

Abstract
Key points
The catechol metabolites of 17β‐oestradiol (E2β), 2‐hydroxyoestradiol (2‐OHE2) and 4‐hydroxyoestradiol (4‐OHE2), stimulate proliferation of pregnancy‐derived ovine uterine artery endothelial cells (P‐UAECs) through β‐adrenoceptors (β‐ARs) and independently of the classic oestrogen receptors (ERs).
Herein we show that activation of ERK1/2, p38 and JNK mitogen activated protein kinases (MAPKs) is necessary for 2‐OHE2‐ and 4‐OHE2‐induced P‐UAEC proliferation, as well as proliferation induced by the parent hormone E2β and other β‐AR signalling hormones (i.e. catecholamines).
Conversely, although 2‐OHE2 and 4‐OHE2 rapidly activate phosphatidylinositol 3‐kinase (PI3K), its activation is not involved in catecholoestradiol‐induced P‐UAEC proliferation.
We also show for the first time the signalling mechanisms involved in catecholoestradiol‐induced P‐UAEC proliferation; which converge at the level of MAPKs with the signalling mechanisms mediating E2β‐ and catecholamine‐induced proliferation.
The present study advances our understanding of the complex signalling mechanisms involved in regulating uterine endothelial cell proliferation during pregnancy.
Abstract
Previously we demonstrated that the biologically active metabolites of 17β‐oestradiol, 2‐hydroxyoestradiol (2‐OHE2) and 4‐hydroxyoestradiol (4‐OHE2), stimulate pregnancy‐specific proliferation of uterine artery endothelial cells derived from pregnant (P‐UAECs), but not non‐pregnant ewes. However, unlike 17β‐oestradiol, which induces proliferation via oestrogen receptor‐β (ER‐β), the catecholoestradiols mediate P‐UAEC proliferation via β‐adrenoceptors (β‐AR) and independently of classic oestrogen receptors. Herein, we aim to further elucidate the signalling mechanisms involved in proliferation induced by catecholoestradiols in P‐UAECs. P‐UAECs were treated with 2‐OHE2 and 4‐OHE2 for 0, 0.25, 0.5, 1, 2, 4, 12 and 24 h, to analyse activation of mitogen activated protein kinases (MAPKs) and phosphatidylinositol 3‐kinase (PI3K)–AKT. Specific inhibitors for ERK1/2 MAPK (PD98059), p38 MAPK (SB203580), JNK MAPK (SP600125), or PI3K (LY294002) were used to determine the involvement of individual kinases in agonist‐induced P‐UAEC proliferation. 2‐OHE2 and 4‐OHE2 stimulated biphasic phosphorylation of ERK1/2, slow p38 and JNK phosphorylation over time, and rapid monophasic AKT phosphorylation. Furthermore, ERK1/2, p38 and JNK MAPKs, but not PI3K, were individually necessary for catecholoestradiol‐induced proliferation. In addition, when comparing the signalling mechanisms of the catecholoestradiols, to 17β‐oestradiol and catecholamines, we observed that convergent MAPKs signalling pathways facilitate P‐UAEC proliferation induced by all of these hormones. Thus, all three members of the MAPK family mediate the mitogenic effects of catecholoestradiols in the endothelium during pregnancy. Furthermore, the convergent signalling of MAPKs involved in catecholoestradiol‐, 17β‐oestradiol‐ and catecholamine‐induced endothelial cell proliferation may be indicative of unappreciated evolutionary functional redundancy to facilitate angiogenesis and ensure maintenance of uterine blood flow during pregnancy.
Keywords: endothelial cell, MAPKs, oestrogens
Key points
The catechol metabolites of 17β‐oestradiol (E2β), 2‐hydroxyoestradiol (2‐OHE2) and 4‐hydroxyoestradiol (4‐OHE2), stimulate proliferation of pregnancy‐derived ovine uterine artery endothelial cells (P‐UAECs) through β‐adrenoceptors (β‐ARs) and independently of the classic oestrogen receptors (ERs).
Herein we show that activation of ERK1/2, p38 and JNK mitogen activated protein kinases (MAPKs) is necessary for 2‐OHE2‐ and 4‐OHE2‐induced P‐UAEC proliferation, as well as proliferation induced by the parent hormone E2β and other β‐AR signalling hormones (i.e. catecholamines).
Conversely, although 2‐OHE2 and 4‐OHE2 rapidly activate phosphatidylinositol 3‐kinase (PI3K), its activation is not involved in catecholoestradiol‐induced P‐UAEC proliferation.
We also show for the first time the signalling mechanisms involved in catecholoestradiol‐induced P‐UAEC proliferation; which converge at the level of MAPKs with the signalling mechanisms mediating E2β‐ and catecholamine‐induced proliferation.
The present study advances our understanding of the complex signalling mechanisms involved in regulating uterine endothelial cell proliferation during pregnancy.
Abbreviations
- 2‐OHE2
2‐hydroxyoestradiol
- 4‐OHE2
4‐hydroxyoestradiol
- β‐AR
β‐adrenoceptor
- ADR
adrenaline
- E2β
oestradiol
- EBM
endothelial basal media
- ERK1/2
extracellular signal‐regulated kinase 1 and 2
- ER‐β
oestrogen receptor β
- FBS
fetal bovine serum
- JNK
c‐Jun NH2‐terminal kinase
- LY
LY294002
- MAPK
mitogen activated protein kinase
- NA
noradrenaline
- PD
PD98059
- PI3K
phosphatidylinositol 3‐kinase
- P‐UAECs
pregnancy‐derived uterine artery endothelial cells
- SB
SB203580
- SP
SP600125
- VEGF
vascular endothelial growth factor
Introduction
Elevated circulating levels of oestrogens during pregnancy play a crucial role in regulating cardiovascular adaptations that facilitate substantial rises in utero‐placental blood flow (Rosenfeld et al. 1976; Magness, 1998; Magness et al. 2005; Osol & Mandala, 2009; Sprague et al. 2009; Magness & Ford, 2014). Oestrogen metabolites are also important regulators of cardiovascular adaptations to pregnancy, such as vasodilatation and angiogenesis, (Dubey et al. 2004; Jobe et al. 2010, 2011, 2013a,b) and thus their aberrant regulation is likely to be implicated in the development of pregnancy disorders such as preeclampsia (Kanasaki et al. 2008; Jobe et al. 2013b). We recently showed that aberrant synthesis, metabolism and accumulation of 17β‐oestradiol (E2β) and its bioactive metabolites 2‐hydroxyoestradiol (2‐OHE2) and 4‐hydroxyoestradiol (4‐OHE2) is associated with pregnancies complicated by preeclampsia (Jobe et al. 2013b), where abnormally low utero‐placental blood flow is observed (Lunell et al. 1982; Palmer et al. 1999). Moreover, Barker and others have shown that insults during fetal development such as insufficient utero‐placental blood flow can result in fetal programming of adult‐onset diseases like hypertension and cardiovascular disease (Barker et al. 2002; Barker, 2007; Jansson & Powell, 2007). Thus, understanding the oestrogen‐regulated mechanisms controlling uterine blood flow during pregnancy is of great clinical importance.
Oestrogens are known to play an important role in endothelial cell proliferation and angiogenesis (Morales et al. 1995; Losordo & Isner, 2001; Jobe et al. 2010, 2011), which is a major component in the regulation of uterine blood flow during pregnancy (Magness, 1998; Pastore et al. 2012; Magness & Ford, 2014). We previously reported that 2‐OHE2 and 4‐OHE2 play a role in uterine angiogenesis in a pregnancy‐specific manner. Specifically, both 2‐OHE2 and 4‐OHE2 stimulate proliferation of uterine artery endothelial cells (UAECs) derived from pregnant (P‐), but not from non‐pregnant ewes (Jobe et al. 2010). In addition, unlike the parent substrate, 17β‐oestradiol, which induces P‐UAEC proliferation through an oestrogen receptor‐β (ERβ)‐dependent mechanism (Jobe et al. 2010), 2‐OHE2 and 4‐OHE2 induce P‐UAEC proliferation specifically via β‐adrenoceptors (β‐ARs) and independently of the classic oestrogen receptors (Jobe et al. 2011). These data indicate a yet‐unexplored signalling mechanism through which elevated levels of oestrogens regulate endothelial cell proliferation and angiogenesis during pregnancy. Consequently, in order to fully understand the role of oestrogens in the regulation of angiogenesis and uterine blood flow during pregnancy, it is important to elucidate the downstream signalling mechanisms involved in P‐UAEC proliferation induced by 2‐OHE2 and 4‐OHE2.
Regulation of cell proliferation is mediated via numerous signal transduction pathways, including the well‐characterized mitogen‐activated protein kinase (MAPK) and the phosphatidylinositol 3‐kinase (PI3K) pathways (Pedram et al. 1998; Zhang & Liu, 2002; Munoz‐Chapuli et al. 2004; Rose et al. 2010). Activation of extracellular signal‐regulated kinase 1 and 2 (ERK1/2), p38 MAPK, c‐Jun NH2‐terminal (JNK) MAPK, and PI3K have been shown to be involved in E2β‐induced endothelial cell proliferation (Geraldes et al. 2002; Liu et al. 2002; Sengupta et al. 2004; Marino et al. 2006; Fu et al. 2007; Parvathaneni et al. 2013). Similarly, activation of ERK1/2 (Iaccarino et al. 2005; Kim et al. 2008), or PI3K (Steinle et al. 2003; Iaccarino et al. 2005) have been shown to mediate endothelial cell proliferation induced via β‐AR signalling initiated by classic beta agonists (catecholamines). To date, no studies have evaluated the involvement of MAPKs or PI3K in catecholoestradiol‐stimulated endothelial cell proliferation; however, based on oestrogenic and β‐AR signalling, it is plausible that activation of ERK1/2, p38, JNK, or PI3K may mediate the post‐receptor signalling of the catecholoestradiols leading to P‐UAEC proliferation.
We hypothesize that post‐receptor activation of MAPKs or PI3K signalling pathways will be intimately involved in catecholoestradiol‐induced P‐UAEC proliferation. In the present study we examined: (1) the precise temporal kinase phosphorylation patterns of ERK1/2, p38, JNK, and PI3K signalling following exposure to 2‐OHE2 and 4‐OHE2; (2) the effect of ERK1/2, p38, JNK, or PI3K inhibitors on P‐UAEC proliferation to determine if activation of any or all of these kinases is necessary for catecholoestradiol‐induced P‐UAEC proliferation. In addition, similarities or differences amongst catecholoestradiols, their parent substrate (E2β), and other or β‐AR‐acting hormones were examined by performing the same experiments under E2β, noradrenaline, and adrenaline stimulation.
Methods
Materials
E2β, 2‐OHE2 and 4‐OHE2 were obtained from Steraloids Inc. (Newport, RI, USA). Noradrenaline and adrenaline were purchased from Sigma‐Aldrich (St Louis, MO, USA). BrdU cell proliferation assays were obtained from EMD Millipore (Bellerica, MA, USA). Click‐It EdU microplate proliferation assays were obtained from Life Technologies (Thermo Fisher, Carlsbad, CA, USA). Rabbit anti‐active MAPK (ERK1/2) was obtained from Promega (Fitchburg WI, USA). Rabbit anti‐total ERK1/2, rabbit anti‐phospho p38, rabbit anti‐total p38, rabbit anti‐total JNK, rabbit anti‐phospho AKT, rabbit anti‐total AKT, and rabbit anti‐β‐actin were obtained from Cell Signalling Technologies (Danvers, MA, USA). mouse anti‐phospho‐JNK, was obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Kinase inhibitors: PD98059 (ERK1/2), SB 203580 (p38), SP 600125 (JNK), and LY294002 (PI3K) were obtained from Tocris Bioscience (Bristol, UK).
Cell preparation and culture
All experiments herein were performed in vitro using primary cell lines previously isolated as described by Bird et al. (2000). Cells were then validated and cultured as described by Jobe et al. (2010). Briefly, uterine arteries were obtained from ewes of mixed Western and Polypay breeds. All procedures were approved by the University of Wisconsin‐Madison School of Medicine and Public Health Research Animal Care Committees for both the Medical School and the College of Agricultural and Life Sciences, following the recommendation from the American Veterinary Medicine Association Guidelines for Humane Treatment and Euthanasia of Laboratory Farm Animals. Ewes were naturally bred and pregnancies confirmed with ultrasound between gestational days 60 and 90. At gestational age 120–130 (term = 147 days), under sodium pentobarbital (50 mg min−1) anaesthesia, ewes were killed through bilateral thoracotomy and exsanguination by cardiac laceration. Then, both left and right uterine arteries from sheep (n = 4) were quickly and aseptically obtained during non‐survival surgery for endothelial cell isolations and further in vitro studies. Uterine artery endothelial cells (UAECs) were isolated from both uterine arteries by collagenase digestion and combined to generate an n value of one (n = 4). Cells were then cultured in growth media: endothelial basal media (EBM) with 20% fetal bovine serum (FBS), 100 mg ml−1 penicillin, and 100 mg ml−1 streptomycin) as described by Bird et al. (2000). Finally, cell validations were conducted on each cell preparation for PECAM‐1 and eNOS expression, acetylated LDL‐uptake and smooth muscle myosin expression (negative control) prior to long term storage and experiments as describe by Jobe et al. (2010). At passage 4 and ∼70% confluence, cells were transferred to 6‐well plates for Western blot analysis and 96 well plates for cell proliferation assays.
Experimental treatments
P‐UAECs were serum starved with endothelial basal media (EBM) for 24 h. Following starvation, fresh EBM media containing 0.1 nm of 2‐OhE2, 4‐OHE2, E2β, noradrenaline and adrenaline was added to replace starvation media. Treatment dose was chosen based on detailed dose and time‐course studies of P‐UAEC proliferation (Jobe et al. 2010, 2011). For Western blot analysis, cells were plated in 6‐well plates and allowed to reach 100% confluence. Cells were then serum starved for 24 h followed by treatment with 0.1 nm 2‐OHE2, 4‐OHE2, E2β, noradrenaline and adrenaline for 0, 0.25, 0.5, 1, 2, 4, 12, 24 h and proteins were collected for protein analysis and Western blotting. For kinase blockade studies, cells plated at about 60% density in 96‐well plates were allowed to attach overnight. Then complete growth media was removed, and cells were washed twice with sterile 1× phosphate buffered saline (PBS), followed by serum starvation with EBM for 24 h. Following starvation, ERK1/2, p38, JNK MAPKs and PI3K were inhibited by pre‐treating P‐UAECs for 1 h with 2.5 or 5 μm of the antagonists PD98059 (ERK1/2 MAPK), SB203580 (p38 MAPK), SP600125 (JNK MAPK), or LY294002 (PI3K), followed by treatment with vehicle or 0.1 nm of E2β, 2‐OHE2, 4‐OHE2, noradrenaline and adrenaline. A subset of wells was treated with only 0.1 nm E2β, 2‐OHE2, and 4‐OHE2, noradrenaline or adrenaline as positive controls. Cell proliferation assays were performed as described below.
Western blotting
Protein extraction was performed on UAECs with 400 μl of lysis buffer (0.004 m Sodium pyrophosphate, 0.05 m HEPES, 0.1 m NaCl, 0.01 m EDTA, 0.01 m Sodium Fluoride, 0.002 m Sodium Orthovanadate, 0.001 m PMSF, 0.1% Triton‐X, 5 μg ml‐1 Leupeptin, 5 μg ml‐1 aprotinin, 1 μg ml‐1 Microcystin). Total protein content was determined using BCA Protein Assay (Thermo Scientific, Rockford, IL, USA). For Western blotting, 20 μg protein per lane were boiled in SDS sample buffer for 5 min and electrophoresed on 4–20% gradient SDS‐PAGE gels (Bio‐Rad, Hercules, CA, USA, and Thermo Scientific, Waltham, MA, USA) for 100 min at 150 V. Separated proteins were then electrically (100 V, 30 min) transferred to a PVDF membrane. Non‐specific binding was blocked with 5% fat‐free milk in TBST (50 mm Tris‐HCl, pH 7.5, 0.15 m NaCl, 0.05% Tween‐20) for 120 min and incubated with primary antibodies (1 μg ml−1; 1:500) in TBST + 1% BSA overnight. Phosphorylated and total ERK1/2, p38, JNK and AKT proteins were detected using antibodies specified above. β‐Actin was utilized as a loading control. After washing, the membrane was incubated with the corresponding peroxidase‐conjugated IgG for 60 min and detected with the Pierce ECL detection kit (Thermo Scientific). Phosphorylated and total kinase expression were normalized to β‐actin, then normalized phosphorylated was divided by normalized total expression. Data are represented as fold change from time zero control.
Cell proliferation assays
In the present study, we measured newly synthesized DNA as an indication of cell proliferation. Given that cell cycle in mammalian cells can last about 30 h, and the S phase up to 10 h, depending on cell type (BNID:103742; Milo et al. 2010) we carried our experiments for 24 h to ensure acquisition of signal. All P‐UAEC cell proliferation experiments were performed in quadruplicate and replicated in at least four P‐UAEC preparations. After 4 h of agonist treatment, 5‐bromodeoxyuridine (BrdU) or 5‐ethynyl‐2′‐deoxyuridine (EdU) label was added for 16 h during the 24 h of hormone treatment as previously described (Jobe et al. 2010, 2011). BrdU and Click‐iT EdU proliferation assay kits were used according to manufacturer's instructions and internal controls were performed to demonstrate that both assays gave comparable results. BrdU proliferation assay kits were used for determining proliferation with 2‐OHE2, 4‐OHE2 and E2β. Due to a manufacturer issue with BrdU kits used to perform proliferation experiments with 2‐OHE2, 4‐OHE2 and E2β, we switched to using Click‐iT EdU proliferation assay kits with noradrenaline and adrenaline because it yielded less variability among replicates. Plates were read using Synergy HT Multi‐Mode Microplate Reader (BioTek, Winooski, VT, USA). Results were expressed as fold change over untreated control after subtracting blank values.
Statistical analysis
Data are expressed as means ± SEM. Analysis of stimulated phosphorylation at each time point within a treatment was performed using one‐way ANOVA followed by Student‐Newman Keuls post hoc test (n = 3 or n = 4). When normality of data sets was not achieved, non‐parametric analysis was performed using a Kruskal‐Wallis test; significance found with this test is indicated in the tables. Two‐way ANOVA with ‘concentration of blocker’ and ‘blocker effect’ as two ‘between’ factors was performed to determine the effect of kinase inhibitor in agonist‐induce proliferation (n = 4 cell lines run in quadruplicate; SigmaPlot 12.5). A level of significance was established a priori at P < 0.05.
Results
Effects of 2‐OHE2, 4‐OHE2, E2β, noradrenaline and adrenaline on MAPKs and PI3K phosphorylation patterns
The comprehensive patterns of ERK1/2 phosphorylation on P‐UAECs obtained with 2‐OHE2 and 4‐OHE2 treatments were similar to that induced by E2β (Fig. 1 and Table 1). We observed biphasic phosphorylation of ERK1/2 at 0.25 and 12 h that was significantly higher at the first peak compared to untreated controls in response to 0.1 nm 2‐OHE2 (7.64‐fold‐of‐control, P = 0.031; 4.91‐fold, P = 0.05, respectively), 4‐OHE2 (7.30‐fold, P = 0.0005; 6.87‐fold, P = 0.08, respectively), and E2β (6.28‐fold‐of‐control, P = 0.04; 4.82‐fold, P = 0.07, respectively). Conversely, noradrenaline and adrenaline both induced only monophasic phosphorylation of ERK1/2 at 0.25 h of treatment, which was significantly higher than untreated control (6.19‐fold, P = 0.01 and 5.25‐fold, P = 0.03, respectively). Expression of total ERK1/2 was not significantly changed in response to, 2‐OHE2, 4‐OHE2, E2β, noradrenaline, or adrenaline.
Figure 1. Time course responses of ERK1/2 MAPK phosphorylation.
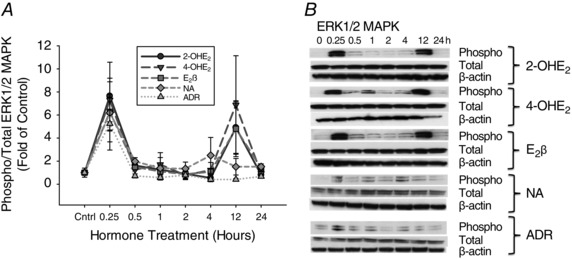
A, phosphorylation of ERK1/2 MAPK in P‐UAECs upon treatment with 2‐OHE2 (n = 4), 4‐OHE2 (n = 4), E2β (n = 4), noradrenaline (NA) (n = 3) and adrenaline (ADR) (n = 3) (all 0.1 nm) over the course of 24 h. Biphasic phosphorylation of ERK1/2 MAPK at 0.25 h and 12 h with 2‐OHE2 and 4‐OHE2, E2β treatment, and monophasic phosphorylation at 0.25 h, with NA, and ADR were observed respectively. Graphs represent the means ± SEM (Student‐Newman‐Keuls test, P < 0.05 was considered statistically significant). B, representative blots of phosphorylated (Phospho), total ERK1/2 MAPK and β‐actin.
Table 1.
Phosphorylation of ERK1/2 MAPK over time, shown as fold change from time zero
| Agonist | Stimulation time (h) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 0.25 | 0.5 | 1 | 2 | 4 | 12 | 24 | |
| 2‐OHE2 | 1.00 ± 0.02 | 7.64 ± 2.26* | 1.62 ± 0.37 | 1.25 ± 0.38 | 0.93 ± 0.38 | 0.53 ± 0.05τ | 4.91 ± 2.23 | 0.80 ± 0.04 |
| 4‐OHE2 | 1.00 ± 0.07 | 7.30 ± 1.89* | 1.25 ± 0.48 | 1.71 ± 1.02 | 0.78 ± 0.40 | 1.22 ± 0.64 | 6.87 ± 4.27 | 0.93 ± 0.21 |
| E2β | 1.00 ± 0.05 | 6.28 ± 2.60* | 1.46 ± 0.23 | 1.10 ± 0.02 | 0.86 ± 0.17 | 1.08 ± 0.27 | 4.82 ± 2.42 | 1.40 ± 0.23 |
| NA | 1.00 ± 0.10 | 6.19 ± 1.57* | 1.95 ± 0.92 | 1.38 ± 0.92 | 1.34 ± 0.57 | 2.04 ± 1.15 | 1.51 ± 0.71 | 1.57 ± 0.25 |
| ADR | 1.00 ± 0.11 | 5.25 ± 2.08* | 0.72 ± 0.17 | 0.57 ± 0.23 | 0.79 ± 0.33 | 0.41 ± 0.19τ | 0.41 ± 0.08τ | 0.67 ± 0.13τ |
*Increased phosphorylation, P < 0.05 vs. 0 h. τDecreased phosphorylation, P < 0.05 vs. 0 h; Student‐Newman‐Keuls test.
Detailed analysis of phosphorylated p38 in P‐UAECs in response to 2‐OHE2, 4‐OHE2, E2β, noradrenaline and adrenaline showed patterns of increased phosphorylation over time (Fig. 2 and Table 2). Maximum p38 phosphorylation was observed at 24 h with 2‐OHE2 (10.2‐fold, P = 0.07) and E2β (9.3‐fold, P = 0.008). While 4‐OHE2 (4.5‐fold, P = 0.01), noradrenaline (4.07‐fold, P = 0.04) and adrenaline (3.35‐fold, P = 0.06) induced maximum phosphorylation at 12 h of treatment. Total p38 MAPK levels were not changed in response to catecholoestradiols, E2β, or catecholamines.
Figure 2. Time course responses of p38 MAPK phosphorylation.
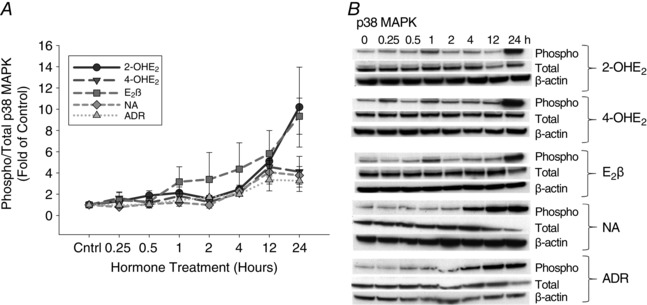
A, phosphorylation of p38 MAPK in P‐UAECs upon treatment with E2β, 2‐OHE2, 4‐OHE2, noradrenaline (NA) and adrenaline (ADR) (all 0.1 nm) over the course of 24 h. Phosphorylation of p38 increased gradually over time with 2‐OHE2, 4‐OHE2, E2β, NA and ADR. Graphs represent the means ± SEM (Student‐Newman‐Keuls test, P < 0.05 was considered statistically significant). B, representative blots of phosphorylated (Phospho), total ERK1/2 MAPK and β‐actin (n = 3).
Table 2.
Phosphorylation of p38 MAPK over time, shown as fold change from time zero
| Agonist | Stimulation time (h) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 0.25 | 0.5 | 1 | 2 | 4 | 12 | 24 | |
| 2‐OHE2 | 1.00 ± 0.05 | 1.33 ± 0.20 | 1.88 ± 0.45 | 2.13 ± 0.55 | 1.58 ± 0.32 | 2.46 ± 0.39 | 5.10 ± 0.82* | 10.2 ± 3.76 |
| 4‐OHE2 | 1.00 ± 0.01 | 1.63 ± 0.59 | 1.18 ± 0.31 | 1.83 ± 0.48 | 1.27 ± 0.13 | 2.09 ± 0.18 | 4.56 ± 0.78*,£ | 4.12 ± 1.44 |
| E2β | 1.00 ± 0.02 | 1.42 ± 0.73 | 1.29 ± 0.46 | 3.17 ± 1.42 | 3.40 ± 2.52 | 4.36 ± 2.47 | 5.81 ± 2.18 | 9.34 ± 1.70* |
| NA | 1.00 ± 0.07 | 0.79 ± 0.11 | 1.07 ± 0.32 | 1.21 ± 0.34 | 0.99 ± 0.30 | 2.30 ± 0.40 | 4.07 ± 1.10*,£ | 3.77 ± 0.83£ |
| ADR | 1.00 ± 0.13 | 0.97 ± 0.30 | 1.05 ± 0.35 | 1.44 ± 0.28* | 1.72 ± 0.15* | 2.00 ± 0.11* | 3.35 ± 1.04£ | 3.26 ± 1.01£ |
*Increased phosphorylation, P < 0.05 vs. 0 h; Student‐Newman‐Keuls test. £ P < 0.05 vs. 0 h; Kruskal‐Wallis one‐way ANOVA.
Overall, there was a modest increase in phosphorylated JNK expression in P‐UAECs over the course of 24 h with 2‐OHE2, 4‐OHE2, E2β, noradrenaline and adrenaline (Fig. 3 and Table 3). Highest phosphorylated JNK expression was observed at either 12 or 24 h of treatment (E2β 3.09‐fold, P = 0.07; 2‐OHE2 2.25‐fold, P = 0.09; 4‐OHE2 2.7‐fold P = 0.29; NA 3.7‐fold, P = 0.02; ADR 2.63‐fold, P = 0.009). Expression of total JNK was unchanged over the 24 h course of treatment.
Figure 3. Time course responses of JNK MAPK phosphorylation.
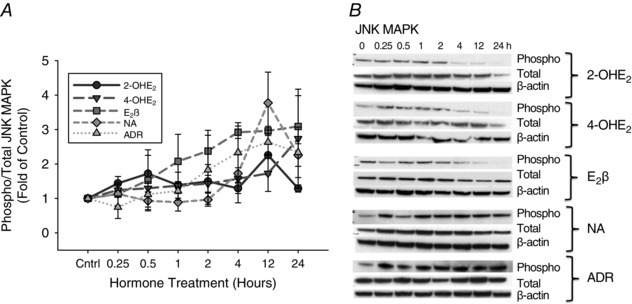
A, phosphorylation of JNK MAPK in P‐UAECs upon treatment with 2‐OHE2, 4‐OHE2, E2β, noradrenaline (NA) and adrenaline (ADR) (all 0.1 nm) over the course of 24 h. Phosphorylation of JNK increased over time with 2‐OHE2 and 4‐OHE2, E2β, NA and ADR. Graphs represent the means ± SEM (Student‐Newman‐Keuls test, P < 0.05 was considered statistically significant). B, representative blots of phosphorylated, total JNK and β‐actin (n = 3).
Table 3.
Phosphorylation of JNK MAPK over time, shown as fold change from time zero
| Agonist | Stimulation time (h) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 0.25 | 0.5 | 1 | 2 | 4 | 12 | 24 | |
| 2‐OHE2 | 1.00 ± 0.04 | 1.44 ± 0.20 | 1.72 ± 0.53 | 1.39 ± 0.38 | 1.49 ± 0.49 | 1.29 ± 0.15 | 2.25 ± 0.57 | 1.29 ± 0.10* |
| 4‐OHE2 | 1.00 ± 0.15 | 1.24 ± 0.17 | 1.3 ± 0.31 | 1.37 ± 0.08* | 1.43 ± 0.24 | 1.57 ± 0.70 | 1.73 ± 0.53 | 2.74 ± 1.44 |
| E2β | 1.00 ± 0.12 | 1.12 ± 0.05 | 1.52 ± 0.88 | 2.08 ± 0.79 | 2.38 ± 0.60 | 2.92 ± 0.18* | 2.96 ± 0.13* | 3.09 ± 0.90 |
| NA | 1.00 ± 0.01 | 1.13 ± 0.33 | 0.93 ± 0.25 | 0.89 ± 0.25 | 0.96 ± 0.19 | 1.73 ± 0.16* | 3.77 ± 0.89* | 2.26 ± 0.33* |
| ADR | 1.00 ± 0.03 | 0.75 ± 0.33 | 1.12 ± 0.37 | 1.22 ± 0.27 | 1.83 ± 1.0 | 2.34 ± 0.87 | 2.63 ± 0.35* | 2.34 ± 0.73 |
*Increased phosphorylation, P < 0.05 vs. 0 h; Student‐Newman‐Keuls test.
The phosphorylation patterns of AKT in P‐UAECs stimulated by catecholoestradiols were different from the patterns obtained with E2β and catecholamine stimulation. Expression of phosphorylated AKT increased sharply at 0.25 h of treatment with 2‐OHE2 (1.27‐fold; P = 0.14) and 4‐OHE2 (1.86‐fold; P = 0.006), followed by decreased phosphorylation over time from 0.5 h until reaching lowest expression at 24 h (0.62‐fold; P = 0.15 and 0.57‐fold; P = 0.13, respectively). By contrast treatment with E2β decreased phosphorylation of AKT over time until reaching lowest expression by 24 h (0.26‐fold, P = 0.007). Noradrenaline (1.64‐fold; P = 0.08) and adrenaline (1.79‐fold; P = 0.24), both induced rapid phosphorylation of AKT at 0.25 h, similar to 2‐OHE2 and 4‐OHE2; however, noradrenaline and adrenaline both induced a second increase in phosphorylated AKT at 2 h (2.04‐fold; P = 0.001 and 1.42‐fold; P = 0.04, respectively) and overall phospho‐AKT remained higher than control until 24 h of treatment (Fig. 4 and Table 4). Levels of total AKT did not change significantly in response to any treatment.
Figure 4. Time course responses of AKT phosphorylation.
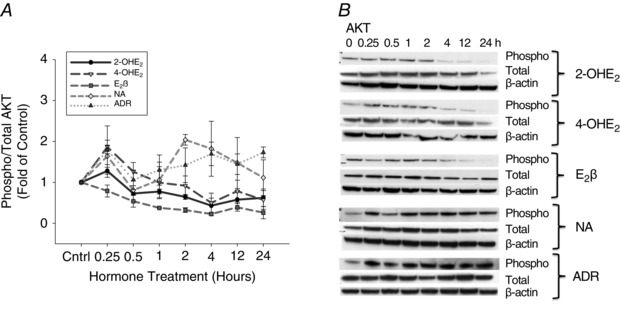
A, phosphorylation of AKT in P‐UAECs upon treatment with 2‐OHE2, 4‐OHE2, E2β, noradrenaline (NA) and adrenaline (ADR) (all 0.1 nm) over the course of 24 h. Different patterns of activation were obtained with the catecholoestradiols, E2β, and the catecholamines. Graphs represent the means ± SEM (Student‐Newman‐Keuls test, P < 0.05 was considered statistically significant). B, representative blots of phosphorylated, total AKT and β‐actin (n = 3).
Table 4.
Phosphorylation of AKT over time, shown as fold change from time zero
| Agonist | Stimulation time (h) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 0.25 | 0.5 | 1 | 2 | 4 | 12 | 24 | |
| 2‐OHE2 | 1.00 ± 0.07 | 1.27 ± 0.16 | 0.72 ± 0.18τ | 0.77 ± 0.05τ | 0.64 ± 0.59 | 0.43 ± 0.18 | 0.57 ± 0.21 | 0.62 ± 0.21 |
| 4‐OHE2 | 1.00 ± 0.02 | 1.86 ± 0.17* | 1.26 ± 0.22 | 0.99 ± 0.39 | 0.91 ± 0.23 | 0.49 ± 0.23 | 0.78 ± 0.26 | 0.57 ± 0.22 |
| E2β | 1.00 ± 0.20 | 0.78 ± 0.14 | 0.53 ± 0.14τ | 0.37±0.01τ | 0.32±0.06τ | 0.23 ± 0.03τ | 0.38±0.09τ | 0.26±0.15τ |
| NA | 1.00 ± 0.05 | 1.64 ± 0.27 | 0.81 ± 0.21 | 1.06 ± 0.46 | 2.04 ± 0.13* | 1.76 ± 0.36 | 1.47 ± 0.62 | 1.11 ± 0.44 |
| ADR | 1.00 ± 0.08 | 1.79 ± 0.59 | 1.06 ± 0.24 | 1.39 ± 0.37 | 1.42 ± 0.42 | 1.69 ± 0.28 | 1.44 ± 0.25 | 1.73 ± 0.13 |
*Increased phosphorylation, P < 0.05 vs. 0 h. τDecreased phosphorylation, P < 0.05 vs. 0 h; Student‐Newman‐Keuls test.
Effects of inhibition of ERK1/2, p38 and JNK MAPK and PI3K pathways on proliferation stimulated by 2‐OHE2, 4‐OHE2, E2β, noradrenaline and adrenaline
Pre‐treatment with the ERK1/2 MAPK specific antagonist, PD98059, at 2.5 and 5.0 μm alone had no effect on P‐UAEC proliferation, but completely abrogated cell proliferation responses to 2‐OHE2, 4‐OHE2, E2β and noradrenaline. Cell proliferation induced by adrenaline was partially inhibited by 2.5 μm PD98059 and completely inhibited by 5.0 μm PD98059 (Fig. 5 A). Treatment with the p38 MAPK inhibitor SB203580 alone had no effect on P‐UAEC proliferation. However, pre‐treatment with 5.0 μm, but not 2.5 μm SB203580 totally inhibited cell proliferation responses to 2‐OHE2, 4‐OHE2, E2β, noradrenaline and adrenaline (Fig. 5 B). Treatment with the JNK inhibitor SP600125 alone had no effect on P‐UAEC proliferation. Nevertheless, SP600125 at both 2.5 μm and 5.0 μm completely abrogated cell proliferation induced by 2‐OHE2, 4‐OHE2, E2β and noradrenaline, while only 5.0 μm, but not 2.5 μm, inhibited proliferation induced by adrenaline (Fig. 5 C). Blockade of PI3K with LY294002 at 2.5 μm and 5.0 μm treatment alone had no effect on P‐UAEC proliferation. In addition, LY294002 also had no effect on E2β‐, 2‐OHE2‐, or 4‐OHE2‐induced P‐UAEC proliferation (P > 0.05). Conversely, pre‐treatment with LY294002 inhibited (P < 0.05) proliferation induced by noradrenaline and adrenaline (Fig. 5 D).
Figure 5. Inhibition of ERK1/2, p38, JNK MAPKs and PI3K.
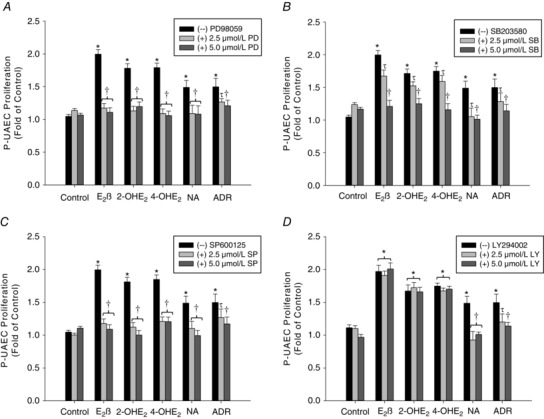
Effects of PD98059 (PD; panel A), SB203580 (SB; panel B), SP600125 (SP; panel C), or LY294002 (LY; panel D) on P‐UAEC proliferation stimulated by 0.1 nm of 2‐OHE2, 4‐OHE2, E2β, noradrenaline (NA) and adrenaline (ADR) alone (–). Treatment (+) with 5.0 μm PD, SB and SP inhibited P‐UAEC proliferation in response to 2‐OHE2, 4‐OHE2, E2β, NA, and ADR. 5.0 μm LY inhibited P‐UAEC proliferation induced by NA and ADR, but had no effect on proliferation induced by E2β, 2‐OHE2 and 4‐OHE2. *Significant increase in proliferation compared to untreated control (P < 0.05). †Complete inhibition (P < 0.05). τPartial inhibition/decrease (n = 4).
Discussion
During pregnancy, elevated levels of oestrogens regulate endothelial cell proliferation and angiogenesis, yet their mechanisms are not fully understood. Evidence suggests that the catechol metabolites of oestrogens are highly involved in the regulation of endothelial cell proliferation via a yet‐unexplored signalling mechanism dependent on β‐adrenoceptors and independently of classic oestrogen receptor signalling. Thus, we evaluated the post‐receptor signalling of catecholoestradiols by determining activation of ERK1/2, p38, JNK MAPKs, or PI3K with acute and prolonged stimulation, as well as the functional contributions of kinases in catecholoestradiol‐induced P‐UAEC proliferation. The results of the present study show for the first time that 2‐OHE2 and 4‐OHE2 induce phosphorylation ERK1/2, p38 and JNK MAPKs, as well as PI3K; however, catecholoestradiol‐induced P‐UAEC proliferation was dependent only on ERK1/2, p38 and JNK, but not PI3K activation. Furthermore, after evaluation of the kinase activation patterns induced by the oestradiol and the catecholamines we showed that although the catecholoestradiols and catecholamines shared the same receptor initiated signalling (β‐AR), the phosphorylation patterns obtained with the catecholoestradiols were more reminiscent of the phosphorylation patterns induced by E2β, despite their signalling through different receptors. These novel data demonstrate convergence of signalling pathways at the level of MAPKs in P‐UAEC proliferation induced by E2β and its catechol metabolites. These previously unknown redundancies in signalling could be a key component of regulating endothelial function and maintaining angiogenesis during pregnancy. This study supports the possibility of crosstalk and redundancies among MAPKs to regulate endothelial cell proliferation, which might be evidence of unappreciated evolutionary functional redundancies in oestrogenic signalling in the uterine endothelium to maintain blood flow during pregnancy.
Catecholoestradiols stimulate activation of ERK1/2, p38, JNK, but not PI3K
2‐OHE2 and 4‐OHE2 stimulated phosphorylation of ERK1/2, p38 and JNK in P‐UAECs. Detailed evaluation of the temporal phosphorylation patterns of each kinase obtained with catecholoestradiols, E2β and catecholamines revealed that ERK1/2 phosphorylation patterns obtained were similar amongst E2β, 2‐OHE2, and 4‐OHE2, with biphasic activation at 15 min and 12 h of stimulation. By contrast, noradrenaline and adrenaline displayed only monophasic ERK1/2 phosphorylation at 15 min of stimulation. Biphasic ERK1/2 activation has long been reported to be intimately involved in the regulation of cell cycle progression and proliferation (see review by Chambard et al. 2007). For example, in Chinese hamster ovary cells, biphasic activation of ERK1/2 was associated with cell cycle entry and transition into S phase (Tamemoto et al. 1992). Similar results have been observed in studies of endothelial cell stimulation with E2β (Kim‐Schulze et al. 1998; Russell et al. 2000; Geraldes et al. 2002; Sengupta et al. 2004) and catecholamines (Steinle et al. 2003; Kim et al. 2008) where either rapid (10–30 min) or delayed (14–20 h) activation of ERK1/2 was studied. Overall, the current results show that ERK1/2 activation by the catecholoestradiols is more reminiscent of oestrogenic ER‐β activation than β‐AR signalling; which could be an intrinsic property of oestrogenic signalling allowing for redundant regulation of endothelial function through different receptors to ensure proper uterine vascular function for fetal growth and survival during pregnancy.
In addition, our results show that although the catecholestradiols and catecholamines stimulate proliferation through β‐ARs, there is a discrepancy in the ERK1/2 and AKT signalling patterns stimulated by these two classes of hormones. Extensive research has been done on ligand‐receptor interactions, which indicate that different agonists can cause different conformational changes of the receptors, thus causing different intracellular signalling cascades and patterns (Ghanouni et al. 2001; Venkatakrishnan et al. 2013). Given the differences in chemical structure and size of the catecholestradiols compared to the catecholamines, it would not be possible for these two types of hormones to interact exactly the same with the β‐ARs, and thus the differences in docking might result in different conformational change of the receptors leading to the different patterns of kinase activation we have observed.
Extensive time course evaluation of phosphorylated p38 and JNK, revealed gradual temporal increases in both p38 and JNK MAPK phosphorylation starting at 15 min with 2‐OHE2, 4‐OHE2, E2β, noradrenaline and adrenaline treatment. Unlike the ERK1/2 MAPK phosphorylation responses, there were no apparent differences in the patterns of either p38 or JNK phosphorylation obtained amongst all agonists studied. These results suggest that both p38 and JNK signalling are likely to play similar roles in oestrogenic and adrenergic regulation of endothelial function under chronic, rather than acute, hormonal stimulation. Temporal activation of p38 in endothelial cells with E2β and β‐AR stimulation has been associated with regulation of angiogenesis through cell migration or proliferation (Razandi et al. 2000; Geraldes et al. 2002; Iaccarino et al. 2005). Conversely, the role of JNK activation in cell proliferation has proven controversial; while some report increased proliferation (Prifti et al. 2001; Ryoo & Bergmann, 2012), others report anti‐angiogenic effects (Fu et al. 2006; Altiok et al. 2007). Still, the phosphorylation patterns, alone, obtained in the current study do not provide sufficient evidence supporting a mitogenic or anti‐angiogenic role of either p38 or JNK in P‐UAECs. Nevertheless, by utilizing the detailed phosphorylation patterns of p38 and JNK in combination with the pharmacological specific inhibition of these kinases, we are able to demonstrate that in P‐UAECs p38 and JNK are involved in regulation of angiogenic processes. Overall, the current data suggest that activation of both p38 and JNK by catecholoestradiols, E2β and catecholamines in P‐UAECs present another level of post‐receptor (ER‐β and β‐AR) convergent or redundant signalling mechanisms in endothelial cells that might be present during pregnancy to provide additional regulatory steps for proper mitogenic function.
In addition, the comprehensive analysis of temporal PI3K–AKT phosphorylation patterns induced by catecholoestradiols and in comparison to E2β, noradrenaline and adrenaline revealed three distinct patterns of phosphorylation with each class of hormones. Both catecholoestradiols induced a rapid increase in phosphorylation followed by a gradual decrease past baseline levels until it was nearly undetectable. While E2β decreased baseline levels of phosphorylated AKT over time until undetectable levels were reached. By contrast, noradrenaline and adrenaline induced biphasic activation of AKT, where the first peak matched the peak observed with the catecholoestradiols and the second peak persisted until the end of stimulation. While the AKT phosphorylation patterns described throughout the literature differ depending on cell type and duration of experimental treatment, some reports show that the catecholoestradiols, E2β and the catecholamines induce activation of PI3K–AKT to mediate cellular processes such as cell proliferation (Steinle et al. 2003; Gao et al. 2004; Simoncini et al. 2004; Kim & Levin, 2006). Thus, the comprehensive patterns of activation obtained with catecholoestradiol, E2β and catecholamine stimulation are indicative of the potential role of PI3K–AKT in P‐UAEC function. Additionally, these data show for the first time the specific PI3K–AKT signalling properties of the catecholoestradiols, which mirror both ER‐β and β‐AR‐mediated signalling and showcase the unappreciated signalling complexity of oestrogens involved in the regulation of uterine endothelium.
ERK1/2, p38, JNK, but not PI3K mediate catecholoestradiol‐induced proliferation
Mitogen activated protein kinases and PI3K belong to the family of Immediate‐early genes, which are genes that can be activated and transcribed within minutes of receptor stimulation (Bahrami & Drablos, 2016). Both, rapid and continued activation of these kinases can lead to cell proliferation. Our time course studies showed different patterns of activation of ERK1/2, p38, JNK and PI3K upon stimulation with catecholoestradiols, E2β and catecholamine; however, the activation these kinases does not always result in cell proliferation (Traverse et al. 1994). Therefore, to investigate the functional role of ERK1/2, p38, JNK MAPKs and PI3K–AKT activation in catecholestradiol‐, E2β‐ and catecholamine‐ induced P‐UAEC proliferation we used specific kinase inhibitors and performed proliferation assays. We determined that activation of ERK1/2 MAPK is needed for catecholoestradiol‐, E2β‐ and catecholamine‐mediated P‐UAEC proliferation, as treatment with PD98059 completely inhibited the proliferation induced by these hormones. These results also indicate that the transient, biphasic, activation of ERK1/2 at 15 min and 12 h of treatment, is sufficient to induce P‐UAEC proliferation. However, further studies are needed to determine if one or both ERK1/2 activation time points are needed for catecholoestradiol‐, E2β‐ and catecholamine‐induced proliferation. Similar to our results, others have reported that activation of ERK1/2 MAPK is required for endothelial cell proliferation stimulated by E2β and β‐AR signalling (Geraldes et al. 2002; Liu et al. 2002; Steinle et al. 2003; Chambard et al. 2007). We also observed that inhibition of p38 MAPK with the specific antagonist SB203580 completely inhibited proliferation induced by 2‐OHE2, 4‐OHE2, E2β and catecholamines, thus indicating that activation of p38 is also independently involved in the regulation of P‐UAEC proliferation stimulated by these hormones. Genetic studies involving p38 MAPK have shown that p38 expression is needed for normal vascular development (Adams et al. 2000), presumably given that p38 phosphorylation is implicated in endothelial cell migration and proliferation (Razandi et al. 2000; Geraldes et al. 2002; Chrzanowska‐Wodnicka et al. 2008). Similarly to ERK1/2 and p38, in the present study, inhibition of JNK MAPK with the specific antagonist SP600125, completely abrogated proliferation induced by 2‐OHE2, 4‐OHE2, E2β, noradrenaline and adrenaline, thus indicating that JNK is also independently involved in mediating the proliferative effects of these hormones on P‐UAECs. Although, JNK signalling has often been linked to endothelial cell stress, there are a few studies indicating that activation of JNK might also be involved in endothelial cell proliferation (Prifti et al. 2001; Ryoo & Bergmann, 2012). Pedram et al. reported that JNK phosphorylation is necessary for endothelial cell proliferation, although this JNK activation was in response to vascular endothelial growth factor (VEGF), rather than E2β or catecholamine signalling (Pedram et al. 1998). Nevertheless, several studies have now determined that some of the angiogenic effects of oestradiol, catecholoestradiols and catecholamines might be mediated via stimulation of VEGF signalling (Gargett et al. 2002; Gao et al. 2004; Lai et al. 2013; Chen et al. 2014). Thus, it is possible that the activation of JNK by catecholoestradiols, E2β and catecholamines, which leads to P‐UAEC proliferation, might be due to their ability to stimulate VEGF or other growth factor signalling. In addition, it has been proposed that significant crosstalk between ERK1/2 and JNK MAPKs might be involved in the process of endothelial cell proliferation (Pedram et al. 1998). Crosstalk between ERK1/2 and JNK is believed to be mediated through the activation of upstream signals like SEK‐1, which then activate JNK. SEK‐1 has also been shown to activate p38 MAPK (Deacon & Blank, 1997). However, further analysis on signalling upstream of MAPKs is needed to demonstrate whether cross‐activation of ERK1/2, p38 and JNK is involved in catecholoestradiol‐mediated proliferation.
Finally, one of the novel findings of this study is that blockade of PI3K did not inhibit catecholoestradiol‐ and E2β‐induced P‐UAEC proliferation, but it inhibited P‐UAEC proliferation induced by catecholamines. These results, along with the detailed phosphorylation patterns obtained in this study, clearly indicate that PI3K activation is not involved in catecholoestradiol‐ or E2β‐induced P‐UAEC proliferation. We therefore conclude that convergent ERK1/2, p38 and JNK MAPK, but not PI3K, signalling pathways are involved in P‐UAEC proliferation mediated by E2β and the catecholoestradiols. However, whether crosstalk among these pathways or simply functional redundancy are at play in the signalling of E2β and its metabolites remain unclear and should be evaluated in future studies.
In conclusion, these data presented herein demonstrate that, despite the absence of common receptors among the catecholoestradiols and their substrate E2β, the post‐receptor signalling mechanisms involved in P‐UAEC proliferation stimulated by these oestrogens are very similar; such that convergent ERK1/2, p38 and JNK MAPKs signalling mediate the regulation of angiogenesis induced by oestrogens during pregnancy. Furthermore, the similarities in uterine artery remodelling and hormonal changes between the ewe and human pregnancy (Griendling et al. 1985; Palmer et al. 1992; Sprague et al. 2009) make our studies relevant to better understanding of human pregnancy adaptations. Thus, the inherent implicit redundancy in the activation of multiple different MAP kinases discovered herein could prove indicative of a multifaceted evolutionary survival mechanism to ensure angiogenesis and elevations in uterine perfusion for fetal survival during a healthy pregnancy.
Additional information
Competing interests
The authors have no conflicts of interest to disclose.
Author contributions
R.V.L.: conception and design; collection and assembly of data; data analysis and interpretation; manuscript writing; final approval of manuscript. S.O.J.: conception and design; collection and assembly of data; data analysis and interpretation; manuscript writing; final approval of manuscript. G.A.‐P.: collection and assembly of data; manuscript writing; final approval of manuscript. G.E.L.: collection and assembly of data; manuscript writing; final approval of manuscript. J.Z.: data analysis and interpretation; manuscript writing; final approval of manuscript. R.R.M.: conception and design; financial support; administrative support; provision of study materials; data analysis and interpretation; manuscript writing; final approval of manuscript. All authors agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed
Funding
This work was supported by National Institutes of Health grants HL49210, HD38843, HL87144 (PI: R. R. Magness), R25‐GM083252 (R. Villalon Landeros and S.O. Jobe) and T32‐HD041921‐07 (S.O. Jobe), F31HD088096 (PI: R. Villalon Landeros). This study is in partial fulfilment for a PhD degree (R. Villalon Landeros) in the Endocrinology and Reproductive Physiology Training Program (www.erp.wisc.edu).
Translational perspective.
We have previously reported that E2β and its metabolites regulate endothelial cell proliferation during pregnancy (Jobe et al. 2010, 2011) and that they might be implicated in the pathology of preeclampsia (Jobe et al. 2013b). The results from the present study demonstrate that the mitogenic actions of E2β and its metabolites, despite signalling through different receptors (ER‐β and β‐AR (Jobe et al. 2011)) on P‐UAECs, are facilitated via the activation of ERK1/2, p38 and JNK MAPKs. We hypothesize that under physiological conditions during pregnancy, E2β and its metabolites play a redundant role in regulating mitogenesis to achieve heightened angiogenesis and high uterine blood flow. Thus, the convergent and/or redundant signal transduction pathways of E2β and its metabolites represent a mechanism of action of oestrogens on endothelial cells to create a cushion of signalling and ensure increased angiogenesis in the event that a signalling pathway malfunctions. Additional studies are required to provide further evidence of the specific role and/or convergence of each kinase signalling pathway in oestrogen‐mediated cell proliferation. Overall, these findings deepen our understanding of oestrogen signalling and its metabolites, which may have important implications for the intricacies of vasculature function during normal and pathological pregnancies.
Acknowledgements
We thank Mayra B. Pastore, Mary Sun, Bryan C. Ampey, Vladimir E. Vargas, Pamela J. Kling, Jason L. Austin, Terrance M. Phernetton, Cindy L. Goss, Jessica H. Youngblood, and Sharon Blohowiak for their help with this research and manuscript preparation.
Linked articles This article is highlighted by a Perspective by Hu & Zhang. To read this Perspective, visit https://doi.org/10.1113/JP274489.
References
- Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, Valladares A, Perez L, Klein R & Nebreda AR (2000). Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol Cell 6, 109–116. [PubMed] [Google Scholar]
- Altiok N, Koyuturk M & Altiok S (2007). JNK pathway regulates estradiol‐induced apoptosis in hormone‐dependent human breast cancer cells. Breast Cancer Res Treat 105, 247–254. [DOI] [PubMed] [Google Scholar]
- Bahrami S & Drablos F (2016). Gene regulation in the immediate‐early response process. Adv Biol Regul 62, 37–49. [DOI] [PubMed] [Google Scholar]
- Barker DJ (2007). The origins of the developmental origins theory. J Intern Med 261, 412–417. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Eriksson JG, Forsen T & Osmond C (2002). Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol 31, 1235–1239. [DOI] [PubMed] [Google Scholar]
- Bird IM, Sullivan JA, Di T, Cale JM, Zhang L, Zheng J & Magness RR (2000). Pregnancy‐dependent changes in cell signaling underlie changes in differential control of vasodilator production in uterine artery endothelial cells. Endocrinology 141, 1107–1117. [DOI] [PubMed] [Google Scholar]
- Chambard JC, Lefloch R, Pouyssegur J & Lenormand P (2007). ERK implication in cell cycle regulation. Biochim Biophys Acta 1773, 1299–1310. [DOI] [PubMed] [Google Scholar]
- Chen H, Liu D, Yang Z, Sun L, Deng Q, Yang S, Qian L, Guo L, Yu M, Hu M, Shi M & Guo N (2014). Adrenergic signaling promotes angiogenesis through endothelial cell‐tumor cell crosstalk. Endocr Relat Cancer 21, 783–795. [DOI] [PubMed] [Google Scholar]
- Chrzanowska‐Wodnicka M, Kraus AE, Gale D, White GC 2nd & Vansluys J (2008). Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b‐deficient mice. Blood 111, 2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon K & Blank JL (1997). Characterization of the mitogen‐activated protein kinase kinase 4 (MKK4)/c‐Jun NH2‐terminal kinase 1 and MKK3/p38 pathways regulated by MEK kinases 2 and 3. MEK kinase 3 activates MKK3 but does not cause activation of p38 kinase in vivo . J Biol Chem 272, 14489–14496. [DOI] [PubMed] [Google Scholar]
- Dubey RK, Tofovic SP & Jackson EK (2004). Cardiovascular pharmacology of estradiol metabolites. J Pharmacol Exp Ther 308, 403–409. [DOI] [PubMed] [Google Scholar]
- Fu XD, Cui YH, Lin GP & Wang TH (2007). Non‐genomic effects of 17β‐estradiol in activation of the ERK1/ERK2 pathway induces cell proliferation through upregulation of cyclin D1 expression in bovine artery endothelial cells. Gynecol Endocrinol 23, 131–137. [DOI] [PubMed] [Google Scholar]
- Fu YC, Yin SC, Chi CS, Hwang B & Hsu SL (2006). Norepinephrine induces apoptosis in neonatal rat endothelial cells via a ROS‐dependent JNK activation pathway. Apoptosis 11, 2053–2063. [DOI] [PubMed] [Google Scholar]
- Gao N, Nester RA & Sarkar MA (2004). 4‐Hydroxy estradiol but not 2‐hydroxy estradiol induces expression of hypoxia‐inducible factor 1α and vascular endothelial growth factor A through phosphatidylinositol 3‐kinase/AKT/FRAP pathway in OVCAR‐3 and A2780‐CP70 human ovarian carcinoma cells. Toxicol Appl Pharmacol 196, 124–135. [DOI] [PubMed] [Google Scholar]
- Gargett CE, Zaitseva M, Bucak K, Chu S, Fuller PJ & Rogers PA (2002). 17β‐estradiol up‐regulates vascular endothelial growth factor receptor‐2 expression in human myometrial microvascular endothelial cells: role of estrogen receptor‐α and ‐β. J Clin Endocrinol Metab 87, 4341–4349. [DOI] [PubMed] [Google Scholar]
- Geraldes P, Sirois MG, Bernatchez PN & Tanguay JF (2002). Estrogen regulation of endothelial and smooth muscle cell migration and proliferation: role of p38 and p42/44 mitogen‐activated protein kinase. Arterioscler Thromb Vasc Biol 22, 1585–1590. [DOI] [PubMed] [Google Scholar]
- Ghanouni P, Steenhuis JJ, Farrens DL & Kobilka BK (2001). Agonist‐induced conformational changes in the G‐protein‐coupling domain of the β2 adrenergic receptor. Proc Natl Acad Sci USA 98, 5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, Fuller EO & Cox RH (1985). Pregnancy‐induced changes in sheep uterine and carotid arteries. Am J Physiol Cell Physiol 248, H658–H665. [DOI] [PubMed] [Google Scholar]
- Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, Cipolletta E, Cerullo V, Cimini V, Altobelli GG, Piscione F, Priante O, Pastore L, Chiariello M, Salvatore F, Koch WJ & Trimarco B (2005). Ischemic neoangiogenesis enhanced by β2‐adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res 97, 1182–1189. [DOI] [PubMed] [Google Scholar]
- Jansson T & Powell TL (2007). Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci 113, 1–13. [DOI] [PubMed] [Google Scholar]
- Jobe SO, Fling SN, Ramadoss J & Magness RR (2011). A novel role for an endothelial adrenergic receptor system in mediating catecholestradiol‐induced proliferation of uterine artery endothelial cells. Hypertension 58, 874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobe SO, Ramadoss J, Koch JM, Jiang Y, Zheng J & Magness RR (2010). Estradiol‐17β and its cytochrome P450‐ and catechol‐O‐methyltransferase‐derived metabolites stimulate proliferation in uterine artery endothelial cells: role of estrogen receptor‐α versus estrogen receptor‐β. Hypertension 55, 1005–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobe SO, Ramadoss J, Wargin AJ & Magness RR (2013a). Estradiol‐17β and its cytochrome P450‐ and catechol‐O‐methyltransferase‐derived metabolites selectively stimulate production of prostacyclin in uterine artery endothelial cells: role of estrogen receptor‐α versus estrogen receptor‐β. Hypertension 61, 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobe SO, Tyler CT & Magness RR (2013b). Aberrant synthesis, metabolism, and plasma accumulation of circulating estrogens and estrogen metabolites in preeclampsia implications for vascular dysfunction. Hypertension 61, 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanasaki K, Palmsten K, Sugimoto H, Ahmad S, Hamano Y, Xie L, Parry S, Augustin HG, Gattone VH, Folkman J, Strauss JF & Kalluri R (2008). Deficiency in catechol‐O‐methyltransferase and 2‐methoxyoestradiol is associated with pre‐eclampsia. Nature 453, 1117–1121. [DOI] [PubMed] [Google Scholar]
- Kim JK & Levin ER (2006). Estrogen signaling in the cardiovascular system. Nuclear Receptor Signaling 4, e013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MO, Na SI, Lee MY, Heo JS & Han HJ (2008). Epinephrine increases DNA synthesis via ERK1/2s through cAMP, Ca2+/PKC, and PI3K/Akt signaling pathways in mouse embryonic stem cells. J Cell Biochem 104, 1407–1420. [DOI] [PubMed] [Google Scholar]
- Kim‐Schulze S, Lowe WL Jr & Schnaper HW (1998). Estrogen stimulates delayed mitogen‐activated protein kinase activity in human endothelial cells via an autocrine loop that involves basic fibroblast growth factor. Circulation 98, 413–421. [DOI] [PubMed] [Google Scholar]
- Lai KB, Sanderson JE & Yu CM (2013). The regulatory effect of norepinephrine on connective tissue growth factor (CTGF) and vascular endothelial growth factor (VEGF) expression in cultured cardiac fibroblasts. Int J Cardiol 163, 183–189. [DOI] [PubMed] [Google Scholar]
- Liu WL, Guo X & Guo ZG (2002). Estrogen prevents bovine aortic endothelial cells from TNF‐α‐induced apoptosis via opposing effects on p38 and p44/42 CCDPK. Acta Pharmacol Sin 23, 213–218. [PubMed] [Google Scholar]
- Losordo DW & Isner JM (2001). Estrogen and angiogenesis: A review. Arterioscler Thromb Vasc Biol 21, 6–12. [DOI] [PubMed] [Google Scholar]
- Lunell NO, Nylund LE, Lewander R & Sarby B (1982). Uteroplacental blood flow in pre‐eclampsia measurements with indium‐113m and a computer‐linked gamma camera. Clin Exp Hypertens B 1, 105–117. [DOI] [PubMed] [Google Scholar]
- Magness RR (1998). Maternal cardiovascular and other physiologic responses to the endocrinology of pregnancy In The Endocrinology of Pregnancy, ed. Bazer FW, pp. 507–539. Humana Press Inc, Totowa, NJ, USA. [Google Scholar]
- Magness RR & Ford SP (2014). Maternal cardiovascular adaptation and uterine circulation – physiology and pathophysiology In Stress and Developmental Programming of Health and Disease: Beyond Phenomenology, eds Zhang LL. & Long LD, pp. 341–374. Nova Science Publishers, Inc, Hauppauge, NY, USA. [Google Scholar]
- Magness RR, Phernetton TM, Gibson TC & Chen DB (2005). Uterine blood flow responses to ICI 182 780 in ovariectomized oestradiol‐17β‐treated, intact follicular and pregnant sheep. J Physiol 565, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M, Galluzzo P & Ascenzi P (2006). Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics 7, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R, Jorgensen P, Moran U, Weber G & Springer M (2010). BioNumbers – the database of key numbers in molecular and cell biology. Nucleic Acids Res 38, D750–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales DE, McGowan KA, Grant DS, Maheshwari S, Bhartiya D, Cid MC, Kleinman HK & Schnaper HW (1995). Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 91, 755–763. [DOI] [PubMed] [Google Scholar]
- Munoz‐Chapuli R, Quesada AR & Angel Medina M (2004). Angiogenesis and signal transduction in endothelial cells. Cell Mol Life Sci 61, 2224–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osol G & Mandala M (2009). Maternal uterine vascular remodeling during pregnancy. Physiology (Bethesda) 24, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer SK, Moore LG, Young D, Cregger B, Berman JC & Zamudio S (1999). Altered blood pressure course during normal pregnancy and increased preeclampsia at high altitude (3100 meters) in Colorado. Am J Obstet Gynecol 180, 1161–1168. [DOI] [PubMed] [Google Scholar]
- Palmer SK, Zamudio S, Coffin C, Parker S, Stamm E & Moore LG (1992). Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet Gynecol 80, 1000–1006. [PubMed] [Google Scholar]
- Parvathaneni K, Grigsby JG, Betts BS & Tsin AT (2013). Estrogen‐induced retinal endothelial cell proliferation: possible involvement of pigment epithelium‐derived factor and phosphoinositide 3‐kinase/mitogen‐activated protein kinase pathways. J Ocul Pharmacol Ther 29, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore MB, Jobe SO, Ramadoss J & Magness RR (2012). Estrogen receptor‐α and estrogen receptor‐β in the uterine vascular endothelium during pregnancy: functional implications for regulating uterine blood flow. Semin Reprod Med 30, 46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A, Razandi M & Levin ER (1998). Extracellular signal‐regulated protein kinase/Jun kinase cross‐talk underlies vascular endothelial cell growth factor‐induced endothelial cell proliferation. J Biol Chem 273, 26722–26728. [DOI] [PubMed] [Google Scholar]
- Prifti S, Mall P, Strowitzki T & Rabe T (2001). Synthetic estrogens‐mediated activation of JNK intracellular signaling molecule. Gynecol Endocrinol 15, 135–141. [PubMed] [Google Scholar]
- Razandi M, Pedram A & Levin ER (2000). Estrogen signals to the preservation of endothelial cell form and function. J Biol Chem 275, 38540–38546. [DOI] [PubMed] [Google Scholar]
- Rose BA, Force T & Wang Y (2010). Mitogen‐activated protein kinase signaling in the heart: angels versus demons in a heart‐breaking tale. Physiol Rev 90, 1507–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CR, Morriss FH Jr, Battaglia FC, Makowski EL & Meschia G (1976). Effect of estradiol‐17β on blood flow to reproductive and nonreproductive tissues in pregnant ewes. Am J Obstet Gynecol 124, 618–629. [DOI] [PubMed] [Google Scholar]
- Russell KS, Haynes MP, Sinha D, Clerisme E & Bender JR (2000). Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc Natl Acad Sci USA 97, 5930–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD & Bergmann A (2012). The role of apoptosis‐induced proliferation for regeneration and cancer. Cold Spring Harb Perspect Biol 4, a008797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta K, Banerjee S, Saxena NK & Banerjee SK (2004). Thombospondin‐1 disrupts estrogen‐induced endothelial cell proliferation and migration and its expression is suppressed by estradiol. Mol Cancer Res 2, 150–158. [PubMed] [Google Scholar]
- Simoncini T, Mannella P, Fornari L, Caruso A, Varone G & Genazzani AR (2004). Genomic and non‐genomic effects of estrogens on endothelial cells. Steroids 69, 537–542. [DOI] [PubMed] [Google Scholar]
- Sprague BJ, Phernetton TM, Magness RR & Chesler NC (2009). The effects of the ovarian cycle and pregnancy on uterine vascular impedance and uterine artery mechanics. Eur J Obstet Gynecol Reprod Biol 144 (Suppl. 1), S170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinle JJ, Booz GW, Meininger CJ, Day JN & Granger HJ (2003). β3‐adrenergic receptors regulate retinal endothelial cell migration and proliferation. J Biol Chem 278, 20681–20686. [DOI] [PubMed] [Google Scholar]
- Tamemoto H, Kadowaki T, Tobe K, Ueki K, Izumi T, Chatani Y, Kohno M, Kasuga M, Yazaki Y & Akanuma Y (1992). Biphasic activation of two mitogen‐activated protein kinases during the cell cycle in mammalian cells. J Biol Chem 267, 20293–20297. [PubMed] [Google Scholar]
- Traverse S, Seedorf K, Paterson H, Marshall CJ, Cohen P & Ullrich A (1994). EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr Biol 4, 694–701. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF & Babu MM (2013). Molecular signatures of G‐protein‐coupled receptors. Nature 494, 185–194. [DOI] [PubMed] [Google Scholar]
- Zhang W & Liu HT (2002). MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12, 9–18. [DOI] [PubMed] [Google Scholar]