

Abstract
Key points
Mutations in genes encoding cardiac troponin I (TNNI3) and cardiac troponin T (TNNT2) caused altered troponin protein stoichiometry in patients with dilated cardiomyopathy.
TNNI3p.98trunc resulted in haploinsufficiency, increased Ca2+‐sensitivity and reduced length‐dependent activation.
TNNT2p.K217del caused increased passive tension.
A mutation in the gene encoding Lamin A/C (LMNA p.R331Q) led to reduced maximal force development through secondary disease remodelling in patients suffering from dilated cardiomyopathy.
Our study shows that different gene mutations induce dilated cardiomyopathy via diverse cellular pathways.
Abstract
Dilated cardiomyopathy (DCM) can be caused by mutations in sarcomeric and non‐sarcomeric genes. In this study we defined the pathogenic effects of three DCM‐causing mutations: the sarcomeric mutations in genes encoding cardiac troponin I (TNNI3p.98truncation) and cardiac troponin T (TNNT2p.K217deletion; also known as the p.K210del) and the non‐sarcomeric gene mutation encoding lamin A/C (LMNAp.R331Q). We assessed sarcomeric protein expression and phosphorylation and contractile behaviour in single membrane‐permeabilized cardiomyocytes in human left ventricular heart tissue. Exchange with recombinant troponin complex was used to establish the direct pathogenic effects of the mutations in TNNI3 and TNNT2. The TNNI3p.98trunc and TNNT2p.K217del mutation showed reduced expression of troponin I to 39% and 51%, troponin T to 64% and 53%, and troponin C to 73% and 97% of controls, respectively, and altered stoichiometry between the three cardiac troponin subunits. The TNNI3p.98trunc showed pure haploinsufficiency, increased Ca2+‐sensitivity and impaired length‐dependent activation. The TNNT2p.K217del mutation showed a significant increase in passive tension that was not due to changes in titin isoform composition or phosphorylation. Exchange with wild‐type troponin complex corrected troponin protein levels to 83% of controls in the TNNI3p.98trunc sample. Moreover, upon exchange all functional deficits in the TNNI3p.98trunc and TNNT2p.K217del samples were normalized to control values confirming the pathogenic effects of the troponin mutations. The LMNAp.R331Q mutation resulted in reduced maximal force development due to disease remodelling. Our study shows that different gene mutations induce DCM via diverse cellular pathways.
Keywords: dilated cardiomyopathy, heart failure, protein phosphorylation, troponin
Key points
Mutations in genes encoding cardiac troponin I (TNNI3) and cardiac troponin T (TNNT2) caused altered troponin protein stoichiometry in patients with dilated cardiomyopathy.
TNNI3p.98trunc resulted in haploinsufficiency, increased Ca2+‐sensitivity and reduced length‐dependent activation.
TNNT2p.K217del caused increased passive tension.
A mutation in the gene encoding Lamin A/C (LMNA p.R331Q) led to reduced maximal force development through secondary disease remodelling in patients suffering from dilated cardiomyopathy.
Our study shows that different gene mutations induce dilated cardiomyopathy via diverse cellular pathways.
Abbreviations
- cTnC
cardiac troponin C
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- DCM
dilated cardiomyopathy
- EC50
[Ca2+] needed to achieve 50% of maximal force
- ExAC
Exome Aggregation Consortium
- Fmax
maximal force
- Fpass
passive force
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- IDCM
idiopathic dilated cardiomyopathy
- KO
knock out
- LDA
length‐dependent activation
- LV
left ventricle
- LVAD
left ventricular assist device
- PKA
protein kinase A
- PKC
protein kinase C
- WT
wild‐type
Introduction
Dilated cardiomyopathy (DCM) is a cardiac disease characterized by dilatation of the left ventricle (LV) and a reduced systolic function. Initially, the prevalence of DCM was determined to be 1:2500 based on phenotypic screening, but recent studies suggested that it could be as high as 1:250 (Hershberger et al. 2013). DCM can be caused by environmental factors (viral infection, alcohol abuse, drug toxicity) or have a genetic basis. With current genetic screening, a genetic cause is found in 20–50% of DCM patients (Hershberger et al. 2010; Herman et al. 2012; van Spaendonck‐Zwarts et al. 2013). Over 30 genes have been found to harbour mutations that are likely to cause DCM (Hershberger et al. 2013). The Exome Aggregation Consortium (ExAC) recently reported that many rare variants in various sarcomeric and non‐sarcomeric genes, which were assumed to be disease‐causing, only have limited pathogenic burden as no or limited excess variation was found in a DCM population compared with ∼60,000 reference samples (Walsh et al. 2017). On the other hand, the presence of rare variants of uncertain significance was reported to be significantly higher in the DCM population compared with the ExAC reference samples indicating an overly conservative estimation of pathogenicity of these variants (Walsh et al. 2017). Among the various genes implicated in DCM are genes encoding sarcomeric proteins such as cardiac troponin I (encoded by TNNI3) (Carballo et al. 2009; van Spaendonck‐Zwarts et al. 2013), cardiac troponin T (encoded by TNNT2) (Hershberger et al. 2009; van Spaendonck‐Zwarts et al. 2013; Walsh et al. 2017) and titin (encoded by TTN) (Herman et al. 2012; Walsh et al. 2017), and genes encoding for non‐sarcomeric proteins such as lamin A/C (encoded by LMNA), a protein involved in nuclear stability (Parks et al. 2008; van Spaendonck‐Zwarts et al. 2013; Walsh et al. 2017). The fact that mutations in proteins of such diverse function can cause DCM implies that multiple pathomechanisms can lead to cardiac dilatation and associated cardiac dysfunction. In this study we defined the pathological effects on cardiomyocyte function of three different DCM‐causing mutations in genes encoding sarcomeric (TNNI3, TNNT2) and non‐sarcomeric (LMNA) proteins.
The troponin complex consists of three different troponin proteins; cardiac troponin T (cTnT), cardiac troponin I (cTnI) and cardiac troponin C (cTnC). The involvement of troponin and tropomyosin in force generation has been described in the three state model of the thin filaments (McKillop & Geeves, 1993). The role of cTnI is to inhibit actin–myosin interaction and, through its interaction with cTnC, plays an important role in the Ca2+‐sensitivity of sarcomere activation (Westfall et al. 1999). The troponin complex can lock tropomyosin in the blocked state (so called B‐state) at low Ca2+ concentrations during which contraction does not occur since tropomyosin sterically hinders the interaction between myosin and actin. During contraction calcium binds to cTnC, which leads to a conformational change that enhances binding of cTnC to cTnI. This results in a large conformational change in cTnI, which leads to displacement of its inhibitory domains away from actin and thereby releasing its inhibitory effect on actin–myosin interaction (Spyracopoulos et al. 1997; Stone et al. 1998). Tropomyosin moves and transits into the closed state (C‐state), which enables myosin to bind to actin and subsequently cause force generation. The open state (M‐state) is the final shift of tropomyosin after myosin has bound and facilitates the formation of a strong cross‐bridge. By binding to both cTnC and tropomyosin (Li et al. 2002), cTnT regulates ATPase activity during contraction, but also serves as an anchor on the thin filaments for the troponin complex. The different troponin proteins as a complex and the location of the mutations studied are shown in a schematic representation in Fig. 1. The 292C→T transition in the TNNI3 gene encoding cTnI is predicted to result in a premature stop codon at amino acid 98. Truncation in this part of the protein would cause loss of the cTnC and two actin‐binding domains of cTnI (Mogensen et al. 2015). The p.K217del (also known as p.K210del; Otten et al. 2010) mutation in the TNNT2 gene has been reported across the world in unrelated families and is associated with high mortality and disease onset at a young age (∼33 years) (Kamisago et al. 2000; Mogensen et al. 2004; Hershberger et al. 2009; Otten et al. 2010). Mutations in the non‐sarcomeric gene LMNA, encoding the inner nuclear protein lamin A/C, have been found in 6% of DCM patients (Parks et al. 2008). Many patients carrying a LMNA mutation show conduction abnormalities and arrhythmias (Parks et al. 2008; Perrot et al. 2009). The LMNAp.R331Q mutation is located in the coil 2B domain, which is important for homodimerization. The LMNAp.R331Q mutation is predicted to cause loss of salt‐bridge interaction and thereby affect lamina stability (Gangemi & Degano, 2013).
Figure 1. Schematic representation of the troponin complex.
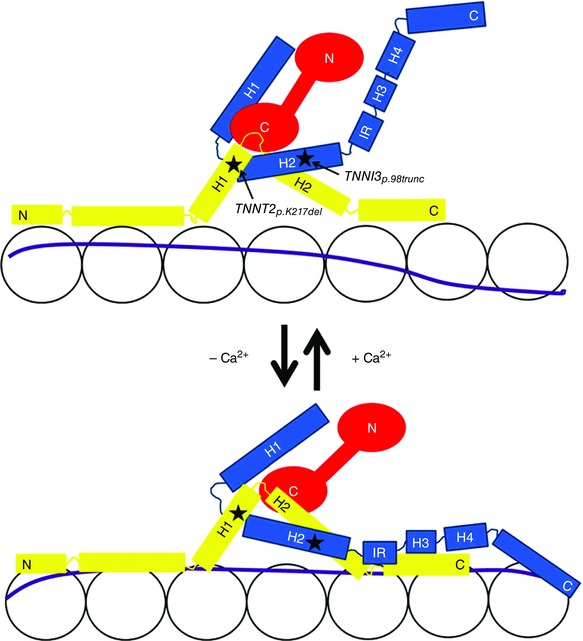
cTnT is shown in yellow, cTnI in blue and cTnC in red. The letters N and C indicate the N‐ and C‐terminus, respectively. The upper diagram shows the troponin complex in the presence of Ca2+ while the lower diagram shows the troponin complex without Ca2+. Location of the studied mutations are indicated with stars and an arrow in the upper panel. The letter H indicates a helix structure. IR, inhibitory region.
Apart from the direct mutation‐mediated changes in cardiac function, secondary disease remodelling plays an important role in DCM pathogenesis (Kötter et al. 2013; Beqqali et al. 2016). Phosphorylation by protein kinase A (PKA) of cTnI can fine‐tune Ca2+‐sensitivity and length‐dependent activation (LDA) of sarcomeres (Konhilas et al. 2003; Sequeira et al. 2013). PKA‐mediated phosphorylation of cTnI upon activation of the β‐adrenergic receptors by adrenaline reduces myofilament Ca2+‐sensitivity and enhances LDA (Konhilas et al. 2003). In heart failure, the β‐adrenergic receptor system is chronically stimulated leading to down‐regulation and desensitization of the β‐adrenergic receptors and subsequently decreased PKA‐mediated phosphorylation (Harding et al. 1994). Therefore, it is important to distinguish between the direct effects of mutations on sarcomere function and the indirect effects through changes in the β‐adrenergic system.
Even though various genes are implicated in DCM, these mutations ultimately result in a dilated heart and cardiac dysfunction. Mutations can induce different cellular changes depending on their effect on protein function. Therefore, patients could have different disease mechanisms leading to DCM. Our studies in human cardiac tissue of DCM patients showed reduced expression of the troponin complex in samples harbouring sarcomeric mutations in TNNI3p.98trunc and TNNT2p.K217del. In the TNNI3p.98trunc sample we did not find a truncated protein, and the haploinsufficiency led to increased Ca2+‐sensitivity and reduced LDA. The TNNT2p.K217del mutation caused increased passive tension (F pass) and a non‐significant mild reduction in Ca2+‐sensitivity. Upon exchange with wild‐type (WT) troponin complex, all parameters normalized to control confirming the pathogenicity of these sarcomeric mutations. The non‐sarcomeric mutation LMNAp.R331Q showed decreased maximal force (F max) development and increased Ca2+‐sensitivity of sarcomeres, which were both attributed to secondary disease remodelling. Also idiopathic DCM (IDCM) samples showed increased myofilament Ca2+‐sensitivity and reduced LDA, which could be attributed to secondary disease remodelling. Therefore, mutations in genes encoding proteins of diverse functions can cause DCM, which implies that changes in different cellular pathways can lead to cardiac dilatation and dysfunction.
Methods
Ethical approval
Left ventricular (LV) tissue was obtained from DCM patients who underwent cardiac transplantation, two samples of patients who carried the LMNAp.R331Q mutation were derived from a biopsy taken prior to LV assist device (LVAD) implantation. The other LMNAp.R331Q sample was derived from a cardiac transplantation of a heart that had been supported by a LVAD prior to transplantation. Most DCM patient samples used in this study were acquired from the Biobank of the University Medical Centre Utrecht, the Netherlands. This study was approved by the Biobank Research Ethics Committee, University Medical Centre Utrecht, Utrecht, the Netherlands (protocol number WARB 12/387). Written informed consent was obtained. Samples were obtained from regions halfway between the atrioventricular valves and the apex. As control samples we used explanted LV heart tissue of healthy donors – people who had died from a non‐cardiac cause, typically motor vehicle accidents. These healthy donor samples and three DCM were acquired from the University of Sydney, with the ethical approval of the Human Research Ethics Committee no. 2012/2814. The control samples used were 3.160, 4.049, 6.042, 3.162, 5.128, 6.020, 7.044, 3.164, 3.141, 6.008, 5.086, 8.004, 7.054, and the DCM samples used were 4.036, 3.107 and 2.082. All samples were stored in liquid nitrogen or at −80ºC until use.
Cardiomyocyte force measurements
F max and F pass of sarcomeres were measured at pCa 4.5 and pCa 9.0, respectively, in single membrane‐permeabilized cardiomyocytes mechanically isolated from heart tissue as previously described (van Dijk et al. 2012). LDA experiments and PKA incubations were performed as previously described (van der Velden et al. 2000). Ca2+‐sensitivity was measured as the [Ca2+] needed to achieve 50% of F max (EC50) and LDA was measured as the shift in EC50 (ΔEC50) at a sarcomere length of 1.8 μm and 2.2 μm.
Protein expression and phosphorylation
Titin
Titin isoforms were separated on a 1% (w/v) agarose gel and stained with SYPRO Ruby protein stain (Invitrogen, Carlsbad, CA, USA) as described previously (Warren et al. 2003) and samples were measured in triplicate. Phosphorylation of titin was assessed as previously described (Kötter et al. 2016). For titin phosphorylation, site‐specific antibodies directed to Ser4010 (N2Bunique sequence (N2Bus) domain; PKA and extracellular signal‐regulated kinase 2 (ERK2) target), and Ser12022 and Ser11878 (PEVK domain; protein kinase C (PKC) and Ca2+/calmodulin‐dependent protein kinase II (CaMKIIδ) target) were used.
Troponin
The troponin proteins were separated by 12% polyacrylamide and 4–15% precast gradient gels (BioRAD, Hercules, CA, USA) gel electrophoresis and Western blots were stained with specific antibodies (cTnI: Abcam, Cambridge, UK, ab10231; cTnT: Sigma, St. Louis, MO, USA, T6277; cTnC: Santa Cruz, Dallas, TX, USA, sc48347) to determine their expression, which was corrected by expression of other cellular proteins (glyceraldehyde 3‐phosphate dehydrogenase (GAPDH): Cell signaling, 2118S, Cell signaling, Danvers, MA, USA; α‐actinin: Sigma, A7811). The TNNI3p98.trunc and TNNT2p.K217del samples were measured in duplicate of which the average is shown. Phosphorylation of cTnI was assessed as previously described (Zaremba et al. 2007).
Troponin exchange
The troponin complex was exchanged in membrane‐permeabilized cardiomyocytes as previously described (Wijnker et al. 2013). The recombinant WT or TNNT2p.K217del troponin complex was added to the cells in a concentration of 1 mg ml−1. The recombinant troponin complexes could be distinguished from highly phosphorylated endogenous troponin since the recombinant troponins were not phosphorylated. Quantification of the exchange rate was performed by phos‐tag analysis in which non‐, mono‐ and bis‐phosphorylated cTnI (Pierce, Rockford IL, USA, MA1‐22700) were separated by polyacrylamide‐bound Mn2+‐phos‐tag gel electrophoresis Western blotting as previously described (Najafi et al. 2015). The percentage of recombinant troponin complex present after exchange was quantified as the percentage of non‐phosphorylated cTnI to the total of non‐, mono‐ and bis‐phosphorylated cTnI levels. Total cTnI levels after exchange were quantified by cTnI (Abcam, ab10231) and corrected for myosin light chain‐2 (MLC2) (Enzo, Farmingdale, NY, USA, ALX‐BC‐1150).
Statistics
Graphpad Prism software was used for statistical analysis. F max of DCM cardiomyocytes compared to control cardiomyocytes was compared with one‐way ANOVA and Tukey's post hoc test. LDA was calculated as ΔEC50. Ca2+‐sensitivity and passive tension in DCM cardiomyocytes were compared to control cardiomyocytes by two‐way ANOVA. All values are shown as mean ± standard error of the mean. A P value <0.05 was considered to represent a significant difference and is indicated with an asterisk in figures. The 95% confidence intervals (CI) of the control group are indicated with a dotted line in the graphs and was used to assess the difference of the sample of interest compared to controls in situations where only a single data point of the sample of interest could be used.
Results
Diverse functional myofilament changes in DCM with sarcomeric and non‐sarcomeric mutations
The 12 control samples used for experiments consisted of four females and eight males with a mean age of 44.5 ± 3.4 years. The patient with TNNI3p.98trunc mutation was a 46‐year‐old male and the patient with TNNT2p.K217del mutation was a 19‐year‐old male. Three patients with LMNAp.R331Q were studied of which two were male and one female with a mean age of 45.3 ± 3.4 years. The IDCM samples consisted of four males and one female with a mean age of 54.6 ± 3.2 years. To determine the functional properties of human DCM samples with sarcomeric and non‐sarcomeric protein mutations, sarcomere function was measured in single isolated membrane‐permeabilized cardiomyocytes at various [Ca2+] to assess passive and active properties of the sarcomeres. No difference in F max (Fig. 2 A) was observed in IDCM, TNNI3p.98trunc and TNNT2p.K217del cardiomyocytes, while F max was significantly lower in cardiomyocytes with the LMNAp.R331Q mutation (17.9 kN m−2, data from Hoorntje et al. 2016) compared to controls (28.2 kN m−2). A previous study showed that this decreased F max was due to decreased myofibril density (Hoorntje et al. 2016). F pass is an important determinant of diastolic function which was measured at low [Ca2+] (pCa 9.0) over a range of sarcomere lengths (Granzier & Irving, 1995). IDCM samples (Fig. 2 B), TNNI3p.98trunc (Fig. 2 C) and LMNAp.R331Q (Fig. 2 E) showed a comparable F pass development over the range of sarcomere lengths compared to controls. The TNNT2p.K217del cardiomyocytes showed a significant increase in F pass compared to controls, which was most pronounced at longer sarcomere lengths (Fig. 2 D). In addition to changes in F pass, stretching of cardiomyocytes during filling of the heart increases active force development. This LDA of myofilaments is the cellular basis of the Frank–Starling mechanism (Sequeira et al. 2013; Beqqali et al. 2016). We measured active force development over a range of [Ca2+] at sarcomere lengths 1.8 μm and 2.2 μm to study LDA. IDCM samples showed an increased Ca2+‐sensitivity and reduced LDA compared to controls (Fig. 2 F). The TNNI3p.98trunc cardiomyocytes showed Ca2+‐sensitivity was increased and LDA was blunted (Fig. 2 G) compared to controls. TNNT2p.K217del cardiomyocytes showed only a minor and non‐significant decrease in Ca2+‐sensitivity and LDA was preserved (Fig. 2 H). The Ca2+‐sensitivity of LMNAp.R331Q cardiomyocytes was significantly increased compared to controls, while LDA was preserved (Fig. 2 I). Overall these data illustrate that changes in passive and active myofilament properties differ between different sarcomeric gene mutations and between sarcomeric and non‐sarcomeric mutations.
Figure 2. Baseline contractile properties.
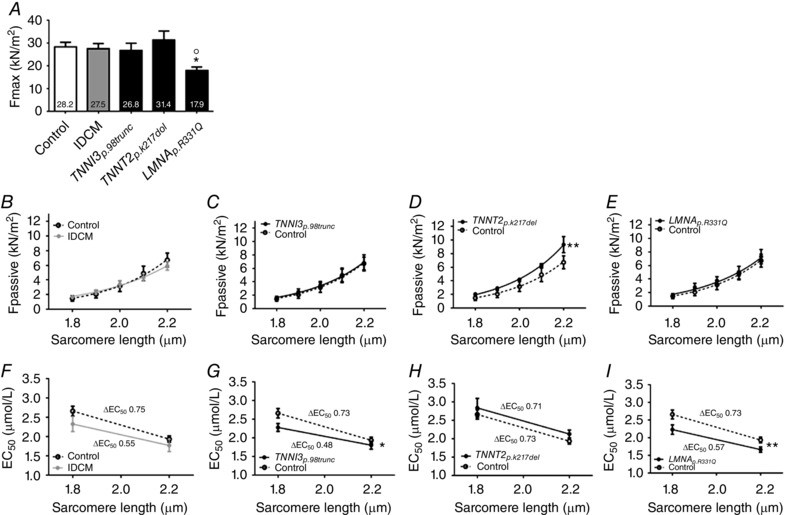
A, F max, measured at pCa 4.5, was significantly decreased (P < 0.05) in LMNAp.R331Q samples (17.9 ± 1.6 kN m−2, N = 3, n = 19) compared to controls (28.2 ± 2.1 kN m−2, N = 6, n = 21) while IDCM (27.5 ± 2.3 kN m−2, N = 5, n = 18), TNNI3p.98trunc (26.8 ± 3.2 kN m−2, N = 1, n = 13) and TNNT2p.K217del (31.4 ± 3.9 kN m−2, N = 1, n = 14) samples showed similar F max as controls. Data obtained from Hoorntje et al. (2016). B, F pass, measured at pCa 9.0, in membrane‐permeabilized cardiomyocytes of IDCM (N = 5, n = 19), were similar compared to control (N = 4, n = 10). C, F pass, measured at pCa 9.0, in membrane‐permeabilized cardiomyocytes of TNNI3p.98trunc sample (N = 1, n = 6), were similar compared to control (N = 4, n = 10). D, F pass, measured at pCa 9.0, in membrane‐permeabilized cardiomyocytes of TNNT2p.K217del patient (N = 1, n = 8), was significantly increased (P < 0.01) compared to control (N = 4, n = 10). E, F pass, measured at pCa 9.0, in LMNAp.R331Q samples (N = 3, n = 12), were similar compared to control (N = 4, n = 10). F, Ca2+‐sensitivity was non‐significantly increased and ΔEC50 was non‐significantly reduced in IDCM (N = 5, n = 11) compared to control (N = 6, n = 13). G, Ca2+‐sensitivity was significantly increased (P < 0.05) in TNNI3p.98trunc patient (N = 1, n = 7) compared to control (N = 6, n = 13), ΔEC50 was non‐significantly reduced. H, Ca2+‐sensitivity was only slightly and non‐significantly reduced compared to controls and ΔEC50 was preserved in TNNT2p.K217del sample (N = 1, n = 7) compared to control (N = 6, n = 13). I, Ca2+‐sensitivity was significantly increased (P < 0.01) in LMNAp.R331Q samples (N = 3, n = 7) compared to control (N = 6, n = 13) while ΔEC50 was preserved. N, number of samples; n, number of total cardiomyocytes measured.
Haploinsufficiency and altered stoichiometry of troponin proteins in DCM with TNNI3p.98trunc and TNNT2p.K217del mutations
Since the troponin complex and the interactions between the various troponin proteins are important for adequate contractile behaviour of the myofilaments, we studied the composition of the troponin complex in the TNNI3p.98trunc and TNNT2p.K217del samples. We observed that cTnI protein level was decreased in both the TNNI3p.98trunc and TNNT2p.K217del samples compared to controls when normalized to the cytoplasmic housekeeping protein GAPDH (Fig. 3 A and B). If the truncated cTnI protein is present in the TNNI3p.98trunc sample an antibody raised against the N‐terminus of cTnI is expected to show two bands: the native protein and the truncated protein. However, only one band was visible at the height of the native cTnI protein (Fig. 3 A). This indicates that the mutant protein is either not expressed or efficiently degraded resulting in cTnI haploinsufficiency. The level of cTnI relative to the sarcomeric housekeeping gene α‐actinin was also decreased in both TNNI3p.98trunc and TNNT2p.K217del samples (Fig. 3 C and D), which indicates that less cTnI is present within the sarcomeres itself. The reduction in cTnI levels was accompanied by a less pronounced decrease in cTnT (Fig. 3 E and F) and near normal cTnC levels (Fig. 3 G and H). This indicates that the TNNI3p.98trunc and TNNT2p.K217del samples have altered stoichiometry of the three troponin proteins since the decrease in cTnI, cTnT and cTnC is not to the same extent.
Figure 3. Expression of troponin in troponin mutants.
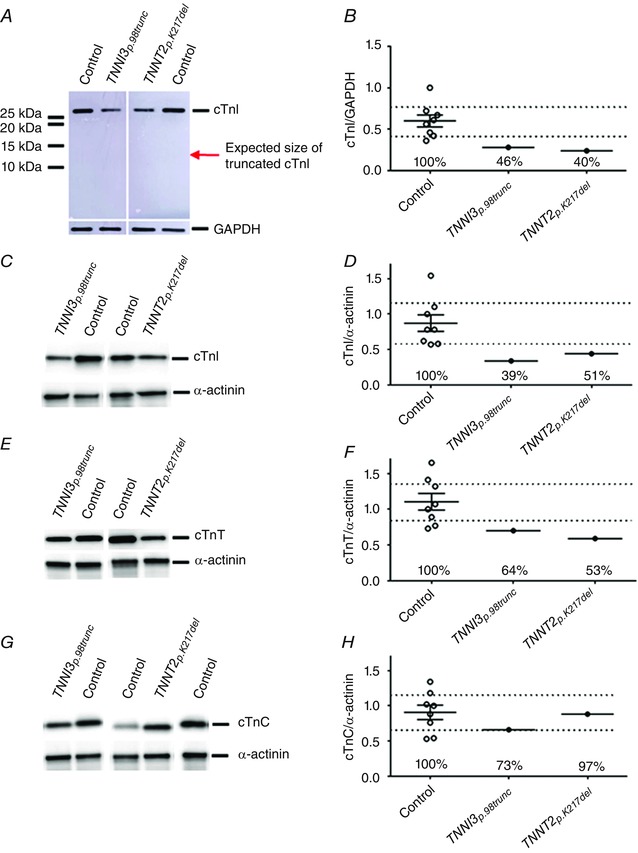
A and B, cTnI levels measured with an antibody directed to the N‐terminal of cTnI and normalized to GAPDH were decreased to 46% in the TNNI3p.98trunc (0.28) and to 40% in TNNT2p.K217del (0.24) samples compared to controls (N = 8, mean = 0.60, CI = 0.43–0.77). A, corresponding gel image showed no additional bands indicative of a truncated cTnI protein. C and D, cTnI levels were decreased to 39% in TNNI3p.98trunc (0.34) and to 51% in TNNT2p.K217del (0.44) samples compared to controls (N = 8, mean = 0.87, CI = 0.59–1.15) when normalized for α‐actinin. E and F, cTnT levels normalized to α‐actinin were also decreased to 64% in TNNI3p.98trunc (0.70) and to 53% in TNNT2p.K217del (0.59) samples compared to controls (N = 8, mean = 1.11, CI = 0.83–1.38). G and H, cTnC levels normalized to α‐actinin were slightly decreased to 73% in TNNI3p.98trunc sample (0.66) but still within the 95% CI of controls (N = 8, mean = 0.91, CI = 0.67–1.15). TNNT2p.K217del showed normal (0.88, 97% of controls) cTnC levels. [Color figure can be viewed at wileyonlinelibrary.com]
Hypophosphorylation of cTnI underlies increased Ca2+‐sensitivity in DCM with the LMNAp.R331Q mutation, but does not underlie myofilament defects in DCM with TNNI3p.98trunc and TNNT2p.K217del mutations
The troponin complex was reduced in both the TNNI3p.98trunc and TNNT2p.K217del samples, while cardiomyocytes from these samples showed different myofilament properties. We therefore studied phosphorylation of cTnI using phos‐tag analysis. Control samples showed prominent bis‐ and mono‐phosphorylated cTnI and low non‐phosphorylated cTnI (Fig. 4 A and B). IDCM samples showed reduced cTnI phosphorylation as is evident by prominent non‐ and mono‐phosphorylated cTnI bands and a weak bis‐phosphorylated cTnI band (Fig. 4 A and B). In the TNNI3p.98trunc and TNNT2p.K217del samples cTnI was highly phosphorylated indicated by an intense bis‐phosphorylated cTnI band and very weak non‐ and mono‐phosphorylated cTnI bands (Fig. 4 A and B). On the contrary, the LMNAp.R331Q samples showed decreased cTnI phosphorylation evident from prominent non‐ and mono‐phosphorylated cTnI bands (Fig. 4 A and B). Low phosphorylation of cTnI has been reported previously in DCM (Wijnker et al. 2014) and may underlie increased Ca2+‐sensitivity (Konhilas et al. 2003; Wijnker et al. 2014) and a blunted LDA (Konhilas et al. 2003; Wijnker et al. 2014). Indeed, after incubation with exogenous PKA we observed normalization of Ca2+‐sensitivity to controls in IDCM (Fig. 4 C) and in the LMNAp.R331Q cardiomyocytes (Fig. 4 D). Incubation with exogenous PKA did not change Ca2+‐sensitivity in controls. Also LDA was restored in IDCM samples compared to controls after incubation with exogenous PKA (Fig. 4 C). However, Ca2+‐sensitivity was still significantly increased compared to controls after incubation with exogenous PKA in the TNNI3p.98trunc cardiomyocytes (Fig. 4 E). These experiments confirm that impaired β‐adrenergic receptor signalling, and subsequent hypophosphorylation of cTnI, is the cause of the increased Ca2+‐sensitivity in IDCM and the LMNAp.R331Q cardiomyocytes, while the observed increased Ca2+‐sensitivity and impaired LDA is a direct mutation effect in the TNNI3p.98trunc cardiomyocytes.
Figure 4. Secondary disease remodelling and direct mutation effects.
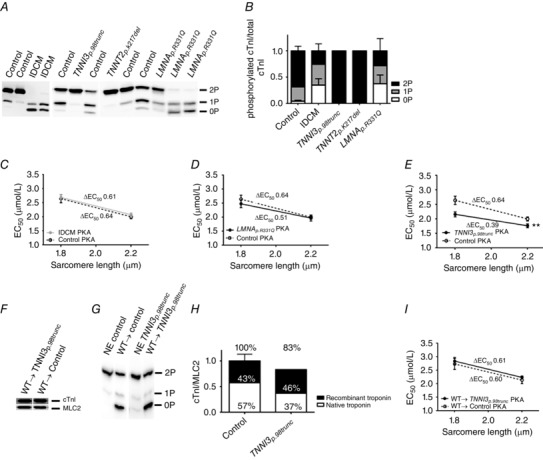
A, phos‐tag analysis showed separation of non‐ (0P), mono‐ (1P) and bis‐ (2P) phosphorylated cTnI. B, phosphorylation of cTnI was increased in TNNI3p.98trunc and TNNT2p.K217del samples compared to controls (N = 7) while cTnI phosphorylation in LMNAp.R331Q (N = 3) and IDCM (N = 3) was decreased compared to controls. C, Ca2+‐sensitivity was normalized in IDCM cardiomyocytes (N = 5, n = 12) compared to control cardiomyocytes (N = 6, n = 14) after incubation with exogenous PKA. D, Ca2+‐sensitivity was normalized in LMNAp.R331Q cardiomyocytes (N = 3, n = 7) compared to control cardiomyocytes (N = 6, n = 14) after incubation with exogenous PKA. E, after incubation with exogenous PKA, Ca2+‐sensitivity in TNNI3p.98trunc cardiomyocytes (N = 1, n = 7) remained significantly increased (P < 0.01) compared to control cardiomyocytes (N = 6, n = 14). F, exchange with WT troponin complex restored cTnI levels in the TNNI3p.98trunc sample to 83% of that of controls exchanged with WT troponin complex (H). G, phos‐tag gel analysis showed high phosphorylation of native troponin complex prior to exchange (NE) and incorporation of unphosphorylated recombinant protein after exchange. H, the 83% was composed of 46% recombinant troponin and 37% native troponin in the TNNI3p.98trunc sample compared with 43% recombinant troponin in the control exchanged with WT troponin complex. I, Ca2+‐sensitivity and LDA were restored in TNNI3p.98trunc cardiomyocytes (N = 1, n = 9) compared to control (N = 2, n = 11) after exchange with WT troponin complex and incubation with exogenous PKA. N, number of samples; n, number of total cardiomyocytes measured.
Correction of high Ca2+‐sensitivity and blunted LDA in DCM with TNNI3p.98trunc by human recombinant WT troponin
Next we aimed to assess whether the observed haploinsufficiency of cTnI, and reduced cTnT and cTnC, caused increased Ca2+‐sensitivity and reduced LDA in the TNNI3p.98trunc cardiomyocytes. We exchanged the endogenous troponin complex with the recombinant WT troponin complex in order to restore total troponin levels. The level of cTnI increased to 83% after exchange (Fig. 4 F and H), relative to the cTnI level in control cells exchanged with exogenous recombinant WT troponin complex. The 83% cTnI in the TNNI3p.98trunc sample after exchange consisted of 46% recombinant troponin complex and 37% native troponin complex, as determined by phos‐tag gel analysis (Fig. 4 G and H). In the control sample 43% of total cTnI levels was derived from the recombinant troponin complex (Fig. 4 G and H). This indicates that we exchanged endogenous troponin complex with recombinant WT complex, but also added additional recombinant WT troponin complex in the exchange process thereby largely overcoming the haploinsufficiency in the TNNI3p.98trunc cardiomyocytes. Since the recombinant troponin complex is unphosphorylated we incubated the exchanged cells with exogenous PKA prior to functional cell measurements. Upon exchange with the WT troponin complex both Ca2+‐sensitivity as well as LDA were normalized to control values in the TNNI3p.98trunc cardiomyocytes (Fig. 4 I).
High passive force in DCM with TNNT2p.K217del is caused by the mutation and not by changes in isoform composition or phosphorylation of titin
We next set out to determine the cause of the increased F pass in the TNNT2p.K217del sample. An important determinant of F pass is titin isoform composition (Makarenko et al. 2004; Nagueh et al. 2004). Titin can exist as a stiff isoform (N2B) or a larger, more compliant isoform (N2BA). All DCM groups showed an increase in compliant titin compared to controls independent of the type of mutation (Fig. 5 A and B). The observed increase in N2BA/N2B ratio cannot explain the high F pass in TNNT2p.K217del. Therefore, we examined phosphorylation of titin at three well‐established phosphorylation sites in the elastic I‐band region. Phosphorylation of Ser4010 on titin, a target of PKA, is known to decrease F pass (Kötter et al. 2013), while PKC‐mediated phosphorylation of Ser12022 and Ser11878 results in increased F pass (Hidalgo et al. 2009). While phosphorylation at Ser4010 was lower in the IDCM, TNNI3p.98trunc and LMNAp.R331Q samples compared to controls, a preserved or even slightly increased Ser4010 phosphorylation was observed in the TNNT2p.K217del sample (Fig. 5 C and D). Ser12022 (Fig. 5 E and F) and Ser11878 (Fig. 5 G and H) phosphorylation, which would increase F pass, was within the 95% CI of controls in IDCM but lower in samples carrying mutations compared to controls. Therefore, the increase in F pass in the TNNT2p.K217del cardiomyocytes is not caused by alterations in titin phosphorylation at the investigated sites. The increase in F pass was not due to impaired PKA‐mediated phosphorylation of titin since F pass remained significantly higher in TNNT2p.K217del cardiomyocytes compared to controls after incubation with exogenous PKA (Fig. 6 A). Exchange with the WT troponin complex led to a 59% incorporation of recombinant troponin complex in the TNNT2p.K217del sample (Fig. 6 B and C). The exchange normalized F pass in the TNNT2p.K217del cardiomyocytes to control level (Fig. 6 D). In addition, after exchange of the mutant TNNT2p.K217del troponin complex into a healthy control sample we observed that only 34% of total troponin present after exchange was recombinant (Fig. 6 B and C). However, this was sufficient to cause a significant increase in F pass (Fig. 6 D). The increase in F pass upon exchange with TNNT2p.K217del in a control sample was not due to impaired PKA‐mediated phosphorylation since after incubation with exogenous PKA F pass remained significantly increased (Fig. 6 E). These results indicate that the TNNT2p.K217del mutant protein itself increases F pass.
Figure 5. Alterations in titin isoform composition and phosphorylation in DCM mutants.
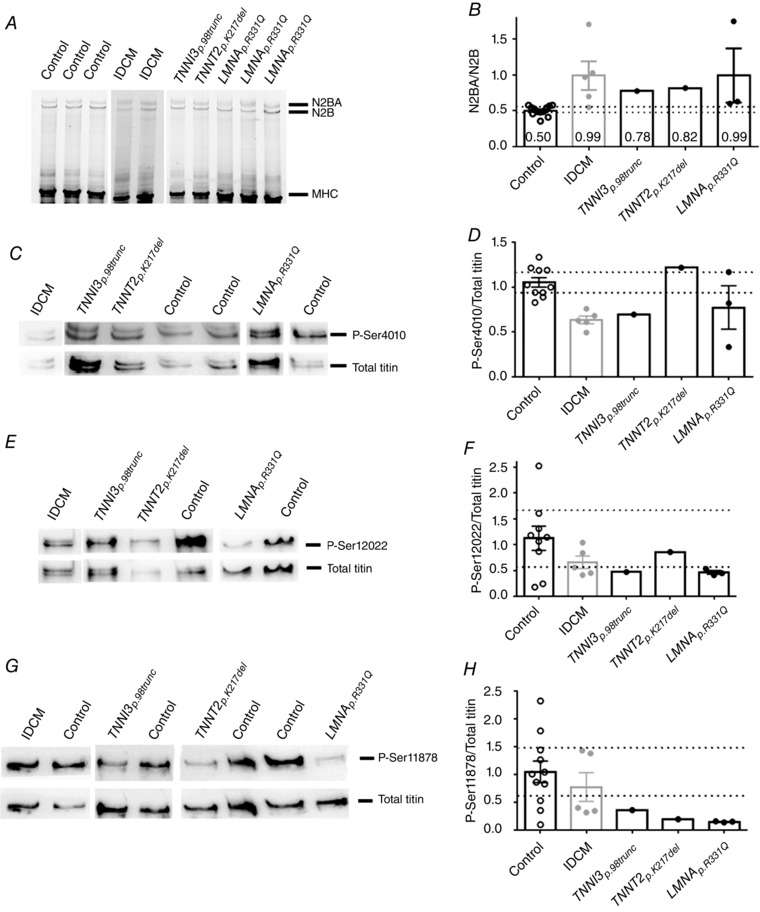
A, titin isoforms, N2BA and N2B, separated by agarose gel electrophoresis. B, titin N2BA/N2B ratios were increased in IDCM (0.99 ± 0.20, N = 5), TNNI3p.98trunc (0.78, N = 1), TNNT2p.K217del (0.82, N = 1) and LMNAp.R331Q (0.99 ± 0.38, N = 3) samples compared to controls (0.50 ± 0.02, N = 12, CI = 0.49–0.55). C, phosphorylated Ser4010 compared to total titin levels. D, titin phosphorylation at Ser4010 was decreased in IDCM (N = 5), TNNI3p.98trunc (N = 1) and LMNAp.R331Q (N = 3) compared to controls (N = 10, CI = 0.93–1.17), while TNNT2p.K217del (N = 1) showed slight increased phosphorylation of Ser4010 compared to control. E, phosphorylated Ser12022 compared to total titin levels. F, titin phosphorylation at Ser12022 was decreased in TNNI3p.98trunc (N = 1), TNNT2p.K217del (N = 1) and LMNAp.R331Q (N = 3), compared to control (N = 9, CI = 0.58–1.66), while phosphorylation at Ser12022 was within the 95% CI of controls in IDCM (N = 5). G, phosphorylated Ser11878 compared to total titin levels. H, titin phosphorylation at Ser11878 was decreased in TNNI3p.98trunc (N = 1), TNNT2p.K217del (N = 1) and LMNAp.R331Q (N = 3), compared to control (N = 11, CI = 0.62–1.49) while phosphorylation at Ser11878 was within the 95% CI of controls in IDCM (N = 5).
Figure 6. TNNT2p.K217del increases passive tension.
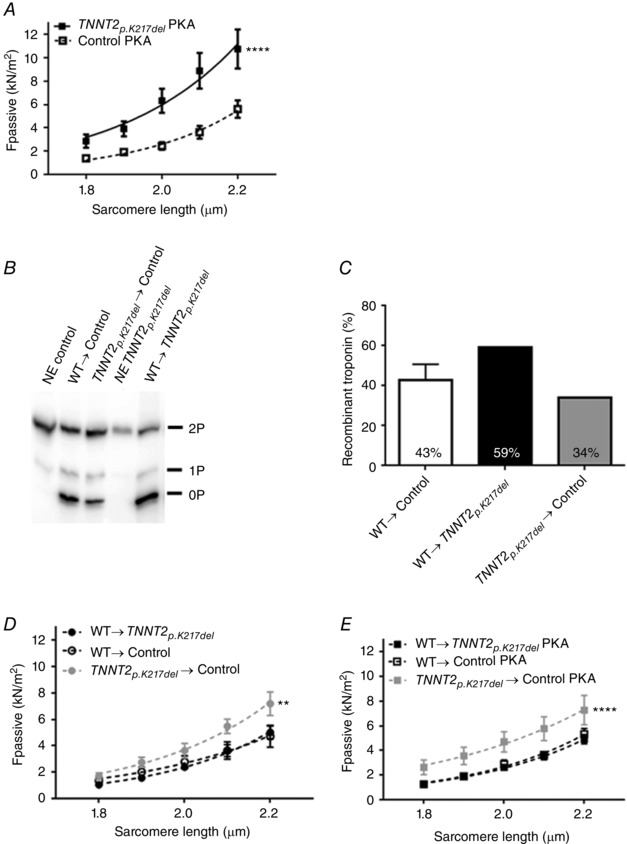
A, F pass remained significantly increased (P < 0.0001) in TNNT2p.K217del cardiomyocytes (N = 1, n = 9) compared to controls (N = 4, n = 13) after incubation with exogenous PKA. B, phos‐tag gel analysis showed high phosphorylation of native troponin complex prior to exchange (NE) and incorporation of unphosphorylated recombinant protein after exchange. C, after exchange 43% of present troponin complex in controls was recombinant WT cTnI while in the TNNT2p.K217del sample this was 59% and in control exchanged with TNNT2p.K217del mutant troponin complex this was 34%. D, upon exchange with WT troponin complex, cardiomyocytes of TNNT2p.K217del (N = 1, n = 11) showed restoration of F pass compared to controls exchanged with WT troponin complex (N = 2, n = 8) while F pass was significantly increased (P = 0.001) in control cardiomyocytes exchanged with mutant TNNT2p.K217del troponin complex (N = 2, n = 7). E, after incubation with exogenous PKA, cardiomyocytes of TNNT2p.K217del exchanged with recombinant WT troponin complex (N = 1, n = 13) showed normalization of F pass compared to control cardiomyocytes exchanged with WT troponin complex (N = 2, n = 9) while F pass was significantly increased (P < 0.0001) in control cardiomyocytes exchanged with mutant TNNT2p.K217del troponin complex (N = 2, n = 7). N, number of samples; n, number of total cardiomyocytes measured.
Discussion
Mutations in various sarcomeric and non‐sarcomeric genes can induce DCM. In this study we aimed to define the pathogenic effects of the sarcomeric TNNI3p98.trunc and TNNT2p.K217del mutations, and the non‐sarcomeric LMNAp.R331Q mutation. Our study provides proof that the two sarcomere mutations cause myofilament dysfunction, while changes in myofilament properties in IDCM and the non‐sarcomeric mutation samples are the result of secondary disease remodelling. One of the LMNAp.R331Q samples showed a smaller decrease in cTnI phosphorylation compared to the other two samples. This was the sample obtained from a patient who used a LVAD prior to transplantation. It is therefore possible that the LVAD has partly reversed the secondary remodelling (Sakamuri et al. 2016). However, this patient did not show deviations from the other two LMNAp.R331Q patients in other protein analyses.
Haploinsufficiency and altered stoichiometry of troponin proteins in human DCM
We show that the TNNI3p.98trunc sample does not lead to a truncated protein but causes haploinsufficiency. This is in line with Kostareva et al. who showed that a truncation in the TNNI3 gene at the 176th amino acid in a patient with restrictive cardiomyopathy did not lead to a truncated protein, but instead to a 50% reduction of cTnI (Kostareva et al. 2009). The decrease in cTnI in our patient was associated with decreased levels of cTnT, while cTnC levels remained near normal. Also in the TNNT2p.K217del sample reduced levels of cTnI and cTnT were observed albeit to a smaller extent than observed in the TNNI3p.98trunc sample. While cTnI was most reduced in both samples, this was accompanied by a less pronounced decrease in cTnT, while cTnC levels remained near normal leading to altered stoichiometry. Since cTnT itself can bind to the thin filaments through tropomyosin and serves as an anchor for the whole troponin complex, this might explain why we observed a smaller decrease in cTnT protein levels compared to cTnI, which is more dependent on the formation of the whole troponin complex to attach to the thin filaments. This is in line with a study by Feng et al. in which they showed that an expression level of 25% of cTnI was accompanied by a 53% decrease in cTnT (Feng et al. 2009).
A TNNI3 truncation mutation in human DCM causes haploinsufficiency, high Ca2+‐sensitivity and impaired LDA of myofilaments
Various mutations in TNNI3 have been shown to increase Ca2+‐sensitivity and subsequently impair cardiac relaxation leading to hypertrophic cardiomyopathy (HCM) (Takahashi‐Yanaga et al. 2001). These observations have been attributed to the possibility that the mutations act as a poison peptide and ‘lock’ tropomyosin in the C‐ or M‐state. They are suggested to increase the stability of the Ca2+‐bound form of the thin filaments or destabilize the Ca2+‐free form of the thin filaments (Kobayashi & Solaro, 2006). In this study, we show that the TNNI3p.98trunc mutation in DCM patient cardiomyocytes also increased Ca2+‐sensitivity and in addition impaired LDA, which could not be corrected with exogenous PKA (Figs 2 G and 4 E) while the increased Ca2+‐sensitivity and impaired LDA in IDCM could be corrected by exogenous PKA (Figs 2 F and 4 C). This is in line with Sequeira et al. who reported that the HCM‐causing TNNI3p.R145W mutation impaired LDA, which could not be rescued with exogenous PKA (Sequeira et al. 2013). However, it has been heavily debated if mutations in TNNI3 and TNNT2 are able to cause DCM or HCM through haploinsufficiency. Homozygous TNNT2 knock out (KO) mice are embryonically lethal, while heterozygous TNNT2 KO mice have reduced cTnT mRNA levels, but normal cTnT protein levels, and show no cardiac phenotype (Ahmad et al. 2008). The TNNT2 gene apparently has robust compensatory mechanisms in order to maintain protein levels. In addition, full TNNI3 KO in mice is lethal around 18 days of age (Liu et al. 2007; Feng et al. 2009), while heterozygous TNNI3 KO mice survive without detectable phenotype (Feng et al. 2009). Feng et al. suggested that a cTnI threshold of 25% WT protein exists for the mice to survive. They also showed that cTnI is likely to be produced in excess amounts under healthy conditions (Feng et al. 2009). TNNI3 KO mice were characterized by impaired diastolic function as an early cardiac phenotype, followed by enlarged cardiac dimensions and overt heart failure (Liu et al. 2007; Feng et al. 2009). In support of impaired diastolic dysfunction, an increased resting tension in isolated ventricular myocytes of TNNI3 KO mice has been found (Huang et al. 1999). However, we did not find any alterations in F pass in the TNNI3p.98trunc cardiomyocytes. In the early postnatal life of TNNI3 KO mice, slow skeletal TnI (ssTnI) production was maintained in order to compensate for the absence of cTnI (Huang et al. 1999; Liu et al. 2007; Feng et al. 2009). Although ssTnI was elevated in KO mice for a longer period than in WT littermates, it also decreased over time and the compensatory effect was gradually lost. Ca2+‐sensitivity decreased in TNNI3 KO mice along with the decrease of ssTnI. However, compared to WT littermates of the same age, the TNNI3 KO mice showed an increased Ca2+‐sensitivity (Huang et al. 1999). This is in line with the increased Ca2+‐sensitivity we observed in the TNNI3p.98trunc cardiomyocytes, and with the increased Ca2+‐sensitivity of ATPase activity in rabbit skeletal muscle upon extraction of TnI that has been reported previously (Shiraishi & Yamamoto, 1994). Using troponin exchange experiments in single human cardiomyocytes, we were able to increase cTnI levels close to control levels and normalize Ca2+‐sensitivity and LDA in the TNNI3p.98trunc cardiomyocytes. Our data prove that the increased Ca2+‐sensitivity and impaired LDA were directly caused by the mutation‐induced haploinsufficiency.
Secondary disease‐related changes in DCM with sarcomeric and non‐sarcomeric mutations
In line with previous reports in human DCM (Makarenko et al. 2004; Nagueh et al. 2004; Beqqali et al. 2016), all DCM patients showed an increase in compliant titin, indicated by a higher N2BA/N2B ratio compared to controls (Fig. 5 A and B). The increase in compliant titin therefore seems to be a general hallmark of DCM and not a specific effect of the mutations studied. Despite the increase in compliant titin, F pass was similar to controls in IDCM, the LMNAp.R331Q and TNNI3p.98trunc cardiomyocytes. Interestingly, PKA‐mediated phosphorylation titin was unaltered in the TNNT2p.K217del sample. In addition, cTnI phosphorylation was also not affected in the TNNT2p.K217del and TNNI3p.98trunc samples suggesting that these specific mutations do not lead to defects in β‐adrenergic receptor signalling. This is contrary to what we observed in the IDCM samples and to what has been reported in other DCM samples (Wijnker et al. 2014) and might indicate that mutations in troponin can impair phosphorylation through local signalling.
The TNNT2p.K217del mutation causes high passive stiffness in human cardiomyocytes
The TNNT2p.K217del cardiomyocytes showed increased F pass (Fig. 2 D). Since the troponin levels in the TNNI3p.98trunc and TNNT2p.K217del sample were reduced in a similar fashion we expect that the poison peptide of TNNT2p.K217del and not reduced troponin complex was the cause of the high F pass. We hypothesize that the mutant TNNT2p.K217del troponin complex is less likely to incorporate in the sarcomeres than the WT troponin complex. Also the observed decreased cTnT levels in TNNT2p.K217del might indicate the mutant protein is not as stable as healthy cTnT. We observed a low exchange rate of the mutant TNNT2p.K217del protein complex in a control sample (34%) and a high exchange rate of the WT troponin complex in the TNNT2p.K217del sample (59%) compared to the exchange rate of WT in a control sample (43%) (Fig. 6 C). Most models have high incorporation of the mutant with values reported to be 79% (Michael et al. 2016) and an estimated incorporation of ∼55% (Morimoto et al. 2002). The limited incorporation of the mutant cTnT in our study in combination with the decrease in total troponin levels we observed have important implications. The mutant protein levels are probably higher in exchange experiments in healthy tissue, knock in (KI) or transgenic mouse models than in human patients. In addition, total troponin levels might not be affected in these models while we show they can be decreased in human patient tissue. Therefore, cardiomyocytes of DCM patients with the TNNT2p.K217del mutation might have different contractile performance than reported in previously published animal models. In support of this, a transgenic mouse model of the TNNT2p.K217del mutation showed that the severity of DCM is related to the ratio of mutant vs. WT transcript (Ahmad et al. 2008). Inoue et al. also showed an increase in F pass in a mouse KI model of this mutation (Inoue et al. 2013). They indicated that part of the F pass increase was titin based, but they did not find an increase in N2B titin. Inoue et al. proposed that the increase in F pass might be due to increased PKC‐mediated phosphorylation of titin although they did not assess titin phosphorylation. We observed an increase in compliant titin and lower PKC‐mediated phosphorylation in the TNNT2p.K217del sample (Fig. 5), neither of which could explain the high F pass. Our troponin exchange experiments provided proof that the TNNT2p.K217del mutation itself causes a significant increase in F pass, irrespective of PKA‐mediated phosphorylation. To our knowledge we are the first to show that the TNNT2p.K217del mutation causes a profound increase in F pass in human heart tissue. The lysine at 217 is part of the H1 helix of cTnT which directly interacts with tropomyosin. The interaction between cTnT and tropomyosin might therefore be affected by the TNNT2p.K217del mutation. The TNNT2p.K217del might cause tropomyosin to be available for residual cross‐bridge interaction even at low calcium concentrations resulting in high F pass. We observed a mild, though non‐significant decrease in Ca2+‐sensitivity in TNNT2p.K217del cardiomyocytes compared to controls. The lysine at 217 in cTnT is believed to be involved in calcium‐sensitive cTnC binding (Tanokura et al. 1983). Decreased Ca2+‐sensitivity has been reported in various studies that exchanged human WT or TNNT2p.K217del in various animal models (Morimoto et al. 2002; Venkatraman et al. 2003; Michael et al. 2016), while another study showed no effect on Ca2+‐sensitivity (Bai et al. 2013). In addition, a KI mouse model (Du et al. 2007; Inoue et al. 2013; Memo et al. 2013) and a heterozygous KO mouse model with transgenic expression of TNNT2p.K217del also showed decreased Ca2+‐sensitivity (Ahmad et al. 2008). An impaired interaction of cTnT with cTnI and cTnC due to the TNNT2p.K217del mutation has been reported (Mogensen et al. 2004), while another study showed no significant difference in the secondary structure of TNNT2p.K217del measured as α‐helical content (Venkatraman et al. 2003). We hypothesize that we only observed a minor decrease in Ca2+‐sensitivity in the TNNT2p.K217del cardiomyocytes due to the reduced level of total troponin complex. In the TNNI3p.98trunc sample we observed an increased Ca2+‐sensitivity, which was corrected upon troponin exchange which increased troponin complex levels. Decreased troponin complex levels combined with the Ca2+‐desensitizing effect of the TNNT2p.K217del mutation might have counteracted each other leading to a negligible effect on myofilament Ca2+‐sensitivity. Reports about F max in TNNT2p.K217del mutants range from no effect (Morimoto et al. 2002; Du et al. 2007; Ahmad et al. 2008; Inoue et al. 2013; Michael et al. 2016) to a decrease (Venkatraman et al. 2003; Bai et al. 2013). In our study we did not find a decrease in F max in the TNNT2p.K217del sample (Fig. 2 A). Inoue et al. also showed a depressed Frank–Starling mechanism (Inoue et al. 2013) which we could not confirm in the human patient tissue and was also not observed in a transgenic mouse model of TNNT2p.K217del (Ahmad et al. 2008). A KI mouse model, transgenic mouse model, or exchange experiments might give rise to different levels of mutant protein in the sarcomeres and explain the different results on force‐generating capacity. Differences in the ability of the body to degrade the mutant protein and to compensate with the healthy allele might cause variable penetrance, age of onset and severity in human patients.
Conclusion
Mutations in different sarcomeric and non‐sarcomeric genes lead to DCM and we have shown that these mutations trigger different pathological routes leading to end‐stage dilated hearts (Fig. 7). In this study we show that the sarcomeric mutations TNNI3p.98trunc and TNNT2p.K217del cause reduced expression of the troponin complex and altered stoichiometry between the troponin subunits. In the TNNI3p.98trunc cardiomyocytes this led to increased Ca2+‐sensitivity, which could not be corrected with exogenous PKA but was normalized to control levels upon exchange with WT troponin complex. The TNNT2p.K217del mutation caused a mild, non‐significant, reduction in Ca2+‐sensitivity and significantly increased F pass, which could not be corrected by PKA but was normalized to control levels upon exchange with WT troponin complex. In addition, incorporation of the TNNT2p.K217del mutant troponin complex in a control sample confirmed the mutant protein itself causes increased F pass. This implies that even mutations in the genes encoding for the troponin proteins have different effects on myofilament function. In contrast, the LMNAp.R331Q mutation caused reduced maximal force development and increased Ca2+‐sensitivity due to secondary disease remodelling. Also IDCM samples showed an increased Ca2+‐sensitivity due to secondary disease remodelling. We show that although DCM patients present general hallmarks, the causative mutations underlie different cellular changes. Based on our studies, we propose that different mutations cause DCM via diverse pathways.
Figure 7. Overview of pathogenic effects of TNNI3p.98trunc, TNNT2p.K217del and LMNAp.R331Q .
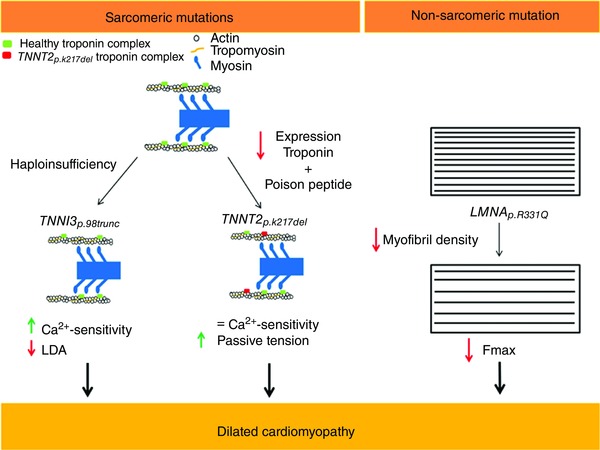
The TNNI3p.98trunc mutation did not result in a truncated protein and instead caused haploinsufficiency leading to increased Ca2+‐sensitivity and impaired LDA. The TNNT2p.K217del mutation might act as a poison peptide and caused decreased Ca2+‐sensitivity as shown by others. We showed that the sample with TNNT2p.K217del mutation resulted in decreased expression of the troponin proteins and in addition has a poison peptide effect. Since the decreased expression of the troponin proteins increased Ca2+‐sensitivity and the poison peptide decreased Ca2+‐sensitivity, there was no significant change in Ca2+‐sensitivity in the TNNT2p.K217del sample. In addition, F pass was increased. The LMNAp.R331Q mutation caused decreased myofibril density and subsequent impaired contractility.
Additional information
Competing interests
None declared.
Author contributions
I.A.E.B., D.W.D.K. and J.V.D.V. conceived, designed and coordinated the study and wrote the paper. I.A.E.B. created Figs 1 and 7 and performed and analysed the experiments shown in Figs 2, 4, 5 and 6. M.S. performed and analysed the experiments shown in Figs 4 and 6. M.H., A.V. and F.W.A. were involved in patient data and material acquisition. J.R.P. created recombinant protein complexes used in Figs 4 and 6. M.K. provided antibodies and supervision for experiments in Fig. 5. Experiments shown in Figs 2, 3, 4, 5A,B and 6 were performed at the Department of Physiology at the VU University Medical Center in Amsterdam, the Netherlands. Experiments shown in Fig 5 C‐H were performed at the Insititute for Cardiovascular Physiology at the Heinrich‐Heine University in Düsseldorf, Germany. All authors critically revised the manuscript, reviewed the results and approved the final version of the manuscript. All authors agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
We acknowledge the support from the Netherlands Cardiovascular Research Initiative, an initiative with the support of the Dutch Heart Foundation, CVON2011‐11 ARENA and CVON2014‐40 DOSIS, and Rembrandt Institute for Cardiovascular Sciences 2013. J.R.P. is supported by the National Heart, Lung and Blood Institute of the National Institutes of Health (Grant HL128683). F.W.A. is supported by a Dekker scholarship (Junior Staff Member 2014T001) of the Dutch Heart Foundation and UCL Hospitals NIHR Biomedical Research Centre.
Acknowledgements
We would like to thank Max Goebel, Ruud Zaremba, Wies Lommen and Sabine Bongardt for technical assistance. We thank Cristobal dos Remedios from the University of Sydney and the Sydney Heart Bank for the control samples used in this study. We would also like to thank Peter van Tintelen and Edgar Hoorntje for assistance with acquisition of patient data and material.
References
- Ahmad F, Banerjee SK, Lage ML, Huang XN, Smith SH, Saba S, Rager J, Conner DA, Janczewski AM, Tobita K, Tinney JP, Moskowitz IP, Perez‐Atayde AR, Keller BB, Mathier MA, Shroff SG, Seidman CE & Seidman JG (2008). The role of cardiac troponin T quantity and function in cardiac development and dilated cardiomyopathy. PLoS One 3, e2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai F, Caster HM, Pinto JR & Kawai M (2013). Analysis of the molecular pathogenesis of cardiomyopathy‐causing cTnT mutants I79N, DeltaE96, and DeltaK210. Biophys J 104, 1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beqqali A, Bollen IA, Rasmussen TB, van den Hoogenhof MM, van Deutekom HW, Schafer S, Haas J, Meder B, Sorensen KE, van Oort RJ, Mogensen J, Hubner N, Creemers EE, van der Velden J & Pinto YM (2016). A mutation in the glutamate‐rich region of RNA‐binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank‐Starling mechanism. Cardiovasc Res 112, 452–463. [DOI] [PubMed] [Google Scholar]
- Carballo S, Robinson P, Otway R, Fatkin D, Jongbloed JD, de Jonge N, Blair E, van Tintelen JP, Redwood C & Watkins H (2009). Identification and functional characterization of cardiac troponin I as a novel disease gene in autosomal dominant dilated cardiomyopathy. Circ Res 105, 375–382. [DOI] [PubMed] [Google Scholar]
- Du CK, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, Lu QW, Wang YY, Zhan DY, Mochizuki M, Kita S, Miwa Y, Takahashi‐Yanaga F, Iwamoto T, Ohtsuki I & Sasaguri T (2007). Knock‐in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res 101, 185–194. [DOI] [PubMed] [Google Scholar]
- Feng HZ, Hossain MM, Huang XP & Jin JP (2009). Myofilament incorporation determines the stoichiometry of troponin I in transgenic expression and the rescue of a null mutation. Arch Biochem Biophys 487, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangemi F & Degano M (2013). Disease‐associated mutations in the coil 2B domain of human lamin A/C affect structural properties that mediate dimerization and intermediate filament formation. J Struct Biol 181, 17–28. [DOI] [PubMed] [Google Scholar]
- Granzier HL & Irving TC (1995). Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68, 1027–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding S, Brown L, Wynne D, Davies C & Poole‐Wilson P (1994). Mechanisms of beta adrenoceptor desensitisation in the failing human heart. Cardiovasc Res 28, 1451–1460. [DOI] [PubMed] [Google Scholar]
- Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJR, Cook SA, Mestroni L, Seidman JG & Seidman CE (2012). Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger RE, Hedges DJ & Morales A (2013). Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10, 531–547. [DOI] [PubMed] [Google Scholar]
- Hershberger RE, Norton N, Morales A, Li D, Siegfried JD & Gonzalez‐Quintana J (2010). Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet 3, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S, Cowan J, Morales A, Parvatiyar MS & Potter JD (2009). Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2, 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S & Granzier H (2009). PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res 105, 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoorntje ET, Bollen IAE, van Tienen FHJ, Vink A, van den Wijngaard A, van der Velden J, Jongbloed JDH, van den Berg MP & van Tintelen JP (2016). LMNA related cardiac disease: a late onset phenotype in a large cohort of patients with a lmna r331q mutation. Eur J Clin Invest 46, 18. [Google Scholar]
- Huang X, Pi Y, Lee KJ, Henkel AS, Gregg RG, Powers PA & Walker JW (1999). Cardiac troponin I gene knockout: a mouse model of myocardial troponin I deficiency. Circ Res 84, 1–8. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kobirumaki‐Shimozawa F, Kagemoto T, Fujii T, Terui T, Kusakari Y, Hongo K, Morimoto S, Ohtsuki I, Hashimoto K & Fukuda N (2013). Depressed Frank–Starling mechanism in the left ventricular muscle of the knock‐in mouse model of dilated cardiomyopathy with troponin T deletion mutation ΔK210. J Mol Cell Cardiol 63, 69–78. [DOI] [PubMed] [Google Scholar]
- Kamisago M, Sharma SD, ePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG & Seidman CE (2000). Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 343, 1688–1696. [DOI] [PubMed] [Google Scholar]
- Kobayashi T & Solaro RJ (2006). Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy‐related mutant cardiac troponin I. J Biol Chem 281, 13471–13477. [DOI] [PubMed] [Google Scholar]
- Konhilas JP, Irving TC, Wolska BM, Jweied EE, Martin AF, Solaro RJ & de Tombe PP (2003). Troponin I in the murine myocardium: influence on length‐dependent activation and interfilament spacing. J Physiol 547, 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostareva A, Gudkova A, Sjöberg G, Mörner S, Semernin E, Krutikov A, Shlyakhto E & Sejersen T (2009). Deletion in TNNI3 gene is associated with restrictive cardiomyopathy. Int J Cardiol 131, 410–412. [DOI] [PubMed] [Google Scholar]
- Kötter S, Gout L, Von Frieling‐Salewsky M, Muller AE, Helling S, Marcus K, Dos Remedios C, Linke WA & Kruger M (2013). Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res 99, 648–656. [DOI] [PubMed] [Google Scholar]
- Kötter S, Kazmierowska M, Andresen C, Bottermann K, Grandoch M, Gorressen S, Heinen A, Moll JM, Scheller J, Gödecke A, Fischer JW, Schmitt JP & Krüger M (2016). Titin‐based cardiac myocyte stiffening contributes to early adaptive ventricular remodeling after myocardial infarction. Circ Res 119, 1017–1029. [DOI] [PubMed] [Google Scholar]
- Li Y, Mui S, Brown JH, Strand J, Reshetnikova L, Tobacman LS & Cohen C (2002). The crystal structure of the C‐terminal fragment of striated‐muscle α‐tropomyosin reveals a key troponin T recognition site. Proc Natl Acad Sci USA 99, 7378–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Du J, Zhang C, Walker JW & Huang X (2007). Progressive troponin I loss impairs cardiac relaxation and causes heart failure in mice. Am J Physiol Heart Circ Physiol 293, H1273–H1281. [DOI] [PubMed] [Google Scholar]
- McKillop DFA & Geeves MA (1993). Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J 65, 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ & Linke WA (2004). Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res 95, 708–716. [DOI] [PubMed] [Google Scholar]
- Memo M, Leung MC, Ward DG, dos Remedios C, Morimoto S, Zhang L, Ravenscroft G, McNamara E, Nowak KJ, Marston SB & Messer AE (2013). Familial dilated cardiomyopathy mutations uncouple troponin I phosphorylation from changes in myofibrillar Ca2+ sensitivity. Cardiovasc Res 99, 65–73. [DOI] [PubMed] [Google Scholar]
- Michael JJ, Gollapudi SK & Chandra M (2016). Interplay between the effects of a protein kinase C phosphomimic (T204E) and a dilated cardiomyopathy mutation (K211Δ or R206W) in rat cardiac troponin T blunts the magnitude of muscle length‐mediated crossbridge recruitment against the β‐myosin heavy chain background. J Muscle Res Cell Motil 37, 83–93. [DOI] [PubMed] [Google Scholar]
- Mogensen J, Hey T & Lambrecht S (2015). A systematic review of phenotypic features associated with cardiac troponin I mutations in hereditary cardiomyopathies. Can J Cardiol 31, 1377–1385. [DOI] [PubMed] [Google Scholar]
- Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM & McKenna WJ (2004). Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 44, 2033–2040. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Lu QW, Harada K, Takahashi‐Yanaga F, Minakami R, Ohta M, Sasaguri T & Ohtsuki I (2002). Ca2+‐desensitizing effect of a deletion mutation ΔK210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc Natl Acad Sci USA 99, 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagueh SF, Shah G, Wu Y, Torre‐Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S & Granzier HL (2004). Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 110, 155–162. [DOI] [PubMed] [Google Scholar]
- Najafi A, Schlossarek S, van Deel ED, van den Heuvel N, Guclu A, Goebel M, Kuster DW, Carrier L & van der Velden J (2015). Sexual dimorphic response to exercise in hypertrophic cardiomyopathy‐associated MYBPC3‐targeted knock‐in mice. Pflugers Arch 467, 1303–1317. [DOI] [PubMed] [Google Scholar]
- Otten E, dit Deprez L, Weis MM, Van Slegtenhorst M, Joosten M, Van der Smagt JJ, de Jonge N, Kerstjens‐Frederiksten WS, Roofthooft MTR, Balk AHMM, van den Berg MM, Ruiter JS & Van Tintelen JP (2010). Recurrent and founder mutations in the Netherlands: mutation p.K217del in troponin T2, causing dilated cardiomyopathy. Neth Heart J 18, 478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS & Hershberger RE (2008). Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J 156, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot A, Hussein S, Ruppert V, Schmidt HH, Wehnert MS, Duong NT, Posch MG, Panek A, Dietz R, Kindermann I, Bohm M, Michalewska‐Wludarczyk A, Richter A, Maisch B, Pankuweit S & Ozcelik C (2009). Identification of mutational hot spots in LMNA encoding lamin A/C in patients with familial dilated cardiomyopathy. Basic Res Cardiol 104, 90–99. [DOI] [PubMed] [Google Scholar]
- Sakamuri SS, Takawale A, Basu R, Fedak PW, Freed D, Sergi C, Oudit GY & Kassiri Z (2016). Differential impact of mechanical unloading on structural and nonstructural components of the extracellular matrix in advanced human heart failure. Transl Res 172, 30–44. [DOI] [PubMed] [Google Scholar]
- Sequeira V, Wijnker PJ, Nijenkamp LL, Kuster DW, Najafi A, Witjas‐Paalberends ER, Regan JA, Boontje N, Ten Cate FJ, Germans T, Carrier L, Sadayappan S, van Slegtenhorst MA, Zaremba R, Foster DB, Murphy AM, Poggesi C, Dos Remedios C, Stienen GJ, Ho CY, Michels M & van der Velden J (2013). Perturbed length‐dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ Res 112, 1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi F & Yamamoto K (1994). The effect of partial removal of troponin I and troponin C on the Ca2+ sensitive ATPase activity of rabbit skeletal myofibrils. J Biochem 115, 171–173. [DOI] [PubMed] [Google Scholar]
- Spyracopoulos L, Li MX, Sia SK, Gagne SM, Chandra M, Solaro RJ & Sykes BD (1997). Calcium‐induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry 36, 12138–12146. [DOI] [PubMed] [Google Scholar]
- Stone DB, Timmins BA, Schneider DK, Krylova I, Ramos CHI, Reinach FC & Mendelson RA (1998). The effect of regulatory Ca2+ on the in situ structures of troponin C and troponin I: a neutron scattering study. J Mol Biol 281, 689–704. [DOI] [PubMed] [Google Scholar]
- Takahashi‐Yanaga F, Morimoto S, Harada K, Minakami R, Shiraishi F, Ohta M, Lu QW, Sasaguri T & Ohtsuki I (2001). Functional consequences of the mutations in human cardiac troponin I gene found in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 33, 2095–2107. [DOI] [PubMed] [Google Scholar]
- Tanokura M, Tawada Y, Ono A & Ohtsuki I (1983). Chymotryptic subfragments of troponin T from rabbit skeletal muscle. Interaction with tropomyosin, troponin I and troponin C. J Biochem 93, 331–337. [DOI] [PubMed] [Google Scholar]
- van der Velden J, de Jong J, Owen VJ, Burton PB & Stienen GJ (2000). Effect of protein kinase A on calcium sensitivity of force and its sarcomere length dependence in human cardiomyocytes. Cardiovasc Res 46, 487–495. [DOI] [PubMed] [Google Scholar]
- van Dijk SJ, Paalberends ER, Najafi A, Michels M, Sadayappan S, Carrier L, Boontje NM, Kuster DW, van Slegtenhorst M, Dooijes D, dos Remedios C, ten Cate FJ, Stienen GJ & van der Velden J (2012). Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ Heart Fail 5, 36–46. [DOI] [PubMed] [Google Scholar]
- van Spaendonck‐Zwarts KY, van Rijsingen IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM, Asselbergs FW, Christiaans I, van Langen IM, Wilde AA, de Boer RA, Jongbloed JD, Pinto YM & van Tintelen JP (2013). Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years’ experience. Eur J Heart Fail 15, 628–636. [DOI] [PubMed] [Google Scholar]
- Venkatraman G, Harada K, Gomes AV, Kerrick WG & Potter JD (2003). Different functional properties of troponin T mutants that cause dilated cardiomyopathy. J Biol Chem 278, 41670–41676. [DOI] [PubMed] [Google Scholar]
- Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, Exome Aggregation Consortium , MacArthur DG, Farrall M, Cook SA & Watkins H (2017). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 19, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CM, Krzesinski PR & Greaser ML (2003). Vertical agarose gel electrophoresis and electroblotting of high‐molecular‐weight proteins. Electrophoresis 24, 1695–1702. [DOI] [PubMed] [Google Scholar]
- Westfall MV, Albayya FP & Metzger JM (1999). Functional analysis of troponin I regulatory domains in the intact myofilament of adult single cardiac myocytes. J Biol Chem 274, 22508–22516. [DOI] [PubMed] [Google Scholar]
- Wijnker PJ, Foster DB, Tsao AL, Frazier AH, dos Remedios CG, Murphy AM, Stienen GJ & van der Velden J (2013). Impact of site‐specific phosphorylation of protein kinase A sites Ser23 and Ser24 of cardiac troponin I in human cardiomyocytes. Am J Physiol Heart Circ Physiol 304, H260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnker PJM, Sequeira V, Foster DB, Li Y, dos Remedios CG, Murphy AM, Stienen GJM & van der Velden J (2014). Length‐dependent activation is modulated by cardiac troponin I bisphosphorylation at Ser23 and Ser24 but not by Thr143 phosphorylation. Am J Physiol Heart Circ Physiol 306, H1171–H1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba R, Merkus D, Hamdani N, Lamers JM, Paulus WJ, Dos Remedios C, Duncker DJ, Stienen GJ & van der Velden J (2007). Quantitative analysis of myofilament protein phosphorylation in small cardiac biopsies. Proteomics Clin Appl 1, 1285–1290. [DOI] [PubMed] [Google Scholar]