

Previous clinical studies have shown that the presence of receptor tyrosine kinase (RTK) FLT3 with activating mutation, FLT3-ITD (internal tandem duplication), translates into poor prognosis in acute myeloid leukemia (AML).1 Several tyrosine kinase inhibitors (TKIs) of constitutively active FLT3 have been developed and tested in clinical trials.1 So far, these small molecule inhibitors have resulted in only modest improvement in AML patients because of, in part, the presence of leukemic blasts showing resistance against FLT3 inhibitors.2 Thus, devising an improved therapeutic strategy to overcome AML resistance to FLT3 inhibitors could improve the likelihood of achieving long-term survival of AML patients.
Tumor suppressor p53 is a critical gatekeeper for cell growth and division, and regulates a large number of downstream targets in response to various oncogenes and genotoxic stresses, ultimately suppressing tumorigenesis.3 Although mutations in the tumor suppressor p53 gene (TP53) are found in only about 5–10% of AML patients, inactivation of wild-type p53 protein frequently occurs through overexpression of its negative regulatory molecule, murine double minute 2 (MDM2), which targets p53 for ubiquitin-mediated degradation.3 On genotoxic stimuli such as irradiation, p53 is phosphorylated and the interaction between p53 and MDM2 is blocked, which leads to an increased level of p53 and eventually inhibition of the growth of tumor cells. Recent studies have shown that FLT3-ITD blocks p53 activation in AML, and restoration of p53 activities sensitizes AML blasts to FLT3 inhibitor treatment.4,5 In addition, BA/F3 cells expressing FLT3-ITD (BA/F3-FLT3-ITD) were resistant to FLT3 inhibitor PKC412 on being transfected with lentivirus encoding shRNA targeting p53.4 This suggests that FLT3-ITD+ AML cells acquire resistance to FLT3 inhibitors, at least in part, by inactivation of p53, which provides a rationale to target p53 and its regulatory network for overcoming AML resistance to FLT3 inhibitors.
First, we tested whether activation of p53 by Nutlin-3 could be exploited for overcoming resistance of FLT3-ITD AML cells to FLT3 inhibitors. Nutlin-3 is a highly specific, non-genotoxic MDM2 antagonist and functions as a competitive inhibitor of the p53-MDM2 interaction.6 As shown in Figure 1a, Nutlin-3 was able to significantly suppress the growth of the AML cell line MOLM13-R-PKC412 that is resistant to the FLT3 inhibitor PKC412. Another PKC412-resistant AML cell line MV4-11R-PKC412 that was generated from PKC412-sensitive MV4-11 harboring mutant form of p53 (ref.7) was resistant to Nutlin-3. Consistently, Nutlin-3 increased the level of p53 and regulated the expression of well-known p53 target genes, p21 and Axl,3 in MOLM13-R-PKC412 (wild-type p53) but not in MV4-11R-PKC412 (mutant p53) (Figure 1b). In primary AML blasts from patients that showed intrinsic resistance to the FLT3 inhibitor PKC412, Nutlin-3 was able to suppress the growth of AML blasts (Figure 1c). In those AML patient blasts, the levels of p53 and p21 were increased by Nutlin-3 (Figure 1d). In addition, bone marrow AML blasts from double knock-in mice co-expressing the partial tandem duplication (PTD) of Mll (PTD/wt) and the Flt3-ITD (ITD/wt)8 have been previously shown to harbor wild-type p53 and to be intrinsically resistant to the FLT3 inhibitor PKC412 (unpublished data). As shown in Figure 1e, Nutlin-3, but not PKC412, could inhibit the growth of bone marrow blasts from the leukemic mouse model. Altogether, these results suggest that Nutlin-3 can abrogate AML resistance to FLT3 inhibition and it requires wild-type p53.
Figure 1.
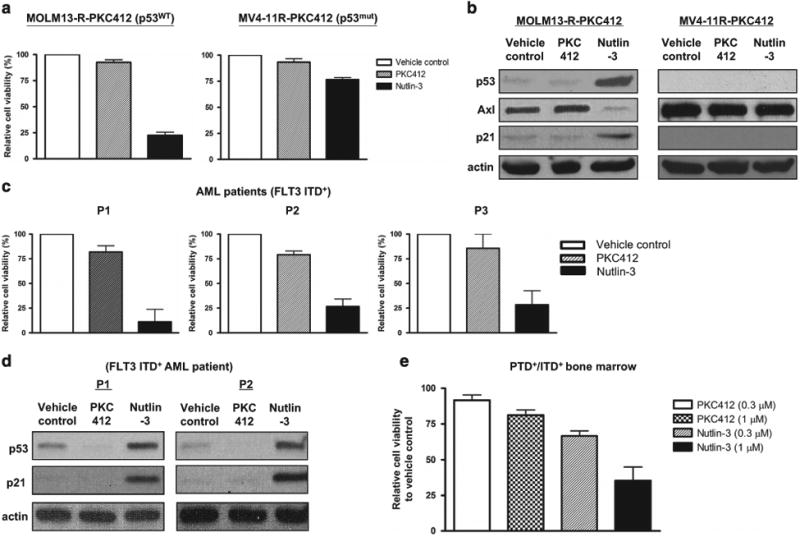
Nutlin-3, an MDM2 antagonist, suppresses the growth of FLT3 inhibitor-resistant AML cells harboring wild-type p53. (a) FLT3 inhibitor PKC412-resistant AML cell line MOLM13-R-PKC412 (left) or MV4-11 R-PKC412 (right) was treated with vehicle control, PKC412 (100 nM) or Nutlin-3 (2.5 μM) for 72 h. Cell viability was measured by MTS Tetrazolium salt cell proliferation assay. Viability of the cells treated with vehicle control was set as 100%. The graph indicates mean ± s.e.m. from three separate experiments. (b) MOLM13-R-PKC412 (left) or MV4-11 R-PKC412 (right) was treated with vehicle control, PKC412 (100 nM) or Nutlin-3 (2.5 μM) for 12 h. Cells were then harvested and subject to immunoblotting to detect p53, Axl and p21. Actin was used as a loading control. This is the representative of two independent experiments. (c) Primary AML blasts from three patients (P1 to P3) were treated with vehicle control, PKC412 (1 μM) or Nutlin-3 (2.5 μM) for 72 h. Cell viability was measured as described above in a. (d) AML cells from two patients (P1 and P2) as described in c were treated with vehicle control, PKC412 (1 μM) or Nutlin-3 (2.5 μM) for 12 h. Cells were then harvested and subject to immunoblotting to detect p53 and p21. (e) Bone marrow AML blasts from double knock-in mice (PTD+/ITD+) co-expressing the PTD of Mll (PTD/wt) and the Flt3-ITD (ITD/wt) were treated with either PKC412 or Nutlin-3 at the indicated concentrations for 72 h. Cell viability was measured and viability of the cells treated with vehicle control was set as 100%. The graph indicates mean ± s.d. from two repeated experiments.
Cbl-b is a member of a highly conserved E3 ubiquitin ligases Cbl family and negatively regulates signaling of activated receptor tyrosine kinases, including FLT3.9 In fact, mice with a loss-of-function mutation of Cbl-b (Cbl-b-C373A) have been shown to exhibit augmented FLT3 signaling and develop a myeloproliferative disease that progresses to leukemia.10 Cbl-b was expressed in MOLM13-R-PKC412 (which was sensitive to Nutlin-3) but not in MV4-11R-PKC412 (which was resistant to Nutlin-3) (Figure 2a). Furthermore, Cbl-b expression was up-regulated in FLT3 inhibitor-resistant MOLM13-R-PKC412 when compared with its parental, FLT3 inhibitor-sensitive MOLM13 (Supplementary Figure S1). These results imply that Cbl-b might be a molecule mediating the therapeutic effects of Nutlin-3. To test the hypothesis, we generated a PKC412-resistant MOLM13-R-PKC412 AML cell line stably transfected with lentivirus encoding shRNA targeting Cbl-b. Although MOLM13-R-PKC412 transfected with control shRNA was sensitive to Nutlin-3, the MOLM13-R-PKC412 transfected with Cbl-b shRNA was largely resistant to Nutlin-3 (Figure 2b). In addition, activation of p53 was almost completely impaired in MOLM13-R-PKC412 transfected with Cbl-b shRNA (Figure 2c). These data indicate that Cbl-b is critical for activation of the p53 pathway by Nutlin-3 and in turn suppression of the growth of FLT3 inhibitor-resistant AML cells.
Figure 2.
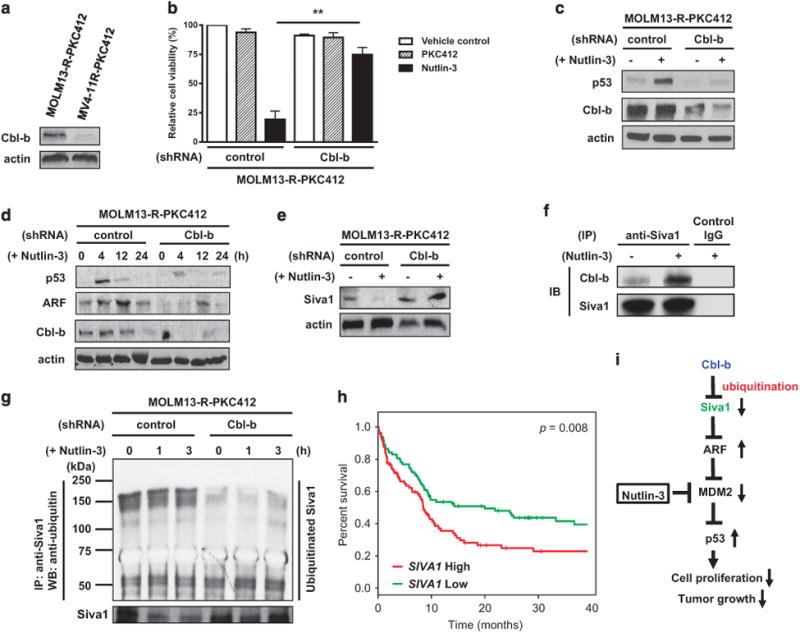
Cbl-b activates the ARF–p53 axis by ubiquitinating Siva1, an ARF E3 ubiquitin ligase. (a) Expression of Cbl-b in MOLM13-R-PKC412 and MV4-11R-PKC412 was detected by immunoblotting. This is representative of two separate experiments. (b) MOLM13-R-PKC412 AML cell line stably transfected with either control or Cbl-b shRNA was treated with vehicle control, PKC412 (100 nM) or Nutlin-3 (2.5 μM) for 72 h. Cell viability was then measured and viability of the cells treated with vehicle control was set as 100%. The graph indicates mean ± s.e.m. from three independent experiments. **P <0.01. (c) MOLM13-R-PKC412 AML cell line stably transfected with either control or Cbl-b shRNA was treated with vehicle control ( − ) or Nutlin-3 (2.5 μM) (+) for 12 h. Cells were then harvested and immunoblotting was performed to detect p53. Cbl-b was used to validate shRNA-mediated gene knockdown. This is the representative of three separate experiments. (d) MOLM13-R-PKC412 AML cell line stably transfected with either control or Cbl-b shRNA was treated with Nutlin-3 (2.5 μM) for the indicated time periods. Cells were then harvested and subject to immunoblotting to detect p53 and ARF. This is the representative of two separate experiments. (e) MOLM13-R-PKC412 AML cell line stably transfected with either control or Cbl-b shRNA was treated with vehicle control or Nutlin-3 (2.5 μM) for 4 h. Cells were then harvested and subject to immunoblotting to detect Siva1. Actin was used as a loading control. This is representative of two separate experiments. (f) MOLM13-R-PKC412 AML cell line was treated with Nutlin-3 (2.5 μM) for 4 h. Harvested cells were lysed with RIPA buffer, which was followed by immunoprecipitation using anti-Siva1 antibody or control IgG. Immunoprecipitated proteins were then subject to immunoblot to detect Cbl-b and Siva1. (g) MOLM13-R-PKC412 AML cell line stably transfected with either control or Cbl-b shRNA was treated with Nutlin-3 (2.5 μM) for the indicated time periods. Cells were lysed and whole cell lysates were immunoprecipitated using anti-Siva1 antibody, which was followed by immunoblotting to assess ubiquitination using anti-ubiquitin antibody. This is representative of two independent experiments. (h) Kaplan–Meier survival curve from human AML data set (GSE12417) generated using SurvExpress and gene expression from the SIVA1 probe 203489_at. SIVA1 expression levels: low, green; high, red. Statistical significance was analyzed by log-rank test. (I) Model of regulation of the Siva1–ARF–p53 axis by E3 ubiquitin ligase Cbl-b in AML. Cbl-b negatively regulates Siva1 by ubiquitination and subsequent degradation of Siva1, which is followed by stabilization of ARF. This in turn downregulates MDM2, thereby induction of p53 and activation of its downstream targets for the suppression of tumor growth
Another tumor suppressor, ARF for alternative reading frame, acts as a major positive regulator of p53 by binding to and rapidly degrading MDM2, thereby stabilizing p53.11 As shown in Figure 2d, the induction of ARF, as well as of p53, was greatly reduced in MOLM13-R-PKC412 lacking Cbl-b. This indicates that Cbl-b is crucial for activation of the ARF–p53 axis in FLT3 inhibitor-resistant AML cells.
Previous studies have shown that ARF is negatively regulated by the E3 ubiquitin ligase Siva1. Siva1 negatively regulates p53 activities by impeding interaction between ARF and MDM2 as well as by ubiquitination and degradation of ARF.12 Knockdown of Siva1 stabilizes ARF and p53, whereas ectopic expression of Siva1 destabilizes ARF and p53.12 As ubiquitin ligases are frequently regulated by other ubiquitin ligases, we hypothesized that E3 ubiquitin ligase Cbl-b may ubiquitinate Siva1 and accelerate its degradation, thereby in turn activating the ARF–p53 axis. In fact, we have demonstrated that Nutlin-3 could downregulate Siva1 protein, and proteasome inhibitor MG132 was able to largely abrogate it (Supplementary Figure S2). This suggests that down-regulation of Siva1 by Nutlin-3 is mediated through the proteasome pathway and subsequent degradation. As shown in Figure 2e, knockdown of Cbl-b led to an increase in the level of the Siva1 protein, which indicates that Cbl-baccelerates degradation of Siva1. Treatment with Nutlin-3 increased the physical association of Cbl-b with Siva1 (Figure 2f) and promoted Lys48-linked, but not Lys63-linked, polyubiquitination of Siva1 (Supplementary Figure S3). In the absence of Cbl-b, ubiquitination of Siva1 was diminished (Figure 2g). Collectively, these results suggest that Cbl-b ubiquitinates Siva1, which is followed by degradation of Siva1.
Our data in this study have demonstrated that Cbl-b down-regulates Siva1 through ubiquitination, which in turn results in induction of p53. It led us to hypothesize that the expression level of Siva1 may be correlated with prognosis of AML patients. Using a publicly available human AML data set, we analyzed the association between the expression of SIVA1 and survival of AML patients. As shown in Figure 2h, AML patients with higher expression level of SIVA1 showed significantly worse survival compared with those with lower expression level of SIVA1. These data suggest that SIVA1 may be an adverse prognostic marker for survival of AML patients.
In this study, we have demonstrated that activation of the p53 pathway by Nutlin-3 was able to overcome AML resistance to FLT3 inhibition. We have also shown that the E3 ubiquitin ligase Cbl-b is crucial for activation of the p53 pathway through ubiquitinating and promoting degradation of Siva1, the E3 ubiquitin ligase targeting ARF, a positive regulator of p53. On the basis of our data presented in the study, we propose the model (Figure 2i) that Cbl-b negatively regulates Siva1 by ubiquitination and subsequent degradation of Siva1, which is followed by stabilization of ARF. This in turn downregulates MDM2, thereby promoting the induction of p53 and activation of its downstream targets.
Although Nutlin-3 targets MDM2 to activate the p53 pathway, previous studies suggest that the sensitivity of tumor cells to Nutlin-3 is largely determined by the expression of tumor suppressor ARF. In tumor cells, knockdown of ARF (p14ARF) attenuated the response to Nutlin-3, which was evidenced by increased tumor cell viability as well as reduction of the expression of p53 target genes.13 These data are corroborated by the data from the p53 knock-in mouse model of lymphoma showing that p53 reactivation strongly selects for the emergence of p53-resistant tumors through inactivation of either p53 or ARF.14 These results may explain our data showing that AML cells lacking Cbl-b were resistant to Nutlin-3. Without Cbl-b, the expression of ARF was greatly diminished and loss of ARF attenuated the sensitivity of AML cells to Nutlin-3 (Figures 2b and d). Furthermore, our data indicate that the occurrence of aberrations in ARF needs to be taken in consideration for the development of therapies harnessing the p53 pathway against AML.
Previous studies have shown that Siva1 negatively regulates p53 expression and regulates cell cycle progression and proliferation in an ARF/p53-dependent manner.12 This suggests that Siva1 promotes tumorigenesis. Indeed, conditional knockout of SIVA1 inhibited tumorigenesis in KRAS-driven non-small cell lung cancer (NSCLC) mouse model, suggesting that Siva1 facilitates tumorigenesis.15 Similarly, SIVA1 knockdown in both mouse and human NSCLC cell lines decreased proliferation and transformation. Consistent with this pro-tumorigenic role for Siva1, a higher level of SIVA1 expression correlated with reduced NSCLC patient survival.15 These data also support the notion that SIVA1 can be an adverse prognostic factor for AML (Figure 2h).
In summary, our study here has shown that reactivation of p53 by Nutlin-3, an MDM2 antagonist, could be effective against FLT3 TKI-resistant AML. RG7112, an MDM2 antagonist that belongs to the Nutlin family of compounds, has been evaluated in a Phase I clinical trial in AML.16 A second generation p53-MDM2 inhibitor, RG7388, is currently undergoing clinical evaluation. If proven to be safe and relatively non-toxic, further evaluation of these agents should be undertaken alone or in combination with FLT3 TKIs in AML patients that showed resistance to FLT3 TKIs, along with assessment of alterations in the Siva1–ARF–p53 axis.
Supplementary Material
Acknowledgments
We thank Drs James D Griffin (Dana-Farber Cancer Institute, Boston, MA, USA) and Christian Thiede (TU Dresden, Germany) for providing MOLM13-R-PKC412 and MV4-11 R-PKC412 AML cell lines, respectively. We also thank the OSUCCC Leukemia Tissue Bank Shared Resource for providing AML patient samples. This work was supported by the National Cancer Institute grants (CA016058, CA09338, CA031946, CA140158, CA89341, R35 CA210087 to MAC and CA138744 to SDB).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Sudhindra A, Smith CC. FLT3 inhibitors in AML: are we there yet? Curr Hematol Malig Rep. 2014;9:174–185. doi: 10.1007/s11899-014-0203-8. [DOI] [PubMed] [Google Scholar]
- 2.Weisberg E, Barrett R, Liu Q, Stone R, Gray N, Griffin JD. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist Updat. 2009;12:81–89. doi: 10.1016/j.drup.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 4.Sasca D, Hahnel PS, Szybinski J, Khawaja K, Kriege O, Pante SV, et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood. 2014;124:121–133. doi: 10.1182/blood-2013-11-538819. [DOI] [PubMed] [Google Scholar]
- 5.Li L, Osdal T, Ho YW, Chun S, McDonald T, Agarwal P, et al. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell. 2014;15:431–446. doi: 10.1016/j.stem.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 7.Zauli G, Celeghini C, Melloni E, Voltan R, Ongari M, Tiribelli M, et al. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica. 2012;97:1722–1730. doi: 10.3324/haematol.2012.062083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zorko NA, Bernot KM, Whitman SP, Siebenaler RF, Ahmed EH, Marcucci GG, et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood. 2012;120:1130–1136. doi: 10.1182/blood-2012-03-415067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liyasova MS, Ma K, Lipkowitz S. Molecular pathways: Cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin Cancer Res. 2015;21:1789–1794. doi: 10.1158/1078-0432.CCR-13-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oshikawa G, Nagao T, Wu N, Kurosu T, Miura O. c-Cbl and Cbl-b ligases mediate 17-allylaminodemethoxygeldanamycin-induced degradation of autophosphorylated Flt3 kinase with internal tandem duplication through the ubiquitin proteasome pathway. J Biol Chem. 2011;286:30263–30273. doi: 10.1074/jbc.M111.232348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen D, Yoon JB, Gu W. Reactivating the ARF-p53 axis in AML cells by targeting ULF. Cell Cycle. 2010;9:2946–2951. doi: 10.4161/cc.9.15.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Zha M, Zhao X, Jiang P, Du W, Tam AY, et al. Siva1 inhibits p53 function by acting as an ARF E3 ubiquitin ligase. Nat Commun. 2013;4:1551. doi: 10.1038/ncomms2533. [DOI] [PubMed] [Google Scholar]
- 13.Van Maerken T, Rihani A, Dreidax D, De Clercq S, Yigit N, Marine JC, et al. Functional analysis of the p53 pathway in neuroblastoma cells using the small-molecule MDM2 antagonist nutlin-3. Mol Cancer Ther. 2011;10:983–993. doi: 10.1158/1535-7163.MCT-10-1090. [DOI] [PubMed] [Google Scholar]
- 14.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Van Nostrand JL, Brisac A, Mello SS, Jacobs SB, Luong R, Attardi LD. The p53 target gene SIVA enables non-small cell lung cancer development. Cancer Discov. 2015;5:622–635. doi: 10.1158/2159-8290.CD-14-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andreeff M, Kelly KR, Yee KW, Assouline SE, Strair R, Popplewell L, et al. Results of the Phase 1 Trial of RG7112, a Small-molecule MDM2 Antagonist in Leukemia. Clin Cancer Res. 2015;22:868–876. doi: 10.1158/1078-0432.CCR-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.