

Abstract
Adipose tissue represents a critical component in healthy energy homeostasis. It fulfills important roles in whole‐body lipid handling, serves as the body's major energy storage compartment and insulation barrier, and secretes numerous endocrine mediators such as adipokines or lipokines. As a consequence, dysfunction of these processes in adipose tissue compartments is tightly linked to severe metabolic disorders, including obesity, metabolic syndrome, lipodystrophy, and cachexia. While numerous studies have addressed causes and consequences of obesity‐related adipose tissue hypertrophy and hyperplasia for health, critical pathways and mechanisms in (involuntary) adipose tissue loss as well as its systemic metabolic consequences are far less understood. In this review, we discuss the current understanding of conditions of adipose tissue wasting and review microenvironmental determinants of adipocyte (dys)function in related pathophysiologies.
Keywords: adipose tissue dysfunction, cachexia, lipodystrophy, metabolic disorders, obesity
Subject Categories: Cancer, Metabolism, Molecular Biology of Disease
Functional adipose tissue is essential for health
The adipose organ can be divided into two distinct types of adipose tissues, white and brown. Brown adipose tissue (BAT) dissipates energy by producing heat (thermogenesis) via the uncoupling of the activity of the mitochondrial electron transport chain through the specific expression of uncoupling protein 1 (UCP1). BAT is composed of brown adipocytes characterized by a high mitochondrial content and endowed with a high capacity of lipid oxidation (Berriel Diaz et al, 2014). In addition to the thermogenic brown adipocytes located in BAT, white adipose tissue (WAT) contains thermogenic fat cells, called “brown‐in‐white” (“brite”) or “beige” adipocytes which are able to burn fat and carbohydrates via non‐shivering thermogenesis (Petrovic et al, 2010; Vegiopoulos et al, 2010).
White adipose tissue is specialized in the storage and release of fat, the balance of which is critical to maintain healthy energy homeostasis (Rohm et al, 2013). Indeed, in humans, a combination of excessive lipid storage and decreased removal leads to obesity and associated comorbidities, including insulin resistance and type 2 diabetes (Langin, 2011), now affecting more than 500 million overweight and/or obese people worldwide (Finucane et al, 2011).
In addition to its lipid‐storing capacity, WAT has been described as an important endocrine organ controlling the systemic handling of energy substrates (Galic et al, 2009). Also, intracellular WAT lipid homeostasis is a key determinant of body weight and insulin sensitivity in both mice and humans (Yu & Ginsberg, 2005). In this respect, excessive lipid load causes adipocyte stress, which in turn accounts for many adverse effects of obesity, particularly alterations in adipocytokine release and a low‐grade inflammatory response, ultimately leading to the development of metabolic dysfunction such as worsened insulin sensitivity and glucose intolerance (Hotamisligil, 2006; Rasouli & Kern, 2008). Due to their critical role in the maintenance of proper adipocyte function, both storage and release of lipids in WAT are physiologically under tight hormonal control: Whereas insulin promotes triglyceride (TG) storage during the postprandial phase, catecholamines trigger the breakdown of TG into glycerol and fatty acids (“lipolysis”) to provide energy substrates for other organs, including liver and skeletal muscle, during fasting (Yu & Ginsberg, 2005). An intriguing but still largely unexplored question is how the local microenvironment determines adipose tissue function and its impact on systemic metabolism.
Furthermore, whereas numerous studies have highlighted mechanisms in obesity predisposition, development, and manifestation, also conditions of absence or scarcity of WAT such as lipodystrophy and cachexia are associated with severe metabolic complications (Shimomura et al, 1998; Petruzzelli & Wagner, 2016). Intriguingly, many of these metabolic conditions, including insulin resistance, glucose intolerance, and inflammation, are commonly shared between both opposing states of WAT mass, that is, scarcity of WAT and obesity, suggesting similar or even identical molecular mechanisms linking these conditions. However, how and whether obesity and adipose tissue deficiency couple at the molecular level, and whether scientists and clinicians can learn from either side to therapeutically counteract the opposite phenotype remain to be solved.
Given the high number of excellent reviews summarizing pathomechanisms in obesity development, this review aimed to highlight our current understanding of both mechanisms of WAT energy handling and also local determinants of WAT functionality as particularly related to lipodystrophy, cancer cachexia, and other states of WAT deficiencies.
Handling of energy substrates determines the adipocyte impact on systemic energy balance
Organismal survival depends on the ability of adipocytes to temporarily store excess substrates and mobilize them upon decreased external nutrient supply. Substrates are stored mainly as triacylglycerols in lipid droplets and are released as fatty acids and glycerol by lipolysis. In the postprandial state, adipocytes take up fatty acids delivered by gut‐derived chylomicrons or lipoprotein particles synthesized in the gut and the liver. Intracellularly, fatty acids are sequentially esterified to glycerol to form triacylglycerols and thus be incorporated as neutral lipids into the lipid droplet. Although the liver has been shown to be the major site of conversion of carbohydrates to lipid, adipocytes are also able to synthesize fatty acids from excess circulating glucose (de novo lipogenesis) (Abel et al, 2001; Mao et al, 2009).
During prolonged fasting or physical activity, the acyl moieties are released from triglycerides through the sequential action of lipases, namely adipose triglyceride lipase (ATGL/Pnpla2), hormone‐sensitive lipase (HSL/Lipe) and monoglyceride lipase (MGL/Mgll). Following transport mainly through the lymph, these fatty acids become systemically available for oxidation and ATP synthesis. In addition, released glycerol can serve as a substrate for gluconeogenesis in the liver.
The regulation of uptake of circulating substrates, triglyceride biosynthesis as well as lipolysis depends on the balance of systemic anabolic and catabolic signals. A complex interplay between multiple endocrine mediators and the sympathetic nervous system has been shown to govern adipocyte metabolism. Among these signals, the pancreatic hormone insulin seems to play a central role as a gauge of short‐term systemic fuel availability. In the postprandial state, insulin promotes glucose and fatty acid uptake as well as lipogenesis and suppresses triglyceride lipolysis (Samuel & Shulman, 2012). The key role of insulin is highlighted by the severe lipodystrophy observed in adipocyte‐specific insulin receptor knockout mice (Lee et al, 2013a; Qiang et al, 2016). However, as discussed below, local signals in the adipose tissue microenvironment can affect adipocyte metabolism with impact on systemic homeostasis.
The chronic availability of excess nutrients leads to enlargement of lipid droplets and adipocyte hypertrophy, culminating in larger fat depots and body weight gain. In fact, the gradual development of overweight as observed in the majority of the Western world population has been estimated to result from an imbalance of 50–100 kcal per day over several years (Mozaffarian et al, 2011). Adipocyte hypertrophy is not only characteristic for adipose tissue in obesity but is also associated with the development of obesity‐associated pathology [(Blüher, 2013; Spalding et al, 2008) and refs therein]. In particular, hypertrophy correlated with dyslipidemia, inflammation, and impaired glucose homeostasis in humans (Hoffstedt et al, 2010; Klöting et al, 2010). Conversely, adipocyte size was shown to be smaller in obese individuals without metabolic disease (“metabolically healthy obese”) compared to obese patients with metabolic complications (Klöting et al, 2010). These clinical findings could be interpreted in two ways: (i) large adipocytes are pathogenic per se and/or (ii) the inability of adipocytes to further expand limits the capacity for excess fatty acid storage with the consequence of systemically elevated lipid levels. A recent report demonstrated that hypertrophic adipocytes had reduced ability for insulin‐stimulated glucose uptake in a cell‐autonomous manner (Kim et al, 2015). However, increased adipocyte size has been shown to correlate with improved systemic glucose homeostasis in certain transgenic mouse models, arguing against a general pathogenic effect of hypertrophic adipocytes (Khan et al, 2009; Abreu‐Vieira et al, 2015). Furthermore, the inability to efficiently store lipid in adipocytes of lipodystrophic patients and mouse models is associated with metabolic dysfunction as discussed below. Thus, it appears more likely that the limited storage capacity and the concomitant increase in ectopic lipids are major contributors to the pathogenesis of metabolic disease, as proposed in the “expandability hypothesis” (Unger & Scherer, 2010; Virtue & Vidal‐Puig, 2010). Specific intermediates of fatty acid metabolism which accumulate under conditions of impaired triglyceride storage seem to play a key role in this process by causing cellular stress in adipocytes and systemically lipotoxicity (Fig 1, Box 1).
Figure 1. Lipid storage and metabolism in healthy and dysfunctional adipocytes.
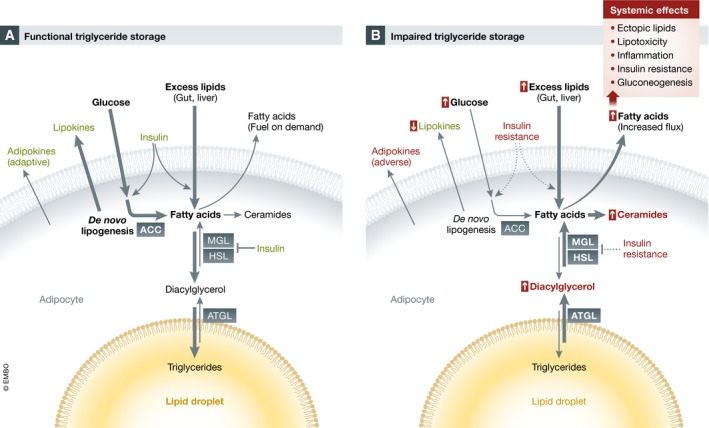
(A) Functional adipocytes metabolize and store excess circulating lipids and glucose in the inert form of triglycerides in the lipid droplet. Fatty acids can be mobilized by lipolysis on demand. Pancreatic insulin promotes de novo lipogenesis as well as the uptake and storage of circulating lipids. The secretion of adipokines and lipokines by functional adipocytes contributes to systemic metabolic regulation. (B) Impairment of triglyceride storage in overloaded or dysfunctional adipocytes is associated with constitutive fatty acid mobilization, reduced glucose uptake and de novo lipogenesis as well as the generation of lipotoxic diacylglycerol and ceramides (these intermediates also accumulate in remote tissues). These metabolic changes are partly due to insulin resistance (see also Fig 4). Dysfunctional adipocytes are characterized by reduced production of certain lipokines as well as an adverse adipokine profile. Overall, the increased flux of fatty acids away from adipocytes promotes systemic metabolic dysfunction. Adaptive and pathogenic effects are shown in green and red, respectively. MGL, monoglyceride lipase; HSL, hormone‐sensitive lipase; ATGL, adipose triglyceride lipase; ACC, acetyl‐CoA carboxylase.
Box 1: Impaired triglyceride storage and lipotoxicity.
The biochemical form in which excess fatty acids exist in cells and tissues is critical for their pathogenicity. Thus, triglycerides stored in lipid droplets represent an inert non‐pathogenic form as demonstrated in mouse models with genetic modifications influencing the final step of triglyceride biosynthesis (Samuel & Shulman, 2012). In line with this, mutations in genes involved in lipid droplet formation cause lipodystrophy and metabolic syndrome (as discussed below). In contrast, various intermediates of fatty acid metabolism have been shown to cause cellular stress and toxicity (lipotoxicity) in adipocytes and other relevant cell types, including myocytes, hepatocytes, and immune cells (Fig 1). A well‐established case is represented by diacylglycerol, an intermediate of lipolysis and fatty acid esterification which can activate members of the protein kinase C (PKC) family to inhibit insulin signaling in mice and humans [reviewed in Shulman (2014)]. Saturated fatty acids have been shown to act as ligands for the Toll‐like receptor 4, thereby promoting inflammation and insulin resistance (Shi et al, 2006; Lackey & Olefsky, 2016). Excess availability of saturated fatty acids is also associated with increased generation of the sphingolipid ceramide. Ceramides can interfere with insulin signaling and mitochondrial oxidation and promote endoplasmic reticulum stress and apoptosis (Chaurasia & Summers, 2015). Importantly, inhibition of ceramide synthesis was protective against insulin resistance in mice (Holland et al, 2007), and others reviewed by Chaurasia and Summers (2015).
Which are the consequences of exceeded adipocyte storage capacity for the function of remote organs? As discussed in Box 1, excess circulating lipid and the concomitant increase in “reactive” lipid metabolites in remote tissues directly impact on insulin sensitivity in liver and muscle cells, resulting in increased glucose output in liver and reduced uptake in muscle, which are important contributors to impaired glucose tolerance and type 2 diabetes (Samuel & Shulman, 2012). In parallel, key nodes of insulin signaling can be inhibited indirectly by several central inflammatory pathways. Indeed, overloaded adipose tissue is associated with the activation of inflammatory and immune cells along with a network of inflammatory mediators locally as well as systemically. Chronic low‐level inflammation results in insulin resistance, as demonstrated in numerous models with genetic inactivation of key signal transducers including, for example, tumor necrosis factor‐alpha (TNF‐α), IκB kinase (IKKβ) and c‐Jun N‐terminal kinase (JNK) [reviewed by Lackey and Olefsky (2016)]. Most recent advances in this direction revealed the role of the lipid mediator leukotriene B4 and the macrophage‐derived Galectin‐3 in the induction of obesity‐associated systemic insulin resistance (Li et al, 2015, 2016). In addition to inflammation‐related cytokines, alterations in secreted adipocyte‐derived polypeptides (adipokines) mediate adipose tissue dysfunction to remote organs, including liver, muscle, and brain, thereby substantially contributing to systemic metabolic deterioration (Box 2). Furthermore, although the local signaling function of lipid mediators is well established, recent studies have revealed products of adipocyte lipid metabolism as beneficial endocrine regulators (“lipokines”) (Yilmaz et al, 2016). In particular, both palmitoleate (C16:1) and palmitic acid‐hydroxy‐stearic acid, deriving from adipocyte de novo lipogenesis, are able to improve systemic glucose metabolism and a reduction in their systemic levels may contribute to metabolic disease in humans (Cao et al, 2008; Yore et al, 2014).
Box 2: Adipokines and the endocrine function of fat.
Beyond the systemic impact of local lipid metabolism, adipocytes substantially affect metabolic regulation of remote tissues through the endocrine action of secreted polypeptides termed adipokines. The plethora of adipokines, identified in the last two decades, essentially influence every organ system mediating physiological effects on metabolism, immunity, behavior, cardiovascular function, and reproduction, often with disease relevance (Fasshauer & Blüher, 2015). A prime example for how aberrant expression of adipokines contributes to systemic metabolic pathogenesis upon adipocyte dysfunction is given by adiponectin. Adiponectin promotes systemic insulin sensitivity and improves glucose and lipid metabolism in liver and muscle as well as pancreatic endocrine function (Stern et al, 2016). Most of its actions appear to be mediated by activation of the adenosine monophosphate‐activated protein kinase (AMPK) and the ceramidase‐mediated reduction in ceramide levels downstream of the AdipoR1 and R2 transmembrane adiponectin receptors. Adiponectin levels are reduced in obesity and show a remarkable inverse correlation with insulin resistance and type 2 diabetes in humans (Spranger et al, 2003; Klöting et al, 2010; Ye & Scherer, 2013). Thus, the loss of protective effects of adiponectin is likely to be a major pathogenic factor in metabolic disease. The prototype of adipokines is probably best represented by leptin (Friedman, 2016). The overtly obese phenotypes of humans and mice with inactivating mutations in the leptin/leptin receptor system illustrates the physiological importance of this pathway (Farooqi et al, 1999; Friedman, 2016). Although the most potent effects of leptin are mediated through the regulation of food intake and energy expenditure in the central nervous system, leptin modulates lipid metabolism and insulin sensitivity in several tissues, either directly or through the central nervous system (Stern et al, 2016). In contrast to adiponectin, leptin levels are increased in obesity but the protective effects of leptin are hindered by resistance to leptin signaling in the central nervous system (leptin resistance). Nevertheless, the potency of the leptin system is highlighted by the exploitation of leptin administration for the treatment of metabolic disorders in the absence of obesity, in particular lipodystrophy (see Box 3), non‐alcoholic fatty liver disease and potentially type 1 diabetes (Wang et al, 2010; Stern et al, 2016).
A newer development relates to the role of secreted fatty acid binding protein 4 (FABP4). Proteins in the FABP family are able to bind lipophilic molecules intracellularly and influence lipid transport and metabolism (Hotamisligil & Bernlohr, 2015). Recently, circulating FABP4, secretion of which seems to be a regulated process, was shown to enhance hepatic glucose production (Cao et al, 2013). Along with accumulating evidence supporting a correlation of circulating FABP4 with obesity and metabolic disease, these findings define FABP4 as a novel disease‐relevant adipokine (Hotamisligil & Bernlohr, 2015).
Whereas the effects of overloaded adipose tissue on insulin sensitivity in liver and muscle are central in our understanding of lipid‐induced systemic metabolic dysfunction, a simpler metabolic component needs to be considered. Impaired uptake of excess fatty acids or increased lipolysis rates in adipocytes result in higher fatty acid flux to the liver. This is a major contributor to the accumulation of lipids in the liver leading to non‐alcoholic fatty liver disease, which can progress to non‐alcoholic steatohepatitis involving inflammation and fibrosis (Donnelly et al, 2005). Importantly, these conditions are strongly associated with cardiometabolic disease and are prominent risk factors for hepatocellular carcinoma (Cornier et al, 2008; Yki‐Jarvinen, 2014). In addition, liver glucose output and thereby blood glucose regulation have been shown to be driven by the control of adipose tissue lipolysis rather than liver insulin action (Rebrin et al, 1996; Mittelman & Bergman, 2000; Titchenell et al, 2016). Using sophisticated in vivo flux analysis, the Shulman laboratory could show that the availability of adipose‐derived fatty acids determined acetyl‐CoA levels in the liver and thereby glucose output and blood glucose regulation, independently of liver insulin sensitivity (Perry et al, 2015). This depended on increased gluconeogenic flux through the allosteric activation of pyruvate carboxylase by acetyl‐CoA.
Beyond the regulation of glucose levels by liver and muscle, newer concepts of how systemic excess lipids affect metabolic homeostasis and promote cardiometabolic disease have emerged. These include hypothalamic and pancreatic lipotoxicity and inflammation, in addition to the well‐established links to atherosclerosis and heart disease (Cornier et al, 2008; Janikiewicz et al, 2015; Kälin et al, 2015). Local toxic and inflammatory effects of excess lipids in the hypothalamus can impair the ability of this key central nervous system site to regulate food intake, systemic energy expenditure, and peripheral metabolism (Kälin et al, 2015). Similarly, ectopic fat and inflammation in the pancreas have been suggested to compromise the endocrine function, in particular insulin secretion (Janikiewicz et al, 2015).
Lipodystrophy
Lipodystrophies (LD) are defined by the partial or complete absence of metabolically active adipose tissue and can be acquired or originated in a genetic defect (Garg & Agarwal, 2009). In line with the idea of a critical functional adipose tissue mass, insulin resistance, dyslipidemia, hypertension, and diabetes often accompany lipodystrophy, and the degree of fat loss determines the degree of metabolic disease, arguing for the “expandability hypothesis” as outlined above (Fig 2) (Unger & Scherer, 2010; Virtue & Vidal‐Puig, 2010). While circulating triglyceride levels are elevated, free fatty acid levels are mostly unaffected in lipodystrophy (Patni & Garg, 2015). Adipose tissue expansion has been shown to have beneficial effects in both LD and obesity: Adipose tissue transplantation into dyslipidemic, insulin‐resistant A‐ZIP/F‐1 “fatless” mice reversed both ectopic fat accumulation and insulin resistance (Kim et al, 2000). Adipocyte progenitor injection had the same effect (Rodeheffer et al, 2008). In addition, adipose tissue expansion by increased adiponectin levels improved metabolic health in leptin‐deficient obese ob/ob mice (Kim et al, 2007). Thus, adipose tissue plays a metaboloprotective role in the face of over‐nutrition by storing excessive supplies. The major factors believed to cause metabolic syndrome in lipodystrophy are ectopic fat or “reactive” lipid species, and alterations in the adipokine profile (see Box 3). Adipose tissue inflammation as typically seen in obesity is not commonly observed in LD, although present in HIV‐related and some types of partial LD (Jan et al, 2004; Gandotra et al, 2011), and a recent study in Fsp27‐deficient lipodystrophic mice shows that hepatic steatosis and insulin resistance are independent of adipose tissue inflammation (Zhou et al, 2003).
Figure 2. Obesity and wasting diseases in light of the adipose tissue expandability hypothesis.
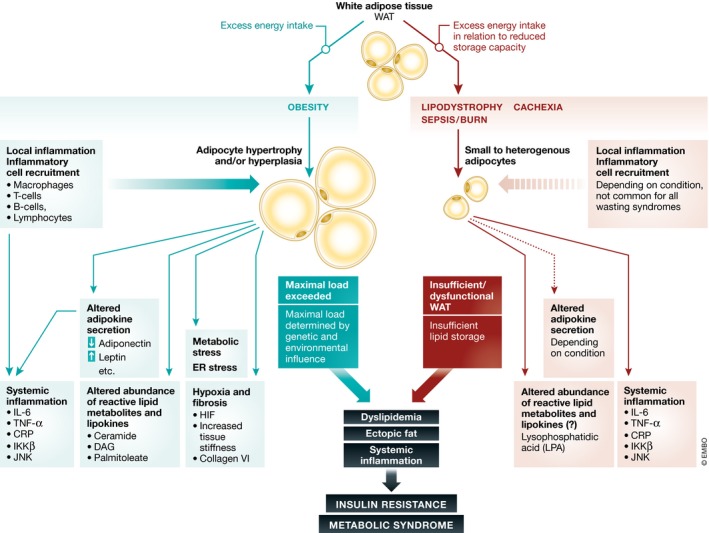
Both obesity and wasting diseases cause adipose tissue dysfunction due to insufficient storage capacity, resulting in dyslipidemia, systemic inflammation and altered adipokine profiles. Combination of these factors favors the development of insulin resistance and the metabolic syndrome, which further impairs adipose tissue function, creating a futile cycle. IKKβ, IκB kinase; HIF, hypoxia‐inducible factor; IL‐6, interleukin‐6; JNK, c‐Jun N‐terminal kinase; TNF‐α, tumor necrosis factor‐alpha; CRP, C‐reactive protein; DAG, diacylglycerol.
Box 3: Lipodystrophy and metabolic disease.
Lipodystrophies can be acquired or genetic diseases and are defined by various degrees of adipose tissue deficiency. They are often accompanied by metabolic complications such as insulin resistance, dyslipidemia, or cardiovascular disease. The severity of metabolic disease in LD depends on the degree of fat loss. The two major factors causing metabolic dysfunction associated with LD are believed to be dyslipidemia and alterations in the adipokine profile. Firstly, a deficit in adipose tissue leads to triglyceride redistribution toward other organs, like skeletal muscle or liver (Garg, 2004), leading to organ damage and reduced whole‐body insulin sensitivity. This is underlined by the observation that over‐expression of Agpat2 in livers of Agpat2−/− mice did not reverse lipodystrophy‐induced fatty liver disease (Agarwal et al, 2011). Hepatic steatosis is a characteristic of CGL (Berardinelli, 1954). Defective adipose tissue storage also leads to elevated circulating levels of “reactive” lipid metabolites, cholesterol, and triglycerides, causing atherosclerosis and cardiovascular disease, as shown in adipose tissue‐deficient Seipin−/− mice (Wang et al, 2015) and patients with CGL (Sanon et al, 2016).
In addition to dyslipidemia, adipokine and cytokine profiles are frequently altered in patients with LD. Both adiponectin and leptin levels are very low in LD, particularly in generalized LD (Haque et al, 2002). Whereas the reduction in adiponectin levels affects lipid metabolism, inflammation and insulin sensitivity in remote organs, low leptin levels increase the systemic lipid load by acting mainly centrally to increase food intake. A mouse study investigating the effect of leptin on liver in Agpat2−/− mice suggests that the major function of leptin is this setting lies in regulation of food intake, as leptin protects Agpat2−/− mice from hepatic steatosis and insulin resistance despite the deletion of the leptin receptor in hepatocytes (Cortes et al, 2014). Elevated energy expenditure has been measured in patients with LD, which is directly proportional to their energy intake. A calorie‐restricted diet not only reversed energy expenditure, but also reversed the adverse metabolic effects of LD (Robbins et al, 1979), further underlining the importance of (leptin‐regulated) food intake. In a mouse model of generalized LD, a combination therapy of adiponectin and leptin fully reversed insulin resistance (Yamauchi et al, 2001). Other studies performed in a range of murine LD models ranging from HAART to CGL have shown improved lipid profiles and insulin sensitivity by adiponectin and leptin therapy (Shimomura et al, 1999; Ebihara et al, 2001; Duntas et al, 2004; Xu et al, 2004). In patients with LD, the synthetic leptin analogue metreleptin (human recombinant methionyl leptin) has first been described to have beneficial effects in patients with LD over a decade ago (Oral et al, 2002) and is now successfully used to treat dyslipidemia and insulin resistance due to CGL (Javor et al, 2005; Ebihara et al, 2007), partial LD (Chong et al, 2010; Diker‐Cohen et al, 2015; Ajluni et al, 2016), and antiretroviral therapy‐induced LD (Lee et al, 2006; Mulligan et al, 2009).
Congenital generalized lipodystrophy (CGL) is an autosomal recessive disorder caused by mutations in genes involved in triglyceride synthesis and lipid droplet formation, namely AGPAT2 (lysophosphatidic acid acyltransferase‐β), BSCL2 (Seipin), CAV1 (Caveolin 1), and PTRF (Cavin‐1) (Magre et al, 2001; Agarwal et al, 2002; Kim et al, 2008; Hayashi et al, 2009). AGPAT2 directly affects adipocyte differentiation via AKT and PPARγ pathways (Patni & Garg, 2015). It catalyzes the second step in the de novo phospholipid synthesis pathway and plays a key role in triglyceride and glycerophospholipid formation from glycerol‐3‐phosphate (Agarwal et al, 2002). Thus, mutations in AGPAT2 lead to dysfunctional triglyceride storage, and in addition cause the accumulation of pathway intermediates such as lysophosphatidic acid (LPA). Increased LPA levels may further decrease adipose tissue mass and functionality by inhibiting adipogenesis and adipose tissue expansion (Rancoule et al, 2014a). Indeed, mice treated with the LPA‐receptor antagonist Ki16425 showed enhanced adipose tissue expansion during high‐fat diet feeding. Secreted LPA has also been implicated in glucose sensitivity (Rancoule et al, 2014a). Interestingly, a recent report also links LPA to increased adipose tissue fibrosis in obesity (Rancoule et al, 2014b), and LPA may thus be involved in lipodystrophy‐induced fibrosis as discussed below. BSCL2, CAV1, and PTRF, which are mutated in other forms of CGL, are mainly involved in vesicle trafficking and affect the formation or maturation of lipid droplets (Patni & Garg, 2015).
Congenital generalized lipodystrophy is characterized by near‐complete lack of body fat present at birth or from a very young age (Patni & Garg, 2015). Interestingly, while these patients have almost no metabolically active storage fat, mechanical fat (i.e., in joints, orbits, palms, and soles) is still present (Garg et al, 1992). Patients develop signs of metabolic syndrome during childhood, including dyslipidemia and ectopic triglyceride accumulation (Berardinelli, 1954) and hyperinsulinemia or type 2 diabetes (Van Maldergem et al, 2002).
Depending on the degree of adipose tissue loss, the same symptoms are observed in partial lipodystrophies, both familial forms [familial partial lipodystrophy, FPLD (Garg, 2000)] and acquired forms, most commonly occurring in patients with human immunodeficiency virus (HIV) receiving highly active antiretroviral therapy (HAART) with protease inhibitors (Blumer et al, 2008). Indeed, it has been reported that up to 79% of lipodystrophic patients on HAART have insulin resistance or type 2 diabetes (Vigouroux et al, 1999). Protease inhibitor treatment leads to reduced levels of key transcription factors involved in regulating adipocyte differentiation and function, such as PPARγ, C/EBPα, ATF‐4, CHOP, and XBP‐1, and increase levels of reactive oxygen species, causing inflammation. This leads to permanent alteration of adipose tissue functionality, so that lipodystrophy persists even after the protease treatment is discontinued (Nolis, 2014). Impaired PPARγ activity has also been reported in patients with FPLD3, where PPARG is mutated (Agarwal & Garg, 2002). Likewise, mutations in AKT2 and PLIN1 impair adipocyte function and cause FPLD, and a mutation in Lamin A/C (LMNA) causes cell death by weakening the nuclear envelope (Nolis, 2014). Unless CGL, FPLD results in partial AT loss mainly from the subcutaneous depots in the extremities and upper trunk region. Other adipose depots, such as those in the facial or vulvar areas, are often enlarged. Why some AT depots are affected by the lipodystrophy while others are preserved remains to be clarified, but likely traces back to alterations in microenvironment, extracellular matrix (ECM) composition, or different progenitor populations.
Cachexia
Adipose tissue insufficiency and dysfunction also occurs in cachexia, a pathological wasting syndrome defined by muscle and adipose tissue loss that cannot be reversed by nutrition (Fearon et al, 2011). The wasting can be caused by a range of chronic diseases including chronic obstructive pulmonary disease, congestive heart failure, rheumatoid arthritis, chronic kidney disease, and HIV infection (Morley et al, 2006). It also occurs in up to 30% of patients with cancer (Morley et al, 2006) and is associated with a significantly increased mortality risk (Utech et al, 2012). Despite the clear clinical relevance, the mechanisms by which cachexia develops are still not well understood. Initially, wasting in cancer cachexia was thought to be caused exclusively by enhanced energy requirements of the tumor (Theologides, 1979). Indeed, tumors require substantial amounts of energy. A study by Lieffers et al estimated that a metastatic colon cancer required up to 17,700 kcal in 3 months (Lieffers et al, 2009). However, tumor size and degree of cachexia do not correlate, and tumors of the same type and degree can cause cachexia in some patients but not in others, just like outcomes in sepsis can be influenced by genetic variations in immunity (Fearon et al, 2012). Thus, the original idea was expanded to the concept of a cachectic metabolism in which tumors secrete factors that alter systemic metabolism, overall favoring catabolism (Fearon et al, 2012). A number of tumor‐secreted or tumor‐induced/host‐secreted factors involved in cachexia development have already been described, including the inflammatory cytokines interleukin 1 and interleukin 6 (IL‐1, IL‐6) (Uehara et al, 1989; Strassmann et al, 1992; Baltgalvis et al, 2008), tumor necrosis factor‐alpha (TNF‐α) (Oliff et al, 1987), and interferon gamma (IFN‐γ) (Acharyya et al, 2004), but also other factors such as zinc‐alpha2‐glycoprotein (ZAG) (Mracek et al, 2011), myostatin (Zimmers et al, 2002), ataxin 10 (Schäfer et al, 2016) and parathyroid hormone‐related protein (PTHrP) (Kir et al, 2014). Despite the advance in identifying cachexia‐inducing agents, treatment success has been limited so far, which can be attributed to a complex interplay between several tumor‐ and/or host‐derived factors responsible for inducing wasting (Schäfer et al, 2016).
Cachexia has long been regarded as a muscle wasting disorder, but recently, adipose tissue wasting has started to gain attention in regard to cachexia (Fearon et al, 2011), especially after noting that fat mass was lost before lean mass during the progression of cancer cachexia (Fouladiun et al, 2005). Increased adipose tissue lipolysis is a key element in the development of cachexia (Zuijdgeest‐van Leeuwen et al, 2000). Indeed, increased levels of free fatty acids and glycerol are often observed in patients with cachexia (Tisdale, 2009), and many of the above‐mentioned cachexokines induce lipolysis (Das et al, 2011). Microarray analysis of adipose tissue from patients with cancer cachexia revealed significant induction of genes involved in energy turnover and fatty acid degradation (Dahlman et al, 2010). The key enzymes of lipolysis, ATGL and HSL, play major roles in disease progression. ATGL and HSL activities are increased in patients with cancer cachexia (Das et al, 2011). Knockout of either of these lipases protected against cancer‐induced adipose tissue loss and partly inhibited lean mass wasting. In line with a central role of lipolysis for disease progression, other cachexokines such as IL‐6, TNF‐α, or ZAG were unchanged in these models (Das et al, 2011), and in vitro studies confirmed that tumor‐derived factor(s) rather than secondary effects were responsible for the induction of lipolysis (Rohm et al, 2016). Thus, disruption of fat catabolism has the potential to cause whole‐body wasting, and therapies targeting adipocyte lipolysis are already under investigation for cachexia treatment (Mayer et al, 2013).
While enhanced adipose tissue lipolysis in cachexia is well established, the influence of lipogenesis on fat wasting is less clear. With dwindling adipose tissue supplies, one would expect reduced lipid synthesis in cachexia, and indeed, early studies in mice suggested that de novo lipogenesis was inhibited in tumor‐bearing animals (Trew & Begg, 1959; Lanza‐Jacoby et al, 1984). However, newer evidence suggests that in addition to lipolysis, lipogenesis is also increased in cachexia (Mulligan & Tisdale, 1991; Rohm et al, 2016). In the latter study, adipocytes showed both markedly enhanced lipolysis and triglyceride synthesis in response to tumor‐derived stimuli, suggesting enhanced substrate cycling. Increased production of triacylglycerol‐bound glycerol and de novo lipid synthesis has also been described in a different model of cancer cachexia (Beck & Tisdale, 2004). Since substrate cycling is an energy costly process, the parallel induction of lipolysis and lipogenesis in the adipocyte leads to enhanced energy demand. In the face of energy undersupply due to cachectic metabolism and anorexia, this contributes to adipose tissue loss in cachexia. In line with this, ATP levels in cachectic adipose tissue are reduced by more than 50% (Rohm et al, 2016).
Energy deprivation usually activates the cellular energy sensor adenosine monophosphate‐activated protein kinase (AMPK) by a conformational change in its gamma subunit and subsequent activating phosphorylation of its alpha subunit (Hardie et al, 2012). Generally speaking, active AMPK promotes energy conservation by activating ATP‐producing catabolic pathways and inhibiting ATP‐consuming anabolic pathways (Hardie et al, 2012). In adipocytes, AMPK activation leads to inhibition of fatty acid synthesis and lipolysis by inhibitory phosphorylation of the key enzymes HSL (Garton & Yeaman, 1990) and acetyl‐CoA carboxylase (ACC) (Sullivan et al, 1994). In cachexia, however, this regulation is dysfunctional: AMPK levels and protein activity in cachectic adipose tissue are low despite energy deprivation (Rohm et al, 2016). Thus, an essential brake to energy wasting is missing. In line with a central role of adipocyte AMPK activity in fat wasting, a previous study on AMPK knockout mice (Prkab1−/−, knock out of AMPK β1 subunit) described reduced body fat content, slightly elevated circulating fatty acid levels, and enhanced adipocyte lipogenesis (Dzamko et al, 2010). Interestingly, adipocyte lipolysis was not altered in this study, arguing for additional regulatory factors. Chronic AMPK activation by the AMP analogue AICAR (5‐aminoimidazole‐4‐carboxamide riboside) has been shown to inhibit lipolysis in both mice and men (Boon et al, 2008; Anthony et al, 2009). Thus, under certain conditions, AMPK inhibition can promote lipid mobilization.
How is AMPK dysregulated in cancer cachexia? We have recently shown that the AMPK dysfunction in cachexia roots in AMPK complex disintegration and subsequent protein degradation caused—at least in part—by interaction with CIDEA (Fig 3) (Rohm et al, 2016). Cell death‐inducing DNA fragmentation factor‐alpha‐like effector A (CIDEA) has previously been reported to interact with the AMPK β1 subunit (Qi et al, 2008), thereby preventing AMPK complex formation and destabilizing the protein. Cidea−/− mice displayed lower concentrations of plasma free fatty acids and dampened fatty acid release from BAT (Zhou et al, 2003). We and others have shown that CIDEA is upregulated in cachectic adipose tissue in both patients and rodents (Laurencikiene et al, 2008; Rohm et al, 2016). In cachexia, CIDEA binds the AMPK β1 subunit, the AMPK complex dissociates, and major brakes in lipolysis and lipogenesis are disabled (Rohm et al, 2016). Under normal conditions, CIDEA is hardly expressed in WAT, particularly in rodents, and is considered to be a brown fat marker (Zhou et al, 2003; Laurencikiene et al, 2008). However, CIDEA expression is induced during browning of WAT. Brite adipose tissue has the potential to create increased energy expenditure and body weight as well as adipose tissue loss (Sidossis & Kajimura, 2015). It has recently been proposed that WAT browning may play a role in the progression of wasting during cancer‐ (Kir et al, 2014; Petruzzelli et al, 2014) and nephrectomy‐induced (Kir et al, 2016) cachexia. These studies showed elevated brown fat markers, including UCP1 and CIDEA, in cachectic WAT and proposed that browning‐induced elevated energy expenditure contributed to wasting. However, elevated resting energy expenditure is often not present in cachexia, both in patients and mouse models (Blum et al, 2011; Rohm et al, 2016), arguing for a different role of CIDEA in cachexia, such as in AMPK regulation. In line with this, cachexia has been shown to develop both under thermoneutral conditions, when the thermogenic program is largely inactive (Cui et al, 2016), and under blockage of beta‐adrenergic signaling (Rohm et al, 2016). Also, UCP1−/− mice injected with cachexia‐inducing Lewis lung cancer (LLC) cells showed no difference in lipolysis, energy expenditure, or loss of body weight, lean mass and adipose tissue mass compared to their LLC‐injected wild‐type littermates (Rohm et al, 2016).
Figure 3. Activation of energy wasting pathways in cancer cachexia.
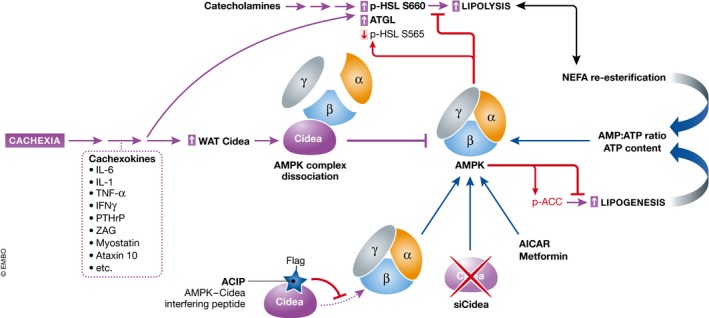
AMPK (adenosine monophosphate‐activated protein kinase) in adipose tissue phosphorylates HSL (hormone‐sensitive lipase) and ACC (acetyl‐CoA carboxylase) at inhibitory sites, blocking lipolysis and lipogenesis. Cachexia‐inducing tumors activate adipocyte lipolysis and lipogenesis by (i) upregulating lipase levels (ATGL, HSL), and (ii) reducing the inhibitory AMPK activity. Cachexia causes reduced AMPK activity due to increased levels and binding to CIDEA (cell death‐inducing DNA fragmentation factor‐alpha‐like effector A) resulting in AMPK complex dissociation. Treatments targeting adipose tissue AMPK function (including ACIP, AMPK‐CIDEA interfering peptide) have proven beneficial in counteracting cachexia. ZAG, zinc‐alpha2‐glycoprotein; PTHrP, parathyroid hormone‐related protein; NEFA, non‐esterified fatty acid.
Thus, there are still many things to be learned about the role of adipose tissue in cachexia. It certainly plays a prominent role in disease progression, as a number of studies over the last few years show (Das et al, 2011; Kir et al, 2014; Petruzzelli et al, 2014; Rohm et al, 2016). It may also be a promising therapeutic target, especially since treatment options for cachexia are limited and altering major clinical outcomes is not possible so far (Fearon et al, 2013). Current lines of treatment involve nutritional supplementation, resistance exercise, steroid hormones, and inhibition of inflammatory mediators, for example using thalidomide or cyclooxygenase inhibitors (such as indomethacin and ibuprofen) (Fearon et al, 2013). Newer approaches include the use of the hunger hormone ghrelin or anti‐myostatin antibodies (Fearon et al, 2013). Cachexia is a multifactorial disease and as such is unlikely to be cured by a single agent. Thus, including modulators of adipose tissue metabolism is a promising future approach to treat the disease. Kir et al (2014) have shown that blocking PTHrP action on adipose tissue prevented cachexia in LLC‐injected mice. Petruzzelli et al (2014) have shown that blocking IL‐6 could maintain adipose tissue integrity and prevented wasting in a cachexia model. Other adipocentric therapies, for example, Atglistatin, which targets lipolysis by inhibiting ATGL, are currently developed (Mayer et al, 2013). We have recently shown that maintaining adipose tissue AMPK functionality by introducing a peptide blocking the CIDEA–AMPK interaction (termed AMPK‐CIDEA interfering peptide, ACIP) prevented excessive lipolysis and prolonged life of cachectic animals (Rohm et al, 2016). In line with this, activating AMPK in human adipocytes using biguanides and thiazolidinediones (TZD) has been shown to inhibit lipolysis (Bourron et al, 2010), and metformin treatment of cachectic rats has proven effective in preventing wasting (Oliveira & Gomes‐Marcondes, 2016).
Burn injury
Novel treatment approaches gained from cachexia research have the potential to improve adipose tissue wasting in other diseases, too. Burn injury, for example, has a strong impact on adipose tissue metabolism. Severe burn injuries, which cover 30% or more of the total body, induce lasting metabolic changes in part by altering adipose tissue function (Rojas et al, 2012). The trauma induces a biphasic metabolic response, with an initial phase of reduced energy expenditure, followed by a prolonged phase of increased resting energy expenditure and hypermetabolism (Breznock, 1980). This second phase can last for years after the actual burn injury and is associated with lean mass and adipose tissue loss, increased cardiac output and metabolic rate as well as poor immune function. This is often accompanied by insulin resistance (Rojas et al, 2012).
A major factor in the development of hypermetabolism due to burn injuries are catecholamines (Wilmore et al, 1974; Kulp et al, 2010). Catecholamine levels can remain elevated for years after the actual injury (Rojas et al, 2012), and a high catecholamine concentration is associated with a more severe form of hypermetabolism (Williams et al, 2009). Blocking catecholamine signaling, for example, by the β‐adrenergic receptor antagonist propranolol, reduces burn‐induced hypermetabolism (Pereira et al, 2005). Catecholamines are also major drivers of adipocyte lipolysis. Lipolysis and fatty acid release into the circulation have been shown to be elevated upon burn injury (Williams et al, 2009). Interestingly, a recent study showed that adipose tissue loss due to septic burn injury could be traced back to a loss in AMPK‐dependent inhibition of lipolysis in the adipose tissue (Diao et al, 2015). Similar to cachexia, burn injury often presents with elevated levels of inflammatory cytokines such as IL‐6 (Abdullahi et al, 2017). Studies in mice have shown that IL‐6 [which in case of burn injury is released from the bone marrow (Abdullahi et al, 2017)] has the potential to induce hypermetabolism, and IL‐6 blockage can prevent adipose tissue defects in burn injury (Abdullahi et al, 2017). Interestingly, burn injury has recently been linked to increased adipose tissue browning (Patsouris et al, 2015; Abdullahi et al, 2017), and multilocular, UCP1‐positive adipocytes have been found in WAT from burn patients (Sidossis et al, 2015). In the latter study, UCP1 mRNA levels, mitochondrial density, and leak respiratory capacity were all increased in adipocytes after burn injury (Sidossis et al, 2015).
Local determinants of adipose tissue functionality
Adipose tissue cellularity (number and size of adipocytes)
The capacity of adipose tissue to store excess nutrients safely and without metabolic overloading of adipocytes depends on the number of adipocytes per depot. The fact that metabolically healthy obese are predicted to have higher adipocyte numbers compared to weight‐matched insulin‐resistant individuals suggest that adipose tissue hyperplasia is beneficial (Klöting et al, 2010). In accordance, insulin sensitization through treatment with the anti‐diabetic TZD is associated with increased numbers of smaller adipocytes (Okuno et al, 1998; Tang et al, 2011). The number of adipocytes depends on the balance between cell death and adipogenesis, that is, the formation of new adipocytes from progenitor cells. Adipocyte progenitors have been recently identified in vivo as immature mesenchymal stromal cells largely associated with the vasculature (Rodeheffer et al, 2008; Tang et al, 2008). The turnover rate in adult humans has been estimated at 10% per year, rather low levels compared to other tissues (Spalding et al, 2008). In any case, the size of fat depots correlates with the number of adipocytes, as shown for some depots in humans (Arner et al, 2013). Importantly, obese individuals have higher total adipocyte numbers, and this becomes evident already in childhood obesity (Spalding et al, 2008). Since the genetic determination of obesity has been demonstrated by GWAS as well as monogenic phenotypes, it is likely that the regulation of adipocyte numbers will have a genetic component (Dahlman et al, 2016). However, this remains to be proven by human studies focusing on adipose tissue cellularity.
How does the nutritional environment influence adipocyte numbers? In a unique study with healthy adults, Tchoukalova et al (2010) could show that 8 weeks of overfeeding increased the number of adipocytes in association with the enlargement of the fat depot by 1.6 kg. In rodents, adipose tissue hyperplasia has been demonstrated in hyperphagic dietary and genetic models of obesity (Johnson & Hirsch, 1972; Klyde & Hirsch, 1979). More recent genetic fate mapping experiments could prove that this occurs through the formation of new adipocytes through differentiation of progenitor cells (Lee et al, 2012; Wang et al, 2013; Vishvanath et al, 2016). Intriguingly, work from the Rodeheffer laboratory revealed that a hypercaloric diet with high fat content (HFD) stimulated a proliferation wave of adipocyte progenitors, which subsequently gave rise to mature adipocytes, thereby contributing substantially to adipose tissue expansion (Jeffery et al, 2015). To which extent progenitor differentiation without preceding cell proliferation contributes to fat hyperplasia remains to be determined.
A common clue from the above studies was the marked fat depot‐specificity of the hyperplastic response to the diet. Thus, increased adipocyte numbers could be detected in the femoral subcutaneous fat depot in humans but not in the upper abdominal subcutaneous depot (Tchoukalova et al, 2010). In male mice, higher rates of proliferation/adipogenesis were shown in the intra‐abdominal gonadal fat depot but not in the posterior subcutaneous depot (Wang et al, 2013; Jeffery et al, 2015). These differences can have substantial impact on the systemic adaptation of metabolism given the pathophysiological relevance of the individual depots. Accumulation of fat in the intra‐abdominal/visceral depots is a central feature of the metabolic syndrome and is associated with insulin resistance and increased risk for diabetes and cardiovascular disease (Cornier et al, 2008; Lee et al, 2013b). In contrast, higher subcutaneous fat mass is associated with improved metabolic parameters including insulin sensitivity and blood lipid profiles. Interestingly, female mice increased progenitor proliferation and adipogenesis in both the intra‐abdominal and the subcutaneous depots in response to HFD (Jeffery et al, 2016). This is in line with the fact that weight gain in premenopausal women generally comes along with preferential subcutaneous fat accumulation, whereas men show increased visceral fat deposition. This differential fat distribution has been suggested to contribute to the relative protection from cardiometabolic disease in women (Karastergiou et al, 2012). Taken together, location‐specific hyperplastic expansion of adipose tissue can affect body fat distribution. Although direct proof is pending, the adaptive increase in adipocyte numbers is likely to contribute to systemic metabolic homeostasis and influence pathogenesis. This notion is supported by a study using stable isotope tracing to model proliferation‐dependent adipocyte turnover in mice (Kim et al, 2014). The authors concluded that HFD‐induced adipogenesis occurred predominantly in juvenile mice and that the reduction in hyperplastic potential in adults could be responsible for the development of insulin resistance. Furthermore, Lotta et al could identify a genetic association of 53 loci with traits related to insulin resistance as well with type 2 diabetes and coronary heart disease (Lotta et al, 2017). Remarkably, genetic predisposition for insulin resistance predicted by these loci was associated with reduced fat deposition in peripheral “metabolically safe” depots. Moreover, the study revealed common links to lipodystrophy‐related insulin resistance, highlighting the importance of adipose tissue function for systemic homeostasis.
The molecular regulation of adipogenesis has been investigated over several decades, and a comprehensive network of signaling, transcriptional, and epigenetic factors has been described (Cristancho & Lazar, 2011). However, with limited exceptions, the links to the regulation of adipocyte number in vivo including the genetic determination of adipose tissue growth in humans have not been established. In particular, the mechanisms by which adipocyte progenitor proliferation and differentiation are controlled by systemic factors and the microenvironment in response to excess nutrients are poorly understood. In a recent report, activation of the Akt2 signal transducer in adipocyte progenitors was shown to be crucial for their HFD‐induced proliferation and adipogenesis (Jeffery et al, 2015). This is consistent with the pro‐adipogenic effect of insulin and insulin‐like growth factor 1 (IGF1) as upstream Akt activators in cell models of adipogenesis (Garten et al, 2012). It remains to be determined to which extent insulin/IGF1 mediates the systemic anabolic state to adipocyte progenitors upon over‐nutrition in vivo.
The role of the microenvironment as the adipocyte progenitor niche in the regulation of tissue plasticity is increasingly attracting attention. Obesity is associated with changes in the extracellular matrix (ECM, discussed below) and manipulation of the ECM has been shown to alter diet‐induced adipose tissue growth (Christiaens et al, 2008). Given the established function of the ECM in the control of cell fate in multiple stem cell systems, it is likely that future studies will reveal similar findings in relation to adipose tissue (Guilak et al, 2009; Cristancho & Lazar, 2011). A further emerging concept is the involvement of transient local inflammation as a trigger for adipose tissue remodeling, both in the context of hyperplastic expansion and browning of white fat depots. Thus, genetic inhibition of central inflammatory pathways in fat resulted in reduced tissue growth (Wernstedt Asterholm et al, 2014). Lee et al (2013c) described the induction of an adipogenic niche by HFD and other triggers with macrophages playing a central role. Generally, according to the current consensus, adipocyte progenitor responses are driven by M2‐type macrophages and type 2 innate immune cells during adaptive tissue remodeling (Brestoff & Artis, 2015; Odegaard & Chawla, 2015).
Inflammation
In contrast to transient adaptive inflammatory responses, chronic inflammation due to adipocyte overloading and lipotoxicity has a major impact on adipocyte metabolism and adipose tissue function. Thus, macrophage infiltration of adipose tissue was a strong predictor of insulin resistance in obese subjects after matching for BMI and other factors (Klöting et al, 2010). Similarly, one of the effects of bariatric surgery is the reduction of macrophage infiltration in conjunction with insulin sensitization (Frikke‐Schmidt et al, 2016). The causal relationship between obesity‐induced chronic inflammation and metabolic dysfunction has been established in numerous mouse models with genetic manipulation of key inflammatory pathways [reviewed in (Lackey & Olefsky, 2016)]. Inflammatory mediators can affect adipocyte metabolism in several ways. The inhibition of insulin signaling compromises the clearance of glucose and fatty acids in the postprandial state. In addition, the stimulation of lipolysis further contributes to fatty acid flux to the liver and other organs, leading to systemic lipotoxicity and insulin resistance as discussed above (Perry et al, 2015). Furthermore, certain cytokines have been shown to inhibit the formation of new adipocytes, thereby possibly limiting the storage capacity of the tissue (Gustafson et al, 2009). It becomes apparent that a futile cycle between impaired lipid storage, inflammation and insulin resistance drives the metabolic syndrome and progression to disease (Fig 4).
Figure 4. Futile cycles driving impaired triglyceride storage and lipolysis.
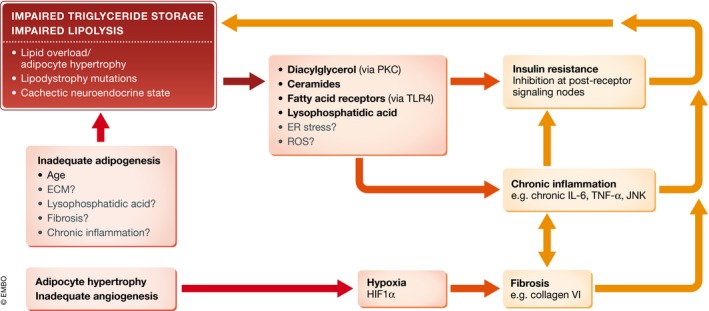
Adipocyte overloading in the absence of compensatory adipocyte formation and angiogenesis or lipodystrophy‐associated impaired triglyceride storage can increase “reactive” lipid intermediates, other cell stressors, and hypoxia and thereby promote inflammation, fibrosis and insulin resistance. These factors interact and synergistically feed back to deteriorate adipocyte metabolism and safe lipid storage. ECM, extracellular matrix; ER, endoplasmic reticulum; HIF1α, hypoxia‐inducible factor 1α; PKC, protein kinase C; ROS, reactive oxygen species; TLR4, Toll‐like receptor 4.
As discussed below, inflammation can also promote fibrosis as a further pathogenic feature in adipose tissue. Finally, beyond inflammation, vascularization and angiogenesis as well as sympathetic innervation have been suggested to be important determinants of adipocyte metabolism and adipose tissue expansion (Bartness & Song, 2007; Cao, 2013).
Extracellular matrix and fibrosis
The ECM plays an important role in regulating tissue integrity and function. In recent years, adipose tissue ECM has emerged as a new regulator of adipose tissue differentiation, metabolism, and inflammation, and as such has important implications for metabolic disease. The ECM consists of a fibrillary network of structural and adhesive proteins, mainly fibronectin and collagens, which provide both mechanical support and create micro‐domains as basis for cellular signaling. Other components include laminin, elastin, proteoglycans, and other polysaccharides, and ECM components are also directly linked to membrane receptors such as integrins (Mariman & Wang, 2010; Sun et al, 2013). Matrix metalloproteinases (MMPs) dynamically degrade and remodel the ECM (Sun et al, 2013). In adipose tissue, ECM components are produced by adipocytes, pre‐adipocytes and inflammatory cells (Buechler et al, 2015). They regulate mechanical properties, adipogenesis, cellular expansion and lipid droplet growth (Nakajima et al, 2002; Alkhouli et al, 2013). For example, studies using different ECM components like fibronectin or laminin for pre‐adipocyte culture demonstrate that ECM remodeling from a fibrillar to a laminar structure is required for adipocyte differentiation (Spiegelman & Ginty, 1983; O'Connor et al, 2003).
Excess accumulation of ECM is termed fibrosis. Fibrosis is a ubiquitous response to chronic inflammation (Wynn, 2007), and increased fibrosis is commonly observed in adipose tissue of obese mice and patients (Strissel et al, 2007; Henegar et al, 2008; Divoux et al, 2010; Sun et al, 2013). Fibrotic areas and collagen accumulation in the interstitial and pericellular space are up to 4 times higher in obese than in lean patients (Divoux et al, 2010). Collagen VI expression in human adipose tissue is increased already after 8 weeks of overfeeding (Pasarica et al, 2009), and Col6 knockout in obese ob/ob or high‐fat diet fed mice reduces weight gain and adiposity (Khan et al, 2009). Interestingly, weight loss after bariatric surgery does not resolve fibrosis in obese adipose tissue, even 2 years after surgery (Cancello et al, 2013). Hypoxia is thought to be one of the main drivers of fibrosis in obesity. Obesity induces reduced capillary density (Spencer et al, 2011) and local hypoxia at the site of hypertrophic adipocytes, which leads to the activation of hypoxia‐inducible factor 1 alpha (HIF1α). HIF1α stimulates ECM synthesis (Halberg et al, 2009) and cross‐linking (Mariman & Wang, 2010), but also inflammatory gene expression, leading to macrophage recruitment. HIF1α overexpression in adipocytes is sufficient to stimulate fibrosis and local inflammation (Halberg et al, 2009). In addition to adipocytes, macrophages and mast cells are also pro‐fibrotic (Divoux et al, 2012; Hirai et al, 2014; Jang et al, 2016), so inflammatory cell recruitment contributes to fibrosis development in obesity. Increased ECM, particularly surrounding adipocytes (Divoux et al, 2010), can further lead to adipocyte necrosis and additional inflammatory cell recruitment. Inflammation also influences adipocyte differentiation and lipid metabolism (Virtue & Vidal‐Puig, 2010). Adipose stiffness due to increased ECM limits adipocyte growth (Abdennour et al, 2014), which in line with the “expandability hypothesis” (Virtue & Vidal‐Puig, 2010) leads to dyslipidemia and metabolic disease. In line with this, mice lacking Col6 show extreme adipocyte hypertrophy without metabolic disease (Khan et al, 2009). Fibrosis and inflammation are tightly linked, but it is unclear so far which comes first in adipose tissue. Fibrosis is a universal response to inflammation, an attempt of wound healing that occurs in multiple tissues upon chronic inflammation. A microarray time course of diet‐induced obesity suggests that inflammation is the primary driver of fibrosis, since in this study, fibrosis is a late event in obesity development (McGregor et al, 2013). On the other hand, fibrosis also causes inflammation: Hypertrophic adipocytes induce hypoxia, thereby activating Hif1α and promoting fibrosis, which then causes adipose tissue stiffness, cell death, and inflammatory cell recruitment. Hypoxia in adipose tissue is one of the first pathological changes that occurs during obesity development (Halberg et al, 2009). In line with this, Halberg et al (2009) showed that fibrosis markers appeared before inflammatory markers upon high‐fat diet feeding. Increased adipocyte necrosis due to tissue restriction, increased deposition of collagens, and integrin signaling attract pro‐inflammatory cells, thereby promoting further fibrosis. In line with this, a recent study has shown that deletion of the integrin CD11b inhibits alternative activation and proliferation of adipose tissue macrophages (McGregor et al, 2013; Zheng et al, 2015). Other factors, such as LPA or the adipokines adiponectin and leptin, have been described to influence fibrosis independent of inflammation (Rancoule et al, 2014b; Saxena & Anania, 2015). In lipodystrophy (discussed below), adipose tissue inflammation is not always present, while fibrosis occurs frequently. Thus, more work is still needed to fully understand the mechanistic links between the two conditions.
Likewise, it is unclear so far whether the increased adipose tissue fibrosis in obesity is linked to metabolic disease, that is, whether it is beneficial or detrimental. The strong correlation with both local inflammation and systemic inflammation (Sun et al, 2013) would suggest that high adipose tissue fibrosis is metabolically unfavorable. Indeed, some studies described a positive correlation between fibrosis and markers of metabolic disease or insulin resistance (Pasarica et al, 2009; Spencer et al, 2011). Tissue stiffness as measured by shear‐wave velocity was also increased in adipose tissue of obese diabetic compared to non‐diabetic patients (Abdennour et al, 2014). However, other studies detected no correlation between total adipose tissue fibrosis and diabetes (Divoux et al, 2010), or provided evidence for reduced fibrosis in obese diabetic patients when compared to metabolically healthy obese (Lackey et al, 2014; Muir et al, 2016). Thus, the relationship between fibrosis and diabetes is not fully understood yet, and variability likely arises through differences in methodology, that is, different fat depots studied, different ways to measure fibrosis, etc., as well as a high variability between individuals, as noted particularly in the obese state by Pasarica et al (2009).
Fibrosis and adipose tissue stiffness in wasting
Transgenic mice with overexpression of a constitutively active TGF‐β develop a phenotype resembling lipodystrophy, with extremely reduced adipose tissue mass, which is mainly attributed to inhibited adipogenesis upon TGF‐β overexpression. They also display fibrosis of the liver, kidney, and adipose tissue, as measured by trichrome staining (Clouthier et al, 1997). Interestingly, this mouse also displayed an altered BAT with white appearance, fibrosis and mostly unilocular adipocytes (Clouthier et al, 1997). In patients with HIV‐induced lipodystrophy, adipose tissue fibrosis presents in combination with inflammation, apoptosis, and decreased adipogenesis (Jan et al, 2004). A different study on patients with partial lipodystrophy (either by LMNA mutation or HIV protease inhibitor treatment‐induced) describes increased fibrosis and a larger number of small adipocytes in the remaining adipose tissues (Bereziat et al, 2011). This was without effects on inflammation or angiogenesis. However, these patients displayed increased UCP1 expression and mitochondrial function in their remaining WAT (Bereziat et al, 2011). Patients with partial lipodystrophy caused by PLIN1 mutations display smaller adipocytes accompanied by macrophage infiltration and marked fibrosis (Gandotra et al, 2011).
In a microarray from cachectic adipose tissue of patients, ECM, actin cytoskeleton and focal adhesion were the most strongly downregulated pathways (Dahlman et al, 2010). The observed gene expression changes were largely reciprocal to those previously associated with obesity (Dahlman et al, 2005), arguing for an important role of ECM plasticity and cytoskeleton for adipose tissue wasting. ECM genes are also strongly downregulated in human adipose tissue after long‐term weight reduction (Kolehmainen et al, 2008). This may represent adaptations of the adipose tissue to reduced lipid droplet volume and nutrient or oxygen availability.
Of note, downregulated expression of ECM‐related proteins does not necessarily equal reduced ECM accumulation, since negative regulators such as MMPs or signaling molecules are also included in this list (Dahlman et al, 2010). The observation that AMPK activation using metformin inhibits fibrotic gene expression and collagen deposition in genetic or diet‐induced obese mice, and interstitial fibrosis is associated with AMPK inactivation in adipose tissue of obese humans (Luo et al, 2016), opens the question whether fibrosis is also involved in adipose tissue wasting during cachexia development, where AMPK function is impaired (Rohm et al, 2016). Studies in this field are rare to date, but initial reports describe increased interstitial space and collagen staining in cachectic adipose tissue from patients (Mracek et al, 2011). Picro Sirius Red staining and expression of collagens Col3a1 and Col6a1 (Alves et al, 2015) as well as tissue remodeling and accumulation of inflammatory cells in fibrotic areas (Batista et al, 2016) was observed in adipose tissue of patients with cancer cachexia. These changes in fibrosis were associated with reduced adipocyte size and adipose tissue atrophy (Batista et al, 2016).
MMP‐2, MMP‐3, and MMP‐14, which degrade components of the ECM and are associated with pathology in models of heart failure and muscular dystrophy, have been shown to be induced in cardiac and skeletal muscle in the Colon26 mouse model of cancer cachexia (Devine et al, 2015). Cardiac fibrosis is also noted. Whether altered MMP expression in cachectic adipose tissue also plays a role in disease progression is not known so far. There certainly is evidence for increased fibrosis in cachectic adipose tissue, both in mice and men (Mracek et al, 2011; Batista et al, 2016; Luo et al, 2016). However, whether this is cause or consequence of the tissue wasting in cachexia remains to be investigated. Of note, patients with burn injury also display a higher collagen content in their adipose tissue, arguing for a role of adipose tissue remodeling in burn injuries, too (Saraf et al, 2016).
Lessons learned
Given its localization across different organ compartments, adipose tissue depots represent key checkpoints in systemic energy homeostasis. At the cellular level, energy handling within the adipocyte largely determines overall adipose tissue functionality and recent studies have highlighted the importance of local, intra‐adipose “niches” for adipocyte integrity. As described above, not only obesity‐related energy excess but also—still “under‐investigated”—conditions of adipose tissue scarcity are characterized by adipocyte and niche dysfunctions which translate into disturbances of systemic energy balance and organismal wasting conditions (Table 1). In this respect, key questions for future research include the role of ECM remodeling and fibrosis for adipose tissue wasting conditions, for example in cancer cachexia, and to which degree the protection of adipose tissue integrity may contribute to the prevention and/or therapy of human wasting disorders. Indeed, high levels of fibrosis have been shown to correlate with little weight loss after bariatric surgery (Divoux et al, 2010), that is, low fibrosis is associated with enhanced weight loss, opening the seemingly paradoxical question whether a high fibrotic burden might even serve a protective role against cachectic conditions in the tumor‐bearing state. In light of the clinical relevance of wasting disorders and the current lack of effective predictive and therapeutic options in humans, a certain re‐focusing of clinical and research efforts onto the maintenance of healthy adipocytes and their niches under these conditions seems to be justified and mandatory in the future.
Table 1.
Comparison of adipose tissue‐specific and systemic effects in obesity, metabolically healthy obesity, lipodystrophy, and wasting disease
| Obesity (insulin resistant) | Obesity (insulin sensitive, metabolically healthy) | Lipodystrophy | Cachexia | References | |
|---|---|---|---|---|---|
| Adipose tissue | |||||
| Storage capacity | Exceeded | Sufficient | Insufficient | Insufficient | Garg & Agarwal (2009); Lotta et al (2017); Unger & Scherer (2010); Virtue & Vidal‐Puig (2010) |
| Relative adipocyte number (Adipogenesis) | Low | High (in particular in subcutaneous depots) | Low | Unchanged | Bereziat et al (2011); Dahlman et al (2010); Hoffstedt et al (2010); Jan et al (2004); Klöting et al (2010); Lotta et al (2017); Okuno et al (1998); Rodeheffer et al (2008); Tang et al (2011); |
| Adipocyte size | Large | Small | Heterogenous | Small | Dahlman et al (2010); Klöting et al (2010); Okuno et al (1998); Spalding et al (2008); Tang et al (2011) |
| Chronic immune cell infiltration and inflammation (type 1) | High | Low | Normal to high (depending on type of LD) | High | Blüher (2013); Gandotra et al (2011); Jan et al (2004); Klöting et al (2010); Lackey & Olefsky (2016) |
| Fibrosis | High | Low | High | High (?) | Abdennour et al (2014); Batista et al (2016); Bereziat et al (2011); Gandotra et al (2011); Jan et al (2004); Khan et al (2009); Lackey et al (2014); Muir et al (2016) |
| Systemic | |||||
| Energy balance (intake – expenditure) | Positive | Positive | Balanced | Negative | Blum et al (2011); Kir et al (2014); Langin (2011); Petruzzelli et al (2014); Rohm et al (2016) |
| Ectopic fat and “reactive” lipids | High | Low | High | High (?) | Unger & Scherer (2010); Samuel & Shulman (2012); Van Maldergem et al (2002); Garg (2004) |
| Inflammation | High | Low | High | High | Batista et al (2016); Fouladiun et al (2005); Gandotra et al (2011); Jan et al (2004); Lackey & Olefsky (2016) |
|
Adipokine profiles Adiponectin Leptin |
Adverse Low High |
Normal High High |
Adverse Low Low |
Adverse High? Low? |
Fasshauer & Blüher (2015); Fouladiun et al (2005); Haque et al (2002); Stern et al (2016) |
| Insulin resistance | Present | Normal | Present | Present | Blüher (2013); Garg & Agarwal (2009); Holroyde (1984); Klöting et al (2010) |
Acknowledgements
We apologize to all colleagues who could not be cited due to space limitations. Our work is supported by the German Research Foundation (DFG) (He3260/8‐1 to S.H.) and the Human Frontier Science Program (RGY0082/2014 to A.V.). M.R. is supported by a Novo Nordisk Postdoctoral Fellowship run in partnership with the University of Oxford.
Conflict of interest
The authors declare that they have no conflict of interest.
The EMBO Journal (2017) 36: 1999–2017
References
- Abdennour M, Reggio S, Le Naour G, Liu Y, Poitou C, Aron‐Wisnewsky J, Charlotte F, Bouillot JL, Torcivia A, Sasso M, Miette V, Zucker JD, Bedossa P, Tordjman J, Clement K (2014) Association of adipose tissue and liver fibrosis with tissue stiffness in morbid obesity: links with diabetes and BMI loss after gastric bypass. J Clin Endocrinol Metab 99: 898–907 [DOI] [PubMed] [Google Scholar]
- Abdullahi A, Chen P, Stanojcic M, Sadri7 AR , Coburn N, Jeschke MG (2017) IL‐6 signal from the bone marrow is required for the browning of white adipose tissue post burn injury. Shock 47: 33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB (2001) Adipose‐selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409: 729–733 [DOI] [PubMed] [Google Scholar]
- Abreu‐Vieira G, Fischer AW, Mattsson C, de Jong JM, Shabalina IG, Ryden M, Laurencikiene J, Arner P, Cannon B, Nedergaard J, Petrovic N (2015) Cidea improves the metabolic profile through expansion of adipose tissue. Nat Commun 6: 7433 [DOI] [PubMed] [Google Scholar]
- Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, Guttridge DC (2004) Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 114: 370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, Barnes RI, Garg A (2002) AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet 31: 21–23 [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Garg A (2002) A novel heterozygous mutation in peroxisome proliferator‐activated receptor‐gamma gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab 87: 408–411 [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Sukumaran S, Cortes VA, Tunison K, Mizrachi D, Sankella S, Gerard RD, Horton JD, Garg A (2011) Human 1‐acylglycerol‐3‐phosphate O‐acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2(−/−) gene lipodystrophic mice. J Biol Chem 286: 37676–37691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajluni N, Dar M, Xu J, Neidert AH, Oral EA (2016) Efficacy and safety of metreleptin in patients with partial lipodystrophy: lessons from an expanded access program. J Diabetes Metab 7: 659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhouli N, Mansfield J, Green E, Bell J, Knight B, Liversedge N, Tham JC, Welbourn R, Shore AC, Kos K, Winlove CP (2013) The mechanical properties of human adipose tissues and their relationships to the structure and composition of the extracellular matrix. Am J Physiol Endocrinol Metab 305: E1427–E1435 [DOI] [PubMed] [Google Scholar]
- Alves M, Neto EM, Maximiliano L, Alcantara P, Otoch J, Batista M, Seelaender M (2015) Adipose tissue extracellular matrix remodelling in cancer cachexia. FASEB J 29: 925–927 [Google Scholar]
- Anthony NM, Gaidhu MP, Ceddia RB (2009) Regulation of visceral and subcutaneous adipocyte lipolysis by acute AICAR‐induced AMPK activation. Obesity (Silver Spring) 17: 1312–1317 [DOI] [PubMed] [Google Scholar]
- Arner P, Andersson DP, Thorne A, Wiren M, Hoffstedt J, Naslund E, Thorell A, Ryden M (2013) Variations in the size of the major omentum are primarily determined by fat cell number. J Clin Endocrinol Metab 98: E897–E901 [DOI] [PubMed] [Google Scholar]
- Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, Carson JA (2008) Interleukin‐6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol 294: R393–R401 [DOI] [PubMed] [Google Scholar]
- Bartness TJ, Song CK (2007) Brain‐adipose tissue neural crosstalk. Physiol Behav 91: 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista ML Jr, Henriques FS, Neves RX, Olivan MR, Matos‐Neto EM, Alcantara PS, Maximiano LF, Otoch JP, Alves MJ, Seelaender M (2016) Cachexia‐associated adipose tissue morphological rearrangement in gastrointestinal cancer patients. J Cachexia Sarcopenia Muscle 7: 37–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck SA, Tisdale MJ (2004) Effect of cancer cachexia on triacylglycerol/fatty acid substrate cycling in white adipose tissue. Lipids 39: 1187–1189 [DOI] [PubMed] [Google Scholar]
- Berardinelli W (1954) An undiagnosed endocrinometabolic syndrome: report of 2 cases. J Clin Endocrinol Metab 14: 193–204 [DOI] [PubMed] [Google Scholar]
- Bereziat V, Cervera P, Le Dour C, Verpont MC, Dumont S, Vantyghem MC, Capeau J, Vigouroux C, Lipodystrophy Study G (2011) LMNA mutations induce a non‐inflammatory fibrosis and a brown fat‐like dystrophy of enlarged cervical adipose tissue. Am J Pathol 179: 2443–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriel Diaz M, Herzig S, Vegiopoulos A (2014) Thermogenic adipocytes: from cells to physiology and medicine. Metabolism 63: 1238–1249 [DOI] [PubMed] [Google Scholar]
- Blüher M (2013) Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract Res Clin Endocrinol Metab 27: 163–177 [DOI] [PubMed] [Google Scholar]
- Blum D, Omlin A, Baracos VE, Solheim TS, Tan BH, Stone P, Kaasa S, Fearon K, Strasser F, European Palliative Care Research C (2011) Cancer cachexia: a systematic literature review of items and domains associated with involuntary weight loss in cancer. Crit Rev Oncol Hematol 80: 114–144 [DOI] [PubMed] [Google Scholar]
- Blumer RM, van Vonderen MG, Sutinen J, Hassink E, Ackermans M, van Agtmael MA, Yki‐Jarvinen H, Danner SA, Reiss P, Sauerwein HP (2008) Zidovudine/lamivudine contributes to insulin resistance within 3 months of starting combination antiretroviral therapy. AIDS 22: 227–236 [DOI] [PubMed] [Google Scholar]
- Boon H, Bosselaar M, Praet SF, Blaak EE, Saris WH, Wagenmakers AJ, McGee SL, Tack CJ, Smits P, Hargreaves M, van Loon LJ (2008) Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 51: 1893–1900 [DOI] [PubMed] [Google Scholar]
- Bourron O, Daval M, Hainault I, Hajduch E, Servant JM, Gautier JF, Ferre P, Foufelle F (2010) Biguanides and thiazolidinediones inhibit stimulated lipolysis in human adipocytes through activation of AMP‐activated protein kinase. Diabetologia 53: 768–778 [DOI] [PubMed] [Google Scholar]
- Brestoff JR, Artis D (2015) Immune regulation of metabolic homeostasis in health and disease. Cell 161: 146–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breznock EM (1980) The systemic response of the traumatized patient: an overview. Vet Clin North Am Small Anim Pract 10: 523–532 [DOI] [PubMed] [Google Scholar]
- Buechler C, Krautbauer S, Eisinger K (2015) Adipose tissue fibrosis. World J Diabetes 6: 548–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancello R, Zulian A, Gentilini D, Mencarelli M, Della Barba A, Maffei M, Vitti P, Invitti C, Liuzzi A, Di Blasio AM (2013) Permanence of molecular features of obesity in subcutaneous adipose tissue of ex‐obese subjects. Int J Obes (Lond) 37: 867–873 [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS (2008) Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 134: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Sekiya M, Ertunc ME, Burak MF, Mayers JR, White A, Inouye K, Rickey LM, Ercal BC, Furuhashi M, Tuncman G, Hotamisligil GS (2013) Adipocyte lipid chaperone AP2 is a secreted adipokine regulating hepatic glucose production. Cell Metab 17: 768–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y (2013) Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab 18: 478–489 [DOI] [PubMed] [Google Scholar]
- Chaurasia B, Summers SA (2015) Ceramides – lipotoxic inducers of metabolic disorders. Trends Endocrinol Metab 26: 538–550 [DOI] [PubMed] [Google Scholar]
- Chong AY, Lupsa BC, Cochran EK, Gorden P (2010) Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 53: 27–35 [DOI] [PubMed] [Google Scholar]
- Christiaens V, Scroyen I, Lijnen HR (2008) Role of proteolysis in development of murine adipose tissue. Thromb Haemost 99: 290–294 [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Comerford SA, Hammer RE (1997) Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy‐like syndrome in PEPCK‐TGF‐beta1 transgenic mice. J Clin Invest 100: 2697–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, Van Pelt RE, Wang H, Eckel RH (2008) The metabolic syndrome. Endocr Rev 29: 777–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes VA, Cautivo KM, Rong S, Garg A, Horton JD, Agarwal AK (2014) Leptin ameliorates insulin resistance and hepatic steatosis in Agpat2‐/‐ lipodystrophic mice independent of hepatocyte leptin receptors. J Lipid Res 55: 276–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristancho AG, Lazar MA (2011) Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol 12: 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Nguyen NL, Zarebidaki E, Cao Q, Li F, Zha L, Bartness T, Shi H, Xue B (2016) Thermoneutrality decreases thermogenic program and promotes adiposity in high‐fat diet‐fed mice. Physiol Rep 4: e12799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman I, Linder K, Arvidsson Nordstrom E, Andersson I, Liden J, Verdich C, Sorensen TI, Arner P (2005) Changes in adipose tissue gene expression with energy‐restricted diets in obese women. Am J Clin Nutr 81: 1275–1285 [DOI] [PubMed] [Google Scholar]
- Dahlman I, Mejhert N, Linder K, Agustsson T, Mutch DM, Kulyte A, Isaksson B, Permert J, Petrovic N, Nedergaard J, Sjolin E, Brodin D, Clement K, Dahlman‐Wright K, Ryden M, Arner P (2010) Adipose tissue pathways involved in weight loss of cancer cachexia. Br J Cancer 102: 1541–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman I, Ryden M, Brodin D, Grallert H, Strawbridge RJ, Arner P (2016) Numerous genes in loci associated with body fat distribution are linked to adipose function. Diabetes 65: 433–437 [DOI] [PubMed] [Google Scholar]
- Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M, Zimmermann R, Vesely P, Haemmerle G, Zechner R, Hoefler G (2011) Adipose triglyceride lipase contributes to cancer‐associated cachexia. Science 333: 233–238 [DOI] [PubMed] [Google Scholar]
- Devine RD, Bicer S, Reiser PJ, Velten M, Wold LE (2015) Metalloproteinase expression is altered in cardiac and skeletal muscle in cancer cachexia. Am J Physiol Heart Circ Physiol 309: H685–H691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao L, Patsouris D, Sadri AR, Dai X, Amini‐Nik S, Jeschke MG (2015) Alternative mechanism for white adipose tissue lipolysis after thermal injury. Mol Med 21: 959–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diker‐Cohen T, Cochran E, Gorden P, Brown RJ (2015) Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab 100: 1802–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, Basdevant A, Guerre‐Millo M, Poitou C, Zucker JD, Bedossa P, Clement K (2010) Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 59: 2817–2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divoux A, Moutel S, Poitou C, Lacasa D, Veyrie N, Aissat A, Arock M, Guerre‐Millo M, Clement K (2012) Mast cells in human adipose tissue: link with morbid obesity, inflammatory status, and diabetes. J Clin Endocrinol Metab 97: E1677–E1685 [DOI] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115: 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duntas LH, Popovic V, Panotopoulos G (2004) Adiponectin: novelties in metabolism and hormonal regulation. Nutr Neurosci 7: 195–200 [DOI] [PubMed] [Google Scholar]
- Dzamko N, van Denderen BJ, Hevener AL, Jorgensen SB, Honeyman J, Galic S, Chen ZP, Watt MJ, Campbell DJ, Steinberg GR, Kemp BE (2010) AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J Biol Chem 285: 115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara K, Ogawa Y, Masuzaki H, Shintani M, Miyanaga F, Aizawa‐Abe M, Hayashi T, Hosoda K, Inoue G, Yoshimasa Y, Gavrilova O, Reitman ML, Nakao K (2001) Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes 50: 1440–1448 [DOI] [PubMed] [Google Scholar]
- Ebihara K, Kusakabe T, Hirata M, Masuzaki H, Miyanaga F, Kobayashi N, Tanaka T, Chusho H, Miyazawa T, Hayashi T, Hosoda K, Ogawa Y, DePaoli AM, Fukushima M, Nakao K (2007) Efficacy and safety of leptin‐replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab 92: 532–541 [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O'Rahilly S (1999) Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 341: 879–884 [DOI] [PubMed] [Google Scholar]
- Fasshauer M, Blüher M (2015) Adipokines in health and disease. Trends Pharmacol Sci 36: 461–470 [DOI] [PubMed] [Google Scholar]
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, Davis M, Muscaritoli M, Ottery F, Radbruch L, Ravasco P, Walsh D, Wilcock A, Kaasa S, Baracos VE (2011) Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 12: 489–495 [DOI] [PubMed] [Google Scholar]
- Fearon KC, Glass DJ, Guttridge DC (2012) Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166 [DOI] [PubMed] [Google Scholar]
- Fearon K, Arends J, Baracos V (2013) Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 10: 90–99 [DOI] [PubMed] [Google Scholar]
- Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim AN, Farzadfar F, Riley LM, Ezzati M (2011) National, regional, and global trends in body‐mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country‐years and 9.1 million participants. Lancet 377: 557–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouladiun M, Korner U, Bosaeus I, Daneryd P, Hyltander A, Lundholm KG (2005) Body composition and time course changes in regional distribution of fat and lean tissue in unselected cancer patients on palliative care–correlations with food intake, metabolism, exercise capacity, and hormones. Cancer 103: 2189–2198 [DOI] [PubMed] [Google Scholar]
- Friedman J (2016) The long road to leptin. J Clin Invest 126: 4727–4734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frikke‐Schmidt H, O'Rourke RW, Lumeng CN, Sandoval DA, Seeley RJ (2016) Does bariatric surgery improve adipose tissue function? Obes Rev 17: 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic S, Oakhill JS, Steinberg GR (2009) Adipose tissue as an endocrine organ. Mol Cell Endocrinol 316: 129–139 [DOI] [PubMed] [Google Scholar]
- Gandotra S, Le Dour C, Bottomley W, Cervera P, Giral P, Reznik Y, Charpentier G, Auclair M, Delepine M, Barroso I, Semple RK, Lathrop M, Lascols O, Capeau J, O'Rahilly S, Magre J, Savage DB, Vigouroux C (2011) Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med 364: 740–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Fleckenstein JL, Peshock RM, Grundy SM (1992) Peculiar distribution of adipose tissue in patients with congenital generalized lipodystrophy. J Clin Endocrinol Metab 75: 358–361 [DOI] [PubMed] [Google Scholar]
- Garg A (2000) Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab 85: 1776–1782 [DOI] [PubMed] [Google Scholar]
- Garg A (2004) Acquired and inherited lipodystrophies. N Engl J Med 350: 1220–1234 [DOI] [PubMed] [Google Scholar]
- Garg A, Agarwal AK (2009) Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta 1791: 507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten A, Schuster S, Kiess W (2012) The insulin‐like growth factors in adipogenesis and obesity. Endocrinol Metab Clin North Am 41: 283–295 [DOI] [PubMed] [Google Scholar]
- Garton AJ, Yeaman SJ (1990) Identification and role of the basal phosphorylation site on hormone‐sensitive lipase. Eur J Biochem 191: 245–250 [DOI] [PubMed] [Google Scholar]
- Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen CS (2009) Control of stem cell fate by physical interactions with the extracellular matrix. Cell Stem Cell 5: 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson B, Gogg S, Hedjazifar S, Jenndahl L, Hammarstedt A, Smith U (2009) Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am J Physiol Endocrinol Metab 297: E999–E1003 [DOI] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt‐Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner‐Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE (2009) Hypoxia‐inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29: 4467–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque WA, Shimomura I, Matsuzawa Y, Garg A (2002) Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab 87: 2395 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, Park YE, Nonaka I, Hino‐Fukuyo N, Haginoya K, Sugano H, Nishino I (2009) Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest 119: 2623–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henegar C, Tordjman J, Achard V, Lacasa D, Cremer I, Guerre‐Millo M, Poitou C, Basdevant A, Stich V, Viguerie N, Langin D, Bedossa P, Zucker JD, Clement K (2008) Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol 9: R14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai S, Ohyane C, Kim YI, Lin S, Goto T, Takahashi N, Kim CS, Kang J, Yu R, Kawada T (2014) Involvement of mast cells in adipose tissue fibrosis. Am J Physiol Endocrinol Metab 306: E247–E255 [DOI] [PubMed] [Google Scholar]
- Hoffstedt J, Arner E, Wahrenberg H, Andersson DP, Qvisth V, Lofgren P, Ryden M, Thorne A, Wiren M, Palmer M, Thorell A, Toft E, Arner P (2010) Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia 53: 2496–2503 [DOI] [PubMed] [Google Scholar]
- Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid‐, saturated‐fat‐, and obesity‐induced insulin resistance. Cell Metab 5: 167–179 [DOI] [PubMed] [Google Scholar]
- Holroyde CP, Skutches CL, Boden G, Reichard GA (1984) Glucose metabolism in cachectic patients with colorectal cancer. Cancer Res 44(12 Pt 1): 5910–5913 [PubMed] [Google Scholar]
- Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444: 860–867 [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Bernlohr DA (2015) Metabolic functions of FABPs–mechanisms and therapeutic implications. Nat Rev Endocrinol 11: 592–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan V, Cervera P, Maachi M, Baudrimont M, Kim M, Vidal H, Girard PM, Levan P, Rozenbaum W, Lombes A, Capeau J, Bastard JP (2004) Altered fat differentiation and adipocytokine expression are inter‐related and linked to morphological changes and insulin resistance in HIV‐1‐infected lipodystrophic patients. Antivir Ther 9: 555–564 [PubMed] [Google Scholar]
- Jang JE, Ko MS, Yun JY, Kim MO, Kim JH, Park HS, Kim AR, Kim HJ, Kim BJ, Ahn YE, Oh JS, Lee WJ, Harris RA, Koh EH, Lee KU (2016) Nitric oxide produced by macrophages inhibits adipocyte differentiation and promotes profibrogenic responses in preadipocytes to induce adipose tissue fibrosis. Diabetes 65: 2516–2528 [DOI] [PubMed] [Google Scholar]
- Janikiewicz J, Hanzelka K, Kozinski K, Kolczynska K, Dobrzyn A (2015) Islet beta‐cell failure in type 2 diabetes–Within the network of toxic lipids. Biochem Biophys Res Commun 460: 491–496 [DOI] [PubMed] [Google Scholar]
- Javor ED, Cochran EK, Musso C, Young JR, Depaoli AM, Gorden P (2005) Long‐term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes 54: 1994–2002 [DOI] [PubMed] [Google Scholar]
- Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS (2015) Rapid depot‐specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol 17: 376–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery E, Wing A, Holtrup B, Sebo Z, Kaplan JL, Saavedra‐Pena R, Church CD, Colman L, Berry R, Rodeheffer MS (2016) The adipose tissue microenvironment regulates depot‐specific adipogenesis in obesity. Cell Metab 24: 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PR, Hirsch J (1972) Cellularity of adipose depots in six strains of genetically obese mice. J Lipid Res 13: 2–11 [PubMed] [Google Scholar]
- Kälin S, Heppner FL, Bechmann I, Prinz M, Tschöp MH, Yi CX (2015) Hypothalamic innate immune reaction in obesity. Nat Rev Endocrinol 11: 339–351 [DOI] [PubMed] [Google Scholar]
- Karastergiou K, Smith SR, Greenberg AS, Fried SK (2012) Sex differences in human adipose tissues – the biology of pear shape. Biol Sex Differ 3: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE (2009) Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 29: 1575–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI (2000) Mechanism of insulin resistance in A‐ZIP/F‐1 fatless mice. J Biol Chem 275: 8456–8460 [DOI] [PubMed] [Google Scholar]
- Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE (2007) Obesity‐associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117: 2621–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O'Rahilly S, Dugail I, Capeau J, Lathrop M, Magre J (2008) Association of a homozygous nonsense caveolin‐1 mutation with Berardinelli‐Seip congenital lipodystrophy. J Clin Endocrinol Metab 93: 1129–1134 [DOI] [PubMed] [Google Scholar]
- Kim SM, Lun M, Wang M, Senyo SE, Guillermier C, Patwari P, Steinhauser ML (2014) Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab 20: 1049–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Huh JY, Sohn JH, Choe SS, Lee YS, Lim CY, Jo A, Park SB, Han W, Kim JB (2015) Lipid‐overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol Cell Biol 35: 1686–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, Spiegelman BM (2014) Tumour‐derived PTH‐related protein triggers adipose tissue browning and cancer cachexia. Nature 513: 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, Hodin RA, Spiegelman BM (2016) PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab 23: 315–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöting N, Fasshauer M, Dietrich A, Kovacs P, Schön MR, Kern M, Stumvoll M, Blüher M (2010) Insulin‐sensitive obesity. Am J Physiol Endocrinol Metab 299: E506–E515 [DOI] [PubMed] [Google Scholar]
- Klyde BJ, Hirsch J (1979) Increased cellular proliferation in adipose tissue of adult rats fed a high‐fat diet. J Lipid Res 20: 705–715 [PubMed] [Google Scholar]
- Kolehmainen M, Salopuro T, Schwab US, Kekalainen J, Kallio P, Laaksonen DE, Pulkkinen L, Lindi VI, Sivenius K, Mager U, Siitonen N, Niskanen L, Gylling H, Rauramaa R, Uusitupa M (2008) Weight reduction modulates expression of genes involved in extracellular matrix and cell death: the GENOBIN study. Int J Obes (Lond) 32: 292–303 [DOI] [PubMed] [Google Scholar]
- Kulp GA, Herndon DN, Lee JO, Suman OE, Jeschke MG (2010) Extent and magnitude of catecholamine surge in pediatric burned patients. Shock 33: 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey DE, Burk DH, Ali MR, Mostaedi R, Smith WH, Park J, Scherer PE, Seay SA, McCoin CS, Bonaldo P, Adams SH (2014) Contributions of adipose tissue architectural and tensile properties toward defining healthy and unhealthy obesity. Am J Physiol Endocrinol Metab 306: E233–E246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey DE, Olefsky JM (2016) Regulation of metabolism by the innate immune system. Nat Rev Endocrinol 12: 15–28 [DOI] [PubMed] [Google Scholar]
- Langin D (2011) In and out: adipose tissue lipid turnover in obesity and dyslipidemia. Cell Metab 14: 569–570 [DOI] [PubMed] [Google Scholar]
- Lanza‐Jacoby S, Lansey SC, Miller EE, Cleary MP (1984) Sequential changes in the activities of lipoprotein lipase and lipogenic enzymes during tumor growth in rats. Cancer Res 44: 5062–5067 [PubMed] [Google Scholar]
- Laurencikiene J, Stenson BM, Arvidsson Nordstrom E, Agustsson T, Langin D, Isaksson B, Permert J, Ryden M, Arner P (2008) Evidence for an important role of CIDEA in human cancer cachexia. Cancer Res 68: 9247–9254 [DOI] [PubMed] [Google Scholar]
- Lee JH, Chan JL, Sourlas E, Raptopoulos V, Mantzoros CS (2006) Recombinant methionyl human leptin therapy in replacement doses improves insulin resistance and metabolic profile in patients with lipoatrophy and metabolic syndrome induced by the highly active antiretroviral therapy. J Clin Endocrinol Metab 91: 2605–2611 [DOI] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Mottillo EP, Granneman JG (2012) In vivo identification of bipotential adipocyte progenitors recruited by beta3‐adrenoceptor activation and high‐fat feeding. Cell Metab 15: 480–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, Kahn BB, Kahn CR (2013a) Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62: 864–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Wu Y, Fried SK (2013b) Adipose tissue heterogeneity: implication of depot differences in adipose tissue for obesity complications. Mol Aspects Med 34: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Granneman JG (2013c) Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab 18: 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Mayoral R, Maris M, Ofrecio JM, Taguchi S, Lu M, Olefsky JM (2015) LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med 21: 239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Liu S, Lu M, Bandyopadhyay G, Oh D, Imamura T, Johnson AM, Sears D, Shen Z, Cui B, Kong L, Hou S, Liang X, Iovino S, Watkins SM, Ying W, Osborn O, Wollam J, Brenner M, Olefsky JM (2016) Hematopoietic‐derived galectin‐3 causes cellular and systemic insulin resistance. Cell 167: 973–984.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieffers JR, Mourtzakis M, Hall KD, McCargar LJ, Prado CM, Baracos VE (2009) A viscerally driven cachexia syndrome in patients with advanced colorectal cancer: contributions of organ and tumor mass to whole‐body energy demands. Am J Clin Nutr 89: 1173–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, De Lucia Rolfe E, Stewart ID, Wheeler E, Willems SM, Adams C, Yaghootkar H, Consortium EP‐I , Cambridge FC, Forouhi NG, Khaw KT et al (2017) Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 49: 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, Nocon A, Fry J, Sherban A, Rui X, Jiang B, Xu XJ, Han J, Yan Y, Yang Q, Li Q, Zang M (2016) AMPK activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes 65: 2295–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magre J, Delepine M, Khallouf E, Gedde‐Dahl T Jr, Van Maldergem L, Sobel E, Papp J, Meier M, Megarbane A, Bachy A, Verloes A, d'Abronzo FH, Seemanova E, Assan R, Baudic N, Bourut C, Czernichow P, Huet F, Grigorescu F, de Kerdanet M et al (2001) Identification of the gene altered in Berardinelli‐Seip congenital lipodystrophy on chromosome 11q13. Nat Genet 28: 365–370 [DOI] [PubMed] [Google Scholar]
- Mao J, Yang T, Gu Z, Heird WC, Finegold MJ, Lee B, Wakil SJ (2009) aP2‐Cre‐mediated inactivation of acetyl‐CoA carboxylase 1 causes growth retardation and reduced lipid accumulation in adipose tissues. Proc Natl Acad Sci USA 106: 17576–17581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariman EC, Wang P (2010) Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci 67: 1277–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer N, Schweiger M, Romauch M, Grabner GF, Eichmann TO, Fuchs E, Ivkovic J, Heier C, Mrak I, Lass A, Höfler G, Fledelius C, Zechner R, Zimmermann R, Breinbauer R (2013) Development of small‐molecule inhibitors targeting adipose triglyceride lipase. Nat Chem Biol 9: 785–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor RA, Kwon EY, Shin SK, Jung UJ, Kim E, Park JH, Yu R, Yun JW, Choi MS (2013) Time‐course microarrays reveal modulation of developmental, lipid metabolism and immune gene networks in intrascapular brown adipose tissue during the development of diet‐induced obesity. Int J Obes (Lond) 37: 1524–1531 [DOI] [PubMed] [Google Scholar]
- Mittelman SD, Bergman RN (2000) Inhibition of lipolysis causes suppression of endogenous glucose production independent of changes in insulin. Am J Physiol Endocrinol Metab 279: E630–E637 [DOI] [PubMed] [Google Scholar]
- Morley JE, Thomas DR, Wilson MM (2006) Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr 83: 735–743 [DOI] [PubMed] [Google Scholar]
- Mozaffarian D, Hao T, Rimm EB, Willett WC, Hu FB (2011) Changes in diet and lifestyle and long‐term weight gain in women and men. N Engl J Med 364: 2392–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mracek T, Stephens NA, Gao D, Bao Y, Ross JA, Ryden M, Arner P, Trayhurn P, Fearon KC, Bing C (2011) Enhanced ZAG production by subcutaneous adipose tissue is linked to weight loss in gastrointestinal cancer patients. Br J Cancer 104: 441–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir LA, Neeley CK, Meyer KA, Baker NA, Brosius AM, Washabaugh AR, Varban OA, Finks JF, Zamarron BF, Flesher CG, Chang JS, DelProposto JB, Geletka L, Martinez‐Santibanez G, Kaciroti N, Lumeng CN, O'Rourke RW (2016) Adipose tissue fibrosis, hypertrophy, and hyperplasia: correlations with diabetes in human obesity. Obesity (Silver Spring) 24: 597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan HD, Tisdale MJ (1991) Lipogenesis in tumour and host tissues in mice bearing colonic adenocarcinomas. Br J Cancer 63: 719–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan K, Khatami H, Schwarz JM, Sakkas GK, DePaoli AM, Tai VW, Wen MJ, Lee GA, Grunfeld C, Schambelan M (2009) The effects of recombinant human leptin on visceral fat, dyslipidemia, and insulin resistance in patients with human immunodeficiency virus‐associated lipoatrophy and hypoleptinemia. J Clin Endocrinol Metab 94: 1137–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima I, Muroya S, Tanabe R, Chikuni K (2002) Positive effect of collagen V and VI on triglyceride accumulation during differentiation in cultures of bovine intramuscular adipocytes. Differentiation 70: 84–91 [DOI] [PubMed] [Google Scholar]
- Nolis T (2014) Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies. J Hum Genet 59: 16–23 [DOI] [PubMed] [Google Scholar]
- O'Connor KC, Song H, Rosenzweig N, Jansen DA (2003) Extracellular matrix substrata alter adipocyte yield and lipogenesis in primary cultures of stromal‐vascular cells from human adipose. Biotechnol Lett 25: 1967–1972 [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A (2015) Type 2 responses at the interface between immunity and fat metabolism. Curr Opin Immunol 36: 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno A, Tamemoto H, Tobe K, Ueki K, Mori Y, Iwamoto K, Umesono K, Akanuma Y, Fujiwara T, Horikoshi H, Yazaki Y, Kadowaki T (1998) Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J Clin Invest 101: 1354–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliff A, Defeo‐Jones D, Boyer M, Martinez D, Kiefer D, Vuocolo G, Wolfe A, Socher SH (1987) Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 50: 555–563 [DOI] [PubMed] [Google Scholar]
- Oliveira AG, Gomes‐Marcondes MC (2016) Metformin treatment modulates the tumour‐induced wasting effects in muscle protein metabolism minimising the cachexia in tumour‐bearing rats. BMC Cancer 16: 418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, Gorden P, Garg A (2002) Leptin‐replacement therapy for lipodystrophy. N Engl J Med 346: 570–578 [DOI] [PubMed] [Google Scholar]
- Pasarica M, Gowronska‐Kozak B, Burk D, Remedios I, Hymel D, Gimble J, Ravussin E, Bray GA, Smith SR (2009) Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab 94: 5155–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patni N, Garg A (2015) Congenital generalized lipodystrophies–new insights into metabolic dysfunction. Nat Rev Endocrinol 11: 522–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsouris D, Qi P, Abdullahi A, Stanojcic M, Chen P, Parousis A, Amini‐Nik S, Jeschke MG (2015) Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Rep 13: 1538–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CT, Murphy KD, Herndon DN (2005) Altering metabolism. J Burn Care Rehabil 26: 194–199 [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, Ruan HB, Yang X, Caprio S, Kaech SM, Sul HS, Birnbaum MJ, Davis RJ, Cline GW, Petersen KF, Shulman GI (2015) Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160: 745–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J (2010) Chronic peroxisome proliferator‐activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1‐containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem 285: 7153–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruzzelli M, Schweiger M, Schreiber R, Campos‐Olivas R, Tsoli M, Allen J, Swarbrick M, Rose‐John S, Rincon M, Robertson G, Zechner R, Wagner EF (2014) A switch from white to brown fat increases energy expenditure in cancer‐associated cachexia. Cell Metab 20: 433–447 [DOI] [PubMed] [Google Scholar]
- Petruzzelli M, Wagner EF (2016) Mechanisms of metabolic dysfunction in cancer‐associated cachexia. Genes Dev 30: 489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, Li P (2008) Downregulation of AMP‐activated protein kinase by Cidea‐mediated ubiquitination and degradation in brown adipose tissue. EMBO J 27: 1537–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang G, Whang Kong H, Xu S, Pham HA, Parlee SD, Burr AA, Gil V, Pang J, Hughes A, Gu X, Fantuzzi G, MacDougald OA, Liew CW (2016) Lipodystrophy and severe metabolic dysfunction in mice with adipose tissue‐specific insulin receptor ablation. Mol Metab 5: 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancoule C, Dusaulcy R, Treguer K, Gres S, Attane C, Saulnier‐Blache JS (2014a) Involvement of autotaxin/lysophosphatidic acid signaling in obesity and impaired glucose homeostasis. Biochimie 96: 140–143 [DOI] [PubMed] [Google Scholar]
- Rancoule C, Viaud M, Gres S, Viguerie N, Decaunes P, Bouloumie A, Langin D, Bascands JL, Valet P, Saulnier‐Blache JS (2014b) Pro‐fibrotic activity of lysophosphatidic acid in adipose tissue: in vivo and in vitro evidence. Biochim Biophys Acta 1841: 88–96 [DOI] [PubMed] [Google Scholar]
- Rasouli N, Kern PA (2008) Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab 93: S64–S73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin K, Steil GM, Mittelman SD, Bergman RN (1996) Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest 98: 741–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins DC, Danforth E Jr, Horton ES, Burse RL, Goldman RF, Sims EA (1979) The effect of diet on thermogenesis in acquired lipodystrophy. Metabolism 28: 908–916 [DOI] [PubMed] [Google Scholar]
- Rodeheffer MS, Birsoy K, Friedman JM (2008) Identification of white adipocyte progenitor cells in vivo . Cell 135: 240–249 [DOI] [PubMed] [Google Scholar]
- Rohm M, Sommerfeld A, Strzoda D, Jones A, Sijmonsma TP, Rudofsky G, Wolfrum C, Sticht C, Gretz N, Zeyda M, Leitner L, Nawroth PP, Stulnig TM, Berriel Diaz M, Vegiopoulos A, Herzig S (2013) Transcriptional cofactor TBLR1 controls lipid mobilization in white adipose tissue. Cell Metab 17: 575–585 [DOI] [PubMed] [Google Scholar]
- Rohm M, Schäfer M, Laurent V, Üstünel BE, Niopek K, Algire C, Hautzinger O, Sijmonsma TP, Zota A, Medrikova D, Pellegata NS, Ryden M, Kulyte A, Dahlman I, Arner P, Petrovic N, Cannon B, Amri EZ, Kemp BE, Steinberg GR et al (2016) An AMP‐activated protein kinase‐stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat Med 22: 1120–1130 [DOI] [PubMed] [Google Scholar]
- Rojas Y, Finnerty CC, Radhakrishnan RS, Herndon DN (2012) Burns: an update on current pharmacotherapy. Expert Opin Pharmacother 13: 2485–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanon VP, Handelsman Y, Pham SV, Chilton R (2016) Cardiac manifestations of congenital generalized lipodystrophy. Clin Diabetes 34: 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraf MK, Herndon DN, Porter C, Toliver‐Kinsky T, Radhakrishnan R, Chao T, Chondronikola M, Sidossis LS (2016) Morphological changes in subcutaneous white adipose tissue after severe burn injury. J Burn Care Res 37: e96–e103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NK, Anania FA (2015) Adipocytokines and hepatic fibrosis. Trends Endocrinol Metab 26: 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer M, Oeing CU, Rohm M, Baysal‐Temel E, Lehmann LH, Bauer R, Volz HC, Boutros M, Sohn D, Sticht C, Gretz N, Eichelbaum K, Werner T, Hirt MN, Eschenhagen T, Müller‐Decker K, Strobel O, Hackert T, Krijgsveld J, Katus HA et al (2016) Ataxin‐10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol Metab 5: 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid‐induced insulin resistance. J Clin Invest 116: 3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL, Brown MS (1998) Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP‐1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev 12: 3182–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL (1999) Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 401: 73–76 [DOI] [PubMed] [Google Scholar]
- Shulman GI (2014) Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 371: 1131–1141 [DOI] [PubMed] [Google Scholar]
- Sidossis L, Kajimura S (2015) Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J Clin Invest 125: 478–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidossis LS, Porter C, Saraf MK, Borsheim E, Radhakrishnan RS, Chao T, Ali A, Chondronikola M, Mlcak R, Finnerty CC, Hawkins HK, Toliver‐Kinsky T, Herndon DN (2015) Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab 22: 219–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Naslund E, Britton T, Concha H, Hassan M, Ryden M, Frisen J, Arner P (2008) Dynamics of fat cell turnover in humans. Nature 453: 783–787 [DOI] [PubMed] [Google Scholar]
- Spencer M, Unal R, Zhu B, Rasouli N, McGehee RE Jr, Peterson CA, Kern PA (2011) Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J Clin Endocrinol Metab 96: E1990–E1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM, Ginty CA (1983) Fibronectin modulation of cell shape and lipogenic gene expression in 3T3‐adipocytes. Cell 35: 657–666 [DOI] [PubMed] [Google Scholar]
- Spranger J, Kroke A, Mohlig M, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF (2003) Adiponectin and protection against type 2 diabetes mellitus. Lancet 361: 226–228 [DOI] [PubMed] [Google Scholar]
- Stern JH, Rutkowski JM, Scherer PE (2016) Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab 23: 770–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassmann G, Fong M, Kenney JS, Jacob CO (1992) Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest 89: 1681–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW II, DeFuria J, Jick Z, Greenberg AS, Obin MS (2007) Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56: 2910–2918 [DOI] [PubMed] [Google Scholar]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK (1994) Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell‐permeable activator of AMP‐activated protein kinase. FEBS Lett 353: 33–36 [DOI] [PubMed] [Google Scholar]
- Sun K, Tordjman J, Clement K, Scherer PE (2013) Fibrosis and adipose tissue dysfunction. Cell Metab 18: 470–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, Graff JM (2008) White fat progenitor cells reside in the adipose vasculature. Science 322: 583–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zeve D, Seo J, Jo AY, Graff JM (2011) Thiazolidinediones regulate adipose lineage dynamics. Cell Metab 14: 116–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchoukalova YD, Votruba SB, Tchkonia T, Giorgadze N, Kirkland JL, Jensen MD (2010) Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc Natl Acad Sci USA 107: 18226–18231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theologides A (1979) Cancer cachexia. Cancer 43: 2004–2012 [DOI] [PubMed] [Google Scholar]
- Tisdale MJ (2009) Mechanisms of cancer cachexia. Physiol Rev 89: 381–410 [DOI] [PubMed] [Google Scholar]
- Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, Chen H, Monks BR, Chen J, Rabinowitz JD, Birnbaum MJ (2016) Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab 23: 1154–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trew JA, Begg RW (1959) In vitro incorporation of acetate‐1‐C14 into adipose tissue from normal and tumor‐bearing rats. Cancer Res 19: 1014–1019 [PubMed] [Google Scholar]
- Uehara A, Sekiya C, Takasugi Y, Namiki M, Arimura A (1989) Anorexia induced by interleukin 1: involvement of corticotropin‐releasing factor. Am J Physiol 257: R613–R617 [DOI] [PubMed] [Google Scholar]
- Unger RH, Scherer PE (2010) Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab 21: 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utech AE, Tadros EM, Hayes TG, Garcia JM (2012) Predicting survival in cancer patients: the role of cachexia and hormonal, nutritional and inflammatory markers. J Cachexia Sarcopenia Muscle 3: 245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Maldergem L, Magre J, Khallouf TE, Gedde‐Dahl T Jr, Delepine M, Trygstad O, Seemanova E, Stephenson T, Albott CS, Bonnici F, Panz VR, Medina JL, Bogalho P, Huet F, Savasta S, Verloes A, Robert JJ, Loret H, De Kerdanet M, Tubiana‐Rufi N et al (2002) Genotype‐phenotype relationships in Berardinelli‐Seip congenital lipodystrophy. J Med Genet 39: 722–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegiopoulos A, Müller‐Decker K, Strzoda D, Schmitt I, Chichelnitskiy E, Ostertag A, Berriel Diaz M, Rozman J, Hrabe de Angelis M, Nüsing RM, Meyer CW, Wahli W, Klingenspor M, Herzig S (2010) Cyclooxygenase‐2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 328: 1158–1161 [DOI] [PubMed] [Google Scholar]
- Vigouroux C, Gharakhanian S, Salhi Y, Nguyen TH, Chevenne D, Capeau J, Rozenbaum W (1999) Diabetes, insulin resistance and dyslipidaemia in lipodystrophic HIV‐infected patients on highly active antiretroviral therapy (HAART). Diabetes Metab 25: 225–232 [PubMed] [Google Scholar]
- Virtue S, Vidal‐Puig A (2010) Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome–an allostatic perspective. Biochim Biophys Acta 1801: 338–349 [DOI] [PubMed] [Google Scholar]
- Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, Gupta RK (2016) Pdgfr beta(+) mural preadipocytes contribute to adipocyte hyperplasia induced by high‐fat‐diet feeding and prolonged cold exposure in adult mice. Cell Metab 23: 350–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MY, Chen L, Clark GO, Lee Y, Stevens RD, Ilkayeva OR, Wenner BR, Bain JR, Charron MJ, Newgard CB, Unger RH (2010) Leptin therapy in insulin‐deficient type I diabetes. Proc Natl Acad Sci USA 107: 4813–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QA, Tao C, Gupta RK, Scherer PE (2013) Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med 19: 1338–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Gao M, Liao J, Han Y, Wang Y, Liu G (2015) Dysfunction of lipid metabolism in lipodystrophic Seipin‐deficient mice. Biochem Biophys Res Commun 461: 206–210 [DOI] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado‐Lopez F, Wang ZV, Scherer PE (2014) Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 20: 103–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams FN, Jeschke MG, Chinkes DL, Suman OE, Branski LK, Herndon DN (2009) Modulation of the hypermetabolic response to trauma: temperature, nutrition, and drugs. J Am Coll Surg 208: 489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmore DW, Long JM, Mason AD Jr, Skreen RW, Pruitt BA Jr (1974) Catecholamines: mediator of the hypermetabolic response to thermal injury. Ann Surg 180: 653–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 117: 524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Yin S, Wong L, Chan KW, Lam KS (2004) Adiponectin ameliorates dyslipidemia induced by the human immunodeficiency virus protease inhibitor ritonavir in mice. Endocrinology 145: 487–494 [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama‐Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K et al (2001) The fat‐derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7: 941–946 [DOI] [PubMed] [Google Scholar]
- Ye R, Scherer PE (2013) Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab 2: 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz M, Claiborn KC, Hotamisligil GS (2016) De novo lipogenesis products and endogenous lipokines. Diabetes 65: 1800–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yki‐Jarvinen H (2014) Non‐alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol 2: 901–910 [DOI] [PubMed] [Google Scholar]
- Yore MM, Syed I, Moraes‐Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, Dhaneshwar AS, Hammarstedt A, Smith U, McGraw TE, Saghatelian A, Kahn BB (2014) Discovery of a class of endogenous mammalian lipids with anti‐diabetic and anti‐inflammatory effects. Cell 159: 318–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YH, Ginsberg HN (2005) Adipocyte signaling and lipid homeostasis: sequelae of insulin‐resistant adipose tissue. Circ Res 96: 1042–1052 [DOI] [PubMed] [Google Scholar]
- Zheng C, Yang Q, Xu C, Shou P, Cao J, Jiang M, Chen Q, Cao G, Han Y, Li F, Cao W, Zhang L, Zhang L, Shi Y, Wang Y (2015) CD11b regulates obesity‐induced insulin resistance via limiting alternative activation and proliferation of adipose tissue macrophages. Proc Natl Acad Sci USA 112: E7239–E7248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Yon Toh S, Chen Z, Guo K, Ng CP, Ponniah S, Lin SC, Hong W, Li P (2003) Cidea‐deficient mice have lean phenotype and are resistant to obesity. Nat Genet 35: 49–56 [DOI] [PubMed] [Google Scholar]
- Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ (2002) Induction of cachexia in mice by systemically administered myostatin. Science 296: 1486–1488 [DOI] [PubMed] [Google Scholar]
- Zuijdgeest‐van Leeuwen SD, van den Berg JW, Wattimena JL, van der Gaast A, Swart GR, Wilson JH, Dagnelie PC (2000) Lipolysis and lipid oxidation in weight‐losing cancer patients and healthy subjects. Metabolism 49: 931–936 [DOI] [PubMed] [Google Scholar]