

Abstract
To counteract damage to our genomes, numerous endo‐ and exonucleases incise the DNA backbone to remove damaged and aberrant DNA structures. It is imperative that such incisions be very tightly controlled, as unwanted DNA breaks are a key source of genome instability. Two new papers in The EMBO Journal shed light on how the activity of one such nuclease—ERCC1‐XPF, an enzyme involved in various DNA repair pathways—is regulated to perform incision in the vicinity of DNA interstrand crosslinks.
Subject Categories: DNA Replication, Repair & Recombination
The maintenance of genome stability involves a bewildering array of pathways that remove chemically altered sites in DNA and resolve obstructive secondary DNA structures from our genomes. One of the most critical, yet also potentially most harmful class of enzymes involved are structure‐specific endonucleases (SSEs; Dehe & Gaillard, 2017). SSEs are often the first enzyme in a repair pathway that covalently alter the primary DNA structure by cleaving the phosphodiester backbone, thus representing the first irreversible step in a given DNA repair pathway. The proper regulation of SSEs is therefore of utmost importance to avoid unlicensed DNA incisions that result in genomic injury. It seems that the solution evolution has found to address this issue is that SSEs are very inefficient enzymes until they undergo specific recruitment and activation to cut appropriate DNA substrates.
One of first known and still most fascinating SSEs is the heterodimeric ERCC1‐XPF (Fig 1A). It is involved in several distinct pathways, including nucleotide excision repair (NER), interstrand crosslink (ICL) repair, the single‐stranded annealing (SSA) branch of homologous recombination, and telomere maintenance; it further has possible backup roles in repairing oxidative damage and DNA breaks with damaged ends. In line with its many functions, mutations in XPF are associated with several distinct human syndromes: Xeroderma pigmentosum (XP), Fanconi anemia (FA), Cockayne syndrome (CS), and a premature aging syndrome termed XFE, while a mutation in ERCC1 can cause cerebro‐oculo‐facio‐skeletal syndrome (COFS). XP, a disorder characterized by an extremely high incidence of skin cancer, is caused by a defect in NER and XP patients are unable to repair UV‐induced DNA lesions in the skin. FA, a complex disorder characterized by congenital abnormalities, bone marrow failure, and cancer predisposition, is caused by an inability to properly handle stalled replication forks, in particular in response to ICLs. While we understand that XP and FA are caused by NER and ICL repair defects, respectively, it is less clear whether CS, COFS, and XFE are caused by a combined defect in both NER and ICL, and/or in any of the additional pathways ERCC1‐XPF contributes to.
Figure 1. Interactions of ERCC1‐XPF in NER and ICL repair.
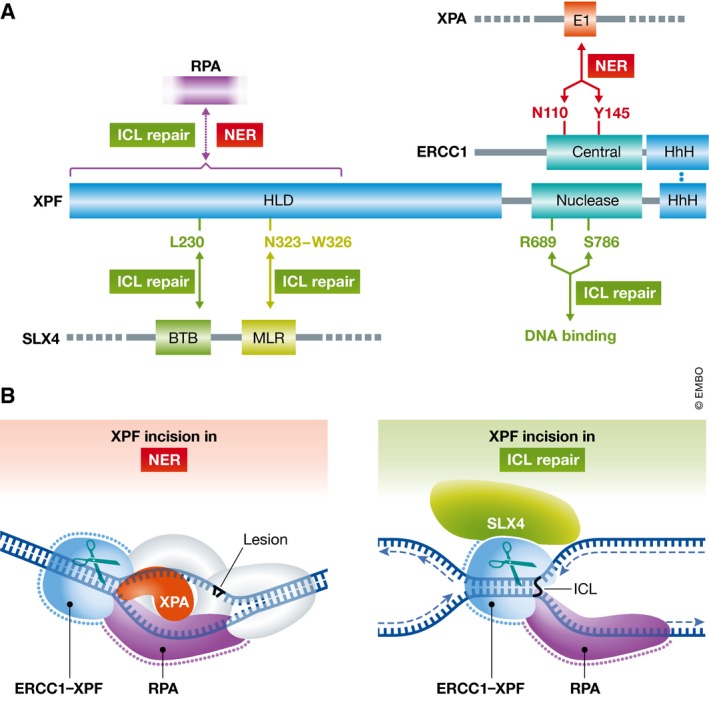
(A) ERCC1‐XPF heterodimerizes via the C‐terminal HhH domains. The central domain of ERCC1 mediates the interaction with XPA in NER and mutations in residues N110 and Y145 compromise the NER activity of ERCC1‐XPF (shown in red). The nuclease domain in XPF contains the active site residues. Mutation of residues R689 and S786 does not affect the catalytic mechanism, but alters DNA binding in the nuclease domain and leads to an ICL repair‐specific defect (shown in green). The helicase‐like domain (HLD) of XPF interacts with SLX4 and likely RPA. Mutation of residue L230 abolishes ICL repair and SLX4 binding; deletion of residues 323–326 abolishes ICL repair, but retains binding affinity for SLX4 (shown in green). This region may mediate an interaction with SLX4 that is needed for the proper positioning of XPF at sites of ICL repair. The interaction with RPA is required for NER and ICL repair, but the exact location of the RPA binding site in the HLD remains to be determined. (B) The XPA protein recruits ERCC1‐XPF to NER complexes, SLX4 to sites of ICL repair. RPA positions ERCC1‐XPF for incision in NER and ICL repair by binding ssDNA with a defined polarity at the junction to position ERCC1‐XPF for incision. The role of RPA is conserved between the two pathways.
XPF was originally described as the nuclease responsible for the incision 5′ to bulky lesions in NER. We have a good understanding of how ERCC1‐XPF is targeted to NER complexes (Orelli et al, 2010), via a specific interaction of the XPA protein with the central domain of ERCC1 (Fig 1). This interaction is specific for NER, as mutations that abolish it affect NER, but not the ICL repair functions of ERCC1‐XPF. The actual incision reaction is a highly coordinated process and also involves an interaction with the single‐stranded DNA protein RPA (de Laat et al, 1998).
By contrast, we know less about how ERCC1‐XPF functions in ICL repair. It is known that an interaction between XPF and the scaffold protein SLX4 is essential for ICL repair. SLX4 interacts with at least three structure‐specific endonucleases—ERCC1‐XPF, MUS81‐EME1, and SLX1—and it has been shown that mutations in two proposed XPF interaction domains in SLX4 (termed MLR and BDB domains) are essential for ICL repair (Guervilly et al, 2015). Conversely, mutations in the N‐terminal helicase‐like domain (HLD) of XPF are associated with FA, suggesting that the HLD mediates an interaction that is important for ICL repair (Bogliolo et al, 2013; Kashiyama et al, 2013).
Recently, using a system in which an ICL‐containing plasmid is subjected to replication‐dependent ICL repair in Xenopus egg extracts, the Knipscheer laboratory demonstrated that ERCC1‐XPF is responsible for at least one and possibly both of the unhooking incisions, during which the ICL is cleaved from one of the two parental strands at a replication fork (Klein Douwel et al, 2014). In their current work, they now study how XPF is recruited to ICL repair complexes (Klein Douwel et al, 2017). Using XPF patient mutations that cause FA and an XPF allele generated in Drosophila and known to have limited interaction with SLX4 in a yeast two‐hybrid system, they show that specific mutations in the HLD of XPF abrogate incisions at ICLs, while they do not affect NER (Fig 1A). Interestingly, two sets of mutations exert their defects by different mechanisms. XPF‐L219R (L230P in humans) is deficient in SLX4 binding, is not recruited to ICLs, and is deficient in ICL repair. By contrast, XPF‐∆312–315 (∆323–326 in humans), although deficient in ICL repair, binds SLX4 and is recruited to ICLs. The authors suggest that these mutations are located in two distinct SLX4‐binding sites in XPF mediating the interaction with the BTB and MLR domains of SLX4, respectively. This suggests that one of these interactions is required for recruitment and the other for proper positioning of XPF for ICL incision. Two mutations in the nuclease active site of XPF—XPF‐R670S and XPF‐S767F (R689S and S786F in humans)—also confer a specific ICL repair defect. As XPF proteins with these mutations are fully active in NER, the catalytic mechanism of XPF appears unaffected, instead the positioning of the catalytic center at ICLs is likely altered, revealing distinct architectures of the substrate binding site in NER and ICL repair.
Now that this study has firmly established the requirement of an interaction of XPF and SLX4 for ICL incision, it would be pertinent to get insights into whether additional proteins may also stimulate the incision of ERCC1‐XPF at ICLs. This issue is addressed in a second new The EMBO Journal paper from the McHugh laboratory (Abdullah et al, 2017), studying the question how the ssDNA binding protein RPA directs and stimulates incision of ERCC1‐XPF at ICLs. It has previously been shown that ERCC1‐XPF can incise an ICL in the duplex region adjacent to a ss/dsDNA junction (Kuraoka et al, 2000). It has furthermore been shown that a protein encompassing the XPF interaction domains of SLX4 can stimulate incisions at an ICL (Hodskinson et al, 2014). Seemingly, unrelated earlier work had shown that the incisions by ERCC1‐XPF on similar model substrates for NER can be greatly stimulated by RPA, and that the polarity of RPA binding is one of the factors directing the NER incisions to the damaged strand (de Laat et al, 1998).
Abdullah et al (2017) thus investigated whether RPA plays a similar role in ICL repair, using substrates mimicking an ICL at a stalled replication fork (Fig 1B). ERCC1‐XPF readily cuts splayed arm substrates, and if they contain an ICL, the incision is made on the distal side of the ICL and junction in the duplex DNA. However, the presence of a nascent “leading strand” paired to ssDNA overhang of the strand (Fig 1B) inhibits the incision. This inhibition is readily overcome by addition of RPA, and several experiments were conducted to show that this is a specific interaction: (i) the Escherichia coli ssDNA binding protein SSB does not stimulate the incision reaction by ERCC1‐XPF; (ii) RPA only overcomes the inhibition by incision with a duplex on the 3′ ssDNA overhang, not with duplex on the 5′ ssDNA overhang or if both overhangs are duplex; (iii) the affinity of RPA is the same for either ssDNA overhang, so the directionality of RPA binding rather than its affinity is key for the stimulation of the incision. It will be interesting to see how SLX4 and RPA may work together to stimulate the incision by ERCC1‐XPF.
The authors furthermore show that the exonuclease SNM1A can be loaded onto the nick and digest past the ICL, thus completing the unhooking of the ICL. It has long been thought that two structure‐specific endonucleases would be needed for unhooking on either side of the ICL. However, no conclusive evidence has supported the involvement of endonucleases with a polarity opposite to that of XPF (cleaving a 5′ overhang), such as SLX1, FAN1 or possibly XPG, FEN1, or EXO1, in the same ICL repair pathway as XPF. The current results alleviate the need for a second endonuclease, as XPF together with the exonuclease SNM1A would be sufficient for this purpose.
Taken together, these findings provide a model for how the unhooking reaction may occur in replication‐coupled ICL repair: Upon approach of the leading strand to the ICL and activation of the FA pathway, SLX4 localizes to the vicinity of the ICL and recruits ERCC1‐XPF through an interaction between its MLR domain, possibly with the aid of additional factors. Licensing of XPF incision at the ICL requires a second XPF interaction with SLX4 through its BTB domain, and a directed interaction with RPA located on the lagging strand opposite to the approached fork (Fig 1B).
The study of larger protein assemblies will be required to fully understand these mechanisms. In a recent tour de force, work from the West laboratory brought us one step closer toward this goal (Wyatt et al, 2017). They expressed and purified the six‐subunit SLX4‐SLX1‐MUS81‐EME1‐XPF‐ERCC1 (SMX) complex and showed that the six‐subunit complex had higher activity on most substrates than any of its subcomplexes. By also purifying complexes with all or individual nucleases inactivated by active site mutation, they found that SLX1 and MUS81 are the predominant nucleases on Holliday junction substrates (HJs) within SMX, while MUS81 is responsible for cleaving replication fork‐like substrates. Interestingly, a catalytically inactive ERCC1‐XPF was able to stimulate incision of HJs by SLX1 and MUS81 in the SMX complex. This suggests that these nucleases can stimulate each other's activities in an allosteric fashion. The authors also provide mechanistic insights into how the MUS81 nuclease is activated within this complex. The N‐terminal region of MUS81 is a DNA‐binding domain that contributes to the substrate specificity of MUS81. This is the same part of the protein that interacts with SLX4, and upon doing so, the binding specificity of MUS81 is altered. It will be interesting to see whether ERCC1‐XPF binding specificity for ICLs is similarly altered within the SMX complex, thereby contributing to the substrate specificity for ICLs.
With the ability to purify such large complexes and the advent of high‐resolution cryo‐EM methods, there is new hope that structural insights into how the activity of nucleases in such larger complexes may be regulated are forthcoming. It is also quite likely that not all the players that influence ERCC1‐XPF incisions are known. For example, in the yeast Saccharomyces cerevisiae, the SSA role of the ERCC1‐XPF homologs Rad10‐Rad1 has been shown to require the SAW1 protein (Li et al, 2013). Does a similar protein exist in humans, and which functions of ERCC1‐XPF might it stimulate? We also know little about how SLX4 is recruited to sites of ICL repair. It is known that FANCD2/FANCI mono‐ubiquitination is required for the incisions at ICL to occur, but whether there is a direct interaction between Ub‐FANCD2/FANCI and the ubiquitin‐binding domain in SLX4 mediates this recruitment is not known. Therefore, while three recent papers bring us closer to understanding of how ERCC1‐XPF might work in ICL repair, we still have lot to learn about the makeup and structures of larger complexes that mediate and control endonuclease reactions.
Acknowledgements
Research in the author's laboratory is supported by the Korean Institute for Basic Science (IBS‐R022‐A1‐2017) and the NIH (P01 CA092584).
See also: D Klein Douwel et al (July 2017)and UB Abdullah et al (July 2017)
References
- Abdullah UB, McGouran JF, Brolih S, Ptchelkine D, El‐Sagheer AH, Brown T, McHugh PJ (2017) RPA activates the XPF‐ERCC1 endonuclease to initiate the processing of DNA interstrand crosslinks. EMBO J 36: 2047–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillon J, Ramirez MJ, Pujol R, Casado JA, Banos R, Rio P, Knies K, Zuniga S, Benitez J, Bueren JA, Jaspers NG, Schärer OD, de Winter JP et al (2013) Mutations in ERCC4, encoding the DNA‐repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet 92: 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehe PM, Gaillard PH (2017) Control of structure‐specific endonucleases to maintain genome stability. Nat Rev Mol Cell Biol 18: 315–330 [DOI] [PubMed] [Google Scholar]
- Guervilly JH, Takedachi A, Naim V, Scaglione S, Chawhan C, Lovera Y, Despras E, Kuraoka I, Kannouche P, Rosselli F, Gaillard PH (2015) The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol Cell 57: 123–137 [DOI] [PubMed] [Google Scholar]
- Hodskinson MR, Silhan J, Crossan GP, Garaycoechea JI, Mukherjee S, Johnson CM, Schärer OD, Patel KJ (2014) Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF‐ERCC1 in DNA crosslink repair. Mol Cell 54: 472–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, Li TS, Yoshiura K, Utani A, Hirano A, Yamashita S, Greenblatt D, Nardo T, Stefanini M, McGibbon D, Sarkany R et al (2013) Malfunction of nuclease ERCC1‐XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet 92: 807–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Raschle M, Walter JC, Knipscheer P (2014) XPF‐ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54: 460–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D, Hoogenboom WS, Boonen RA, Knipscheer P (2017) Recruitment and positioning determine the specific role of the XPF‐ERCC1 endonuclease in interstrand crosslink repair. EMBO J 36: 2034–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD (2000) Repair of an interstrand DNA crosslink initiated by ERCC1‐XPF repair/recombination nuclease. J Biol Chem 275: 26632–26636 [DOI] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NG, Hoeijmakers JH (1998) DNA‐binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev 12: 2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Dong J, Eichmiller R, Holland C, Minca E, Prakash R, Sung P, Yong Shim E, Surtees JA, Eun Lee S (2013) Role of Saw1 in Rad1/Rad10 complex assembly at recombination intermediates in budding yeast. EMBO J 32: 461–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelli B, McClendon TB, Tsodikov OV, Ellenberger T, Niedernhofer LJ, Schärer OD (2010) The XPA‐binding domain of ERCC1 is required for nucleotide excision repair but not other DNA repair pathways. J Biol Chem 285: 3705–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt HD, Laister RC, Martin SR, Arrowsmith CH, West SC (2017) The SMX DNA Repair Tri‐nuclease. Mol Cell 65: 848–860 [DOI] [PMC free article] [PubMed] [Google Scholar]