

Abstract
Key points
The role of trimeric intracellular cation (TRIC) channels is not known, although evidence suggests they may regulate ryanodine receptors (RyR) via multiple mechanisms. We therefore investigated whether Tric‐a gene knockout (KO) alters the single‐channel function of skeletal RyR (RyR1).
We find that RyR1 from Tric‐a KO mice are more sensitive to inhibition by divalent cations, although they respond normally to cytosolic Ca2+, ATP, caffeine and luminal Ca2+.
In the presence of Mg2+, ATP cannot effectively activate RyR1 from Tric‐a KO mice.
Additionally, RyR1 from Tric‐a KO mice are not activated by protein kinase A phosphorylation, demonstrating a defect in the ability of β‐adrenergic stimulation to regulate sarcoplasmic reticulum (SR) Ca2+‐release.
The defective RyR1 gating that we describe probably contributes significantly to the impaired SR Ca2+‐release observed in skeletal muscle from Tric‐a KO mice, further highlighting the importance of TRIC‐A for normal physiological regulation of SR Ca2+‐release in skeletal muscle.
Abstract
The type A trimeric intracellular cation channel (TRIC‐A) is a major component of the nuclear and sarcoplasmic reticulum (SR) membranes of cardiac and skeletal muscle, and is localized closely with ryanodine receptor (RyR) channels in the SR terminal cisternae. The skeletal muscle of Tric‐a knockout (KO) mice is characterized by Ca2+ overloaded and swollen SR and by changes in the properties of SR Ca2+ release. We therefore investigated whether RyR1 gating behaviour is modified in the SR from Tric‐a KO mice by incorporating native RyR1 into planar phospholipid bilayers under voltage‐clamp conditions. We find that RyR1 channels from Tric‐a KO mice respond normally to cytosolic Ca2+, ATP, adenine, caffeine and to luminal Ca2+. However, the channels are more sensitive to the inactivating effects of divalent cations, thus, in the presence of Mg2+, ATP is inadequate as an activator. Additionally, channels are not characteristically activated by protein kinase A even though the phosphorylation levels of Ser2844 are similar to controls. The results of the present study suggest that TRIC‐A functions as an excitatory modulator of RyR1 channels within the SR terminal cisternae. Importantly, this regulatory action of TRIC‐A appears to be independent of (although additive to) any indirect consequences to RyR1 activity that arise as a result of K+ fluxes across the SR via TRIC‐A.
Keywords: Ca2+ release, ryanodine receptor, sarcoplasmic reticulum
Key points
The role of trimeric intracellular cation (TRIC) channels is not known, although evidence suggests they may regulate ryanodine receptors (RyR) via multiple mechanisms. We therefore investigated whether Tric‐a gene knockout (KO) alters the single‐channel function of skeletal RyR (RyR1).
We find that RyR1 from Tric‐a KO mice are more sensitive to inhibition by divalent cations, although they respond normally to cytosolic Ca2+, ATP, caffeine and luminal Ca2+.
In the presence of Mg2+, ATP cannot effectively activate RyR1 from Tric‐a KO mice.
Additionally, RyR1 from Tric‐a KO mice are not activated by protein kinase A phosphorylation, demonstrating a defect in the ability of β‐adrenergic stimulation to regulate sarcoplasmic reticulum (SR) Ca2+‐release.
The defective RyR1 gating that we describe probably contributes significantly to the impaired SR Ca2+‐release observed in skeletal muscle from Tric‐a KO mice, further highlighting the importance of TRIC‐A for normal physiological regulation of SR Ca2+‐release in skeletal muscle.
Abbreviations
- DM
n‐decyl‐β‐d‐maltopyranoside
- ER/SR
endoplasmic/sarcoplasmic reticulum
- KO
knockout
- MS
mass spectrometry
probability density function
- PKA
protein kinase A
- PP1
protein phosphatase 1
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- TRIC
trimeric intracellular cation channel
- WT
wild‐type
Introduction
There are two subtypes of trimeric intracellular cation channel (TRIC), termed TRIC‐A and TRIC‐B, and both are found on the endoplasmic/sarcoplasmic reticulum (ER/SR) and the nuclear membranes of most cell types (Yazawa et al. 2007). The conductance and gating properties of purified recombinant TRIC channels reconstituted into artificial membranes are similar to those of the monovalent cation selective SR K+ channels first observed from preparations of isolated rabbit skeletal SR vesicles (Labarca et al. 1980; Yazawa et al. 2007; Pitt et al. 2010). The TRIC channels have been shown to be trimeric in structure, each formed from three individual monomers of ∼30 kDa in molecular mass (Yazawa et al. 2007; Kasuya et al. 2016; Yang et al. 2016). It was assumed that the SR K+ channel fulfils the essential role of a counter‐ion pathway, allowing rapid charge compensation for the SR Ca2+ release via ryanodine receptor (RyR) channels (Miller & Rosenberg 1979; Somlyo et al. 1981; Fink & Veigel 1996). It was subsequently suggested that the RyR channels may be able to pass most or all of their own counter‐current (Gillespie & Fill 2008; Gillespie et al. 2009). If this is so, then the necessity for the SR K+ channel to pass counter‐ion flux is not as critical as first assumed, although equilibration of K+ across the SR will still be important. Gene knockout (KO) studies, however, demonstrate that TRIC is essential for the normal functioning of many tissues. For example, the Tric‐a/Tric‐b double KO mouse dies in heart failure before birth (Yazawa et al. 2007). The Tric‐b KO mouse dies immediately after birth in respiratory failure and the Tric‐a KO mouse exhibits an abnormal SR ultrastructure and unstable contractile behaviour under stress in skeletal muscle (Yamazaki et al. 2009; Zhao et al. 2010). More recently, mutations in TRIC‐B have been associated with the disease osteogenesis imperfecta (Volodarsky et al. 2013; Rubinato et al. 2014). The absolute requirement in some tissues for the presence of TRIC perhaps indicates additional roles of the TRIC channels in addition to their capacity to act as pathway for monovalent cation flux across the SR. Investigation of TRIC:RyR stoichiometry in various tissues indicates that, in excitable tissues such as cardiac and skeletal muscle, the SR is packed with many more TRIC‐A channels than RyR and TRIC‐B channels (Pitt et al. 2010; Zhao et al. 2010). RyR and TRIC channels have not been co‐purified in previous biochemical studies (Yazawa et al. 2007); however, reversible protein–protein interactions between the densely packed ion channels in the SR may provide an important regulatory influence on RyR activity and SR Ca2+ release. Indeed, a protein termed SPR‐27 but subsequently discovered to be the same protein as TRIC‐A was previously suggested to form part of the RyR macromolecular complex (Bleunven et al. 2008). Because the Tric‐a KO mouse survives until adulthood, we isolated SR membranes from the mature skeletal muscle and incorporated them into bilayers to investigate whether the gating or conductance of the RyR channels are modified by the absence of TRIC‐A. The results obtained show that the RyR from Tric‐a KO mice exhibit modified gating properties that prevent the channels from responding normally to activators such as ATP or to phosphorylation by protein kinase A (PKA).
Methods
Ethical approval
All experiments in the present study were conducted with the approval of the Animal Research Committee in accordance with the regulations on animal experimentation at Kyoto University (Agreement no. 11‐6).
Isolation of membrane fractions from mouse skeletal muscle
Isolated membrane vesicles were prepared from wild‐type (WT) and Tric‐a KO mouse skeletal muscle using methods described previously (Venturi et al. 2013) with some modifications. Mouse skeletal muscle was dissected and snap‐frozen in liquid N2. Frozen tissue was pulverized and finely homogenised in a buffer containing 300 mm sucrose and 20 mm PIPES (pH 7.4) and supplemented with a protease inhibitor cocktail (Sigma‐Aldrich, Poole, UK), 1 mm phenylmethane sulphonyl fluoride and 2.5 mm dithiothreitol. The tissue homogenate was centrifuged at 6000 g for 20 min at 4°C. The supernatant obtained was collected and the pellets were re‐homogenized, resuspended in the same buffer and centrifuged at 6000 g for 20 min at 4°C. The supernatants obtained were filtered through a cheesecloth and spun at 100 000 g for 1 h at 4°C. The pellets, containing the membrane fractions, were resuspended in 400 mm sucrose, 5 mm HEPES, 2.5 mm DTT (pH 7.2), aliquoted, snap‐frozen in liquid N2 and stored at −80°C. Isolated membrane fractions were used in single‐channel and [3H]ryanodine binding experiments.
Purification of TRIC‐A
A stable Chinese hamster ovary cell line overexpressing mouse TRIC‐A was generated. The cDNA encoding the full‐length mouse TRIC‐A was fused with a PA‐tag at the N‐terminal and subcloned into the pcDNA3 expression vector (Invitrogen, Carlsbad, CA, USA). Cells expressing TRIC‐A were cultured in α‐MEM (Gibco, Gaithersburg, MD, USA) with 10% FBS (Sigma), 1:200 penicillin–streptomycin (Sigma) and 200 μg ml−1 G418 (Sigma). Cells were cultured in 25 × 175 cm2 flasks, collected and homogenized with a dounce homogenizer in hypotonic buffer containing 10 mm HEPES (pH 7.4). Solubilisation was achieved by the addition of an equal volume of 2x binding buffer containing 0.5 m sucrose, 0.6 m NaCl, 10 mm HEPES (pH 7.4) and 2% (w/v) n‐decyl‐β‐d‐maltopyranoside (DM) to the cell lysate followed by a re‐homogenization step. Insoluble material was pelleted by a high‐speed centrifugation step (200 000 g for 30 min at 4°C) when the supernatant containing soluble proteins was collected. The supernatant was diluted to reduce the detergent concentration to 0.5% by adding an equal volume of 1x binding buffer (0.25 m sucrose, 0.3 m NaCl, 10 mm HEPES, pH 7.4). Anti‐PA tag antibody beads (Wako Chemicals GmbH, Neuss, Germany) were added to the supernatant and the mixture was then incubated with continuous stirring for 2 h at 4°C. The beads were then transferred into a centrifuge column (Thermo Fisher Scientific, Waltham, MA, USA) and washed five times in a washing buffer containing 10% (v/v) glycerol, 0.4 m NaCl, 1 mm EDTA, 20 mm Tris‐HCl, pH 7.4 and 0.1% DM. Fractions containing purified TRIC‐A proteins were eluted by supplementing the washing buffer with 0.2 mg ml−1 PA‐tag peptide (Wako Chemicals GmbH). All purification steps were carried out at 4°C. The buffers used for the purification were supplemented with 1 mm DTT and a protease inhibitor cocktail (Sigma). Western blot using an anti‐PA tag antibody (dilution 1:2000; Wako Chemicals GmbH) was used to confirm protein purification and enrichment.
Reconstitution of purified TRIC‐A into liposomes
Phosphatidylcholine (Avanti Polar Lipids, Alabaster, AL, USA) in chloroform solution was dried under a nitrogen stream, resuspended in reconstitution buffer (100 mm NaCl, 20 mm HEPES, pH 7.4) at a concentration of 10 mg ml−1 and sonicated until the lipids formed a cloudy homogeneous suspension. Liposomes were disrupted by adding 35 mm 3‐[(3‐cholamidopropyl)dimethylammonio]‐1‐propanesulphonate. Purified TRIC‐A protein was added to the clear suspension at a protein to lipid ratio of 1:1 (v:v). An equal volume of washing buffer containing 0.1% DM was added to the lipid to produce empty control liposomes. The mixture was then dialysed in a 10 kDa‐cut‐off Slide‐A‐Lyzer cassette (Thermo Fisher Scientific) against 1 litre of reconstitution buffer for 6 h with buffer exchange every hour at 4°C. Liposomes containing TRIC‐A or empty liposomes were added to the cytosolic side of RyR1 channels from Tric‐a KO mice gating in bilayers with Ca2+ as the permeant ion (see below).
Single‐channel recordings
Single‐channel recordings of RyR channels obtained from WT and Tric‐a KO skeletal muscles were performed as described previously (Sitsapesan et al. 1991). RyR current fluctuations were recorded under voltage clamp conditions using K+ or Ca2+ as the permeant ion. Isolated membrane vesicles containing RyR channels always incorporated in a fixed orientation such that the cis chamber corresponded to the cytosol, whereas the trans chamber corresponded to the SR lumen. For experiments with Ca2+ as the permeant ion, recording solutions were 250 mm HEPES, 80 mm Tris and 10 μm free Ca2+ (pH 7.2) on the cis side and 250 mm glutamic acid and 10 mm HEPES (pH to 7.2) with Ca(OH)2 (free [Ca2+] ∼50 mm) on the trans side of the bilayer. The trans chamber was voltage clamped at ground. For experiments with K+ as the permeant ion, symmetrical solutions of 210 mm KPIPES (pH 7.2) were used and luminal and cytosolic free [Ca2+] was adjusted as required. PKA‐dependent phosphorylation of RyR was achieved by incubating the cytosolic side of the channels with 10 units of the catalytic subunit of PKA (Sigma‐Aldrich) in the presence of 10 μm free Ca2+, 3 mm ATP and 1 mm free Mg2+ for 10 min. Single RyR channels were treated with 5 units of protein phosphatase 1 (PP1) (New England Biolabs, Beverly, MA, USA) in presence of Mn2+ for 10 min. After the PKA or PP1 incubation, the cytosolic chamber was washed back to control conditions. Experiments were performed at room temperature (22 ± 2°C). The free [Ca2+] and pH of the solutions were maintained constant during the experiment and were determined using a Ca2+ electrode (Orion 93‐20; Thermo Fisher Scientific) and a Ross‐type pH electrode (Orion 81‐55; Thermo Fisher Scientific) as described previously (Sitsapesan et al. 1991).
Single‐channel analysis
Single‐channel recordings were digitized at 20 kHz and recorded on a computer hard drive using pClamp (Molecular Devices, Sunnyvale, CA, USA). Before idealization, traces were filtered at 800 Hz (−3 db) in experiments where Ca2+ was the permeant ion or at 4 kHz where K+ was the permeant ion. The open and closed channel levels were assessed using manually controlled cursors. Open probability (Po) was determined over 3 min of continuous recording using the 50% threshold method (Colquhoun & Sigworth 1983) at 0 mV, when Ca2+ was the permeant ion or at potentials relative to ground in K+‐containing solutions. Lifetime distributions were calculated from idealizations where only a single channel was gating in the bilayer. Events shorter than 1 ms (where Ca2+ was the permeant ion) or 0.2 ms (where K+ was the permeant ion) were stripped from the idealized event sequences using Clampfit, version 10.2 (Molecular Devices). Individual time constants were fitted with an exponential log probability density function (pdf) in Clampfit, using maximum‐likelihood fitting (Colquhoun & Sigworth 1983). The optimal number of time constants for each distribution was determined using a log‐likelihood ratio test at a confidence level of P = 0.95 (Blatz & Magleby 1986).
RyR1 immunoprecipitation and immunoblotting
RyR1 was immunoprecipitated from 400 μg of mouse skeletal mixed membrane preparations using an anti‐RyR antibody (34C; Abcam, Cambridge, UK) and Protein G Dynabeads (Life Technologies, Oslo, Norway) by overnight incubation at 4°C with continuous mixing in 0.4 ml of homogenization buffer [20 mm HEPES, 150 mm NaCl, 5 mm EDTA, 20% (v/v) glycerol, protease inhibitors (Roche Diagnostics Limited, Burgess Hill, UK), Triton‐X 0.5%, pH 6.8]. Protein immunocomplexes were separated magnetically (DynaMag‐2 magnet; Thermo Fisher) and beads were washed three times with homogenization buffer. For PKA phosphorylation of RyR1, beads were incubated with 1 U of the catalytic subunit of PKA (Sigma‐Aldrich) per μg protein for 10 min at 37°C in a solution containing 50 mm HEPES, 16 mm Tris, 5 mm Mg2+ and 5 mm NaF (pH 7.2). After PKA treatment, the supernatant was removed by magnetic separation and samples were resuspended in 50 μl of Laemmeli sample buffer containing 5% β‐mercaptoethanol and incubated at 95°C for 5 min. Samples were then used for western blotting as described previously (Carter et al. 2011). Immunoblots were probed with anti‐RyR antibody (34C; dilution 1:1000) and Phospho‐(Ser/Thr) PKA Substrate Antibody #9621 (dilution 1:1000; Cell Signaling Technology, Leiden, The Netherlands). RyR1 protein and phosphorylation levels were quantified by densitometry.
Mass spectrometry (MS) methods
Microsomes from WT and Tric‐a KO mouse skeletal muscle were treated with PKA as described previously (Carter et al. 2011). Microsomal proteins were separated on a 6% SDS‐PAGE and either stained with Coomassie Brilliant Blue for visualization or transferred to a nitrocellulose membrane and probed with RyR1 antibody as described above. The corresponding bands containing RyR1 were cut from the Coomassie Brilliant Blue stained gel and subjected to in‐gel tryptic digestion for MS analysis as described previously (Shevchenko et al. 2007). The peptides generated were then separated by nanoflow reversed‐phase liquid chromatography coupled to Q Exactive Hybrid Quadrupole‐Orbitrap mass spectrometer (Thermo Fisher Scientific). Peptides were loaded on a C18 PepMap100 pre‐column (inner diameter 300 μm × 5 mm, 3 μm C18 beads; Thermo Fisher Scientific) and separated on a 50 cm reversed‐phase C18 column (inner diameter 75 μm, 2 μm C18 beads). Separation was conducted with a linear gradient of 7–30% of B for 30 min at a flow rate of 200 nl min−1 (A: 0.1% formic acid, B: 0.1% formic acid in acetonitrile). All data were acquired in a data‐dependent mode, automatically switching from MS to collision‐induced dissociation MS/MS on the top 10 most abundant ions with a precursor scan range of 350–1650 m/z. MS spectra were acquired at a resolution of 70 000 and MS/MS scans at 17 000. Dynamic exclusion was enabled with an exclusion duration of 40 s. The raw data files generated were processed using MaxQuant, version 1.5.0.35, integrated with the Andromeda search engine as described previously (Cox & Mann 2008; Cox et al. 2011). The MS/MS spectra were searched against the mouse proteome (UniProt 2013/04/03), precursor mass tolerance was set to 20 ppm with variable modifications defined as phosphorylation (S, T and Y). Enzyme specificity was set to trypsin with a maximum of two missed cleavages. Protein and peptide spectral matches false discovery rate was set at 0.01 and a minimum score of 40 and localization probability of > 0.7 for phosphopeptides. Match between runs was applied. The ratio of phosphorylated (S2844) to unphosphorylated KISQTAQTYDPR peptide intensities was calculated for three biological replicates of WT and Tric‐a KO under control and PKA treatment.
[3H]ryanodine binding
Binding of [3H]ryanodine (PerkinElmer Inc., Waltham, MA, USA) to skeletal membrane vesicles was measured at 37°C for 90 min with constant shaking in buffer consisting of 200 μg protein ml–1, 5 nm [3H]ryanodine, 250 mm KCl, 25 mm PIPES and 200 μm AEBSF (pH 7.2). RyR1 channel modulators Ca2+, Mg2+, caffeine and adenine were included in specific experiments as described where appropriate. Non‐specific binding was determined in the presence of a 1000‐fold excess unlabelled ryanodine. Bound and free ligand were separated by rapid filtration through Whatman GF/B glass microfibre filters (GE Healthcare Life Sciences, Little Chalfont, UK). [3H]ryanodine retained in filters was quantified by liquid scintillation spectrometry using a scintillation counter. Measurements were performed in triplicate and each experiment was performed using at least three independent skeletal muscle preparations.
Statistical analysis
Data are expressed as the mean ± SD where n = 3 or the mean ± SEM where n ≥ 4. Differences between mean values were assessed using Student's t test. P < 0.05 was considered statistically significant.
Materials
All chemicals were purchased from VWR (Lutterworth, UK), Sigma‐Aldrich (UK) or as otherwise stated. All solutions were prepared in deionized water (Millipore, Feltham, UK) and those used in bilayer experiments were filtered through a membrane with 0.45 μm pore diameter (Millipore).
Results
Evidence suggests that luminal Ca2+ is higher than normal in skeletal muscle from Tric‐a KO tissue (Zhao et al. 2010) yet Ca2+‐release is impaired, so we initially performed experiments using K+ as the permeant ion so that we could investigate whether the luminal Ca2+ sensitivity of the single RyR channels was modified. Figure 1, showing top traces from WT and Tric‐a KO tissue, demonstrates that the Po was similar for channels from both WT and Tric‐a KO tissue when the cytosolic and luminal [Ca2+] was maintained at 10 μm. Adding 1 mm ATP to the cytosolic channel side led to similar increases in Po in channels from WT and Tric‐a KO tissue (second trace), indicating that the response of RyR1 to ATP was not altered in Tric‐a KO tissue. Subsequently, increasing the luminal [Ca2+] to 100 μm and 1 mm also increased Po to levels that were comparable in both groups of channel, therefore providing no evidence for impairment of luminal Ca2+ sensitivity in RyR1 channels from Tric‐a KO tissue. Mean Po data are also shown (Fig. 1 C and D).
Figure 1. The effects of cytosolic ATP and luminal Ca2+ on RyR1 channels from Tric‐a KO mice with K+ as the permeant ion.
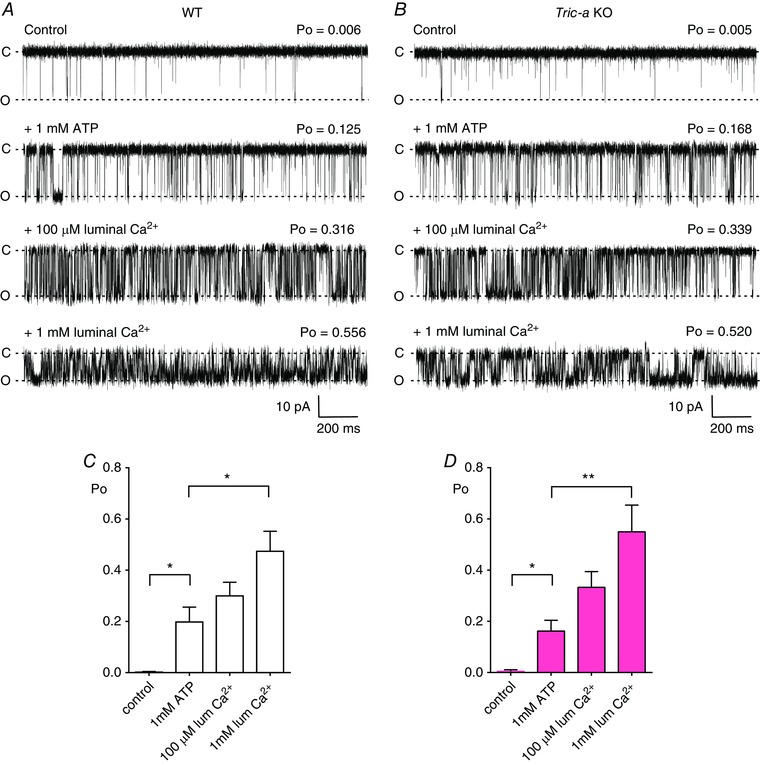
Representative single‐channel recordings of RyR1 from WT (A) and Tric‐a KO (B) mice under control conditions (10 μm cytosolic and luminal Ca2+), after subsequent addition of 1 mm cytosolic ATP (second traces) and subsequent increasing concentrations of luminal Ca2+ (100 μm and 1 mm as indicated). The bar charts below illustrate the mean data for RyR1 channels from WT (white) (C) and Tric‐a KO (pink) (D) skeletal muscle under control conditions, in the presence of ATP and increasing concentrations of luminal Ca2+. Values are the mean ± SEM (n = 6–10; * P < 0.05, ** P < 0.01). The holding potential was −30 mV. O and C indicate the open and closed channel levels, respectively.
We used [3H]ryanodine binding to the SR from WT and Tric‐a KO mice to examine the responses of populations of RyR1 channels in their native membranes to regulatory ligands (Fig. 2). The [Ca2+] concentration response relationship showed no difference in sensitivity to activating levels of Ca2+ but suggested that channels from Tric‐a KO mice are more sensitive to inhibition by high [Ca2+] (Fig. 2 A). Caffeine sensitizes RyR channels to activation by cytosolic Ca2+ (Holmberg & Williams 1990; Sitsapesan & Williams 1990) and we found that caffeine stimulated [3H]ryanodine binding to a similar extent in SR vesicles isolated from Tric‐a KO and WT tissue (Fig. 2 B). To examine the sensitivity of RyR to adenine nucleotides more thoroughly, we investigated the effects of adenine on [3H]ryanodine binding to SR vesicles. Adenine binds to the same sites as ATP on RyR channels (Rousseau et al. 1988; Chan et al. 2000; 2003) but cannot phosphorylate proteins and so the use of this compound allows an investigation of the response of the RyR channels to the direct effects of an agent binding to the adenine nucleotide‐binding sites on RyR without the complication of phosphorylation. SR vesicles contain a mix of many kinases that can be activated by ATP; thus, [3H]ryanodine binding studies cannot distinguish between the action of ATP as a reversible activator of RyR (by direct interaction with the adenine nucleotide binding sites on RyR) and the action of ATP with respect to inducing the phosphorylation of RyR or closely associated proteins. Adenine stimulated [3H]ryanodine binding to WT and Tric‐a KO SR to a similar extent (Fig. 2 C), confirming the results shown in Fig. 1 A and indicating that the interactions of ATP/adenine nucleotides with RyR are not altered in Tric‐a KO mice. We did find, however, that Mg2+ was significantly more effective at inhibiting the binding of [3H]ryanodine to Tric‐a KO SR than to WT SR (Fig. 2 D).
Figure 2. The effects of Ca2+, caffeine, adenine and Mg2+ on [3H]ryanodine binding to WT and Tric‐a KO skeletal muscle membrane vesicles.
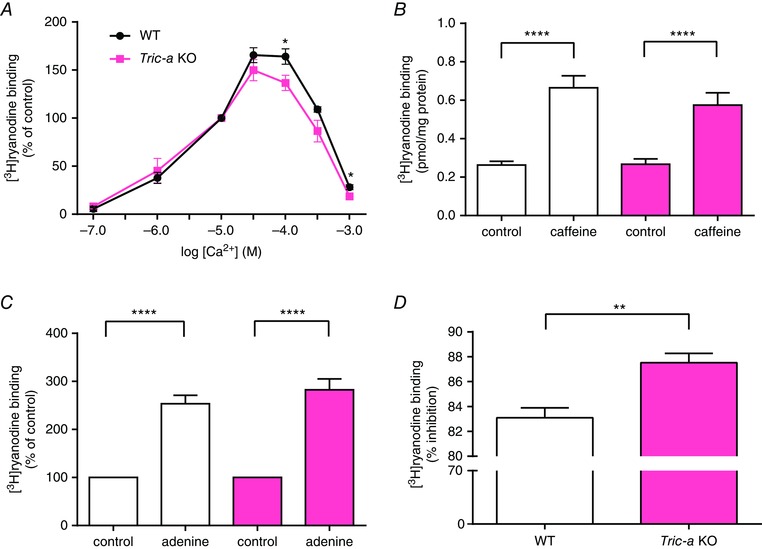
(A), stimulation of [3H]ryanodine binding to WT and Tric‐a KO membrane vesicles by Ca2+. Free [Ca2+] was adjusted by EGTA and CaCl2 solutions according to the Maxchelator software (http://maxchelator.stanford.edu). Each point is the mean ± SEM (n = 7–9; * P < 0.05). Stimulation of [3H]ryanodine binding by caffeine (10 mm; n = 5 or 6) (B) and adenine (1 mm; n = 8) (C) was similar for WT and Tric‐a KO skeletal muscle membrane vesicles; n = 4; **** P < 0.0001). The results in (A) and (C) are expressed as a percentage of the control binding at 10 μm Ca2+ for each genotype. (D), percentage inhibition of binding at 100 μm Ca2+ by 1 mm Mg2+. Mg2+ was significantly more effective at inhibiting [3H]ryanodine binding to skeletal muscle membrane vesicles from Tric‐a KO than from WT mice (n = 7; ** P < 0.01). Tric‐a KO data are shown in pink.
We therefore investigated whether altered regulation of RyR by Mg2+ was also manifest at the single‐channel level. With cytosolic Ca2+ as the sole activator, RyR Po is extremely low and variable, making comparisons of Mg2+ inhibition between the two groups of channels difficult and so we examined Mg2+ inhibition in the presence of ATP where Po is higher because we have shown that the response to adenine nucleotides is not affected in channels from Tric‐a KO mice. We used millimolar luminal Ca2+ (50 mm) as the permeant ion to induce optimum channel activity and the Po of RyR from WT and Tric‐a KO tissue with 10 μm free Ca2+ as sole activator was similar (Fig. 3 A and B, top traces), in agreement with the experiments with K+ as the permeant ion (Fig. 1 A and B). The representative traces show that the Po of RyR from Tric‐a KO mice was much lower than that of WT channels after adding 1 mm Mg2+ in the presence of 3 mm ATP, confirming the hypothesis that Mg2+ inhibition is more pronounced. Washout of Mg2+/ATP from the cytosolic chamber reversed Po back to control levels in both groups of channel (Fig. 3, bottom traces) demonstrating that the ATP‐induced increase in Po was not caused by phosphorylation of the channels by an endogenous kinase. The mean data are shown in Fig. 3 C.
Figure 3. Comparison of the effects of Mg2+/ATP on the gating of RyR1 from WT and Tric‐a KO mice with Ca2+ as the permeant ion.
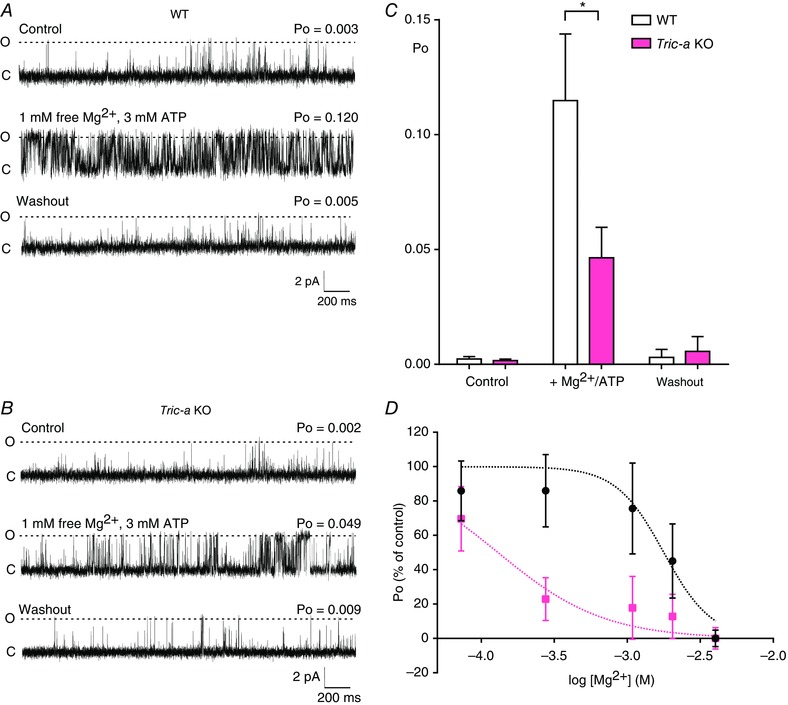
Representative RyR single‐channel recordings obtained from WT (A) and Tric‐a KO (B) mice under control conditions (top traces, 10 μm cytosolic Ca2+) in the presence of 1 mm free Mg2+/3 mm ATP/10 μm cytosolic Ca2+ (middle traces) and after washout of the Mg2+/ATP back to control conditions (bottom traces). The holding potential was 0 mV. O and C indicate the open and closed channel levels, respectively. C, mean Po values for RyR1 derived from WT (white) and Tric‐a KO mice (pink) under control conditions and in the presence of and after washout of Mg2+/ATP (n = 14 for WT; n = 22 for KO; * P < 0.05). D, single RyR1 channels from WT (black) or Tric‐a KO (pink) mice were activated with 10 μm cytosolic free Ca2+ and 1 mm ATP. The inhibition of Po with increasing concentrations of cytosolic Mg2+ was monitored and expressed as a percentage of the initial Po. Data points indicate the mean ± SEM Po for n = 12–18 for WT and n = 5–11 for Tric‐a KO channels. Dashed lines are the Hill equation fits to the data. IC50 values were 1.77 mm for WT and 0.13 mm for Tric‐a KO.
To investigate whether the affinity of Mg2+ for RyR1 was altered in the Tric‐a KO mice, we activated single RyR1 channels with ATP first and then increased cytosolic [Mg2+]. The data shown in Fig. 3 D demonstrate that much lower concentrations of Mg2+ (WT: IC50 = 1.77 mm; Tric‐a KO: IC50 = 0.13 mm) were required to inhibit the channels from Tric‐a KO than from WT skeletal muscle, indicating that RyR1 affinity for Mg2+ is increased in Tric‐a KO skeletal muscle.
It was previously reported that a change in Mg2+ inhibition of RyR1 can result when the channels are phosphorylated by PKA (Hain et al. 1994). We therefore examined whether there is a pre‐existing increased level of phosphorylation of RyR from Tric‐a KO tissue that could affect Po by investigating whether the phosphatase, PP1, could alter the gating of RyR from Tric‐a KO muscle. PP1 was added in buffer containing Mn2+, which could affect RyR activity. Therefore, after incorporation of channels into the bilayer with 10 μm Ca2+ as the sole channel activator, PP1 (5 units) was added to the cytosolic channel side and incubated for 10 min before washout of the PP1 and buffer back to control conditions (Fig. 4 A and B). There was no significant effect on channel gating either for channels from WT or Tric‐a KO tissue, indicating that no extra pre‐existing phosphorylation of these channels influenced the action of Mg2+/ATP on RyR from Tric‐a KO mice. The lack of effect of PP1 was not the result of an inadequate experimental protocol or inactive PP1 preparation because the same protocol was used to reverse the effects of phosphorylation of RyR (Fig. 5).
Figure 4. Effect of PP1 incubation on the gating of RyR1 from WT and Tric‐a KO mice with Ca2+ as the permeant ion.
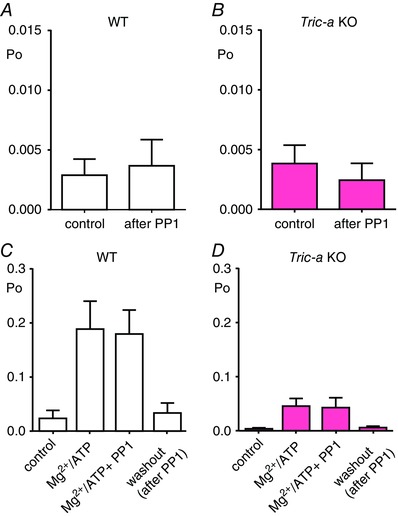
RyR1 channel activity from WT (white) (A) and Tric‐a KO mice (pink) (B) in the presence of 10 μm cytosolic Ca2+ as sole channel activator before (control) and after 5 units of PP1 was added to the cytosolic chamber in the buffer from the supply company (New England Biolabs) for 10 min before washout of the cytosolic chamber back to control conditions in 10 μm cytosolic Ca2+ (after PP1). In addition, 5 units of PP1 was also added to RyR1 channels from WT (white) (C) and Tric‐a KO mice (pink) (n = 5) (D) in the presence of 1 mm free Mg2+/3 mm ATP/10 μm cytosolic Ca2+. The bar charts are labelled as control (10 μm cytosolic Ca2+ as sole channel activator), Mg2+/ATP (in the presence of 1 mm free Mg2+/3 mm ATP/10 μm cytosolic Ca2+), Mg2+/ATP + PP1 (5 units of PP1 was added to the cytosolic chamber in the buffer from New England Biolabs for 10 min in the presence of 1 mm free Mg2+/3 mm ATP/10 μm cytosolic Ca2+) and washout, after PP1 (10 μm cytosolic Ca2+ only) (n = 8 or 9).
Figure 5. Effect of PKA‐dependent phosphorylation on the Ca2+‐dependence of gating of RyR1 from WT and Tric‐a KO mice with Ca2+ as the permeant ion.
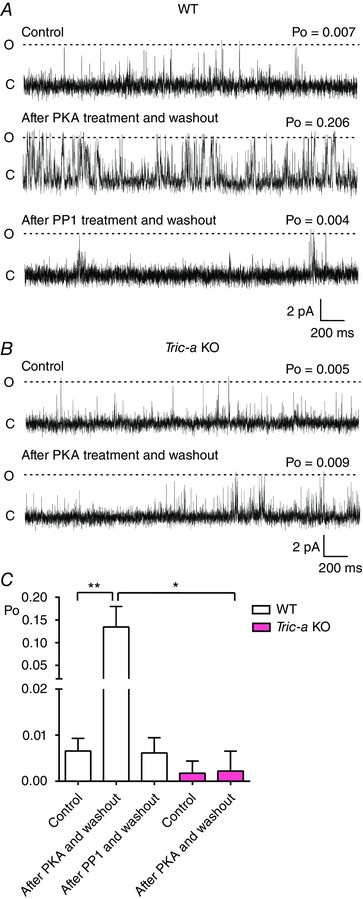
RyR1 channel activity from WT (A) and Tric‐a KO (B) mice in the presence of 10 μm cytosolic Ca2+ as sole channel activator (Control, top traces), after washout of a 10 min treatment of 10 units of PKA, 3 mm ATP and 1 mm free Mg2+ (after PKA treatment and washout, second traces), and after washout of a 10 min treatment with 5 units of PP1 added to the cytosolic chamber in the buffer from New England Biolabs (after PP1 treatment and washout). Holding potential was 0 mV. O and C indicate the open and closed channel levels, respectively. C, mean Po of the RyR1 channels from WT (white) and Tric‐a KO mice (pink) under control conditions (control), after washout of PKA (after PKA and washout) and after washout of PP1 (after PP1 and washout) (n = 10–15; * P < 0.05, ** P < 0.01).
We also investigated whether PP1 could affect the reversible activation of RyR caused by Mg2+/ATP. Figure 4 C and D demonstrate that, in the presence of PP1, the blunted ability of RyR from Tric‐a KO muscle to respond to Mg2+/ATP is unchanged, providing further evidence that there is no pre‐existing altered phosphorylation state of RyR channels from Tric‐a KO mice that affects gating.
We next examined whether the response of RyR to phosphorylation was affected in mice devoid of Tric‐a. We recorded Po with 10 μm Ca2+ as the sole channel activator (Fig. 5). PKA (10 units), 3 mm ATP and 1 mm free Mg2+ were then added to the cytosolic chamber with the free [Ca2+] maintained at 10 μm. After 10 min of incubation, we then perfused away the PKA, Mg2+ and ATP back to the control conditions with 10 μm Ca2+ as sole activator. The typical response to phosphorylation is shown in the second traces. Note the irreversible increase in Po in the channels from WT mice, whereas the channels from Tric‐a KO mice exhibit no observable change in Po compared to controls. Figure 5 C shows the mean data. To test whether the increased Po occurring after PKA incubation was a result of phosphorylation of the channels, we added PP1 to the cytosolic chamber. PP1 reversed the actions of PKA incubation, demonstrating that the activation caused by PKA in WT channels was indeed caused by phosphorylation.
There is no commercially available antibody that recognizes specific phosphorylatable residues of RyR1. Accordingly, to investigate whether RyR1 from Tric‐a KO mice show abnormal levels of phosphorylation, we used MS to identify specific phosphorylated residues and a general phospho‐(Ser/Thr) PKA substrate antibody to observe any changes to phosphorylation of the RyR1 protein. MS identified RyR1‐Ser2844 as being phosphorylated and Fig. 6 A shows that the ratio of phosphorylated:non‐phosphorylated peptides containing Ser2844 was similar for WT and Tric‐a KO samples under basal conditions and was similarly increased following incubation with PKA. The general anti‐phospho‐(Ser/Thr) antibody also detected PKA‐dependent increases in phosphorylation that were similar for WT and Tric‐a KO samples (Fig. 6 B and C).
Figure 6. Comparison of phosphorylation of RyR1 from WT and Tric‐a KO mice before and after PKA treatment.
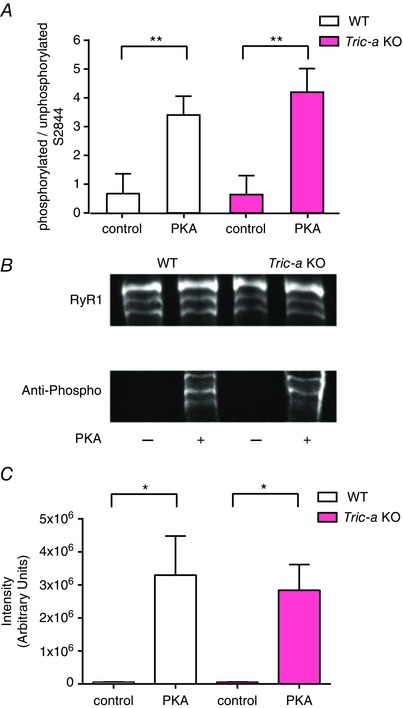
(A), ratio of phosphorylated: unphosphorylated MS intensities of RyR1 peptides containing S2844 for WT and Tric‐a KO before and after PKA treatment (n = 3 preparations for WT and n = 3 preparations for Tric‐a KO; ** P < 0.01). (B), representative immunoblots of immunoprecipitated RyR1 showing total RyR1 and PKA‐dependent phosphorylation of RyR1 in WT and Tric‐a KO mice. (C), quantification of the anti‐phospho antibody signal relative to the total RyR1 signal (34C antibody) by densitometry of the immunoblots shown in (B). Data are from four independent experiments and are presented as the mean ± SEM (* P < 0.05).
In the experiments where only a single RyR was gating in the bilayer, lifetime analysis could be performed to investigate the mechanism by which phosphorylation increased Po. With cytosolic Ca2+ as sole channel activator, the main mechanism for increasing RyR1 Po is an increase in frequency of channel opening, with little change in the duration of the open states (Smith et al. 1986); hence, an agent that sensitizes the channel to cytosolic Ca2+ will primarily increase the frequency of channel opening. Before incubation with PKA, with 10 μm cytosolic Ca2+ as sole activator, there were few events and the open lifetimes were always extremely brief. Mean open times were similar for WT (0.47 ± 0.03 ms, SEM, n = 16) and for RyR from Tric‐a KO (0.46 ± 0.04 ms, SEM, n = 10) mice. Mean closed times were more variable (i.e. because the frequency of opening determines Po under these conditions) but were also comparable: 202.4 ± 84.9 ms (SEM, n = 14) for WT and 304.8 ± 124.4 ms (SEM, n = 5) for RyR from Tric‐a KO mice. It is interesting that, although there were no significant changes in the mean open (0.56 ± 0.13 ms, SEM, n = 6) or closed (101.2 ± 54.6 ms, SEM, n = 5) times for the RyR from Tric‐a KO mice after incubation with PKA, the mean open time of the channels from WT mice was increased significantly (1.47 ± 0.40 ms, SEM, n = 12, P < 0.05) with a non‐significant trend towards a reduction in mean closed time (46.12 ± 21.26 ms, SEM, n = 12). We investigated these changes further with lifetime analysis. The representative open and closed lifetime distributions of a typical RyR channel derived from WT mice, before and after phosphorylation by PKA are illustrated in Fig. 7. Table 1 shows the time constants and areas of the pdfs fitted to the distributions for all the single channels. We found that phosphorylation of RyR channels from WT mice led to a change in the distribution of open times, such that longer openings were observed and additional long open time components were resolved. The closed lifetime distributions shifted towards an increased proportion of short closings reflecting the increased frequency of opening. No changes in lifetime distributions were observed in the channels from Tric‐a KO mice following incubation with PKA (Fig. 7). As the significant change in gating behaviour observed with RyR from WT mice was an increase in the duration of open lifetimes, this suggests that phosphorylation does not just simply sensitize the channels to cytosolic Ca2+ (where we would observe little change in open lifetime durations) but that the channel can open in a Ca2+ independent manner, similar to the effects of PKA phosphorylation for RyR2 from cardiac muscle (Carter et al. 2011). This effectively means that the phosphorylated RyR channels derived from WT skeletal muscle possess an additional mechanism for channel activation that is independent of (and additional to) the cytosolic [Ca2+], whereas the RyR from Tric‐a KO mice do not.
Figure 7. Effects of PKA‐dependent phosphorylation on open and closed lifetime distributions.
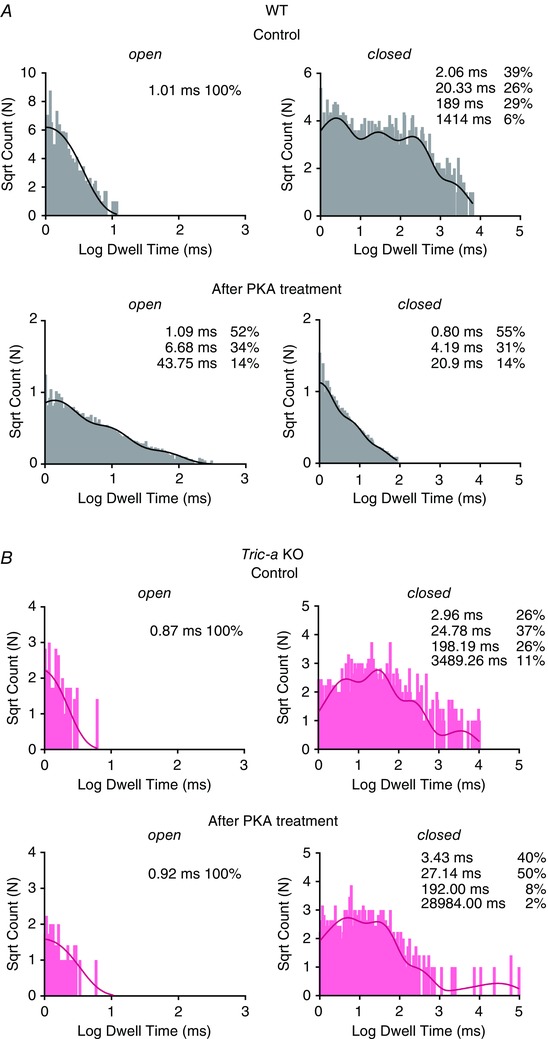
The open and closed lifetime distributions and pdfs for a typical single RyR1 channel from WT (grey) (A) and Tric‐a KO mice (pink) (B) in the presence of 10 μm cytosolic Ca2+ as sole channel activator before and after 10 min of treatment with 10 U of PKA. The best fits to the data were obtained by the method of maximum likelihood and the resulting time constants and percentage areas are shown.
Table 1.
The effect of PKA‐dependent phosphorylation on lifetime parameters
| Open | Closed | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T1 (ms) | A1 (%) | T2 (ms) | A2 (%) | T3 (ms) | A3 (%) | T1 (ms) | A1 (%) | T2 (ms) | A2 (%) | T3 (ms) | A3 (%) | T4 (ms) | A4 (%) | |
| Control WT | ||||||||||||||
| Ch. 1 | 0.67 | 100 | – | – | – | – | 0.53 | 33 | 2.90 | 32 | 23.87 | 26 | 132.00 | 9 |
| Ch. 2 | 1.01 | 100 | – | – | – | – | 2.06 | 39 | 20.33 | 26 | 189.00 | 29 | 1414.00 | 6 |
| Ch. 3 | 0.77 | 100 | – | – | – | – | 0.50 | 31 | 12.39 | 24 | 135 | 35 | 1370 | 9 |
| Ch. 4 | 0.91 | 100 | – | – | – | – | 2.15 | 72 | 14.26 | 28 | 375.42 | 1 | – | – |
| Control Tric‐a KO | ||||||||||||||
| Ch. 1 | 0.78 | 100 | – | – | – | – | 1.45 | 27 | 17.31 | 27 | 172.56 | 29 | 1613.32 | 17 |
| Ch. 2 | 0.87 | 100 | – | – | – | – | 2.96 | 26 | 24.78 | 37 | 198.19 | 26 | 3489.26 | 11 |
| Ch. 3 | 1.42 | 100 | – | – | – | – | 2.29 | 34 | 19.22 | 39 | 152.31 | 27 | – | – |
| Ch. 4 | 1.68 | 100 | – | – | – | – | 1.88 | 40 | 16.55 | 37 | 142.73 | 16 | 5789.07 | 7 |
| PKA WT | ||||||||||||||
| Ch. 1 | 1.09 | 52 | 6.68 | 34 | 43.7 | 14 | 0.80 | 55 | 4.19 | 31 | 20.9 | 14 | – | – |
| Ch. 2 | 1.1 | 89 | 7.12 | 11 | – | – | 1.31 | 65 | 6.17 | 32 | 41.60 | 3 | – | – |
| Ch. 3 | 0.65 | 80 | 3.71 | 18 | 35.9 | 2 | 0.73 | 53 | 3.51 | 32 | 19.88 | 14 | 731 | 1 |
| Ch. 4 | 0.84 | 95 | 6.73 | 5 | – | – | 0.95 | 29 | 10.31 | 21 | 60.74 | 31 | 303.32 | 19 |
| Ch. 5 | 0.8 | 94 | 5.95 | 6 | – | – | 1.26 | 80 | 7.32 | 19 | 342 | 1 | – | – |
| PKA Tric‐a KO | ||||||||||||||
| Ch. 1 | 0.81 | 100 | – | – | – | – | 4.36 | 54 | 41.26 | 30 | 369.00 | 9 | – | – |
| Ch. 2 | 0.92 | 100 | – | – | – | – | 3.43 | 40 | 27.14 | 50 | 192 | 8 | 28984 | 2 |
| Ch. 3 | 0.51 | 92 | 3.18 | 8 | – | – | 1.63 | 50 | 8.95 | 41 | 69.22 | 8 | 2533 | 1 |
Time constants (T1, T2, T3, T4) and percentage areas (A1, A2, A3, A4) are shown as obtained from maximum likelihood fitting of pdfs to open and closed lifetime distributions of single RyR channels from WT and Tric‐a KO muscle before and after PKA‐dependent phosphorylation.
We next performed a series of experiments where we added back purified TRIC‐A after incorporating RyR1 from Tric‐a KO skeletal muscle into bilayers to investigate whether the RyR1 response to Mg2+/ATP could be reversed back to that of the WT RyR1 channels. Figure 8 shows that this did not produce any significant increase in RyR1 Po, suggesting that Mg2+ inhibition was not relieved.
Figure 8. The effects of adding back purified TRIC‐A to the single‐channel function of RyR1 from Tric‐a KO mice.
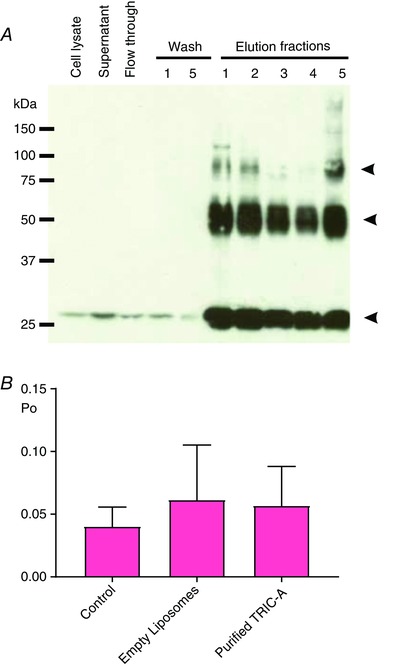
(A), immunoblot illustrating the purification of recombinant TRIC‐A overexpressed in Chinese hamster ovary (CHO) cells. Lanes, from the left: crude cell lysate, supernatant containing solubilized TRIC‐A, PA‐tag column flow through, first and fifth washing steps of the PA‐tag column and all five elution fractions containing recombinant TRIC‐A protein. TRIC‐A protein from ‘Elution 5’ was reconstituted into phosphatidylcholine liposomes for the bilayer experiments. The membrane was probed with an anti‐PA tag antibody. The arrowheads indicate the monomeric, dimeric and trimeric form of TRIC‐A. (B), Po of RyR1 channels from Tric‐a KO mice in the presence of 1 mm free Mg2+/3 mm ATP/10 μm cytosolic free Ca2+ (Control, n = 17), after addition of empty phosphatidylcholine liposomes (empty liposomes, n = 8), or after addition of phosphatidylcholine liposomes containing purified TRIC‐A (purified TRIC‐A, n = 9).
Discussion
The results of the present study demonstrate that the RyR channels derived from Tric‐a KO skeletal muscle exhibit specific gating abnormalities. In the absence of Mg2+, the response of RyR from Tric‐a KO skeletal muscle to activating cytosolic ligands such as Ca2+, ATP, adenine and caffeine does not appear to be altered, nor is the sensitivity to luminal [Ca2+] affected. However, two specific abnormalities can be observed. First, Mg2+ exerts a greater inhibitory effect on RyR from Tric‐a KO skeletal muscle than on RyR from WT muscle as indicated by both [3H]ryanodine binding and single‐channel experiments. The [3H]ryanodine binding experiments also indicate that RyR from Tric‐a KO tissue is more readily inhibited by high [Ca2+] without any significant change in sensitivity to activation by low [Ca2+]. It therefore appears that the channels have become more sensitive to inhibition via the low affinity divalent cation binding sites.
The second major abnormality is that PKA, in the presence of Mg2+/ATP, can phosphorylate RyR derived from WT mice causing an increase in Po, although there is no increase in the Po of RyR from Tric‐a KO mice, even though the phosphorylation levels of both groups of channels are similar before and after PKA‐dependent phosphorylation (Fig. 6). In the RyR from WT mice, we were able to distinguish the activating effects of phosphorylation from the reversible effects of the ATP present in the incubation medium because, even with washout of the Mg2+/ATP/PKA back to the control conditions, where 10 μm Ca2+ is sole activator (Fig. 5), Po remained high. In all cases, incubation with PP1 then reversed the increase in Po back to control levels, confirming that phosphorylation was the cause of increase in Po. Thus, phosphorylation of the channels from Tric‐a KO mice cannot be translated into an increase in Po.
The altered functional properties of the RyR from Tric‐a KO mice that we describe could provide an explanation for the disrupted skeletal muscle function that is prevalent in Tric‐a KO mice (Zhao et al. 2010). A pathologically high level of SR Ca2+ is indicated by electron dense Ca2+ deposits within the SR and the high proportion of large vacuoles that are present (Zhao et al. 2010). The high SR Ca2+ content was confirmed by the use of caffeine, which caused a larger Ca2+ transient from flexor digitorum brevis fibres isolated from Tric‐a KO mice (Zhao et al. 2010). The reduced ability of RyRs to respond to activators or phosphorylation that we observe could lead to increased levels of SR Ca2+ because the release process would be markedly inhibited. This could also explain the reduced frequency of Ca2+ sparks observed in the muscle cells from Tric‐a KO mice (Zhao et al. 2010). Because Zhou et al. 2004 have shown that increasing [Mg2+] causes a reduction in spark frequency in mammalian skeletal muscle cells (Zhou et al. 2004), the increased inhibitory effect of Mg2+ on RyR opening that we observe in channels from Tric‐a KO mice would be expected to reduce spark frequency. In the presence of Mg2+, ATP is significantly more effective at reducing the mean closed time of RyR from WT (mean closed time = 7.72 ± 3.38 ms; n = 14) than from Tric‐a KO mice (mean closed time = 90.22 ± 30.46 ms; n = 10; P < 0.01). This indicates that the frequency of opening of RyR from Tric‐a KO mice does not increase effectively in response to ATP, a factor probably influencing spark number. Accordingly, as the level of Ca2+ within the SR becomes excessively high, and, presumably, because luminal Ca2+ sensitivity does not appear to be affected, this could lead to the reported ‘alternans’ contractile behaviour in tetanic stimuli to Tric‐a KO skeletal muscle (Zhao et al. 2010). If the luminal [Ca2+] content rises sufficiently high, the increased positive influence of luminal Ca2+ on RyR Po could just outweigh the extra Mg2+ inhibition and failure of the channels to respond to phosphorylation. The higher SR/cytosol Ca2+ concentration gradient would also lead to increased Ca2+ flux during each RyR opening, thus initiating the alternans pattern. In line with this thinking, it is interesting that, although we observed dampened activity of RyR1 and increased SR Ca2+ content in the skeletal muscle of Tric‐a KO mice, the opposite effect on SR Ca2+ content was observed in a mouse model with the malignant hyperthermia mutation Y522S (Manno et al. 2013). This mutation causes leaky RyR1 channels and the measured resting SR Ca2+ content in the malignant hyperthermia skeletal muscle cells was much lower than in the WT cells.
There is widespread opinion that SR K+ channels provide the necessary charge compensation to fully balance the rapid loss of Ca2+ from the SR during the Ca2+ release process in skeletal muscle (Miller & Rosenberg 1979; Somlyo et al. 1981; Fink & Veigel 1996). This possible role was first suggested as early as 1979 (Miller & Rosenberg 1979). More recently, mathematical modelling of the ionic fluxes through RyR channels indicated that K+ currents through RyR should be amply able to compensate for the charge movements carried by Ca2+ during the Ca2+ release process (Gillespie & Fill 2008). Thus, the SR K+ channels may serve to balance monovalent cation concentration across the SR without majorly contributing to charge compensation. The results of the present study cannot shed light on the degree of counter‐current contributed by SR K+ channels, although they suggest that there may be additional mechanisms by which they could influence SR Ca2+ release. TRIC‐B is present in most cells at low levels but TRIC‐A is found at high levels in cardiac and skeletal muscle (Yazawa et al. 2007). It is calculated that, for every RyR1 in the junctional regions of skeletal muscle cells, there are approximately five TRIC‐A and one TRIC‐B channels (Pitt et al. 2010; Zhao et al. 2010). Thus, RyR and TRIC channels will jostle together in close proximity, allowing an opportunity for direct physical interactions that could modulate RyR gating in a reversible and dynamic manner. Figure 9 provides a model of the possible organisation of SR cation channels within the terminal cisternae and illustrates the void left by knocking out Tric‐a. In Tric‐a KO skeletal muscle, there is no evidence for up‐regulation of TRIC‐B to compensate (Venturi et al. 2013). At this point, it is worth considering that experiments performed in vascular smooth muscle cells and in HEK293 cells overexpressing RyR2 suggest that TRIC‐A modulates RyR channels, whereas TRIC‐B regulates inositol‐trisphosphate receptor channels (Yamazaki et al. 2011; Zhou et al. 2014). However, when we added back purified TRIC‐A channels after incorporating RyR1 from Tric‐a KO skeletal muscle into bilayers, the RyR1 response to Mg2+/ATP was not reversed back to that of the WT RyR1 channels (Fig. 8). It is possible that the interactions between TRIC‐A and RyR1 in the bilayer are delicate and require additional linking proteins or specific lipids. For example, the recently published structures of the Caenorhabditis elegans TRIC‐B channel (Yang et al. 2016), the bacterial TRIC protein (RsTRIC from Rhodobacter sphaeroides) and archaeal TRIC protein (SsTRIC from Sulfolobus solfataricus) (Kasuya et al. 2016) show that different lipids are integrated into the channels in different positions depending on the isoform.
Figure 9. Model of proposed TRIC‐A modulation of RyR in skeletal muscle.
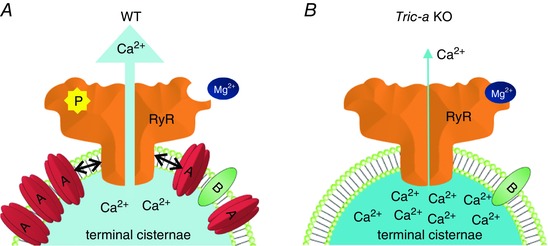
The terminal cisternae membrane of WT skeletal muscle (A) is densely packed with TRIC‐A and RyR (approximate ratio of 1:5:1 for RyR tetramer:TRIC‐A trimer:TRIC‐B trimer) providing ample opportunity for physical interactions between TRIC‐A and RyR. Tric‐a KO terminal cisternae membranes (B) are less sparsely populated with ion channels. The presence of TRIC‐A in the terminal cisternae of skeletal muscle causes conformational changes to RyR that promote dissociation of Mg2+, thus relieving the Mg2+‐induced suppression of the frequency of RyR channel opening. In the absence of TRIC‐A, Mg2+ inhibition of RyR is more pronounced and physiological activators such as ATP and luminal Ca2+ are less effective. Additionally, β‐adrenergic activation of PKA would not lead to an increase in RyR Po, as would occur in WT muscle.
In Tric‐a KO mice, RyR has only the possibility to interact with TRIC‐B (rather than TRIC‐A), although it is feasible that TRIC‐B cannot influence RyR function, as suggested previously (Zhou et al. 2014), or that any interactions influence RyR gating differently. Our experiments shown in Fig. 1 were conducted with K+ as the permeant ion. Recording RyR current fluctuations in this manner is not easy because of the multiple K+ channels that fuse with the bilayer and the excessive K+ currents can break the bilayer. Because it is rare to observe native RyR channel current fluctuations without simultaneous SR K+ channel currents, we can assume that, in those experiments where Ca2+ is the permeant ion (and so we cannot visualize K+ channel openings), there are probably also many K+ channels present in the bilayer that could influence RyR channel function. It is worth noting that there will be no ionic currents flowing through the many SR K+ channels (even when they open) in these experiments and so ionic flux through SR K+ channels cannot be a factor influencing RyR channel opening (unlike in the SR in situ) and, because the membrane is voltage clamped, counter‐current movement is irrelevant.
In summary, the experiments conducted in the present study suggest that TRIC proteins may influence RyR channel behaviour in ways additional to providing movement of monovalent cation current across the SR. The model shown in Fig. 9 summarises the potential influence of TRIC‐A on RyR gating and Ca2+ homeostasis in skeletal muscle. TRIC channels are expressed in high levels in the junctional SR and nuclear membranes and may affect the molecular architecture of these organelles and/or functionally interact with nearby proteins to directly influence SR Ca2+ movements. Thus, the TRIC proteins may influence SR Ca2+ movements by multiple mechanisms and further investigations are required to delineate the full extent of the interactions of TRIC‐A and TRIC‐B with closely positioned proteins.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
TI, MN and HT produced and characterized Tric‐a KO mice and provided tissue. SE, EV, MB, ADW, CL, DE and FO'B performed the single‐channel experiments. SE and EV analysed data and produced the figures. KW performed the [3H]ryanodine binding experiments and analysed the immunoblots. SE, EV, KW and DE isolated SR membrane vesicles. RS wrote the article. All authors discussed the results and commented on the article. All authors approved the final version of the manuscript submitted for publication.
Funding
This work was supported by the British Heart Foundation (RG/10/14/28576, PG/13/76/30353, FS/11/31/28790 and awards through the BHF Oxford 4‐Year Studentship scheme), the Oxford BHF Centre of Research Excellence (RE/08/004), and the Japan Society for the Promotion of Science (Core‐to‐core program).
References
- Blatz AL & Magleby KL (1986). Correcting single channel data for missed events. Biophys J 49, 967–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleunven C, Treves S, Jinyu X, Leo E, Ronjat M, De Waard M, Kern G, Flucher BE & Zorzato F (2008). SRP‐27 is a novel component of the supramolecular signalling complex involved in skeletal muscle excitation‐contraction coupling. Biochem J 411, 343–349. [DOI] [PubMed] [Google Scholar]
- Carter S, Pitt SJ, Colyer J & Sitsapesan R (2011). Ca2+‐dependent phosphorylation of RyR2 can uncouple channel gating from direct cytosolic Ca2+ regulation. J Membr Biol 240, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WM, Welch W & Sitsapesan R (2000). Structural factors that determine the ability of adenosine and related compounds to activate the cardiac ryanodine receptor. Br J Pharmacol 130, 1618–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WM, Welch W & Sitsapesan R (2003). Structural characteristics that govern binding to, and modulation through, the cardiac ryanodine receptor nucleotide binding site. Mol Pharmacol 63, 174–182. [DOI] [PubMed] [Google Scholar]
- Colquhoun D & Sigworth FJ (1983). Fitting and statistical analysis of single‐channel recording In Single‐channel recording, ed. Sakmann B. & Neher E, pp. 191–263. Plenum, New York, NY. [Google Scholar]
- Cox J & Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotech 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV & Mann M (2011). Andromeda: a peptide search engine integrated into the maxquant environment. J Proteome Res 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- Fink RH & Veigel C (1996). Calcium uptake and release modulated by counter‐ion conductances in the sarcoplasmic reticulum of skeletal muscle. Acta Physiol Scand 156, 387–396. [DOI] [PubMed] [Google Scholar]
- Gillespie D & Fill M (2008). Intracellular calcium release channels mediate their own countercurrent: the ryanodine receptor case study. Biophys J 95, 3706–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie D, Giri J & Fill M (2009). Reinterpreting the anomalous mole fraction effect: the ryanodine receptor case study. Biophys J 97, 2212–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hain J, Nath S, Mayrleitner M, Fleischer S & Schindler H (1994). Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from skeletal muscle. Biophys J 67, 1823–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg SRM & Williams AJ (1990). The cardiac sarcoplasmic reticulum calcium‐release channel: modulation of ryanodine binding and single‐channel activity. Biochim Biophys Acta 1022, 187–193. [DOI] [PubMed] [Google Scholar]
- Kasuya G, Hiraizumi M, Maturana AD, Kumazaki K, Fujiwara Y, Liu K, Nakada‐Nakura Y, Iwata S, Tsukada K, Komori T, Uemura S, Goto Y, Nakane T, Takemoto M, Kato HE, Yamashita K, Wada M, Ito K, Ishitani R, Hattori M & Nureki O (2016). Crystal structures of the TRIC trimeric intracellular cation channel orthologues. Cell Res 26, 1288–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarca P, Coronado R & Miller C (1980). Thermodynamic and kinetic studies of the gating behaviour of a K+‐selective channel from the sarcoplasmic reticulum membrane. J Gen Physiol 76, 397–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno C, Figueroa L, Royer L, Pouvreau S, Lee CS, Volpe P, Nori A, Zhou J, Meissner G, Hamilton SL & Rios E (2013). Altered Ca2+ concentration, permeability and buffering in the myofibre Ca2+ store of a mouse model of malignant hyperthermia. J Physiol 591, 4439–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C & Rosenberg RL (1979). A voltage‐gated cation conductance channel from fragmented sarcoplasmic reticulum. Effects of transition metal ions. Biochemistry 18, 1138–1145. [DOI] [PubMed] [Google Scholar]
- Pitt SJ, Park KH, Nishi M, Urashima T, Aoki S, Yamazaki D, Ma J, Takeshima H & Sitsapesan R (2010). Charade of the SR K+‐channel: two ion‐channels, TRIC‐A and TRIC‐B, masquerade as a single K+‐channel. Biophys J 99, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau E, LaDine J, Liu QY & Meissner G (1988). Activation of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum by caffeine and related compounds. Arch Biochem Biophys 267, 75–86. [DOI] [PubMed] [Google Scholar]
- Rubinato E, Morgan A, D'Eustacchio A, Pecile V, Gortani G, Gasparini P & Faletra F (2014). A novel deletion mutation involving TMEM38B in a patient with autosomal recessive osteogenesis imperfecta. Gene 545, 290–292. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV & Mann M (2007). In‐gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protocols 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- Sitsapesan R, Montgomery RAP, MacLeod KT & Williams AJ (1991). Sheep cardiac sarcoplasmic reticulum calcium release channels: modification of conductance and gating by temperature. J Physiol 434, 469–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R & Williams AJ (1990). Mechanisms of caffeine activation of single calcium‐release channels of sheep cardiac sarcoplasmic reticulum. J Physiol 423, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Coronado R & Meissner G (1986). Single channel measurements of the calcium release channel from skeletal muscle sarcoplasmic reticulum. J Gen Physiol 88, 573–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AV, Gonzalez‐Serratos H, Shuman H, McClellan G & Somlyo AP (1981). Calcium release and ionic changes in the sarcoplasmic reticulum of tetanized muscle: an electron probe study. J Cell Biol 90, 577–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi E, Matyjaszkiewicz A, Pitt S, Tsaneva‐Atanasova K, Nishi M, Yamazaki D, Takeshima H & Sitsapesan R (2013). TRIC‐B channels display labile gating: evidence from the TRIC‐A knockout mouse model. Pflügers Arch 465, 1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volodarsky M, Markus B, Cohen I, Staretz‐Chacham O, Flusser H, Landau D, Shelef I, Langer Y & Birk OS (2013). A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum Mutat 34, 582–586. [DOI] [PubMed] [Google Scholar]
- Yamazaki D, Komazaki S, Nakanishi H, Mishima A, Nishi M, Yazawa M, Yamazaki T, Taguchi R & Takeshima H (2009). Essential role of the TRIC‐B channel in Ca2+ handling of alveolar epithelial cells and in perinatal lung maturation. Development 136, 2355–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki D, Tabara Y, Kita S, Hanada H, Komazaki S, Naitou D, Mishima A, Nishi M, Yamamura H, Yamamoto S, Kakizawa S, Miyachi H, Miyata T, Kawano Y, Kamide K, Ogihara T, Hata A, Umemura S, Soma M, Takahashi N, Imaizumi Y, Miki T, Iwamoto T & Takeshima H (2011). TRIC‐A channels in vascular smooth muscle contribute to blood pressure maintenance. Cell Metab 14, 231–241. [DOI] [PubMed] [Google Scholar]
- Yang H, Hu M, Guo J, Ou X, Cai T & Liu Z (2016). Pore architecture of TRIC channels and insights into their gating mechanism. Nature 538, 537–541. [DOI] [PubMed] [Google Scholar]
- Yazawa M, Ferrante C, Feng J, Mio K, Ogura T, Zhang M, Lin PH, Pan Z, Komazaki S, Kato K, Nishi M, Zhao X, Weisleder N, Sato C, Ma J & Takeshima H (2007). TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 448, 78–82. [DOI] [PubMed] [Google Scholar]
- Zhao X, Yamazaki D, Park KH, Komazaki S, Tjondrokoesoemo A, Nishi M, Lin P, Hirata Y, Brotto M, Takeshima H & Ma J (2010). Ca2+ overload and sarcoplasmic reticulum instability in tric‐a null skeletal muscle. J Biol Chem 285, 37370–37376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Launikonis BS, Ríos E & Brum G (2004). Regulation of Ca2+ sparks by Ca2+ and Mg2+ in mammalian and amphibian muscle. An RyR isoform‐specific role in excitation–contraction coupling? J Gen Physiol 124, 409–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lin P, Yamazaki D, Park KH, Komazaki S, Chen SRW, Takeshima H & Ma J (2014). Trimeric intracellular cation channels and sarcoplasmic/endoplasmic reticulum calcium homeostasis. Circ Res 114, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]