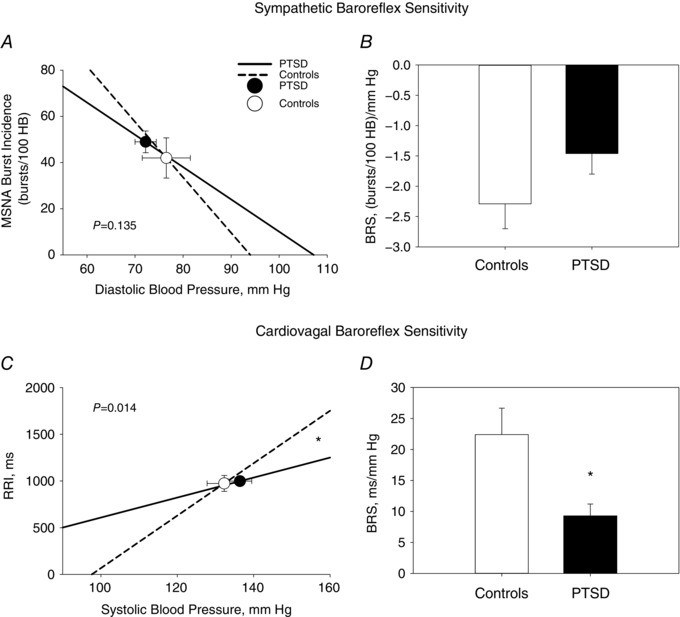
The linear relationship between diastolic blood pressure (DSB) and MSNA burst incidence (A) and between systolic blood pressure and R‐R interval (RRI) (C) are shown during concomitant VRCE, and were derived from the mean slope values from individual linear regressions performed during baroreflex testing in PTSD (continuous line) vs. Controls (dashed line). There was no significant difference between the groups in the mean slope of the individual linear regressions between DBP and MSNA that represents mean sympathetic BRS (A and B). The mean slope of the individual linear regressions between systolic blood pressure and RRI represents cardiovagal BRS and was significantly less steep in PTSD (9.3 ± 1.9 ms mmHg–1; R 2 = 0.75 ± 0.02) vs. Controls (22.4 ± 4.3 ms mmHg–1; R 2 = 0.75 ± 0.03; P = 0.014) (C and D). The operating point around which the baroreflex operates is signified by the closed circles (PTSD) and open circles (Controls). * P < 0.05 between PTSD and Control groups.
Inflammatory biomarkers
PTSD patients had significantly elevated hsCRP levels (PTSD vs. Controls, 2.1 ± 0.4 vs. 1.0 ± 0.3 mg L−1, P = 0.047) (Fig. 6 A). There was no significant difference in IL‐2 or IL‐6 levels between the groups (Fig. 6 B and C). There was a significant linear relationship between hsCRP levels and MSNA total activity response to VRCE (P = 0.011, R = 0.556). There were no significant correlations between hsCRP and MSNA responses during MA, nor to cardiovagal or sympathetic BRS.
Figure 6. Inflammatory biomarkers.
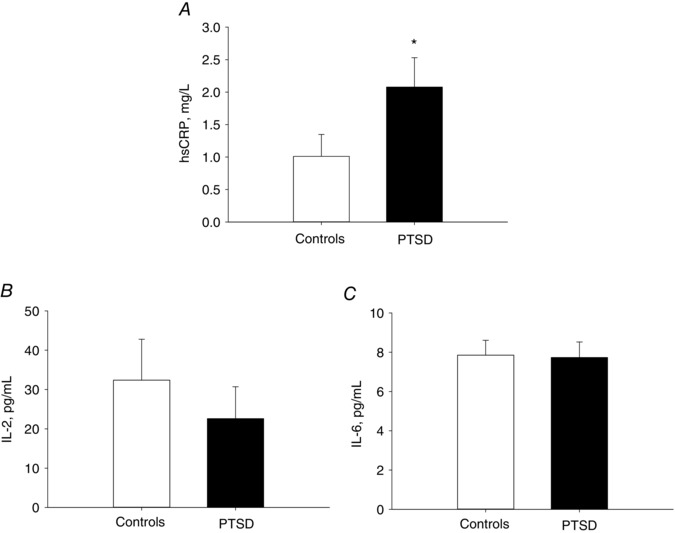
Highly sensitivity hsCRP (A), interleukin 2 (IL‐2) (B) and interleukin 6 (IL‐6) and (C) levels in PTSD patients vs. Controls. * P < 0.05 between PTSD and Control groups.
Discussion
The present study focused on a relatively young population of Veterans from the current combat operations in Iraq and Afghanistan without hypertension or CV disease. The aim was to identify pre‐existing biological mechanisms that are present in young individuals who are currently healthy but at increased risk of CV disease and hypertension by virtue of having PTSD (Cohen et al. 2009). More than 2 000 000 OEF/OIF/OND service members have been deployed to Iraq and Afghanistan over the past 15 years and are returning with high rates of PTSD, estimated at up to 20% post‐deployment (Hoge et al. 2004). PTSD is also common in the civilian population (Bedi & Arora, 2007) and is associated with significant impairment, making PTSD a major public health problem (Kessler, 2000). One less well‐recognized but highly significant consequence of PTSD is an increased risk of hypertension and CV disease (Brudey et al. 2015). In a prospective study that included 562 twins discordant for PTSD, those with PTSD had more than two‐fold greater risk of developing coronary heart disease than their counterparts without PTSD, and this association remained significant even after controlling for lifestyle factors, comorbidities and major depression (Vaccarino et al. 2013). Data from the US National Comorbidity Survey showed that PTSD patients had a 2.9‐fold greater risk for developing hypertension (Kibler et al. 2009) and multivariate analyses that included over 300 000 OEF/OIF Veterans showed that those with PTSD had a 59% higher chance of developing hypertension (Cohen et al. 2009). In a prospective study of over 4000 Vietnam Veterans without CV disease at baseline, those with PTSD had a 2.2‐fold greater risk of CV mortality determined using a multivariate model that adjusted for comorbid conditions, lifestyle and combat exposure (Boscarino, 2008). In older Veterans without CV disease at baseline, PTSD was associated with a significantly greater risk of developing CV disease, chronic heart failure, myocardial infarction and peripheral vascular disease (Beristianos et al. 2016). Thus, multiple large epidemiological studies demonstrate that PTSD is significantly and independently associated with an increased risk of hypertension, CV disease and mortality.
The mechanisms underlying the association between PTSD and CV risk have not been fully determined. PTSD patients have chronic ongoing mental stress that may lead to biological changes as a result of allostasis. Allostasis (Bedi & Arora, 2007) refers to physiological adjustments made to meet demands from external challenges. Such allostatic responses are engaged during times of stress and include activation of the SNS, which is initially adaptive. However, we hypothesized that exposure to extreme stress with subsequent development of PTSD results in chronic physiological changes as a result of high ‘allostatic load’, including chronic elevations of SNS activity, which is maladaptive. High allostatic load also leads to inflammation and oxidative stress, which in turn could lead to SNS overactivity directly via central effects, and indirectly via arterial baroreflex dysfunction. SNS overactivity has a major role in causing and sustaining hypertension (Grassi, 2009), which is a major risk factor for CV disease, and also leads to a number of deleterious effects on end organs that are independent of blood pressure, including the development of heart failure (Julius, 1993), arrhythmias (Grassi et al. 2004), atherogenesis (Erami et al. 2002), insulin resistance (Jamerson et al. 1993) and renal sclerosis (Amann et al. 2000).
Prior studies have provided indirect evidence that PTSD may be characterized by SNS overactivity. PTSD patients have a higher resting HR and BP, as well as decreased HR variability, suggesting a state of chronic SNS overactivity (Buckley & Kaloupek, 2001; Bedi & Arora, 2007). Studies have shown that PTSD patients have decreased low‐frequency and high‐frequency HR variability (Cohen et al. 1998; Chang et al. 2013; Shah et al. 2013), suggestive of increased sympathetic and decreased parasympathetic tone to the heart. In addition, urinary noradrenaline levels have been shown to be significantly elevated in PTSD patients compared to Controls without PTSD (Wingenfeld et al. 2015). In the present study, we present the first direct assessments of sympathetic nerve activity using microneurography in PTSD patients compared to age‐matched Controls. Although we did not observe baseline differences between the groups in MSNA or haemodynamics at rest, PTSD patients had significantly augmented MSNA responses during both combat‐related and non‐combat‐related mental stress. These data suggest that SNS drive in these patients is exacerbated by mental stress associated with PTSD symptoms, such as that experienced during flashbacks, as well as non‐related mental stressors.
Previous studies assessing sympathetic and haemodynamic reactivity during mental stress in PTSD have shown mixed results. Although some have reported that PTSD patients had greater increases in HR, BP and noradrenaline levels during mental stress (Bedi & Arora, 2007), others found that PTSD was characterized by blunted haemodynamic responses during mental stress induced by recall of traumatic events (Cohen et al. 1998). A unique feature of the present studies is the use of direct sympathetic nerve measurements and continuous beat to beat haemodynamics during two different types of mental stress: (i) combat‐related mental stress (i.e. mental stress that is associated with PTSD symptoms) and (ii) non‐combat‐related mental stress that is not related to PTSD symptoms. We showed that young Veterans with PTSD have significantly greater increases in MSNA and HR during VRCE compared to Controls, suggesting that PTSD patients have heightened sympathetic and haemodynamic responses during combat‐related mental stress. PTSD patients also had augmented MSNA and diastolic BP responses during mental arithmetic, suggesting that these exaggerated haemodynamic and sympathetic responses also extend to non‐combat related mental stress in PTSD. However, there was no difference in the haemodynamic and MSNA responses during the CPT, suggesting that augmented haemodynamic and sympathetic responses are not generalized to all sympathoexcitatory stimuli, nor to non‐baroreflex‐mediated stimuli. Thus, the augmented SNS and haemodynamic responses in PTSD are specific to mental stress, both related and unrelated to PTSD symptoms. Exaggerated increases in SNS activity during mental stress could contribute to an increased risk of hypertension and CV disease by increasing the burden of SNS activity. Moreover, exaggerated SNS responses during mental stress have been shown to be associated with an increased risk of hypertension (Matthews et al. 1993) and CV disease (Proietti et al. 2011).
VRCE is considered a neutral stimulus in those without combat‐related PTSD; therefore, it is not surprising that there was minimal MSNA and BP reactivity observed during VRCE amongst the Control group. However, in PTSD patients, the MSNA response was significantly heightened during VRCE but, despite the heightened MSNA response, the BP responses were not augmented. By contrast, MSNA responses during MA were also significantly augmented in PTSD, although they were accompanied by concomitant augmentation of diastolic BP responses. These observations suggest that there may be differences in the degree of vasoconstriction and therefore increase in blood pressure in response to sympathetic nerve activation (i.e. neurovascular transduction) during combat‐related vs. non‐combat‐related mental stress in PTSD. Although not tested in the present study, the differences in neurovascular transduction observed between stress protocols could be a result of differences in the release of noradrenaline at sympathetic nerve terminals, the generation of vasodilators and alternative vasoconstrictors, and adrenergic receptor density and responsiveness. In particular, chronic elevations in sympathetic activation during stress related to PTSD symptoms, could lead to downregulation of α‐1 adrenergic receptors, as has been suggested in other states characterized by high sympathetic activity (Feng et al. 1999; Dinenno et al. 2002), and this may lead to blunted BP responses relative to the degree of sympathetic activation. These and other mechanisms underlying neurovascular transduction during mental stress in PTSD should be evaluated in future studies.
One potential mechanism underlying SNS overactivation in PTSD is decreased arterial BRS. The arterial baroreceptors are important in the moment‐to‐moment control of BP by buffering acute fluctuations in arterial BP during postural, physiological or mental stress. In addition, arterial baroreceptors tonically inhibit SNS activity to maintain BP around a set point. BRS is diminished or blunted if increases and decreases in BP via the baroreflex arc fail to elicit the appropriate changes in SNS activity (sympathetic BRS) and parasympathetic activity (cardiovagal BRS) to maintain appropriate levels of arterial BP, or are chronically reset to higher BP levels as in chronic hypertension. Using the Modified Oxford technique to assess arterial baroreflex sensitivity, we showed that baseline sympathetic and cardiovagal BRS was indeed blunted in PTSD patients compared to Controls, and also that cardiovagal BRS was blunted during concomitant combat‐related mental stress, suggesting that impaired BRS may contribute to abnormal SNS responses during mental stress in PTSD. Decreased BRS contributes to the pathogenesis of many conditions characterized by SNS overactivity, including chronic heart failure (Grassi et al. 1995), obesity (Grassi et al. 1998) and hypertension (Matsukawa et al. 1991a), and the data obtained in the present study demonstrate that impaired BRS is present in Veterans with PTSD. Decreased BRS could be one contributing mechanisms underlying the link between PTSD and cardiovascular disease risk.
Chronic inflammation may contribute to blunted BRS in PTSD because several studies have demonstrated that immune dysfunction is present in this population (von Kanel et al. 2007; Spitzer et al. 2010; Plantinga et al. 2013) and, in experimental animal models of CV disease, anti‐inflammatory treatments have been shown to affect autonomic and baroreceptor function (Marvar et al. 2016). PTSD is associated with higher circulating levels of inflammatory markers (von Kanel et al. 2007; Spitzer et al. 2010; Plantinga et al. 2013) including CRP, IL‐4, IL‐6 and intercellular adhesion molecule‐1, higher brain angiotensin II levels (Matsumura et al. 1998; Saavedra et al. 2011), and greater arterial stiffness (Walczewska et al. 2011). In the population in the present study, we found that young OEF/OIF/OND patients with PTSD had significantly elevated hsCRP levels compared to age‐matched Controls, suggesting that chronic inflammation may play a contributory role in impaired BRS and heightened SNS responses in PTSD. Our finding that hsCRP levels correlate with heightened MSNA responses during VRCE suggest that heightened basal levels of inflammation may be linked to enhanced sympathetic reactivity in PTSD. CRP levels are correlated with increased risk of cardiovascular disease in multiple large epidemiological studies (Ridker et al. 1998; Corrado & Novo, 2007; Greenland et al. 2010; Kaptoge et al. 2010) and the results of the present study provide additional support indicating that basal levels of inflammation are heightened in PTSD and may be linked to enhanced sympathetic activity and baroreflex dysfunction. In addition, there is evidence that, prior to the development of overt CV disease, chronic inflammation could lead to BRS dysfunction at vascular nerve endings or along its tract and within the brainstem (Chapleau et al. 2001; Zubcevic et al. 2011; McBryde et al. 2013). In the present study, although we observed elevated basal hsCRP levels in PTSD, we did not observe a significant correlation between hsCRP and baroreflex sensitivity. However, these analyses may have been limited by a small sample size and the relatively small range of hsCRP values.
We recognize several other limitations to these studies. First, our study population primarily comprised African‐American males with combat‐related PTSD. Whether these findings can be generalized to females, other races and ethnicities or PTSD related to non‐military trauma is unclear. Second, we used a CAPS‐IV score ≥ 45 to confirm the diagnosis of PTSD in patients, and a score of < 45 to exclude the presence of PTSD in Controls (Weathers & Ruscio, 1999; Weathers et al. 2001). The Control group comprised OEF/OIF/OND Veterans without PTSD and had a mean CAPS‐IV score of 38, which is below the threshold for PTSD diagnosis but was within the range for subthreshold PTSD (CAPS‐IV score range 20–39). Indeed, all Control participants had a CAPS‐IV score within the subthreshold PTSD range. Recent investigations of service members with subthreshold PTSD have also revealed physiological alterations (Costanzo et al. 2014). A more robust difference in physiological measures may have been demonstrated if the Controls included only those without any symptoms of PTSD (score range 0–19) or non‐Veterans without combat exposure. Third, although we observed a significantly lower cardiovagal and sympathetic BRS at baseline, and significantly lower cardiovagal BRS in the PTSD group during VRCE, there was a non‐significant trend towards reduced sympathetic BRS in the PTSD group during VRCE. The study may have been underpowered to detect differences in sympathetic BRS during concomitant VRCE. Fourth, although we observed significantly higher levels of basal hsCRP levels in PTSD, IL‐2 and IL‐6 levels were not elevated in this cohort. Although we did not observe a difference in basal levels of IL‐2 and IL‐6, this does not exclude the possibility that these cytokines may be enhanced in PTSD during periods of acute stress or exposure to fear stimuli, and should be tested in future studies. Lastly, it is unclear from this study whether augmented sympathetic responses to mental stress and decreased BRS, are a result of having PTSD, or are present prior to the development of PTSD and associated with a higher propensity for developing PTSD. Because SNS responses to mental stress are variable in the general population, those with higher SNS responses to stress and blunted BRS may be at greater risk of developing PTSD in the setting of high allostatic load. Studies assessing physiological responses to mental stress before and after the development of PTSD, or long‐term prospective studies evaluating the incidence of PTSD amongst those with high vs. low SNS reactivity to mental stress, represent potential strategies for determining whether increased SNS reactivity is associated with an increased risk of developing PTSD.
In conclusion, PTSD is a highly prevalent and debilitating illness that is associated with an increased risk of CV disease and hypertension. Augmented sympathetic and haemodynamic responses to mental stress not only associated with PTSD symptoms, but also generalized to other types of mental stress, may lead to an overall increased burden of sympathetic activity that leads to a greater risk of developing hypertension and CV disease. Blunted sympathetic and cardiovagal BRS may contribute to increased sympathetic and decreased parasympathetic activation in PTSD, and chronic inflammation may play a causative role in decreasing BRS and increasing central sympathetic output. Identifying physiological mechanisms that contribute to an increased CV risk in PTSD patients will allow for the development of interventions aiming to modify CV risk in these patients. Future studies testing therapies to improve SNS reactivity, BRS and inflammation should be conducted to evaluate potential long‐term benefits on CV risk in PTSD patients.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
JP, PM and BR contributed to the conception or design of the study. JP, PL, MK, SN, RD, SA, NL and BR contributed to the acquisition, analysis or interpretation of data. JP, PM, PL, MK, SN, RD, SM, NL and BR contributed to drafting the manuscript or revising it critically for important intellectual content. All authors approve of the final version of the manuscript, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved, and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by Merit Review Award number I01CX001065 (to J. Park) from the United States (US) Department of Veterans Affairs Clinical Sciences Research and Development Program; American Heart Association National Affiliate, Collaborative Sciences Award 15CSA24340001; resources and the use of facilities at the Clinical Studies Centre of the Atlanta VA Medical Centre; the Atlanta Research and Education Foundation; National Institutes of Health Grant K23 HL‐098744 (to J. Park); National Institutes of Health training grants T32 DK‐00756 and K12 GM‐000680; Satellite Health Care, a not‐for‐profit renal care provider; the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Clinical Studies Centre, Decatur, Georgia; the Atlanta Research and Education Foundation; University Research Council grant from Emory University; and the Atlanta Clinical and Translational Science Institute supported by Public Health Service Grant UL1 RR‐025008 from the Clinical and Translational Science Award program, National Institutes of Health, National Centre for Research Resources.
Acknowledgements
We especially thank Albert ‘Skip’ Rizzo for developing and providing the virtual reality combat exposure video clips for use in the present study. A preliminary version of these findings was presented as an abstract at the 2016 Experimental Biology Meeting in San Diego, California.
Linked articles This article is highlighted by a Perspective by Carter. To read this Perspective, visit https://doi.org/10.1113/JP274512.
References
- Amann K, Rump LC, Simonaviciene A, Oberhauser V, Wessels S, Orth SR, Gross ML, Koch A, Bielenberg GW, Van Kats JP, Ehmke H, Mall G & Ritz E (2000). Effects of low dose sympathetic inhibition on glomerulosclerosis and albuminuria in subtotally nephrectomized rats. J Am Soc Nephrol 11, 1469–1478. [DOI] [PubMed] [Google Scholar]
- Bedi US & Arora R (2007). Cardiovascular manifestations of posttraumatic stress disorder. J Natl Med Assoc 99, 642–649. [PMC free article] [PubMed] [Google Scholar]
- Beristianos MH, Yaffe K, Cohen B & Byers AL (2016). PTSD and risk of incident cardiovascular disease in aging veterans. Am J Geriatr Psychiatry 24, 192–200. [DOI] [PubMed] [Google Scholar]
- Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS & Keane TM (1995). The development of a clinician‐administered PTSD scale. J Trauma Stress 8, 75–90. [DOI] [PubMed] [Google Scholar]
- Blanchard EB, Jones‐Alexander J, Buckley TC & Forneris CA (1996). Psychometric properties of the PTSD Checklist (PCL). Behav Res Ther 34, 669–673. [DOI] [PubMed] [Google Scholar]
- Boscarino JA (2008). A prospective study of PTSD and early‐age heart disease mortality among Vietnam veterans: implications for surveillance and prevention. Psychosom Med 70, 668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudey C, Park J, Wiaderkiewicz J, Kobayashi I, Mellman TA & Marvar PJ (2015). Autonomic and inflammatory consequences of posttraumatic stress disorder and the link to cardiovascular disease. Am J Physiol Regul Integr Comp Physiol 309, R315–R321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley TC & Kaloupek DG (2001). A meta‐analytic examination of basal cardiovascular activity in posttraumatic stress disorder. Psychosom Med 63, 585–594. [DOI] [PubMed] [Google Scholar]
- Carter JR, Kupiers NT & Ray CA (2005). Neurovascular responses to mental stress. J Physiol 564, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter JR & Ray CA (2009). Sympathetic neural responses to mental stress: responders, nonresponders and sex differences. Am J Physiol Heart Circ Physiol 296, H847–H853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter JR, Schwartz CE, Yang H & Joyner MJ (2013). Fish oil and neurovascular reactivity to mental stress in humans. Am J Physiol Regul Integr Comp Physiol 304, R523–R530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HA, Chang CC, Tzeng NS, Kuo TB, Lu RB & Huang SY (2013). Decreased cardiac vagal control in drug‐naive patients with posttraumatic stress disorder. Psychiatry Invest 10, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapleau MW, Li Z, Meyrelles SS, Ma X & Abboud FM (2001). Mechanisms determining sensitivity of baroreceptor afferents in health and disease. Ann NY Acad Sci 940, 1–19. [DOI] [PubMed] [Google Scholar]
- Cohen BE, Marmar C, Ren L, Bertenthal D & Seal KH (2009). Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. JAMA 302, 489–492. [DOI] [PubMed] [Google Scholar]
- Cohen H, Kotler M, Matar MA, Kaplan Z, Loewenthal U, Miodownik H & Cassuto Y (1998). Analysis of heart rate variability in posttraumatic stress disorder patients in response to a trauma‐related reminder. Biol Psychiatry 44, 1054–1059. [DOI] [PubMed] [Google Scholar]
- Corrado E & Novo S (2007). Evaluation of C‐reactive protein in primary and secondary prevention. J Invest Med 55, 430–438. [DOI] [PubMed] [Google Scholar]
- Costanzo ME, Leaman S, Jovanovic T, Norrholm SD, Rizzo AA, Taylor P & Roy MJ (2014). Psychophysiological response to virtual reality and subthreshold posttraumatic stress disorder symptoms in recently deployed military. Psychosom Med 76, 670–677. [DOI] [PubMed] [Google Scholar]
- Cui J, Wilson TE & Crandall CG (2002). Baroreflex modulation of muscle sympathetic nerve activity during cold pressor test in humans. Am J Physiol Heart Circ Physiol 282, H1717–H1723. [DOI] [PubMed] [Google Scholar]
- Delius W, Hagbarth KE, Hongell A & Wallin BG (1972a). General characteristics of sympathetic activity in human muscle nerves. Acta Physiol Scand 84, 65–81. [DOI] [PubMed] [Google Scholar]
- Delius W, Hongell A, Hongell A & Wallin BG (1972b). Manoeuvres affecting sympathetic outflow in human muscle nerves. Acta Physiol Scand 84, 82–94. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Dietz NM & Joyner MJ (2002). Aging and forearm postjunctional alpha‐adrenergic vasoconstriction in healthy men. Circulation 106, 1349–1354. [DOI] [PubMed] [Google Scholar]
- Dishman RK, Nakamura Y, Jackson EM & Ray CA (2003). Blood pressure and muscle sympathetic nerve activity during cold pressor stress: fitness and gender. Psychophysiology 40, 370–380. [DOI] [PubMed] [Google Scholar]
- Edmondson D, Kronish IM, Shaffer JA, Falzon L & Burg MM (2013). Posttraumatic stress disorder and risk for coronary heart disease: a meta‐analytic review. Am Heart J 166, 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erami C, Zhang H, Ho JG, French DM & Faber JE (2002). Alpha(1)‐adrenoceptor stimulation directly induces growth of vascular wall in vivo. Am J Physiol Heart Circ Physiol 283, H1577–H1587. [DOI] [PubMed] [Google Scholar]
- Fagius J, Karhuvaara S & Sundlof G (1989). The cold pressor test: effects on sympathetic nerve activity in human muscle and skin nerve fascicles. Acta Physiol Scand 137, 325–334. [DOI] [PubMed] [Google Scholar]
- Feng Q, Sun X, Lu X, Edvinsson L & Hedner T (1999). Decreased responsiveness of vascular postjunctional alpha1‐, alpha2‐adrenoceptors and neuropeptide Y1 receptors in rats with heart failure. Acta Physiol Scand 166, 285–291. [DOI] [PubMed] [Google Scholar]
- Grassi G ( 2009). Assessment of sympathetic cardiovascular drive in human hypertension: achievements and perspectives. Hypertension 54, 690–697. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Cattaneo BM, Lanfranchi A, Vailati S, Giannattasio C, Del Bo A, Sala C, Bolla GB & Pozzi M (1995). Sympathetic activation and loss of reflex sympathetic control in mild congestive heart failure. Circulation 92, 3206–3211. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Colombo M, Bolla G, Cattaneo BM, Cavagnini F & Mancia G (1998). Body weight reduction, sympathetic nerve traffic, and arterial baroreflex in obese normotensive humans. Circulation 97, 2037–2042. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Dell'Oro R, Facchini A, Ilardo V & Mancia G (2004). Sympathetic and baroreflex function in hypertensive or heart failure patients with ventricular arrhythmias. J Hypertens 22, 1747–1753. [DOI] [PubMed] [Google Scholar]
- Greenland P, Alpert JS, Beller GA, Benjamin EJ, Budoff MJ, Fayad ZA, Foster E, Hlatky MA, Hodgson JM, Kushner FG, Lauer MS, Shaw LJ, Smith SC, Jr. , Taylor AJ, Weintraub WS, Wenger NK, Jacobs AK & American College of Cardiology Foundation/American Heart Asscoiation Tasl Force on Practice G (2010). 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 122, 2748–2764. [DOI] [PubMed] [Google Scholar]
- Hoge CW, Castro CA, Messer SC, McGurk D, Cotting DI & Koffman RL (2004). Combat duty in Iraq and Afghanistan, mental health problems, and barriers to care. N Engl J Med 351, 13–22. [DOI] [PubMed] [Google Scholar]
- Ilies C, Bauer M, Berg P, Rosenberg J, Hedderich J, Bein B, Hinz J & Hanss R (2012). Investigation of the agreement of a continuous non‐invasive arterial pressure device in comparison with invasive radial artery measurement. Br J Anaesth 108, 202–210. [DOI] [PubMed] [Google Scholar]
- Jamerson KA, Julius S, Gudbrandsson T, Andersson O & Brant DO (1993). Reflex sympathetic activation induces acute insulin resistance in the human forearm. Hypertension 21, 618–623. [DOI] [PubMed] [Google Scholar]
- Jeleazcov C, Krajinovic L, Munster T, Birkholz T, Fried R, Schuttler J & Fechner J (2010). Precision and accuracy of a new device (CNAPTM) for continuous non‐invasive arterial pressure monitoring: assessment during general anaesthesia. Br J Anaesth 105, 264–272. [DOI] [PubMed] [Google Scholar]
- Julius S ( 1993). Corcoran Lecture. Sympathetic hyperactivity and coronary risk in hypertension. Hypertension 21, 886–893. [DOI] [PubMed] [Google Scholar]
- Kang HK, Bullman TA & Taylor JW (2006). Risk of selected cardiovascular diseases and posttraumatic stress disorder among former World War II prisoners of war. Ann Epidemiol 16, 381–386. [DOI] [PubMed] [Google Scholar]
- Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R & Danesh J (2010). C‐reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta‐analysis. Lancet 375, 132r140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC (2000). Posttraumatic stress disorder: the burden to the individual and to society. J Clin Psychiatry 61 (Suppl 5), 4–12. [PubMed] [Google Scholar]
- Kibler JL, Joshi K & Ma M (2009). Hypertension in relation to posttraumatic stress disorder and depression in the US National Comorbidity Survey. Behav Med 34, 125–132. [DOI] [PubMed] [Google Scholar]
- Kubzansky LD, Koenen KC, Jones C & Eaton WW (2009). A prospective study of posttraumatic stress disorder symptoms and coronary heart disease in women. Health Psychol 28, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubzansky LD, Koenen KC, Spiro A, 3rd , Vokonas PS & Sparrow D (2007). Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Arch Gen Psychiatry 64, 109‐116. [DOI] [PubMed] [Google Scholar]
- Kuniyoshi FH, Trombetta IC, Batalha LT, Rondon MU, Laterza MC, Gowdak MM, Barretto AC, Halpern A, Villares SM, Lima EG & Negrao CE (2003). Abnormal neurovascular control during sympathoexcitation in obesity. Obes Res 11, 1411–1419. [DOI] [PubMed] [Google Scholar]
- Mano T, Iwase S & Toma S (2006). Microneurography as a tool in clinical neurophysiology to investigate peripheral neural traffic in humans. Clin Neurophysiol 117, 2357–2384. [DOI] [PubMed] [Google Scholar]
- Marvar PJ, Hendy EB, Cruise TD, Walas D, DeCicco D, Vadigepalli R, Schwaber JS, Waki H, Murphy D & Paton JF (2016). Systemic leukotriene B4 receptor antagonism lowers arterial blood pressure and improves autonomic function in the spontaneously hypertensive rat. J Physiol 594, 5975–5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa T, Gotoh E, Hasegawa O, Shionoiri H, Tochikubo O & Ishii M (1991a). Reduced baroreflex changes in muscle sympathetic nerve activity during blood pressure elevation in essential hypertension. J Hypertens 9, 537–542. [DOI] [PubMed] [Google Scholar]
- Matsukawa T, Gotoh E, Uneda S, Miyajima E, Shionoiri H, Tochikubo O & Ishii M (1991b). Augmented sympathetic nerve activity in response to stressors in young borderline hypertensive men. Acta Physiol Scand 141, 157–165. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Averill DB & Ferrario CM (1998). Angiotensin II acts at AT1 receptors in the nucleus of the solitary tract to attenuate the baroreceptor reflex. Am J Physiol 275, R1611–R1619. [DOI] [PubMed] [Google Scholar]
- Matthews KA, Woodall KL & Allen MT (1993). Cardiovascular reactivity to stress predicts future blood pressure status. Hypertension 22, 479–485. [DOI] [PubMed] [Google Scholar]
- McBryde FD, Abdala AP, Hendy EB, Pijacka W, Marvar P, Moraes DJ, Sobotka PA & Paton JF (2013). The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat Commun 4, 2395. [DOI] [PubMed] [Google Scholar]
- McLay RN, Wood DP, Webb‐Murphy JA, Spira JL, Wiederhold MD, Pyne JM & Wiederhold BK (2011). A randomized, controlled trial of virtual reality‐graded exposure therapy for post‐traumatic stress disorder in active duty service members with combat‐related post‐traumatic stress disorder. Cyberpsychol Behav Soc Netw 14, 223–229. [DOI] [PubMed] [Google Scholar]
- Noll G, Wenzel RR, Schneider M, Oesch V, Binggeli C, Shaw S, Weidmann P & Luscher TF (1996). Increased activation of sympathetic nervous system and endothelin by mental stress in normotensive offspring of hypertensive parents. Circulation 93, 866–869. [DOI] [PubMed] [Google Scholar]
- Norrholm SD, Jovanovic T, Gerardi M, Breazeale KG, Price M, Davis M, Duncan E, Ressler KJ, Bradley B, Rizzo A, Tuerk PW & Rothbaum BO (2016). Baseline psychophysiological and cortisol reactivity as a predictor of PTSD treatment outcome in virtual reality exposure therapy. Behav Res Ther 82, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Middlekauff HR & Campese VM (2012). Abnormal sympathetic reactivity to the cold pressor test in overweight humans. Am J Hypertens 25, 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantinga L, Bremner JD, Miller AH, Jones DP, Veledar E, Goldberg J & Vaccarino V (2013). Association between posttraumatic stress disorder and inflammation: a twin study. Brain Behav Immun 30, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proietti R, Mapelli D, Volpe B, Bartoletti S, Sagone A, Dal Bianco L & Daliento L (2011). Mental stress and ischemic heart disease: evolving awareness of a complex association. Future Cardiol 7, 425–437. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Glynn RJ & Hennekens CH (1998). C‐reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 97, 2007–2011. [DOI] [PubMed] [Google Scholar]
- Rizzo A, Parsons TD, Lange B, Kenny P, Buckwalter JG, Rothbaum B, Difede J, Frazier J, Newman B, Williams J & Reger G (2011). Virtual reality goes to war: a brief review of the future of military behavioral healthcare. J Clin Psychol Med Settings 18, 176–187. [DOI] [PubMed] [Google Scholar]
- Rizzo AS, Difede J, Rothbaum BO, Reger G, Spitalnick J, Cukor J & McLay R (2010). Development and early evaluation of the Virtual Iraq/Afghanistan exposure therapy system for combat‐related PTSD. Ann NY Acad Sci 1208, 114–125. [DOI] [PubMed] [Google Scholar]
- Rothbaum BO, Price M, Jovanovic T, Norrholm SD, Gerardi M, Dunlop B, Davis M, Bradley B, Duncan EJ, Rizzo A & Ressler KJ (2014). A randomized, double‐blind evaluation of D‐cycloserine or alprazolam combined with virtual reality exposure therapy for posttraumatic stress disorder in Iraq and Afghanistan War veterans. Am J Psychiatry 171, 640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbaum BO, Rizzo AS & Difede J (2010). Virtual reality exposure therapy for combat‐related posttraumatic stress disorder. Ann NY Acad Sci 1208, 126–132. [DOI] [PubMed] [Google Scholar]
- Roy MJ, Costanzo ME, Jovanovic T, Leaman S, Taylor P, Norrholm SD & Rizzo AA (2013). Heart rate response to fear conditioning and virtual reality in subthreshold PTSD. Stud Health Technol Inform 191, 115–119. [PubMed] [Google Scholar]
- Rudas L, Crossman AA, Morillo CA, Halliwill JR, Tahvanainen KU, Kuusela TA & Eckberg DL (1999). Human sympathetic and vagal baroreflex responses to sequential nitroprusside and phenylephrine. Am J Physiol Heart Circ Physiol 276, H1691–H1698. [DOI] [PubMed] [Google Scholar]
- Saavedra JM, Sanchez‐Lemus E & Benicky J (2011). Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: Therapeutic implications. Psychoneuroendocrinology 36, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AJ, Lampert R, Goldberg J, Veledar E, Bremner JD & Vaccarino V (2013). Posttraumatic stress disorder and impaired autonomic modulation in male twins. Biol Psychiatry 73, 1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer C, Barnow S, Volzke H, Wallaschofski H, John U, Freyberger HJ, Lowe B & Grabe HJ (2010). Association of posttraumatic stress disorder with low‐grade elevation of C‐reactive protein: evidence from the general population. J Psychiatr Res 44, 15–21. [DOI] [PubMed] [Google Scholar]
- Vaccarino V, Goldberg J, Rooks C, Shah AJ, Veledar E, Faber TL, Votaw JR, Forsberg CW & Bremner JD (2013). Post‐traumatic stress disorder and incidence of coronary heart disease: a twin study. J Am Coll Cardiol 62, 970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE, Torebjork HE & Wallin BG (1979). Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev 59, 919–957. [DOI] [PubMed] [Google Scholar]
- Victor RG, Leimbach WN Jr., Seals DR, Wallin BG & Mark AL (1987). Effects of the cold pressor test on muscle sympathetic nerve activity in humans. Hypertension 9, 429–436. [DOI] [PubMed] [Google Scholar]
- von Kanel R, Hepp U, Kraemer B, Traber R, Keel M, Mica L & Schnyder U (2007). Evidence for low‐grade systemic proinflammatory activity in patients with posttraumatic stress disorder. J Psychiatr Res 41, 744–752. [DOI] [PubMed] [Google Scholar]
- Walczewska J, Rutkowski K, Wizner B, Cwynar M & Grodzicki T (2011). Stiffness of large arteries and cardiovascular risk in patients with post‐traumatic stress disorder. Eur Heart J 32, 730–736. [DOI] [PubMed] [Google Scholar]
- Wallin BG & Fagius J (1988). Peripheral sympathetic neural activity in conscious humans. Annu Rev Physiol 50, 565–576. [DOI] [PubMed] [Google Scholar]
- Weathers FW, Keane TM & Davidson JR (2001). Clinician‐administered PTSD scale: a review of the first ten years of research. Depress Anxiety 13, 132–156. [DOI] [PubMed] [Google Scholar]
- Weathers FW & Ruscio AM (1999). Psychometric Properties of Nine Scoring Rules for the Clinician‐Administered Posttraumatic Stress Disorder Scale. Psychol Assess 11, 124–133. [Google Scholar]
- Wingenfeld K, Whooley MA, Neylan TC, Otte C & Cohen BE (2015). Effect of current and lifetime posttraumatic stress disorder on 24‐h urinary catecholamines and cortisol: results from the Mind Your Heart Study. Psychoneuroendocrinology 52, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DL, Sheps SG, Elveback LR & Schirger L (1984). Cold pressor test as a predictor of hypertension. Hypertension 6, 301–306. [DOI] [PubMed] [Google Scholar]
- Zubcevic J, Waki H, Raizada MK & Paton JF (2011). Autonomic‐immune‐vascular interaction: an emerging concept for neurogenic hypertension. Hypertension 57, 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]